

Abstract
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity in both civilian life and the battlefield worldwide. Survivors of TBI frequently experience long-term disabling changes in cognition, sensorimotor function and personality. Over the past three decades, animal models have been developed to replicate the various aspects of human TBI, to better understand the underlying pathophysiology and to explore potential treatments. Nevertheless, promising neuroprotective drugs, which were identified to be effective in animal TBI models, have all failed in phase II or phase III clinical trials. This failure in clinical translation of preclinical studies highlights a compelling need to revisit the current status of animal models of TBI and therapeutic strategies.
Traumatic brain injury (TBI) is defined as damage to the brain resulting from an external mechanical force, such as that caused by rapid acceleration or deceleration, blast waves, crush, impact, or penetration by a projectile, and can lead to temporary or permanent impairment of cognitive, physical and psychosocial functions1. TBI is the leading cause of death and disability for people under the age of 45 years2. Indeed, worldwide, 10 million deaths and/or hospitalizations annually are directly attributable to TBI and an estimated 57 million people have experienced such brain injury2.
TBI is not a single pathophysiological event but a complex disease process3 (BOX 1), and causes structural damage and functional deficits that are due to both primary and secondary injury mechanisms12. The primary injury is the result of the immediate mechanical disruption of brain tissue that occurs at the time of exposure to the external force and includes contusion, damage to blood vessels (hemorrhage), and axonal shearing, in which the axons of neurons are stretched and torn13,14. Secondary injury evolves over minutes to months after the primary injury, and is the result of cascades of metabolic, cellular and molecular events that ultimately lead to brain cell death, tissue damage and atrophy15-17.
Box 1. Simplified pathophysiology of traumatic brain injury.
In traumatic brain injury, the primary injury, which is the direct result of the external force, involves mechanical tissue deformation and causes diffuse neuronal depolarization and release of excitatory neurotransmitters including glutamate and aspartate4,5, which bind to glutamate receptors and induce a massive influx of calcium6. Calcium activates calcium-dependent phospholipases, proteases and endonucleases that degrade lipids, proteins and nucleic acids (not shown). Calcium sequestration in mitochondria leads to calcium disturbance, energy deficits, free radical formation, and initiation of apoptosis7,8. Increased formation of oxygen and nitrogen reactive species oxidizes lipids, proteins and nuclei acids after TBI9. TBI up-regulates many transcription factors, inflammatory mediators, and neuroprotective genes but down-regulates neurotransmitter receptors and release mechanisms10. Increased expression of detrimental cytokines and chemokines induces brain oedema, blood–brain barrier damage, and cell death11. The result of these complex cascades after TBI eventually leads to cell damage and death, which causes functional deficits. Substantial experimental and clinical data have accumulated over the past decade indicating that the adult brain is capable of substantial structural and functional reorganization after injury, which may contribute to spontaneous functional recovery. Interventions targeting secondary injury mechanisms and modulating neuroplasticity promote functional recovery in animal models of TBI. Red line: Main effects; dotted black line: possible effects; black line with arrows: possible interactions.
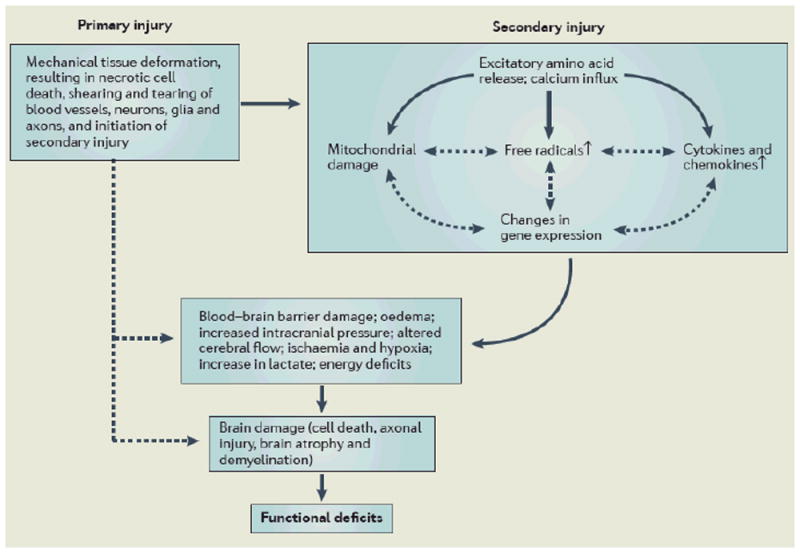
Many biochemical derangements responsible for secondary injury have been identified, including glutamate excitotoxicity, perturbation of cellular calcium homeostasis, increased free radical generation and lipid peroxidation, mitochondrial dysfunction, inflammation, apoptosis, and diffuse axonal injury (DAI)18. As highlighted above, collectively, the cascade of secondary injury events culminates in neuronal, endothelial and glial cell death and white matter degeneration15,19. Cell death occurs within minutes after injury and extends over a period of days to months19,20. Necrotic and apoptotic cell death have been identified in contused areas, the injury boundary zone and subcortical regions20,21, and apoptosis coincides with progressive atrophy of gray and white matter following TBI15.
The relative contributions of cell death and sublethal neurobiological derangements to posttraumatic morbidities remain to be determined. Many sublethal cellular events and systemic insults, including hypoxia and hypotension, may in concert ultimately lead to cell death without timely intervention. Both acute cell death and delayed apoptosis have an important role in mediating functional deficits after TBI. However, even mild TBI without notable cell death can lead to cognitive deficits, which are probably associated with TBI-induced DAI, as indicated by studies in humans22,23, rodents24 and swine25. These findings suggest that multifocal axonal and myelin abnormalities also contribute to posttraumatic cognitive impairments.
Since primary injury occurs immediately after the moment of trauma, it can only be preventable (for example, through use of a seat belt or helmet). By contrast, the elongated nature of secondary injury development provides a window of opportunity for therapeutic intervention, which may prevent and/or reduce secondary brain damage and improve long-term patient outcome. To date, however, promising results from preclinical studies of potential TBI treatments have not been translated into successful outcomes in clinical trials. The pathophysiological heterogeneity observed in patients with TBI, the lack of sufficient pharmacokinetic analysis for determination of optimal dose, and the compounds given outside of the therapeutic window may have led to the clinical trial failures26.
The pathophysiological heterogeneity observed in patients with TBI may arise from the location, nature and severity of the primary injury and preexisting factors and conditions, including but not restricted to age, health, gender, medication, alcohol and drug use, and genetics27. Animal models of TBI are each designed to produce a relatively homogeneous type of injury, with age, gender, genetic background and the injury parameters all well controlled. Thus, any one animal model may not be able to fully recapitulate all aspects of secondary injury development observed in human TBI, and this may explain in part why drugs that showed promise in preclinical studies failed in clinical studies 17. Undoubtedly, however, animal models are essential for studying the biomechanical, cellular and molecular aspects of human TBI that cannot be addressed in the clinical setting, as well as for developing and characterizing novel therapeutic interventions. To develop new therapeutic strategies, new and existing animal models of TBI need to be developed and modified, respectively, to traverse the therapeutic gap between preclinical studies and patient medical care.
This review aims to provide a broad overview of current knowledge of animal models of TBI, to identify the issues and challenges of therapeutic strategies in preclinical studies, and to highlight research strategies for improving animal models and therapeutic efficacy.
Animal models of TBI
In view of the heterogeneous nature of the clinical situation in TBI, numerous animal models of such injury have been developed. Although larger animals are closer in size and physiology to humans, rodents are mostly used in TBI research due to their modest cost, small size and standardized outcome measurements, among other reasons (BOX 2). Whereas early models of TBI addressed the biomechanical aspects of brain injury45-47, more-recent models have been targeted at improving our understanding of the detrimental, complex molecular cascades that are initiated by head trauma. Among them, four specific models are widely used in research: fluid percussion injury (FPI)48, cortical impact injury (CCI)49,50, weight drop–impact acceleration injury51, and blast injury52,53 (FIG 1; TABLE 1,2).
Box 2. Functional outcome testing in animal models of traumatic brain injury.
Motor function is mediated by a complex system of neural networks originating in the brain cortex and terminating in skeletal muscles28. The association cortex, sensorimotor cortex, subcortical nuclei, cerebellum, and brainstem all communicate with each other to send a signal through the spinal cord to coordinate movement28. Brain injury-induced disruption in any or all parts of these pathways will cause motor deficits. Few, if any, purely motor behavioural tasks exist. Deficits caused by traumatic brain injury (TBI) result from disruption of complex motor pathways and sensorimotor integration, and therefore most of the described tests for assessing the outcome of such injury in animal models are sensorimotor in nature28. Widely used sensorimotor function tests include the cylinder test, Rotarod test, grip strength tests, skilled forelimb reach and staircase tests28. The neurological severity score (NSS) composed of motor functions and behavior is widely used for closed head injury in rodents29,30. In addition, the modified neurological severity score (mNSS) is a very useful tool to evaluate neurological functional deficits in rodents after unilateral brain injury31,32.
TBI in humans often leads to cognitive dysfunction, the degree of which often depends on the injury severity33. Cognitive dysfunction has been described in the CCI, lateral and midline FPI, blast, and impact acceleration animal models of TBI24,29,34-37. Commonly used tests of cognition in rodents include the Morris water maze, freezing response test, memory task and object recognition test28. Some more complex behavioural tests have also been developed in experimental TBI research to mimic the complex personality and psychological disturbances in patients with TBI. Anxiety-like tests include the elevated plus maze, emotional and exploratory activity, and the open field tests38-41. Depression, a common clinical problem after TBI, has not been fully studied in animal models, although there are a few reports using the forced swimming test to assess depression-like behaviour41,42.
Functional tests have been rarely developed or performed in large animals after TBI. Recently, a wide range of neurobehavioural functions including open field testing (executive function), glass barrier task (visual-based problem solving), food cover task (olfactory-based problem solving), and balance beam (motor) has been performed in the neonatal pig following closed head injury43,44. Further development or use of functional tests in large animals is warranted to verify the safety and efficacy of promising treatments that are effective in small animal models of TBI before clinical trials are initiated.
Figure 1. Experimental set ups for the animal models of traumatic brain injury.
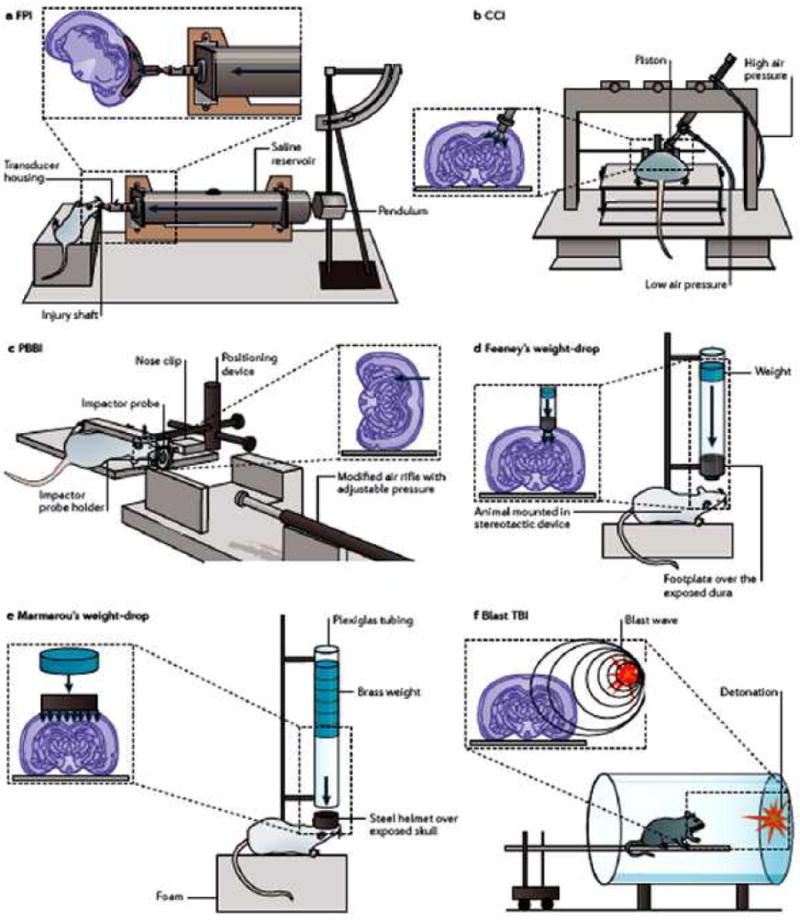
a ∣ The fluid percussion injury (FPI) device uses rapid injection of a fluid pulse into the epidural space. b ∣ The controlled cortical impact (CCI) model uses an air or electromagnetic driven piston to penetrate the brain at a known distance and velocity. c ∣ The penetrating ballistic-like brain injury (PBBI) involves the transmission of projectiles with high energy of a metal rod or expansion of the probe’s elastic balloon. d ∣ In the Feeney weight-drop model, a free weight is released directly onto the exposed dura. e ∣ In the Marmarou weight-drop model, a metal disk is placed over the skull to prevent bone fracture. f ∣ The blast brain injury caused by primary injury of blast or other mechanisms, e.g., thoracic effect. Panels a,b,d,e are modified, with permission, from REF54. [54]© 2003 Mary Ann Liebert, Inc. Publishers. Panel c is modified, with permission, from REF55 [55]© 2007 Mary Ann Liebert, Inc. Publishers.
Table 1.
Commonly used animal models of TBI
| Model | Type of injury | Strengths | Weaknesses | Species |
|---|---|---|---|---|
| CCI | Mainly focal | Highly reproducible | Need for craniotomy | Ferret50, mouse56, rat49, swine57, monkey58 |
| Weight drop | ||||
| Feeney | Mainly focal | Injury mechanism close to human TBI | Need for craniotomy; high mortality rate | Rat59 |
| Shohami | Mainly focal | Easy operation, with immediate neurological severity scoring at 1h | Not highly reproducible | Rat60, mouse29 |
| Marmarou | Mainly diffuse | Injury mechanism close to human TBI; well characterized | Not highly reproducible, high mortality without ventilation | Rat51, mouse61 |
| Maryland model | Mainly diffuse | Injury mechanism close to human TBI | Needs further characterization | Rat62 |
| FPI | ||||
| Middle | Mixed | Highly reproducible with fine-tuning | Need for craniotomy, high mortality | Cat63, rabbit64, rat65, dog and sheep66, swine67 |
| Lateral | Mixed | Highly reproducible with fine-tuning | Need for caniotomy, high mortality | Rat68, mouse69, swine70 |
| Blast | Mainly diffuse | Injury mechanism close to military TBI | Need standardization | Rat52, mouse71, swine72 |
| Repeated mild | Mainly diffuse | Injury mechanism close to sports TBI | Need further characterization | Rat73, mouse74, swine43 |
| PBBI | Mainly focal | Injury mechanism close to human TBI | Need standardization | Cat75, rat76 |
CCI, controlled cortical impact; FPI, fluid percussion injury; PBBI, penetrating ballistic-like brain injury; TBI, traumatic brain injury. The Table is modified, with permission, from REF77. [77] © 2010 BioMed Central.
Table 2. Major pathological features of animal models that are seen in human TBI.
| TBI model | Concussion | Contusion | Traumatic axonal injury | Hemorrhage | Skull fracture | References |
|---|---|---|---|---|---|---|
| Shohami’s and Marmarou’s weight drop models | ++ | + | ++ | + | +/- | 29, 51, 60, 126, 212 |
| Feeney’s weight drop model | + | ++ | + | + | - | 59 |
| Maryland’s model | + | - | + | + | - | 62 |
| Fluid percussion injury | + | ++ | + | + | - | 16, 45, 63-70, 83, 84, 88, 154, 155 |
| Controlled cortical impact | + | ++ | + | ++ | - | 38, 49, 50, 56-58, 93, 99, 103-110 |
| Blast | ++ | + | ++ | + | +/- | 52, 71, 72,74, 132-145 |
|
| ||||||
| Penetrating ballistic-like brain injury | + | ++ | + | ++ | +/- | 37, 55, 75, 76, 111-119 |
-, does not duplicate the condition observed in humans; +/- may duplicate the condition observed in humans; + duplicates the human condition to some degree; ++ shows high fidelity to the human condition.
This review will cover the main types of animal models of TBI and will not discuss cerebellar injury models. Direct cerebellar injury is a relatively uncommon phenomenon79. For animal models of traumatic cerebellar injury, please see the review by Potts et al79.
Fluid percussion injury models
In FPI models, the insult is inflicted by a pendulum striking the piston of a reservoir of fluid to generate a fluid pressure pulse to the intact dura through a craniotomy, which is made either centrally around the midline65, or laterally over the parietal bone68, between bregma and lambda. The percussion produces brief displacement and deformation of brain tissue, and the severity of injury depends on the strength of the pressure pulse68.
FPI models replicate clinical TBI without skull fracture16. Moderate to severe TBI in humans is often associated with skull fracture and contusions across multiple gyri80, features that cannot be replicated in this model. FPI can replicate, however, intracranial hemorrhage, brain swelling and progressive gray matter damage — all pathophysiological hallmarks of human TBI81.
Based on the position of the craniotomy away from the sagittal suture, FPI models can be divided into midline (centered on the sagittal suture), parasagittal (<3.5 mm lateral to midline) and lateral models (>3.5 mm lateral to midline; LFPI)68,82-84. The midline FPI model of TBI was initially developed for use in cats and rabbits63,64,85, subsequently adapted for use in rats48,65 and then modified to produce the LFPI model in rodents68,69. FPI has also been used for studying TBI pathophysiology and pharmacology in cats86, rabbit64, dogs and sheep66, rats87, mice69,88, and pigs67,89.
The LFPI model is one of the most widely used TBI animal models16. In rats, LFPI produces a combination of focal cortical contusion and diffuse subcortical (such as hippocampus and thalamus) neuronal injury, which occurs within minutes of the impact, progresses to a loss of neurons by 12 h, and does not markedly expand into other brain regions by 7 days post injury90 The contused cortex beneath the injury site enlarges over weeks to become a cavity lined with glia and continues to expand up to one year post-injury due to ongoing cell death91. Over days to months, progressive degenerative cascades persist in selectively vulnerable brain regions, including the ipsilateral hippocampus, thalamus, medial septum, striatum and amygdala16,90,92. LFPI produces neurobehavioural and cognitive deficits such as difficulties with movement and memory that are commonly seen in patients with TBI93,94. Cognitive dysfunction and neurological impairments persist for more than a year following severe LFPI95. However, FPI models have high mortality compared with other models, probably due to the brainstem-compromised prolonged apnea13.
The site of craniotomy is crucial in determining the extent and location of tissue injury produced by LFPI in rats83. Indeed, careful attention should be paid to where the craniotomy is conducted to increase the reliability and reproducibility of this model. The LFPI model inflicts primarily unilateral cortical damage, rarely involving the contralateral cortices and brainstem, whereas midline and parasagittal FPI causes bilateral cortical alterations associated with direct axial movement of the lower brainstem13. The degree of cortical damage highly depends on both craniotomy position and injury severity84.
The FPI model allows minimal biomechanical control of the insult, with the height of pendulum as the only adjustable mechanical parameter. To improve reproducibility, Kabadi and colleagues developed a microprocessor-controlled, pneumatically driven instrument to address operational concerns associated with the use of the standard FPI device in rats96. With this new device, the impact pressure and dwell time can be precisely controlled, thus reducing variation between trials. This approach produced acute and chronic TBI features similar to that observed in the LFPI literature, as quantified by histological changes, structural changes seen on MRI and chronic behavioural sequelae. One final point to note is that although the LFPI model has been popular for studying neuronal cell death mechanisms in TBI, there is a recent resurgence of interest in midline FPI because of the increased interest in diffuse brain injury associated with sport and blasts97,98.
Controlled cortical impact injury model
The CCI model uses a pneumatic or electromagnetic impact device to drive a rigid impactor onto the exposed intact dura, and mimics cortical tissue loss, acute subdural hematoma, axonal injury, concussion, blood–brain barrier (BBB) dysfunction and even coma49,50,56,93,99. It has been applied to ferrets50, rats49, mice56, swine57 and monkeys58. The controlled impact is delivered to the intact dura through a unilateral craniotomy most often between bregma and lambda, causing deformation of the underlying cortex49. Hall et al. performed a comprehensive neuropathological evaluation of the CCI TBI model and reported that the associated damage can be widespread, including acute cortical, hippocampal and thalamic degeneration100.
The advantage of this injury model over other TBI models is the ease at which mechanical factors, such as time, velocity and depth of impact, can be controlled; thus, it may be more useful than the FPI model for biomechanical studies of TBI13,101,102. An additional strength of the CCI model, when compared with models involving gravity-driven devices, is the lack of risk of a rebound injury. The histopathological severity of CCI rises with increasing cortical deformation and impact velocity, permitting adjustment of the injury severity appropriate for specific experimental requirements103,104. The functional deficits such as cognitive impairments measured in the Morris water maze test are highly related to both the depth of deformation and the velocity of the impact in mice and rats38,105,106. The cognitive deficits persist up to a year post CCI and may be associated with brain atrophy107,108 and progressive decline in cerebral blood flow109. CCI also caused deficits in emotional behavior as quantified in the forced swim test, elevated-plus maze, and prepulse inhibition of acoustic startle in mice109. Cognitive deficits increase in relation to injury severity, but emotional deficits do not, suggesting that the threshold for emotional changes after experimental TBI is low38. The swine CCI model generates a reproducible injury with pathological features similar to human TBI57,110. Despite its cost and complexity, this large animal model offers the opportunity to collect physiological data following brain injury in an environment similar to the intensive care unit and thus may facilitate translation of animal data into clinical practice.
Penetrating ballistic-like brain injury models
Penetrating ballistic-like brain injury (PBBI) is caused by transmission of projectiles with high energy and a leading shockwave, which produces a temporary cavity in the brain that is many times the size of the projectile itself111. Outcome in this model is directly related to the projectile’s anatomical path and degree of energy transfer76,111,112. In the past, the experimental PBBI studies most relevant to gunshot were done in cats using a penetrating missile model75,113. A rat model of penetrating brain injury has been characterized and shown to produce cognitive impairment111,114. It induces marked white and grey matter damage, brain swelling, seizures, cortical spreading depression and neuroinflammation with a resulting sensorimotor impairment112,115. Therapeutic treatments including dextromethorphan and human amnion-derived multipotent progenitor cells have been recently evaluated in this model116,117.
Recently, several new PBBI rodent models have been developed37,55,118. A novel non-fatal model for low velocity PBBI has been established, involving a modified air rifle that accelerates a pellet55. This PBBI rat model causes cavity formation, white matter degeneration, hemorrhage, oedema, and gliosis. Bullets or shrapnel that penetrate the brain with high energy produce a temporary cavity in the brain126. To mimic the ballistic effect of PBBI, a PBBI rat model has been established to characterize immediate and subacute (out to 7 days) changes in intracranial pressure (ICP)118. BBB permeability, brain oedema formation, enduring motor and cognitive deficits have been identified in a unilateral frontal PBBI in rats37,119. Neurofunctional assessments revealed that motor (balance beam and rotarod tasks) and cognitive deficits (spatial learning in the MWM test) correlated with the degree of injury severity.
Several pathophysiological characteristics of PBBI are similar to those reported in other brain trauma models, including the presence of hemispheric swelling, increased ICP, remote white matter injury, and neuroinflammation76,115. However, compared with other TBI models, PBBI causes extensive intracerebral hemorrhage throughout the primary lesion, due to the penetrating nature of the injury and the temporary cavity that it forms. The PBBI model captures several unique temporal aspects of a ballistic brain injury and may serve as a highly relevant model of moderate-to-severe brain trauma for mechanistic studies and for evaluation of therapeutic intervention.
Weight drop TBI models
In weight drop models, the skull is exposed (with or without a craniotomy) to a free falling, guided weight93. Injury severity in these models can be altered by adjusting the mass of the weight and the height from which it falls.
In Feeney’s weight-drop model, the weight is delivered to the intact dura through a craniotomy and causes a cortical contusion59. Morphologically, these injuries progress from hemorrhages in white matter directly under the contused cortex during the first hours after injury to the development of a necrotic cavity by 24 hours. The cavitation appears to expand over the subsequent two weeks59,120. Although most functional recovery occurs in the first two weeks after trauma in rats, with severe contusions, deficits can persist beyond 90 days59,121.
Shohami’s group later introduced a rodent model for closed head injury (CHI) using a weight-drop impact delivered to one side of the unprotected skull in rat30,60 and mouse29,122,123, with the head being placed on a hard surface. A mouse CHI model was described in detail, with a standardized weight-drop device inducing a focal blunt injury over the unprotected skull123. The resulting impact caused neurological impairment and breakdown of the BBB. Neurological severity score (NSS) was performed to evaluate the neurological impairment in motor function, alertness, and seeking behavior. The neurological impairment highly correlates with the severity of brain injury. Recently, neurobehavioral deficits, activation of microglia and astrocytes, neurodegeneration, and morphological changes assessed by MRI were demonstrated in this mouse CHI model71, indicating this model resembles the clinical conditions of human CHI61.
Marmarou et al.51 developed a model of DAI — Marmarou’s impact acceleration model — to mimic human diffuse TBI caused by falls or motor vehicle accidents51. DAI is common in humans and experimental animals124,125, and in this model, the trauma device consists of a sectioned brass weight set that falls freely from a designated height through a Plexiglas tube. In anaesthetized rats with skull exposure made by a midline incision, a stainless steel disc is mounted with glue to the skull midline between lambda and bregma to prevent skull fracture. The rats are then placed on a foam bed and subjected to the impact by dropping the brass weight onto the stainless steel disc. Death is primarily caused by respiratory depression, and mechanical ventilation after the impact greatly reduces the mortality rate after severe injury51,126. The Marmarou model causes widespread and bilateral damage of neurons, axons, dendrites, and microvasculature as well as extensive DAI, particularly in the corpus callosum, internal capsule, optic tracts, cerebral and cerebellar peduncules, and the long tracts in the brainstem126. It also induces motor and cognitive deficits such as difficulties with beam walking and memory127,128, similar to those observed after FPI and CCI, and these deficits correlate with injury severity30,51,129. One disadvantage of the weight-drop models is the relatively high variability in injury severity. However, it is inexpensive, easy to perform, and capable of producing graded DAI that closely mimics that seen in human TBI.
Previously available rodent models of CHI do not reproduce the frontal impact commonly encountered in motor vehicle and sports accidents62. A new rat model of CHI has been developed, by modification of the Marmarou impact-acceleration model, to investigate these scenarios62. In the new model (called the Maryland model), the impact force is applied to the anterior part of the cranium and produces TBI by causing anterior–posterior plus sagittal rotational acceleration of the brain inside the intact cranium62. The animals are characterized by an absence of cortical contusions, skull fractures, prolonged apnea, and an absence of mortality, but demonstrate petechial hemorrhages and DAI. Neurobehavioural dysfunction, manifesting as reduced spontaneous exploration, persists for more than 1 week. Additional study will be needed to further characterize this model.
Models of blast TBI
Many military personnel exposed to a blast but without external injuries have been diagnosed with TBI130,131. To elucidate the effects of primary blast waves on the central nervous system, various animal models of blast TBI have been established, mainly in rodents52,74,132-134 and swine72,135. Using a compression-driven shock tube to simulate blast effects, Long and colleagues assessed the physiological, neuropathological, and neurobehavioural consequences of blast exposure, and also evaluated the effect of a Kevlar thoracic protective vest on acute mortality in rats and on the frequency of TBI and DAI in those that survived134. The Kevlar vest, which encased the thorax and part of the abdomen, greatly reduced air blast mortality, and also ameliorated the widespread axonal fiber degeneration, indicating that shock tube-generated blast causes TBI in rats, in part through systemic effects, including hypotension and hypoxemia, possibly evoked by blast-induced lung injury and/or hemorrhage136.
Reneer and colleagues developed a blast-induced TBI rat model to mimic real blast mild TBI seen in recent military conflicts137. Non-impact blast injury exhibits an interesting pathophysiology that is characterized by diffuse cerebral brain oedema, extreme hyperemia and a delayed vasospasm seen in animal and human blast brain injury138. DAI was the most prominent feature during the initial 2 weeks following blast exposure in rats with body shielding139. Kuehn et al. found that exposure of the head alone to severe explosive blast predisposes to causes significant neurological dysfunction140. Importantly, even exposure of rats to low level blast increases ICP and causes cognitive deficits141.
Although functional deficits due to blast exposure represent the principal health problem in modern warfare, the majority of available blast models focus on tissue destruction rather than functional deficits136,137,139,142. A recent report indicates that even mild blast brain injury caused prolonged behavioural and motor abnormalities in mice, including deficits in social recognition, spatial memory and motor coordination, and shielding of the torso ameliorated axonal injury and behavioural deficits143. Clearly, further research is necessary to address whether and how blast TBI, in particular multiple exposures to low-level blast, can lead to long-term functional deficits.
Goldstein et al.71 demonstrated that blast-exposed mice show phosphorylated tauopathy, myelinated axonopathy, microvasculopathy, chronic neuroinflammation and neurodegeneration in the absence of macroscopic tissue damage or hemorrhage. Head immobilization during blast exposure prevented blast-induced learning and memory deficits, indicating that head rotation may play an important role in generating these deficits. It has become increasingly clear that brain pathology, the underlying mechanisms and potential biomarkers associated with primary blast exposures may be different from those imposed by focal mechanical head trauma144. It should be noted that animal placement locations along the length of the shock tube (that is, inside, outside or near the exit) have an important role in the biomechanical loading on the animal and thereby alter the injury type, its severity and the probability of lethality145. Considering the variations in the current blast injury models, comparison of the results between different laboratories is virtually impossible. Thus, the further design, characterization, and implementation of relevant standard experimental blast models is of particular importance for the elucidation of the mechanisms of blast injury, the identification of biomarkers and, eventually, the development of strategies for mitigating blast-induced brain injury.
Mild TBI models
Mild TBI constitutes most of the 1.7 million TBIs reported in the US annually146. Repeated mild TBI, a form of CHI, commonly occurs in contact sports (for example, boxing, hockey, soccer and American football), child abuse victims, and modern military personnel147,148. Growing evidence indicates that repeated brain concussion can result in cumulative and long-term behavioural symptoms, neuropathological changes and neurodegeneration146,149. Several models have been developed to mimic the clinical consequences of repeated mild TBI148, including the weight drop model150,151, blast TBI model in mouse74, the FPI model in rat73, and the CHI model in swine152.
Kane and colleagues modified Marmarou’s impact acceleration model to allow repeated head impacts in lightly anaesthetized mice153. This method does not require scalp incision and protective skull helmets. Mice spontaneously recover the righting reflex without evidence of seizures and paralysis, and skull fractures and intracranial bleeding are rare. Minor deficits in motor coordination and locomotor hyperactivity recover over time. Mild astrocytic reactivity and increased phospho-tau levels occur without BBB disruption, oedema and microglial activation. This new animal model is suitable for screening of new therapies for mild concussive injuries.
A single mild LFPI induces short-term behavioural and neuropathological changes in the rat154, whereas repeated mild LFPI in rats causes cumulative long-term behavioural impairments, neuroinflammation and cortical neuron loss155. Interestingly, sub-concussive brain injury induces acute neuroinflammation in the absence of behavioural impairments in the rat after TBI156. In an immature large animal model of TBI in neonatal piglets, two head rotations following injury led to poorer outcomes, as assessed by neuropathology and neurobehavioral functional outcomes, than did a single rotation43. White matter injury increased in the repeat rotation group compared with the single injury group. More importantly, an increase in injury severity and mortality was observed when the head rotations occur 24 hours apart compared with 7 days apart. Worsening performance on cognitive composite score was associated with increasing severity of white matter axonal injury.
These findings in animal models suggest that repeated mild TBI occurring within a short period can be catastrophic or fatal, and are consistent with findings in human patients who have experienced repeated brain concussions. These models will provide further insight into sports- and combat-related repeated concussions to help healthcare providers to make better decisions about allowing individuals with TBI to return to their duties and to identify people who may be at enhanced risk for TBI.
Limitations of current animal models
Physiological differences
Although there is substantial similarity in the physiology between non-human mammals (in particular rodents) and human brains, it is clear that notable differences exist between these groups in terms of brain structure and function, including brain geometry, craniospinal angle, gyral complexity, and white to gray matter ratio93,157. These structural characteristics may lead to substantially different responses to trauma of comparable severity or type from species to species158. This situation becomes even more complex, as a number of investigations have described profound differences between behavioural and histopathological responses to TBI among different rat strains159,160 and mouse strains161.
There is also evidence for sex differences in outcome after TBI in animals and humans162,163. Female sex is often associated with a lower rate of comorbidities and complications after TBI than male sex 163, and experimental animal studies suggest that female sex hormones may have a neuroprotective effect163,164. Current clinical evidence indicates that the female hormone progesterone improves the neurological outcome of patients with TBI165. However, controversy exists regarding the sex differences in clinical TBI outcome163,166. In addition to sex hormones, many other differences between sexes, including preinjury comorbidities, brain function and metabolism, may affect outcome166. As most experimental TBI studies have been conducted in male animals, further studies on sex differences in response to TBI and treatment are clearly warranted.
Many investigators studying TBI models do not rigorously measure physiological variables before and after TBI including PCO2, PO2, pH, blood pressure and brain temperature. These variables are extremely important in determining pathophysiological responses to injury and therapy. Indeed, this is one of the shortcomings of the TBI field and should be strengthened because of the importance of these variables on acute and long-term outcomes.
Injury severity measurement
Acute assessment of injury severity is critical for diagnosis, management and prognosis of TBI. Currently in TBI clinical trials, the Glasgow Coma Scale (GCS) is the primary means for patient selection, and the Glasgow Outcome Scale (GOS), or its eight point extended version (GOSe), remains a primary method for assessing outcome167,168. Although the severity of injury can be determined by the neurological severity score (NSS) evaluated at 1 h after CHI in mice and rats123,169, there has been no common scoring system for injury severity that has been widely adopted for animals based on a brief neurological examination like the GCS in patients with TBI. Thus, the mechanical injury parameters in combination with histological evidence and functional tests are the most reliable measurements for classification of experimental TBI into mild, moderate and severe levels16,38,123,170. Scoring systems based on mechanical parameters may be specific only for a particular laboratory since most injury devices are custom-made and show subtle differences in design and operation. Additionally, small shifts in craniotomy position produce differences in cognitive performance, hippocampal cell loss and reactive astrogliosis in rats after LFPI84, and this variability makes the comparison of experimental findings from different laboratories challenging. Moreover, the posttraumatic sequelae after mild TBI without overt morphological damage and severe TBI with high mortality have not been comprehensively studied in animal models.
MRI is especially useful for non-invasively detecting white matter reorganization after brain injury171,172. Advanced MRI can detect subtle changes in brain activity and morphology related to impairment in cognitive or emotional function even in mild clinical TBI173,174 and in animal models of TBI171,175,176. Despite notable advances in diagnostic neuroimaging, accurate and early evaluation of the severity of TBI and prediction of long-term outcome are difficult. This calls for a concerted effort to search for sensitive and reliable biomarkers of TBI. Unique biochemical, neuroimaging and genetic biomarkers may be identified in response to different injury severities and different types of injuries. To ensure biomarkers in animal models of TBI genuinely reflect those associated with TBI in humans, the biomarkers measured in humans with TBI should also be measured in TBI animals and vice versa, so that clinically relevant biomarkers can be identified. A common biomarker if identified will facilitate translation of findings from the laboratory (for example, evaluation of the efficacy of preclinical therapeutic treatments) to the clinic.
Improving translation from animals to the clinic
Rigorous testing of therapeutic approaches in animal TBI models
To improve the translation of preclinical findings into successful clinical treatments, various factors need to be considered in future preclinical studies. Prior to the translation of a preclinical therapy into TBI clinical trials, ideally, sufficient preclinical data should be obtained from multiple experiments, preferably in several TBI models (in small and large animals) with different injury severities, on optimal administration routes, dose-response, therapeutic windows, single dose versus multiple dose, bolus dose versus continuous infusion.
In addition, the effective progression of strategies into clinical trials may require multifunctional agents and/or combination therapies. These potential combinations could include single pharmaceutical agents together either with cells (for example, somatic or stem cells, or genetically modified derivatives) or with other approaches (biomaterial materials, physical or electrical stimulation) for reduction of secondary damage and increase in neuroplasticity. Of note, the interaction of agents used in a combination therapy should be fully addressed in preclinical studies prior to their assessment in clinical trials. The importance of this point is illustrated by erythropoietin (EPO), which showed promise as a treatment for ischemic stroke in small clinical trials177 but failed in a recent stroke clinical trial where it was combined with the thromobolytic drug tissue plasminogen activator (tPA)178. Recent preclinical data demonstrated that there is a previously unknown interaction of tPA with EPO, suggesting that EPO may not be suitable as a stroke treatment after tPA induces thrombolysis179,180. Multiple drugs are often used to treat the TBI patients with polytrauma or complications such as higher ICP, infection and seizure. This may increase risks of potential interactions of those drugs with a drug tested in the clinical trial. Thus, preclinical studies are needed to rigorously address drug safety and efficacy to guide subsequent clinical trials, especially of combination therapies for TBI. One other point is that many agents entered into clinical trials for TBI were rarely assessed in pharmacokinetic and pharmacodynamic studies or in terms of brain penetration and distribution (for systemically administered drugs) in TBI models27. Extensive studies on these issues are warranted in preclinical studies.
The therapeutic approach tested in preclinical studies has to reflect the clinical scenario. Neuroprotection approaches have historically been dominated by targeting neuron-based injury mechanisms, either as the primary or even exclusive focus of the therapeutic strategy181. In the vast majority of animal models of TBI, the prospective neuroprotective compounds have been administered either early after TBI or, frequently, before the injury is delivered106, which is not clinically relevant. The early administration of a compound by prehospital care personnel may be problematic because of the difficulty in obtaining informed consent182. Thus, it is eminently reasonable to test compounds that can be administered late after onset of TBI, which have neurorestorative effects. The essential difference between neuroprotective and neurorestorative treatments is that the former mainly target the lesion and the latter treat the intact tissue183. Thus, neurorestorative treatments can be made available for targeting a larger portion of patients with TBI. It is essential to rigorously test neurorestorative therapies in addition to neuroprotective therapies in animal models of TBI (Box 3).
Box 3. Neuroprotection and neurorestoration.
Acute neuroprotective therapies aim to block the molecular cascade of injury following traumatic brain injury (TBI). Although neuroprotection is an important strategy for the treatment of such injury184, to date, no effective neuroprotective agents have been identified from TBI clinical trials. The disappointing clinical trials may be due to variability in both treatment approaches and heterogeneity of the population of TBI patients184-187. Another important aspect is that most clinical trial strategies have used drugs that target a single pathophysiological mechanism, despite the fact that many mechanisms are involved in secondary injury after TBI185. Testing multiple functional agents or combination therapy targeting complex mechanisms is an important research direction in animal models of TBI. A major limitation of neuroprotection strategies is the short time window. As such, an efficacy of therapies can be expected only within several minutes to hours after TBI onset.
Subacute neurorestoration therapies enhance neuroplasticity and brain reorganization following TBI. Recent preclinical studies from us and others have revealed that TBI induces many neurorestorative processes including neurogenesis, axonal sprouting, synaptogenesis, oligodendrogenesis and angiogenesis, which may contribute to spontaneous functional recovery188-193. In addition, agents and treatments that promote these neurorestorative processes have been demonstrated to improve functional recovery after brain injury183,194. However, clinical trials in TBI have primarily targeted neuroprotection, and trials directed specifically at neurorestoration have not been fully investigated in animal models and are rarely conducted in TBI patients. Unlike neuroprotection, restorative therapies are aimed at remodeling brain tissue rather than solely against cell death or lesion volume. The extended 24 hour window for treatment which improves neurological recovery, without altering cortical lesion volume, is a major benefit of this novel neurorestorative therapy in TBI animals195,196. Thus, neurorestorative therapy potentially will have a high clinical impact. Further investigation of neurorestorative agents in animal models is warranted to increase the therapeutic window and target an expanded population of patients with TBI.
Long-term versus short-term studies
To date, most of studies in animal models of TBI have focused primarily on short-term survival times, in the range of hours to days and rarely extend beyond one month after injury106. These short-term studies have provided abundant information on the pathophysiology and functional outcomes during the acute stage after TBI. The histological and behavioural data obtained at the early time points post injury may not provide a valid assessment of long-term outcome and cannot be used to assess clinical therapies for long-lasting efficacy. To verify whether early changes can predict long-term outcome, more studies evaluating injury response and functional deficits over longer time periods (3 months up to 1 year after TBI) are warranted. A small number of experimental TBI studies have followed outcomes of animals beyond 1 month after injury28,108,109,197-199. However, several studies have demonstrated that long-term functional and structural changes take place up to 1 year after TBI28,91,109,198. These findings suggest that therapeutic window may not be limited to the first few hours after TBI and may extend far beyond this period. In addition, the delayed progression of brain damage over periods of months and even years suggests that to reduce brain damage, early treatment is necessary but may not be sufficient to promote long-term recovery; continued treatment may be needed for long-term functional recovery. Delayed treatment may benefit patients with TBI who miss the early window of neuroprotection therapy. Previous studies in animals have provided a proof of principle for improvement of functional recovery with delayed neurorestorative treatments initiated 24 hr 195,200 or beyond201 after TBI.
Although long-term behavioural deficits can be detected in rodent models of TBI, it seems that cognitive deficits are more robust and persistent than sensorimotor deficits, and different focal impact sites produce dissociable patterns of cognitive deficits in rats108,202,203, consistent with the observations that cognitive deficits are the most common disabling sequelae of human TBI106,204. These findings suggest that rodents can be used to model different subgroups of patients with TBI. Given that the therapeutic potential of novel treatments may be limited to specific injury types, and even to specific behavioural deficits205, the use of a variety of injury types and a comprehensive battery of long-term behavioural tests is highly recommended for future preclinical studies.
TBI models with comorbidities
Despite modern intensive care, death and disability in polytrauma patients with concomitant TBI remain unacceptably high206. TBI in the clinical setting is a heterogeneous injury with a combination of hematomas, contusion, DAI, subarachnoid hemorrhage, hypoxia, and ischemia and is often accompanied by medication or substance use13. To better mimic clinical situations, some of these factors have been integrated into the animal TBI models. The CCI and impact acceleration models have been combined with hypoxia207, hypoxia and hypotension208,209 and the LFPI model has been combined with hypoxia210 and/or hypotension211. These systemic insults were shown to exacerbate histological and behavioural outcomes in these models212. However, these factors are understudied in animal models of TBI. Further development of more clinically relevant animal models of TBI is necessary to incorporate hypoxia, ischemia, and other potentially relevant factors that influence clinical head injury to reproduce the complete pathobiology of human TBI and to test potential therapies targeting these factors.
Multiple injuries can result in a complex pathophysiological and immunological response213. Indeed, LFPI combined with a tibial fracture initiates a robust systemic inflammatory response in rats214. Pharmacological treatment should be evaluated in TBI models with multiple injuries because injury to other organs may significantly change drug biodistribution, bioavailability and metabolism, which together may affect drug efficacy and toxicity. In addition, identification of unique neurochemical mediators and mechanisms following multiple injuries will help determine effective therapeutic interventions in individual patients with TBI.
Age is another important comorbidity factor affecting outcomes of TBI. TBI is the leading cause of death in children. Survivors of childhood TBI are at risk for developing and sustaining behavioural impairments54. Clinical and experimental studies demonstrate that the developing brain may be more vulnerable to traumatic injury than the adult brain215. Animal models have shown that developmental TBI results in different acute injury responses and recovery54,216. Among the developmental animal models of TBI, CCI, FPI and Marmarou weight drop models are most commonly used in immature rodents and pigs54 while rapid non-impact inertial head injury are used in immature pigs152,217. Relative to adult TBI, our understanding and management of pediatric TBI is still in its infancy. More comprehensive studies in this area will strengthen our understanding of the complex interactions between brain maturation and recovery from injury and will provide critical ground work for addressing unique responses of this specific age group to TBI.
With increasing age comes an increased risk for sustaining TBI218. Elderly individuals with TBI differ from younger adults with TBI in several ways, including their incidence rates, aetiology of injury, nature of complications, lengths of hospitalization, functional outcomes, and mortality219. Adults older than 75 have the highest rates of TBI-related hospitalization and death, with falls as the leading cause of TBI220. Older age is known to negatively affect outcome after TBI221. However, the effect of age is rarely studied in animal models. To address this important public health issue, age is an important factor to consider in pre-clinical efficacy studies218,220. In addition, therapeutic doses of a treatment identified to be effective in young animals with TBI may have no effect even worsen outcome in aged TBI rats218. This finding suggests that it is not sufficient to simply study the effects of age on TBI and novel therapies must be evaluated in aged populations of animals with TBI. Given the high incidence of TBI in the aged population, much more preclinical research is needed in this area.
Conclusions and perspective
Overcoming the lack of drugs with proven clinical efficacy in TBI is a major challenge for the neuroscience research community and the pharmaceutical industry. Studies employing various animal models, in vitro models and computational modeling of TBI have contributed to the current understanding of the posttraumatic sequelae. Among these approaches, the animal models remain necessary to address complex physiological and pathophysiological mechanisms associated with this condition, test new therapeutic agents, and ensure that clinical trials are safe and, ultimately, successful. A variety of rodent models of TBI have been developed to model different injury mechanisms associated with human TBI. The rodent models by Marmarou51 and Shohami29 are widely used for CHI and they reproduce predominantly diffuse and focal brain injury, respectively. Probably due to the excellent reproducibility, LFPI and CCI are the most widely used rodent models for TBI. There is an increasing research in blast TBI and sports-related concussions, especially repeated mild TBI.
Although small animal models have been used in TBI research to investigate the basic mechanisms and pathology of TBI and to test therapeutic efficacy, successful TBI investigations in small animal models have not resulted in marked improvements in clinical outcomes of patients with such injury. One of the major barriers to crossing the translational gap is that, due to ethical and financial issues, researchers rarely use clinically meaningful large animal models of TBI to monitor clinically relevant physiological parameters and long-term functional and/or cognitive outcomes, and to test the efficacy of new treatments. Thus, it is extremely important to further develop and increasingly use higher species with brains that are more anatomically and functionally closer to man. At least, before initiation of clinical trials, an effective treatment in rodents should be tested, with its efficacy confirmed, in large animal models that closely mimic the complex pathogenesis of TBI in humans.
Numerous promising treatment options have emerged in recent years, including neuroprotective, neurorestorative and anti-inflammatory agents186 These drugs should be subjected to a rigorous preclinical dose–response analysis of their efficacy on the target mechanism and the ability to reduce posttraumatic neurodegeneration and to improve behavioural and neurological recovery. This endeavor would facilitate the transition of TBI therapies from the bench to the bedside.
The failure to achieve a therapeutic breakthrough in TBI may not result only from limitations of the animal models per se. Poor clinical study design is also a factor in why therapeutic translation has not occurred. For example, early therapeutic hypothermia is beneficial in many experimental models of TBI. Hypothermia appears to improve outcome in TBI patients undergoing craniotomy for hematoma only when it is applied before or within 1.5 hours after craniotomy, but does not improve the outcome of patients with diffuse brain injury222. This implies that optimal timing of combined treatments has a critical role in beneficial outcome in a specific population of patients with TBI. Thus, the continued translation of new findings from the bench to the bedside and then back to the bench will ultimately teach us a lot about the relevance of our animal models. Most importantly, these types of back and forth exchange of observations and ideas will help us determine which pathophysiological mechanisms are most important to target in specific patient populations. The lack of success of translating preclinical effective treatments to clinical TBI is complex and may result both from the multifaceted issues of suboptimal animal models and inadequate design and implementation of clinical trials, as described by us in this review and by others22,35,36,192. In addition, as we have discussed, the pathophysiological heterogeneity of patients with TBI, the lack of sufficient pharmacokinetic analysis for determination of optimal dose, the compounds given outside of the therapeutic window, and insensitive outcome measures may limit proof of clinical efficacy223-226. The ongoing international effort to develop an improved classification system for individuals with TBI may enable selection of more homogenous patient cohorts in future clinical trials to facilitate multicentre comparisons225.
In conclusion, current animal models mimic some but not all types of human brain injury. To achieve a therapeutic breakthrough in TBI will probably require a multifaceted approach, combining innovations in clinical trial design, the development of new clinically relevant models, refinements of established models and functional tests, consideration of systemic insults and multimodality monitoring, searching for specific and sensitive biomarkers, and optimization of therapeutic dosing and timing of single and combination treatment. In addition, more research into the effect of age, sex and species or strain on the outcome of TBI is necessary. One final important issue is that the majority of drugs tested to date cannot cross the BBB to effectively target the injured brain. Additional studies in improving brain drug delivery systems and monitoring of target drug levels and drug effects are warranted in both animal models and the clinical setting.
Acknowledgments
We thank three anonymous referees for their excellent comments, and we apologize to those researchers whose work has not been cited due to space limitations. This work was supported by National Institutes of Health grants RO1 NS062002 (Y.X.), PO1 NS042345 (A.M. and M.C.) and PO1 NS023393 (M.C.).
Glossary
- Diffuse axonal injury (DAI)
DAI is characterized by impaired axoplasmic flow that progresses to axotomy, and is typically identified by immunohistochemical staining of amyloid-β precursor protein
- Erythropoietin (EPO)
EPO is a glycoprotein hormone secreted by the kidney in the adult and by the liver in the fetus, which acts on stem cells of the bone marrow to stimulate red blood cell production (that is, erythropoiesis)
- Tissue plasminogen activator (tPA)
tPA is an enzyme that catalyzes the conversion of plasminogen to plasmin, used to dissolve blood clots rapidly and selectively, especially in the treatment of heart attacks and ischaemic stroke
- Phosphorylated tauopathy
This is the accumulation of hyperphosphorylated tau protein (a highly soluble microtubule-associated protein), which causes the formation of neurofibrillary tangles, a pathological hallmark of tauopathies, a group of diseases including Alzheimer’s disease, frontal temporal dementia with Parkinsonism and corticobasal degeneration
- Glasgow coma scale (GCS)
The GCS is a standardized scale used to measure level of consciousness, to assess the degree of brain impairment and to identify the seriousness of injury in relation to outcome after TBI. Scoring is determined by summing the ratings assigned to three factors depending on whether and how the patient responds to certain standard stimuli by opening the eyes, giving a verbal response, and giving a motor response. A high score of 13 to 15 indicates a mild brain injury. A score of 9 to 12 reflects a moderate brain injury and a score of 3 to 8 reflects a severe brain injury
- Glasgow outcome scale (GOS)
The GOS is a 5-point score given to victims of TBI for classifying the outcome that rates patient status into one of five categories: Dead, Vegetative State, Severe Disability, Moderate Disability or Good Recovery. The Extended GOS (GOSe) provides more detailed categorization into eight categories by subdividing the categories of severe disability, moderate disability and good recovery into a lower and upper category
- Neurological severity score (NSS)
The NSS is a reliable tool for evaluating neurological damage in closed head trauma in mice and rats and assess both motor function and behavior
- Modified neurological severity score (mNSS)
The mNSS is a composite of motor, sensory, reflex, and balance tests in rats. It is graded on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18). One point is awarded for inability to perform the tasks or for lack of a tested reflex: 13–18, severe injury; 7–12, moderate injury; 1–6, mild injury
- Biomarker
A specific biochemical, molecular, anatomic and physiologic characteristic that is used to measure or indicate the presence or progress of disease or the effects of treatment
References
- 1.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 4.Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med. 2010;14:2381–2392. doi: 10.1111/j.1582-4934.2010.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Lee LL, Galo E, Lyeth BG, Muizelaar JP, Berman RF. Neuroprotection in the rat lateral fluid percussion model of traumatic brain injury by SNX-185, an N-type voltage-gated calcium channel blocker. Exp Neurol. 2004;190:70–78. doi: 10.1016/j.expneurol.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma. 1997;14:23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- 8.McCall JM, Braughler JM, Hall ED. Lipid peroxidation and the role of oxygen radicals in CNS injury. Acta Anaesthesiol Belg. 1987;38:373–379. [PubMed] [Google Scholar]
- 9.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raghavendra Rao VL, Dhodda VK, Song G, Bowen KK, Dempsey RJ. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- 11.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis AE. Mechanisms of traumatic brain injury: biomechanical, structural and cellular considerations. Crit Care Nurs Q. 2000;23:1–13. doi: 10.1097/00002727-200011000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. This article comprehensively reviews animal models of TBI and addresses key factors in development of animal models and neuroprotection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 15.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 16.Thompson HJ, et al. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. This article reviews the lateral fluid percussion model of TBI in small animal species with particular emphasis on its validity, clinical relevance and reliability. [DOI] [PubMed] [Google Scholar]
- 17.Marklund N, Bakshi A, Castelbuono DJ, Conte V, McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12:1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 18.Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555–564. doi: 10.1089/neu.1995.12.555. This article provides a review of those factors involved in the pathogenesis of traumatically induced axonal injury in both animals and man. [DOI] [PubMed] [Google Scholar]
- 19.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17:927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- 21.Yakovlev AG, et al. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439–7446. doi: 10.1523/JNEUROSCI.21-19-07439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niogi SN, et al. Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. AJNR Am J Neuroradiol. 2008;29:967–973. doi: 10.3174/ajnr.A0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipton ML, et al. Multifocal white matter ultrastructural abnormalities in mild traumatic brain injury with cognitive disability: a voxel-wise analysis of diffusion tensor imaging. J Neurotrauma. 2008;25:1335–1342. doi: 10.1089/neu.2008.0547. [DOI] [PubMed] [Google Scholar]
- 24.Rubovitch V, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol. 2011;232:280–289. doi: 10.1016/j.expneurol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browne KD, Chen XH, Meaney DF, Smith DH. Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma. 2011;28:1747–1755. doi: 10.1089/neu.2011.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care. 2007;13:134–142. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- 27.Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26:925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimoto ST, et al. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–378. doi: 10.1016/j.neubiorev.2004.06.002. This article describes and evaluates the tests employed to assess functional outcome after TBI and provides an overview of aspects of cognitive, sensory, and motor function that may be suitable targets for therapeutic intervention. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma. 1996;13:557–568. doi: 10.1089/neu.1996.13.557. [DOI] [PubMed] [Google Scholar]
- 30.Shapira Y, et al. Experimental closed head injury in rats: mechanical, pathophysiologic, and neurologic properties. Crit Care Med. 1988;16:258–265. doi: 10.1097/00003246-198803000-00010. This article presents a model of closed head injury in rats developed using a calibrated weight-drop device. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 32.Mahmood A, Lu D, Chopp M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J Neurotrauma. 2004;21:33–39. doi: 10.1089/089771504772695922. [DOI] [PubMed] [Google Scholar]
- 33.Levin HS, Eisenberg HM, Wigg NR, Kobayashi K. Memory and intellectual ability after head injury in children and adolescents. Neurosurgery. 1982;11:668–673. doi: 10.1227/00006123-198211000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Fox GB, Faden AI. Traumatic brain injury causes delayed motor and cognitive impairment in a mutant mouse strain known to exhibit delayed Wallerian degeneration. J Neurosci Res. 1998;53:718–727. doi: 10.1002/(SICI)1097-4547(19980915)53:6<718::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 35.Hamm RJ, et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- 36.Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- 37.Shear DA, et al. Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J Neurotrauma. 2010;27:1911–1923. doi: 10.1089/neu.2010.1399. [DOI] [PubMed] [Google Scholar]
- 38.Washington PM, et al. The Effect of Injury Severity on Behavior: A phenotypic study of cognitive and emotional deficits after mild, moderate and severe controlled cortical impact injury in mice. J Neurotrauma. 2012 doi: 10.1089/neu.2012.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarzbold ML, et al. Effects of traumatic brain injury of different severities on emotional, cognitive, and oxidative stress-related parameters in mice. J Neurotrauma. 2010;27:1883–1893. doi: 10.1089/neu.2010.1318. [DOI] [PubMed] [Google Scholar]
- 40.Pandey DK, Yadav SK, Mahesh R, Rajkumar R. Depression-like and anxiety-like behavioural aftermaths of impact accelerated traumatic brain injury in rats: a model of comorbid depression and anxiety? Behav Brain Res. 2009;205:436–442. doi: 10.1016/j.bbr.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 41.Jones NC, et al. Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J Neurotrauma. 2008;25:1367–1374. doi: 10.1089/neu.2008.0641. [DOI] [PubMed] [Google Scholar]
- 42.Milman A, Rosenberg A, Weizman R, Pick CG. Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J Neurotrauma. 2005;22:1003–1010. doi: 10.1089/neu.2005.22.1003. [DOI] [PubMed] [Google Scholar]
- 43.Friess SH, et al. Repeated traumatic brain injury affects composite cognitive function in piglets. J Neurotrauma. 2009;26:1111–1121. doi: 10.1089/neu.2008.0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friess SH, et al. Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp Neurol. 2007;204:234–243. doi: 10.1016/j.expneurol.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurdjian ES, Lissner HR, Webster JE, Latimer FR, Haddad BF. Studies on experimental concussion: relation of physiologic effect to time duration of intracranial pressure increase at impact. Neurology. 1954;4:674–681. doi: 10.1212/wnl.4.9.674. [DOI] [PubMed] [Google Scholar]
- 46.Walker AE. The physiological basis of concussion: 50 years later. J Neurosurg. 1994;81:493–494. doi: 10.3171/jns.1994.81.3.0493. [DOI] [PubMed] [Google Scholar]
- 47.Denny-Brown DE, Russell WR. Experimental Concussion: (Section of Neurology) Proc R Soc Med. 1941;34:691–692. doi: 10.1177/003591574103401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dixon CE, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. This study systematically examines the effects of varying magnitudes of FPI in the rat on neurological, systemic physiological and histopathological changes. [DOI] [PubMed] [Google Scholar]
- 49.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 50.Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. This study introduces a new experimental model of mechanical brain injury produced in the laboratory ferret using a stroke-constrained pneumatic impactor. [DOI] [PubMed] [Google Scholar]
- 51.Marmarou A, et al. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. This study describes the development of an experimental head injury model capable of producing diffuse brain injury in the rodent. [DOI] [PubMed] [Google Scholar]
- 52.Cernak I, et al. Involvement of the central nervous system in the general response to pulmonary blast injury. J Trauma. 1996;40:S100–104. doi: 10.1097/00005373-199603001-00023. [DOI] [PubMed] [Google Scholar]
- 53.Leung LY, et al. Blast related neurotrauma: a review of cellular injury. Mol Cell Biomech. 2008;5:155–168. [PubMed] [Google Scholar]
- 54.Prins ML, Hovda DA. Developing experimental models to address traumatic brain injury in children. J Neurotrauma. 2003;20:123–137. doi: 10.1089/08977150360547053. [DOI] [PubMed] [Google Scholar]
- 55.Plantman S, Ng KC, Lu J, Davidsson J, Risling M. Characterization of a novel rat model of penetrating traumatic brain injury. J Neurotrauma. 2012;29:1219–1232. doi: 10.1089/neu.2011.2182. [DOI] [PubMed] [Google Scholar]
- 56.Smith DH, et al. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 57.Manley GT, et al. Controlled cortical impact in swine: pathophysiology and biomechanics. J Neurotrauma. 2006;23:128–139. doi: 10.1089/neu.2006.23.128. [DOI] [PubMed] [Google Scholar]
- 58.King C, et al. Brain temperature profiles during epidural cooling with the ChillerPad in a monkey model of traumatic brain injury. J Neurotrauma. 2010;27:1895–1903. doi: 10.1089/neu.2009.1178. [DOI] [PubMed] [Google Scholar]
- 59.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 60.Shohami E, Shapira Y, Cotev S. Experimental closed head injury in rats: prostaglandin production in a noninjured zone. Neurosurgery. 1988;22:859–863. [PubMed] [Google Scholar]
- 61.Albert-Weissenberger C, Varrallyay C, Raslan F, Kleinschnitz C, Siren AL. An experimental protocol for mimicking pathomechanisms of traumatic brain injury in mice. Exp Transl Stroke Med. 2012;4:1. doi: 10.1186/2040-7378-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kilbourne M, et al. Novel model of frontal impact closed head injury in the rat. J Neurotrauma. 2009;26:2233–2243. doi: 10.1089/neu.2009.0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayes RL, et al. A new model of concussive brain injury in the cat produced by extradural fluid volume loading: II. Physiological and neuropathological observations. Brain Inj. 1987;1:93–112. doi: 10.3109/02699058709034449. [DOI] [PubMed] [Google Scholar]
- 64.Hartl R, Medary M, Ruge M, Arfors KE, Ghajar J. Blood-brain barrier breakdown occurs early after traumatic brain injury and is not related to white blood cell adherence. Acta Neurochir Suppl. 1997;70:240–242. doi: 10.1007/978-3-7091-6837-0_74. [DOI] [PubMed] [Google Scholar]
- 65.McIntosh TK, Noble L, Andrews B, Faden AI. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma. 1987;4:119–134. doi: 10.1089/cns.1987.4.119. [DOI] [PubMed] [Google Scholar]
- 66.Millen JE, Glauser FL, Fairman RP. A comparison of physiological responses to percussive brain trauma in dogs and sheep. J Neurosurg. 1985;62:587–591. doi: 10.3171/jns.1985.62.4.0587. [DOI] [PubMed] [Google Scholar]
- 67.Pfenninger EG, Reith A, Breitig D, Grunert A, Ahnefeld FW. Early changes of intracranial pressure, perfusion pressure, and blood flow after acute head injury. Part 1: An experimental study of the underlying pathophysiology. J Neurosurg. 1989;70:774–779. doi: 10.3171/jns.1989.70.5.0774. [DOI] [PubMed] [Google Scholar]
- 68.McIntosh TK, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 69.Carbonell WS, Maris DO, McCall T, Grady MS. Adaptation of the fluid percussion injury model to the mouse. J Neurotrauma. 1998;15:217–229. doi: 10.1089/neu.1998.15.217. [DOI] [PubMed] [Google Scholar]
- 70.Armstead WM. Vasopressin-induced protein kinase C-dependent superoxide generation contributes to atp-sensitive potassium channel but not calcium-sensitive potassium channel function impairment after brain injury. Stroke. 2001;32:1408–1414. doi: 10.1161/01.str.32.6.1408. [DOI] [PubMed] [Google Scholar]
- 71.Goldstein LE, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. This study develops a mouse model of blast TBI that recapitulated chronic traumatic encephalopathy-linked neuropathology 2 weeks after exposure to a single blast. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bauman RA, et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma. 2009;26:841–860. doi: 10.1089/neu.2008.0898. [DOI] [PubMed] [Google Scholar]
- 73.DeRoss AL, et al. Multiple head injuries in rats: effects on behavior. J Trauma. 2002;52:708–714. doi: 10.1097/00005373-200204000-00017. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y, et al. Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J Neurotrauma. 2011;28:2171–2183. doi: 10.1089/neu.2011.1990. [DOI] [PubMed] [Google Scholar]
- 75.Carey ME, Sarna GS, Farrell JB. Brain edema following an experimental missile wound to the brain. J Neurotrauma. 1990;7:13–20. doi: 10.1089/neu.1990.7.13. [DOI] [PubMed] [Google Scholar]
- 76.Williams AJ, Hartings JA, Lu XC, Rolli ML, Tortella FC. Penetrating ballistic-like brain injury in the rat: differential time courses of hemorrhage, cell death, inflammation, and remote degeneration. J Neurotrauma. 2006;23:1828–1846. doi: 10.1089/neu.2006.23.1828. [DOI] [PubMed] [Google Scholar]
- 77.Albert-Weissenberger C, Siren AL. Experimental traumatic brain injury. Exp Transl Stroke Med. 2010;2:16. doi: 10.1186/2040-7378-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gennarelli TA. Animate models of human head injury. J Neurotrauma. 1994;11:357–368. doi: 10.1089/neu.1994.11.357. [DOI] [PubMed] [Google Scholar]
- 79.Potts MB, Adwanikar H, Noble-Haeusslein LJ. Models of traumatic cerebellar injury. Cerebellum. 2009;8:211–221. doi: 10.1007/s12311-009-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hardman JM, Manoukian A. Pathology of head trauma. Neuroimaging Clin N Am. 2002;12:175–187. vii. doi: 10.1016/s1052-5149(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 81.Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- 82.Sanders MJ, Dietrich WD, Green EJ. Cognitive function following traumatic brain injury: effects of injury severity and recovery period in a parasagittal fluid-percussive injury model. J Neurotrauma. 1999;16:915–925. doi: 10.1089/neu.1999.16.915. [DOI] [PubMed] [Google Scholar]
- 83.Vink R, Mullins PG, Temple MD, Bao W, Faden AI. Small shifts in craniotomy position in the lateral fluid percussion injury model are associated with differential lesion development. J Neurotrauma. 2001;18:839–847. doi: 10.1089/089771501316919201. [DOI] [PubMed] [Google Scholar]
- 84.Floyd CL, Golden KM, Black RT, Hamm RJ, Lyeth BG. Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J Neurotrauma. 2002;19:303–316. doi: 10.1089/089771502753594873. [DOI] [PubMed] [Google Scholar]
- 85.Stalhammar D, et al. A new model of concussive brain injury in the cat produced by extradural fluid volume loading: I. Biomechanical properties. Brain Inj. 1987;1:73–91. doi: 10.3109/02699058709034448. [DOI] [PubMed] [Google Scholar]
- 86.Sullivan HG, et al. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45:521–534. [PubMed] [Google Scholar]
- 87.Dixon CE, Lighthall JW, Anderson TE. Physiologic, histopathologic, and cineradiographic characterization of a new fluid-percussion model of experimental brain injury in the rat. J Neurotrauma. 1988;5:91–104. doi: 10.1089/neu.1988.5.91. [DOI] [PubMed] [Google Scholar]
- 88.Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S. Lateral fluid percussion: model of traumatic brain injury in mice. J Vis Exp. 2011 doi: 10.3791/3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zink BJ, Walsh RF, Feustel PJ. Effects of ethanol in traumatic brain injury. J Neurotrauma. 1993;10:275–286. doi: 10.1089/neu.1993.10.275. [DOI] [PubMed] [Google Scholar]
- 90.Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- 91.Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- 92.Liu YR, et al. Progressive metabolic and structural cerebral perturbations after traumatic brain injury: an in vivo imaging study in the rat. J Nucl Med. 2010;51:1788–1795. doi: 10.2967/jnumed.110.078626. [DOI] [PubMed] [Google Scholar]
- 93.Morales DM, et al. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 94.Hamm RJ. Neurobehavioral assessment of outcome following traumatic brain injury in rats: an evaluation of selected measures. J Neurotrauma. 2001;18:1207–1216. doi: 10.1089/089771501317095241. [DOI] [PubMed] [Google Scholar]
- 95.Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 96.Kabadi SV, Hilton GD, Stoica BA, Zapple DN, Faden AI. Fluid-percussion-induced traumatic brain injury model in rats. Nat Protoc. 2010;5:1552–1563. doi: 10.1038/nprot.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lifshitz J, Kelley BJ, Povlishock JT. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol. 2007;66:218–229. doi: 10.1097/01.jnen.0000248558.75950.4d. [DOI] [PubMed] [Google Scholar]
- 98.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225C:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lighthall JW, Goshgarian HG, Pinderski CR. Characterization of axonal injury produced by controlled cortical impact. J Neurotrauma. 1990;7:65–76. doi: 10.1089/neu.1990.7.65. [DOI] [PubMed] [Google Scholar]
- 100.Hall ED, et al. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 101.Mao H, Zhang L, Yang KH, King AI. Application of a finite element model of the brain to study traumatic brain injury mechanisms in the rat. Stapp Car Crash J. 2006;50:583–600. doi: 10.4271/2006-22-0022. [DOI] [PubMed] [Google Scholar]
- 102.Wang HC, Ma YB. Experimental models of traumatic axonal injury. J Clin Neurosci. 2010;17:157–162. doi: 10.1016/j.jocn.2009.07.099. [DOI] [PubMed] [Google Scholar]
- 103.Goodman JC, Cherian L, Bryan RM, Jr, Robertson CS. Lateral cortical impact injury in rats: pathologic effects of varying cortical compression and impact velocity. J Neurotrauma. 1994;11:587–597. doi: 10.1089/neu.1994.11.587. [DOI] [PubMed] [Google Scholar]
- 104.Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 105.Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 106.Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br J Pharmacol. 2011;164:1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. This review describes the utility of animal models of TBI for preclinical evaluation of pharmacological compounds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dixon CE, et al. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor Neurol Neurosci. 1999;14:285–294. [PubMed] [Google Scholar]
- 108.Dixon CE, et al. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 109.Kochanek PM, et al. Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J Neurotrauma. 2002;19:1029–1037. doi: 10.1089/089771502760341947. [DOI] [PubMed] [Google Scholar]
- 110.Alessandri B, Heimann A, Filippi R, Kopacz L, Kempski O. Moderate controlled cortical contusion in pigs: effects on multi-parametric neuromonitoring and clinical relevance. J Neurotrauma. 2003;20:1293–1305. doi: 10.1089/089771503322686094. [DOI] [PubMed] [Google Scholar]
- 111.Williams AJ, et al. Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma. 2005;22:313–331. doi: 10.1089/neu.2005.22.313. This study characterizes the pathophysiology of a new rat model of penetrating brain injury that simulates the large temporary cavity caused by energy dissipation from a penetrating bullet round. [DOI] [PubMed] [Google Scholar]
- 112.Williams AJ, Ling GS, Tortella FC. Severity level and injury track determine outcome following a penetrating ballistic-like brain injury in the rat. Neurosci Lett. 2006;408:183–188. doi: 10.1016/j.neulet.2006.08.086. [DOI] [PubMed] [Google Scholar]
- 113.Carey ME, Sarna GS, Farrell JB, Happel LT. Experimental missile wound to the brain. J Neurosurg. 1989;71:754–764. doi: 10.3171/jns.1989.71.5.0754. [DOI] [PubMed] [Google Scholar]
- 114.Davis AR, Shear DA, Chen Z, Lu XC, Tortella FC. A comparison of two cognitive test paradigms in a penetrating brain injury model. J Neurosci Methods. 2010;189:84–87. doi: 10.1016/j.jneumeth.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 115.Williams AJ, Wei HH, Dave JR, Tortella FC. Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation. 2007;4:17. doi: 10.1186/1742-2094-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shear DA, Williams AJ, Sharrow K, Lu XC, Tortella FC. Neuroprotective profile of dextromethorphan in an experimental model of penetrating ballistic-like brain injury. Pharmacol Biochem Behav. 2009;94:56–62. doi: 10.1016/j.pbb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 117.Chen Z, et al. Human amnion-derived multipotent progenitor cell treatment alleviates traumatic brain injury-induced axonal degeneration. J Neurotrauma. 2009;26:1987–1997. doi: 10.1089/neu.2008.0863. [DOI] [PubMed] [Google Scholar]
- 118.Wei G, Lu XC, Yang X, Tortella FC. Intracranial pressure following penetrating ballistic-like brain injury in rats. J Neurotrauma. 2010;27:1635–1641. doi: 10.1089/neu.2010.1378. [DOI] [PubMed] [Google Scholar]
- 119.Shear DA, et al. Severity profile of penetrating ballistic-like brain injury on neurofunctional outcome, blood-brain barrier permeability, and brain edema formation. J Neurotrauma. 2011;28:2185–2195. doi: 10.1089/neu.2011.1916. [DOI] [PubMed] [Google Scholar]
- 120.Dail WG, Feeney DM, Murray HM, Linn RT, Boyeson MG. Responses to cortical injury: II. Widespread depression of the activity of an enzyme in cortex remote from a focal injury. Brain Res. 1981;211:79–89. doi: 10.1016/0006-8993(81)90068-8. [DOI] [PubMed] [Google Scholar]
- 121.Gasparovic C, Arfai N, Smid N, Feeney DM. Decrease and recovery of N-acetylaspartate/creatine in rat brain remote from focal injury. J Neurotrauma. 2001;18:241–246. doi: 10.1089/08977150151070856. [DOI] [PubMed] [Google Scholar]
- 122.Stahel PF, et al. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–380. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- 123.Flierl MA, et al. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- 124.Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- 125.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- 127.Heath DL, Vink R. Impact acceleration-induced severe diffuse axonal injury in rats: characterization of phosphate metabolism and neurologic outcome. J Neurotrauma. 1995;12:1027–1034. doi: 10.1089/neu.1995.12.1027. [DOI] [PubMed] [Google Scholar]
- 128.Schmidt RH, Scholten KJ, Maughan PH. Cognitive impairment and synaptosomal choline uptake in rats following impact acceleration injury. J Neurotrauma. 2000;17:1129–1139. doi: 10.1089/neu.2000.17.1129. [DOI] [PubMed] [Google Scholar]
- 129.Shohami E, Shapira Y, Sidi A, Cotev S. Head injury induces increased prostaglandin synthesis in rat brain. J Cereb Blood Flow Metab. 1987;7:58–63. doi: 10.1038/jcbfm.1987.8. [DOI] [PubMed] [Google Scholar]
- 130.Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil. 2006;21:398–402. doi: 10.1097/00001199-200609000-00004. [DOI] [PubMed] [Google Scholar]
- 131.Benzinger TL, et al. Blast-related brain injury: imaging for clinical and research applications: report of the 2008 st. Louis workshop. J Neurotrauma. 2009;26:2127–2144. doi: 10.1089/neu.2009.0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheng J, et al. Development of a rat model for studying blast-induced traumatic brain injury. J Neurol Sci. 2010;294:23–28. doi: 10.1016/j.jns.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 133.Risling M, Davidsson J. Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Front Neurol. 2012;3:30. doi: 10.3389/fneur.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Long JB, et al. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma. 2009;26:827–840. doi: 10.1089/neu.2008.0748. [DOI] [PubMed] [Google Scholar]
- 135.de Lanerolle NC, et al. Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol. 2011;70:1046–1057. doi: 10.1097/NEN.0b013e318235bef2. [DOI] [PubMed] [Google Scholar]
- 136.DeWitt DS, Prough DS. Blast-induced brain injury and posttraumatic hypotension and hypoxemia. J Neurotrauma. 2009;26:877–887. doi: 10.1089/neu.2007.0439. [DOI] [PubMed] [Google Scholar]
- 137.Reneer DV, et al. A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J Neurotrauma. 2011;28:95–104. doi: 10.1089/neu.2010.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab. 2010;30:255–266. doi: 10.1038/jcbfm.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garman RH, et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma. 2011;28:947–959. doi: 10.1089/neu.2010.1540. This study identifies diffuse axonal injury as the most prominent feature during the initial 2 weeks following blast exposure in rats with body shield. [DOI] [PubMed] [Google Scholar]
- 140.Kuehn R, et al. Rodent model of direct cranial blast injury. J Neurotrauma. 2011;28:2155–2169. doi: 10.1089/neu.2010.1532. [DOI] [PubMed] [Google Scholar]
- 141.Saljo A, Svensson B, Mayorga M, Hamberger A, Bolouri H. Low-level blasts raise intracranial pressure and impair cognitive function in rats. J Neurotrauma. 2009;26:1345–1352. doi: 10.1089/neu.2008-0856. [DOI] [PubMed] [Google Scholar]
- 142.Cernak I, et al. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis. 2011;41:538–551. doi: 10.1016/j.nbd.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 143.Koliatsos VE, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol. 2011;70:399–416. doi: 10.1097/NEN.0b013e3182189f06. [DOI] [PubMed] [Google Scholar]
- 144.Bhattacharjee Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science. 2008;319:406–408. doi: 10.1126/science.319.5862.406. [DOI] [PubMed] [Google Scholar]
- 145.Sundaramurthy A, et al. Blast-induced biomechanical loading of the rat: experimental and anatomically accurate computational blast injury model. J Neurotrauma. 2012 doi: 10.1089/neu.2012.2413. [DOI] [PubMed] [Google Scholar]
- 146.Daneshvar DH, et al. Long-term consequences: effects on normal development profile after concussion. Phys Med Rehabil Clin N Am. 2011;22:683–700. ix. doi: 10.1016/j.pmr.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sports-related recurrent brain injuries--United States. Centers for Disease Control and Prevention. Int J Trauma Nurs. 1997;3:88–90. [PubMed] [Google Scholar]
- 148.Weber JT. Experimental models of repetitive brain injuries. Prog Brain Res. 2007;161:253–261. doi: 10.1016/S0079-6123(06)61018-2. [DOI] [PubMed] [Google Scholar]
- 149.Daneshvar DH, Nowinski CJ, McKee AC, Cantu RC. The epidemiology of sport-related concussion. Clin Sports Med. 2011;30:1–17. vii. doi: 10.1016/j.csm.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.DeFord SM, et al. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19:427–438. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- 151.Creeley CE, Wozniak DF, Bayly PV, Olney JW, Lewis LM. Multiple episodes of mild traumatic brain injury result in impaired cognitive performance in mice. Acad Emerg Med. 2004;11:809–819. doi: 10.1111/j.1553-2712.2004.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 152.Raghupathi R, Margulies SS. Traumatic axonal injury after closed head injury in the neonatal pig. J Neurotrauma. 2002;19:843–853. doi: 10.1089/08977150260190438. [DOI] [PubMed] [Google Scholar]
- 153.Kane MJ, et al. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. 2012;203:41–49. doi: 10.1016/j.jneumeth.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shultz SR, MacFabe DF, Foley KA, Taylor R, Cain DP. A single mild fluid percussion injury induces short-term behavioral and neuropathological changes in the Long-Evans rat: support for an animal model of concussion. Behav Brain Res. 2011;224:326–335. doi: 10.1016/j.bbr.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 155.Shultz SR, et al. Repeated mild lateral fluid percussion brain injury in the rat causes cumulative long-term behavioral impairments, neuroinflammation, and cortical loss in an animal model of repeated concussion. J Neurotrauma. 2012;29:281–294. doi: 10.1089/neu.2011.2123. [DOI] [PubMed] [Google Scholar]
- 156.Shultz SR, MacFabe DF, Foley KA, Taylor R, Cain DP. Sub-concussive brain injury in the Long-Evans rat induces acute neuroinflammation in the absence of behavioral impairments. Behav Brain Res. 2012;229:145–152. doi: 10.1016/j.bbr.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 157.Laurer HL, McIntosh TK. Experimental models of brain trauma. Curr Opin Neurol. 1999;12:715–721. doi: 10.1097/00019052-199912000-00010. [DOI] [PubMed] [Google Scholar]
- 158.Povlishock JT, Hayes RL, Michel ME, McIntosh TK. Workshop on animal models of traumatic brain injury. J Neurotrauma. 1994;11:723–732. doi: 10.1089/neu.1994.11.723. [DOI] [PubMed] [Google Scholar]
- 159.Reid WM, et al. Strain-related differences after experimental traumatic brain injury in rats. J Neurotrauma. 2010;27:1243–1253. doi: 10.1089/neu.2010.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tan AA, Quigley A, Smith DC, Hoane MR. Strain differences in response to traumatic brain injury in Long-Evans compared to Sprague-Dawley rats. J Neurotrauma. 2009;26:539–548. doi: 10.1089/neu.2008.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fox GB, LeVasseur RA, Faden AI. Behavioral responses of C57BL/6, FVB/N, and 129/SvEMS mouse strains to traumatic brain injury: implications for gene targeting approaches to neurotrauma. J Neurotrauma. 1999;16:377–389. doi: 10.1089/neu.1999.16.377. [DOI] [PubMed] [Google Scholar]
- 162.Roof RL, Hall ED. Estrogen-related gender difference in survival rate and cortical blood flow after impact-acceleration head injury in rats. J Neurotrauma. 2000;17:1155–1169. doi: 10.1089/neu.2000.17.1155. [DOI] [PubMed] [Google Scholar]
- 163.Berry C, et al. The effect of gender on patients with moderate to severe head injuries. J Trauma. 2009;67:950–953. doi: 10.1097/TA.0b013e3181ba3354. [DOI] [PubMed] [Google Scholar]
- 164.Stein DG. Progesterone in the treatment of acute traumatic brain injury: a clinical perspective and update. Neuroscience. 2011;191:101–106. doi: 10.1016/j.neuroscience.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 165.Junpeng M, Huang S, Qin S. Progesterone for acute traumatic brain injury. Cochrane Database Syst Rev. 2011 doi: 10.1002/14651858.CD008409.pub2. CD008409. [DOI] [PubMed] [Google Scholar]
- 166.Farace E, Alves WM. Do women fare worse: a metaanalysis of gender differences in traumatic brain injury outcome. J Neurosurg. 2000;93:539–545. doi: 10.3171/jns.2000.93.4.0539. [DOI] [PubMed] [Google Scholar]
- 167.Maas AI, Lingsma HF. New approaches to increase statistical power in TBI trials: insights from the IMPACT study. Acta Neurochir Suppl. 2008;101:119–124. doi: 10.1007/978-3-211-78205-7_20. [DOI] [PubMed] [Google Scholar]
- 168.Lu J, Marmarou A, Lapane K, Turf E, Wilson L. A method for reducing misclassification in the extended Glasgow Outcome Score. J Neurotrauma. 2010;27:843–852. doi: 10.1089/neu.2010.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Beit-Yannai E, Zhang R, Trembovler V, Samuni A, Shohami E. Cerebroprotective effect of stable nitroxide radicals in closed head injury in the rat. Brain Res. 1996;717:22–28. doi: 10.1016/0006-8993(95)01492-6. [DOI] [PubMed] [Google Scholar]
- 170.Markgraf CG, et al. Injury severity and sensitivity to treatment after controlled cortical impact in rats. J Neurotrauma. 2001;18:175–186. doi: 10.1089/08977150150502604. [DOI] [PubMed] [Google Scholar]
- 171.Jiang Q, et al. MRI evaluation of axonal reorganization after bone marrow stromal cell treatment of traumatic brain injury. NMR Biomed. 2011;24:1119–1128. doi: 10.1002/nbm.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li L, et al. MRI Measurement of Angiogenesis and the Therapeutic Effect of Acute Marrow Stromal Cell Administration on Traumatic Brain Injury. J Neurotrauma. 2012 doi: 10.1038/jcbfm.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kou Z, et al. The role of advanced MR imaging findings as biomarkers of traumatic brain injury. J Head Trauma Rehabil. 2010;25:267–282. doi: 10.1097/HTR.0b013e3181e54793. [DOI] [PubMed] [Google Scholar]
- 174.Benson RR, et al. Global white matter analysis of diffusion tensor images is predictive of injury severity in traumatic brain injury. J Neurotrauma. 2007;24:446–459. doi: 10.1089/neu.2006.0153. [DOI] [PubMed] [Google Scholar]
- 175.Li L, et al. Transplantation of marrow stromal cells restores cerebral blood flow and reduces cerebral atrophy in rats with traumatic brain injury: in vivo MRI study. J Neurotrauma. 2011;28:535–545. doi: 10.1089/neu.2010.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Immonen RJ, Kharatishvili I, Grohn H, Pitkanen A, Grohn OH. Quantitative MRI predicts long-term structural and functional outcome after experimental traumatic brain injury. Neuroimage. 2009;45:1–9. doi: 10.1016/j.neuroimage.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 177.Ehrenreich H, et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med. 2002;8:495–505. [PMC free article] [PubMed] [Google Scholar]
- 178.Ehrenreich H, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- 179.Jia L, Chopp M, Zhang L, Lu M, Zhang Z. Erythropoietin in combination of tissue plasminogen activator exacerbates brain hemorrhage when treatment is initiated 6 hours after stroke. Stroke. 2010;41:2071–2076. doi: 10.1161/STROKEAHA.110.586198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zechariah A, ElAli A, Hermann DM. Combination of tissue-plasminogen activator with erythropoietin induces blood-brain barrier permeability, extracellular matrix disaggregation, and DNA fragmentation after focal cerebral ischemia in mice. Stroke. 2010;41:1008–1012. doi: 10.1161/STROKEAHA.109.574418. [DOI] [PubMed] [Google Scholar]
- 181.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. This article critically reviews strategies for developing experimental neuroprotective therapies and proposes criteria for improving the probability of successful clinical translation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Menon DK. Unique challenges in clinical trials in traumatic brain injury. Crit Care Med. 2009;37:S129–135. doi: 10.1097/CCM.0b013e3181921225. [DOI] [PubMed] [Google Scholar]
- 183.Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Narayan RK, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. This article provides very informative and insightful views on previous and future TBI trials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Stein DG, Wright DW. Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin Investig Drugs. 2010;19:847–857. doi: 10.1517/13543784.2010.489549. [DOI] [PubMed] [Google Scholar]
- 186.Xiong Y, Mahmood A, Chopp M. Emerging treatments for traumatic brain injury. Expert Opin Emerg Drugs. 2009;14:67–84. doi: 10.1517/14728210902769601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med. 2008;14:731–740. doi: 10.2119/2008-00050.Beauchamp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lu D, et al. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma. 2005;22:1011–1017. doi: 10.1089/neu.2005.22.1011. [DOI] [PubMed] [Google Scholar]
- 189.Lu D, et al. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004;21:21–32. doi: 10.1089/089771504772695913. [DOI] [PubMed] [Google Scholar]
- 190.Zhang Y, et al. Sprouting of corticospinal tract axons from the contralateral hemisphere into the denervated side of the spinal cord is associated with functional recovery in adult rat after traumatic brain injury and erythropoietin treatment. Brain Res. 2010;1353:249–257. doi: 10.1016/j.brainres.2010.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Oshima T, et al. TNF-alpha contributes to axonal sprouting and functional recovery following traumatic brain injury. Brain Res. 2009;1290:102–110. doi: 10.1016/j.brainres.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 192.Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurg Clin N Am. 2007;18:169–181. xi. doi: 10.1016/j.nec.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 193.Xiong Y, et al. Delayed administration of erythropoietin reducing hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome following traumatic brain injury in rats: comparison of treatment with single and triple dose. J Neurosurg. 2010;113:598–608. doi: 10.3171/2009.9.JNS09844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xiong Y, Mahmood A, Chopp M. Neurorestorative treatments for traumatic brain injury. Discov Med. 2010;10:434–442. [PMC free article] [PubMed] [Google Scholar]
- 195.Meng Y, et al. Dose-dependent neurorestorative effects of delayed treatment of traumatic brain injury with recombinant human erythropoietin in rats. J Neurosurg. 2011;115:550–560. doi: 10.3171/2011.3.JNS101721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Xiong Y, et al. Treatment of traumatic brain injury with thymosin beta(4) in rats. J Neurosurg. 2011;114:102–115. doi: 10.3171/2010.4.JNS10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mahmood A, Lu D, Qu C, Goussev A, Chopp M. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J Neurosurg. 2006;104:272–277. doi: 10.3171/jns.2006.104.2.272. [DOI] [PubMed] [Google Scholar]
- 198.Smith DH, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 199.Ning R, et al. Erythropoietin promotes neurovascular remodeling and long-term functional recovery in rats following traumatic brain injury. Brain Res. 2011;1384:140–150. doi: 10.1016/j.brainres.2011.01.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mahmood A, Lu D, Yi L, Chen JL, Chopp M. Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg. 2001;94:589–595. doi: 10.3171/jns.2001.94.4.0589. [DOI] [PubMed] [Google Scholar]
- 201.Xiong Y, et al. Delayed transplantation of human marrow stromal cell-seeded scaffolds increases transcallosal neural fiber length, angiogenesis, and hippocampal neuronal survival and improves functional outcome after traumatic brain injury in rats. Brain Res. 2009;1263:183–191. doi: 10.1016/j.brainres.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lindner MD, et al. Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. J Neurotrauma. 1998;15:199–216. doi: 10.1089/neu.1998.15.199. [DOI] [PubMed] [Google Scholar]
- 203.Shear DA, et al. Neural progenitor cell transplants promote long-term functional recovery after traumatic brain injury. Brain Res. 2004;1026:11–22. doi: 10.1016/j.brainres.2004.07.087. [DOI] [PubMed] [Google Scholar]
- 204.Salmond CH, Sahakian BJ. Cognitive outcome in traumatic brain injury survivors. Curr Opin Crit Care. 2005;11:111–116. doi: 10.1097/01.ccx.0000155358.31983.37. [DOI] [PubMed] [Google Scholar]
- 205.Sinson G, Voddi M, McIntosh TK. Nerve growth factor administration attenuates cognitive but not neurobehavioral motor dysfunction or hippocampal cell loss following fluid-percussion brain injury in rats. J Neurochem. 1995;65:2209–2216. doi: 10.1046/j.1471-4159.1995.65052209.x. [DOI] [PubMed] [Google Scholar]
- 206.Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- 207.Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: analysis of axonal injury and glial responses. J Neurotrauma. 2010;27:1997–2010. doi: 10.1089/neu.2009.1245. [DOI] [PubMed] [Google Scholar]
- 208.Robertson CL, et al. No long-term benefit from hypothermia after severe traumatic brain injury with secondary insult in rats. Crit Care Med. 2000;28:3218–3223. doi: 10.1097/00003246-200009000-00017. [DOI] [PubMed] [Google Scholar]
- 209.Stiefel MF, Tomita Y, Marmarou A. Secondary ischemia impairing the restoration of ion homeostasis following traumatic brain injury. J Neurosurg. 2005;103:707–714. doi: 10.3171/jns.2005.103.4.0707. [DOI] [PubMed] [Google Scholar]
- 210.Bramlett HM, Dietrich WD, Green EJ. Secondary hypoxia following moderate fluid percussion brain injury in rats exacerbates sensorimotor and cognitive deficits. J Neurotrauma. 1999;16:1035–1047. doi: 10.1089/neu.1999.16.1035. [DOI] [PubMed] [Google Scholar]
- 211.Matsushita Y, Bramlett HM, Kuluz JW, Alonso O, Dietrich WD. Delayed hemorrhagic hypotension exacerbates the hemodynamic and histopathologic consequences of traumatic brain injury in rats. J Cereb Blood Flow Metab. 2001;21:847–856. doi: 10.1097/00004647-200107000-00010. [DOI] [PubMed] [Google Scholar]
- 212.Sawauchi S, Marmarou A, Beaumont A, Signoretti S, Fukui S. Acute subdural hematoma associated with diffuse brain injury and hypoxemia in the rat: effect of surgical evacuation of the hematoma. J Neurotrauma. 2004;21:563–573. doi: 10.1089/089771504774129892. [DOI] [PubMed] [Google Scholar]
- 213.Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. doi: 10.1016/j.injury.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 214.Maegele M, et al. Differential immunoresponses following experimental traumatic brain injury, bone fracture and “two-hit”-combined neurotrauma. Inflamm Res. 2007;56:318–323. doi: 10.1007/s00011-007-6141-3. [DOI] [PubMed] [Google Scholar]
- 215.Potts MB, et al. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx. 2006;3:143–153. doi: 10.1016/j.nurx.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Giza CC, Kolb B, Harris NG, Asarnow RF, Prins ML. Hitting a moving target: Basic mechanisms of recovery from acquired developmental brain injury. Dev Neurorehabil. 2009;12:255–268. doi: 10.3109/17518420903087558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ibrahim NG, Ralston J, Smith C, Margulies SS. Physiological and pathological responses to head rotations in toddler piglets. J Neurotrauma. 2010;27:1021–1035. doi: 10.1089/neu.2009.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hoane MR, Lasley LA, Akstulewicz SL. Middle age increases tissue vulnerability and impairs sensorimotor and cognitive recovery following traumatic brain injury in the rat. Behav Brain Res. 2004;153:189–197. doi: 10.1016/j.bbr.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 219.Flanagan SR, Hibbard MR, Gordon WA. The impact of age on traumatic brain injury. Phys Med Rehabil Clin N Am. 2005;16:163–177. doi: 10.1016/j.pmr.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 220.Thompson HJ, McCormick WC, Kagan SH. Traumatic brain injury in older adults: epidemiology, outcomes, and future implications. J Am Geriatr Soc. 2006;54:1590–1595. doi: 10.1111/j.1532-5415.2006.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Depreitere B, Meyfroidt G, Roosen G, Ceuppens J, Grandas FG. Traumatic brain injury in the elderly: a significant phenomenon. Acta Neurochir Suppl. 2012;114:289–294. doi: 10.1007/978-3-7091-0956-4_56. [DOI] [PubMed] [Google Scholar]
- 222.Clifton GL, et al. Early induction of hypothermia for evacuated intracranial hematomas: a post hoc analysis of two clinical trials. J Neurosurg. 2012;117:714–720. doi: 10.3171/2012.6.JNS111690. [DOI] [PubMed] [Google Scholar]
- 223.Bullock MR, et al. Outcome measures for clinical trials in neurotrauma. Neurosurg Focus. 2002;13:ECP1. doi: 10.3171/foc.2002.13.1.6. [DOI] [PubMed] [Google Scholar]
- 224.Maas AI, et al. IMPACT recommendations for improving the design and analysis of clinical trials in moderate to severe traumatic brain injury. Neurotherapeutics. 2010;7:127–134. doi: 10.1016/j.nurt.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Saatman KE, et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Doppenberg EM, Choi SC, Bullock R. Clinical trials in traumatic brain injury: lessons for the future. J Neurosurg Anesthesiol. 2004;16:87–94. doi: 10.1097/00008506-200401000-00019. [DOI] [PubMed] [Google Scholar]