

Abstract
Directed evolution relies on iterative cycles of randomization and selection. The outcome of an artificial evolution experiment is crucially dependent on (i) the numbers of variants that can be screened and (ii) the quality of the assessment of each clone that forms the basis for selection. Compartmentalization of screening assays in water-in-oil emulsion droplets provides an opportunity to screen vast numbers of individual assays with good signal quality. Microfluidic systems have been developed to make and sort droplets, but the operator skill required precludes their ready implementation in nonspecialist settings. We now establish a protocol for the creation of monodisperse double-emulsion droplets in two steps in microfluidic devices with different surface characteristics (first hydrophobic, then hydrophilic). The resulting double-emulsion droplets are suitable for quantitative analysis and sorting in a commercial flow cytometer. The power of this approach is demonstrated in a series of enrichment experiments, culminating in the successful recovery of catalytically active clones from a sea of 1 000 000-fold as many low-activity variants. The modular workflow allows integration of additional steps: the encapsulated lysate assay reactions can be stopped by heat inactivation (enabling ready control of selection stringency), the droplet size can be contracted (to concentrate its contents), and storage (at −80 °C) is possible for discontinuous workflows. The control that can be thus exerted on screening conditions will facilitate exploitation of the potential of protein libraries compartmentalized in droplets in a straightforward protocol that can be readily implemented and used by protein engineers.
Directed evolution is arguably the dominant approach to alter and improve the activity and stability of protein biocatalysts.1−3 Experimentally, directed evolution relies upon iterative rounds of creation of novel protein variants by introduction of random mutations into the target gene and selection of individuals with desirable characteristics. The size of the gene libraries that can be obtained from these experiments easily exceeds the throughput of any screening system, implying that screening is the bottleneck in the exploration of sequence space. The ability to ease this bottleneck depends largely on the resources that are available—in typical academic research laboratories where screening is carried out on agar or microtiter plates, library sizes are limited to around 104 variants, whereas advanced robotic facilities can increase the throughput to the 106 range, although this increase in throughput comes at significant cost.4 As mutations that improve the function of a biocatalyst are rare (i.e., most mutations either do not change the activity or are deleterious), many mutants have to be screened to at least have a chance of finding desired “hits”. To improve the efficiency of screening efforts, the development of user-friendly, low-cost, and high-throughput screening techniques capable of screening larger libraries and selecting rare variants with improved activity are crucial.
Screening of an enzyme activity in individual intact cells, typically using cell survival for essential reactions, or flow cytometry (FACS; fluorescence-activated cell-sorting) if a fluorescent readout of activity is available, is a particularly efficient approach to library screening, but it also has particular restrictions. Specifically, the reaction substrate must be able to diffuse into the cells, and in the case of FACS the reaction product must be unable to leave the cell by diffusion or alternatively the product should be displayable on the cell surface to provide a fluorescent readout.5 As these conditions are not met for most reactions, alternative approaches are needed. One emerging technology that shows promise for screening libraries with remarkable efficiency is miniaturization of the directed evolution assay into artificial reaction compartments with cell-like dimensions. Use of water-in-oil microdroplets typically reduces assay volumes to the picoliter or femtoliter range, representing a reduction in sample volume of up to 100 000-fold (compared to robotic screening systems with volumes >0.1 μL per sample).6−12 The droplet boundary traps reaction products of multiple enzymatic turnovers within the compartment to provide a readout of reaction progress and also allows maintenance of the genotype–phenotype linkage.8 Maintenance of this linkage is necessary during selections to relate the functional trait of a protein (such as catalytic activity) to the nucleic acid sequence encoding it. Thus, the linkage gives access to the identity of a library member after selection.
The simplest approach to production of water-in-oil droplets makes use of bulk emulsion methods in which an aqueous phase and surfactant-bearing oil phase are vigorously mixed to produce an emulsion.13−15 This is a simple and rapid method of droplet formation, but it has the significant disadvantage of producing droplets that are highly polydisperse in size. The cubic dependence of volume on diameter—for example, a doubling of droplet diameter leads to an 8-fold increase in volume—leads to massive variations in enzyme concentration between droplets and potential for substrate limitation in smaller droplets.16 These factors preclude the use of polydisperse droplets for quantitative or comparative applications.
Microfluidic devices have been used to generate monodisperse water-in-oil emulsion droplets of picolitre volumes17,18 that can be filled with single species (i.e., cells4,19,20 or genes).6,21−24 Such droplets are typically made at a rate of 1–10 kHz, although recently it was shown that very small monodisperse droplets (diameter ∼4 μm) can be produced at frequencies of up to 1.3 MHz.25 Monodisperse emulsions have found broad utility in analytical applications such as digital PCR,7 single cell analysis,26 sizing of organelles or nanoparticles,27 or compound screening28 to name but a few.10,29
While straightforward interrogation of water-in-oil droplets by fluorescence microscopy or on microfluidic chips equipped with fluorescence detection allows their use in analytical applications, directed evolution experiments depend on the ability to sort positive droplets from the more numerous negative population. Microfluidic chips and rigs capable of measuring fluorescence and sorting of monodisperse water-in-oil droplets have been developed30,31 that perform at frequencies between 0.3 and 2 kHz, as demonstrated for yeast displaying a peroxidase,4 in vitro expressed proteins,22 or cell lysates (to screen for hydrolases).20 As impressive an advance as these droplet sorters are, they are technically challenging to set up, requiring knowledge of not just microfluidics, but also optics, electronics, and software coding to assemble and control the detection and electrosorting instrumentation that connects to the chip. Due to the complexity of these systems, they are unfortunately suited only to specialist laboratories; common use by a wider community would be facilitated if standard equipment rather than custom-made devices4,20 could be used.
One standard technique that could be used for sorting in place of a microfluidic droplet sorter is FACS. Modern FACS instruments are a mature technology that are user-friendly, high-throughput, widely available, and have low running costs. Furthermore, they have the advantage of being multiparametric and routinely have the ability to detect several different fluorophores in parallel.32 Unfortunately, FACS instruments are incompatible with nonaqueous suspensions, so to sort a water-in-oil emulsion, it is necessary to carry out a further emulsification to produce a water-in-oil-in-water double emulsion. The resulting sample, now dispersed in an aqueous phase, is amenable to FACS sorting.
Double emulsions have in fact been prepared and sorted by FACS previously; however, such attempts involved highly polydisperse bulk emulsions generated by vortexing with a tissue homogenizer or extruder.33−40 Indeed, the polydispersity is exacerbated by the combined effect of the two emulsification steps necessary to generate the double emulsion.34,37,41 Polydisperse emulsions give rise to a situation in which droplets carrying genes encoding proteins with the same activity can exhibit dramatically different assay outcomes depending on their size, although selections in polydisperse droplets may still be successful if the activity difference between positive hits and the rest of the library is very large. Some researchers have addressed the polydispersity problem by introducing external markers,42 such as coexpression of GFP,41 but the inclusion of markers complicates the biological setup and does not fully remedy the problem of varying catalyst concentration and the volume dependence of fluorescence intensity.
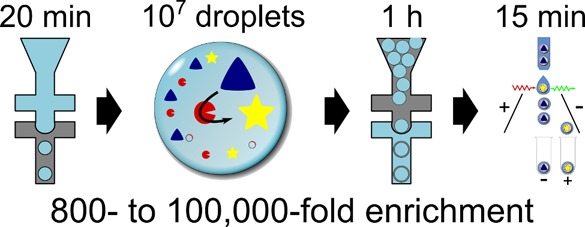
As an alternative to microfluidic droplet sorting, we introduce a straightforward method to convert a directed evolution assay previously conducted in water-in-oil emulsion droplets20 into double emulsions in two separate microfluidic devices at a rate of about 107 droplets per hour. The screening procedure consists of the encapsulation of single cells, their lysis, and enzymatic assay of the cell lysate and sorting of double emulsion droplets in a subsequent step with a standard cytometric sorter (Figure 1). The throughput and suitability of this method for directed evolution is demonstrated by enrichment experiments that recover hits from a sea of 106-fold as many alternative droplets.
Figure 1.

Generating double emulsions on two chips and selection of active biocatalysts. The workflow for one cycle of directed evolution consists of the following steps: (i) Gene libraries are generated from an enzyme-encoding plasmid. (ii) E. coli cells produce the biocatalyst of interest in liquid culture. (iii) In a first microfluidic device (with hydrophobic, fluorocarbon-coated channel walls), single cells are compartmentalized in droplets together with substrate and lysis agents. (iv) After cell lysis, substrate and cytoplasmically expressed enzyme react to yield a fluorescent product. (v) The reaction is allowed to proceed for a desired incubation period (in our case up to 24 h, but droplets are stable for at least one month). The reaction progress can be stopped simultaneously in all water-in-oil droplets by heat inactivation, so that the time required for double emulsion formation and sorting does not extend the assay period. (vi) Next, primary droplets are transformed into double emulsions in a second device with identical design to the one used in (iii) but with hydrophilic coating. (vii) Variants exhibiting the highest activity are identified and sorted in a standard flow cytometer. The recovered DNA can be used for further rounds of evolution without PCR amplification when a high-copy plasmid is used. The procedure takes little time: droplet formation (steps iii and vi) takes place at a frequency of 6–12 kHz, so that a library of 107 double emulsion droplets is produced in 90 min. Sorting 107 droplets at a rate of 10–15 kHz takes about 15 min.
Results and Discussion
Formation of Monodisperse Double Emulsion Droplets
Primary emulsion droplets were formed in a fluorocarbon-coated chip (Figure 2A) in which a surfactant-containing fluorous oil carrier phase meets an aqueous stream at a flow-focusing junction (Figure 2B; see Supporting Information for notes on the choice of oil phase). The aqueous stream is itself produced by mixing the flow from two separate channels (one carrying cell suspension, and the other containing lysis agents and enzyme substrate) immediately prior to droplet formation. This sequence leaves sufficient time for cell encapsulation in droplets prior to lysis, so that the genotype–phenotype linkage is maintained, and also controls the initiation of the enzyme assay.20 After formation, the stable droplets (Figure 2C) are stored temporarily in a syringe (Figure 2D) before injection into a second chip (Figure 2E) along with a surfactant-containing aqueous carrier phase to form a double emulsion. This second chip has a hydrophilic surface to promote wetting of the channel walls with the aqueous carrier phase and prevent droplet adherence to the walls.43 Immediately prior to double emulsion formation, the water-in-oil droplets are spaced out with fluorinated oil to prevent double occupation in double emulsion droplets (Figure 2F). These double emulsion droplets are monodisperse based on inspection of images of 150 droplets that show only a 2.5% standard deviation of the measured diameter. Double emulsion droplets thus obtained (Figure 2G) are stable for at least 1 year when stored submerged in aqueous buffer at room temperature, without any coalescence observed by microscopy. Further manipulation of the double emulsion droplets is possible: they maintain their structural integrity despite heating, freezing, or shrinking or expanding by osmosis, and they are amenable to sorting in a standard FACS instrument (described below).
Figure 2.
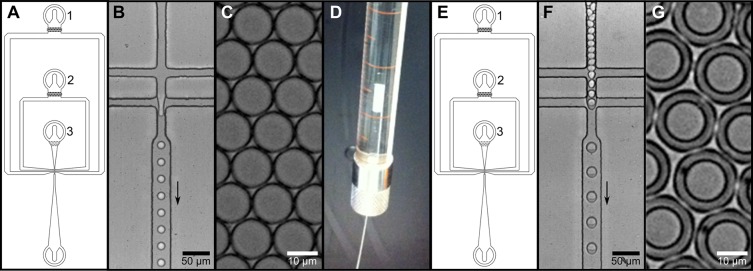
Formation of double emulsion droplets using a two-chip system. (A) Design of the device used in steps (iii) and (vi) in Figure 1. Fluorinated oil (inlet 1), lysis reagent/substrate (inlet 2), and cell suspension (inlet 3) are injected into a microfluidic flow-focusing device from syringes. (B) The aqueous samples (originating from inlets 2 and 3) are first mixed, then primary droplets are formed in the flow-focusing junction; the arrow indicates the direction of flow. (C) Image of the monodisperse water-in-oil droplets formed in this procedure. (D) The emulsion droplets are taken up in a syringe, overlaid with mineral oil, and cushioned with a bottom layer of fluorinated oil. The top mineral oil layer serves to reduce the dead volume of the tubing connecting the syringe and the microfluidic chip. (E) A device with identical design to the first emulsification device, but different surface coating is used for formation of double emulsions. Aqueous carrier phase, spacing oil, and water-in-oil emulsion are injected (inlets 1, 2, and 3, respectively) into a second, hydrophilic chip. (F) Image showing the production of water–oil–water double emulsion. (G) The double emulsion droplets produced in the previous steps are monodisperse. Movies showing single and double emulsion formation are available in the SI.
In contrast to previous double emulsion generation methods carried out in a single step on one microfluidic chip,18,43−45 the system described here uses two separate chips. Disassembly of the two emulsification steps considerably simplifies the process of double emulsion production. Double emulsion formation on a single chip requires careful adjustment of the flow rates for the sample components and both carrier phases to prevent single droplets being split or double emulsions with multiple inner droplets being produced. Use of two separate chips replaces the need for flow rate balancing with two straightforward emulsion procedures and also allows greater control over droplet size by enabling the use of chips with different channel widths to control the thickness of the oil layer of the double emulsion. Importantly, the fabrication of the chips used in this two-step method is more straightforward than production of chips able to produce double emulsion directly on a single chip. To prepare a single chip for double emulsion formation, different sections of the chip must be differently coated (either fluorophilically or hydrophilically) to ensure wetting with the appropriate carrier phase.43 During the application of these surface coatings, the complementary channels have to be blocked with air to maintain their surface properties. The two-chip system described here breaks down these single chip features into separate modules,46 facilitating its operation by researchers with less experience in microfluidics. The device manufacturing remains simple, in contrast to a much more complicated dual-layer device that has recently been used to create double emulsions by coaxial flow-focusing.47
Highly Efficient Identification of “Hits” Measured by Enrichment Analysis
The ability to isolate droplets containing an active enzyme that produces a fluorescent product was tested by measuring the enrichment of hits from an overwhelming majority of droplets containing an inactive variant. The model enzyme used for this experiment was a member of the alkaline phosphatase superfamily, the promiscuous arylsulfatase from Pseudomonas aeruginosa (PAS),48−50 that has previously been evolved on-chip to improve its promiscuous phosphonate hydrolase activity.20 PAS is a well-characterized sulfatase,51 which exhibits hydrolytic activity toward the substrate fluorescein disulfate and releases fluorescein to give a fluorescent readout of reaction progress. To mimic a library sorting experiment, expression of both the active wild-type enzyme and the low activity H211A variant (∼105-fold reduced kcat/KM; see Table S-1 for details) was performed in separate liquid cultures and cells were mixed prior to compartmentalization into droplets to produce a range of active to inactive ratios (Table 1). To minimize doubly occupied droplets, the number of compartmentalized cells was 10-fold lower than the number of droplets produced. According to a Poisson distribution,52 this ensured that ∼95% of occupied droplets contained a single cell. Ten minutes after compartmentalization, droplets enclosing the active PAS variant were highly fluorescent (indicating product formation), whereas empty droplets and droplets containing H211A showed a low level of background fluorescence arising from cell lysis prior to emulsion formation (Figure 3A). In the subsequent FACS sorting step, the highly fluorescent population was collected to obtain active variants (Figure 3B).
Table 1. Enrichment of Active Wild-Type Arylsulfatase (PAS) versus Low Activity Mutant H211Aa.
| percentage active cells in starting population | cells per droplet | enrichment (n-fold) |
|---|---|---|
| 0.1% | 0.1 | 800 |
| 0.01% | 0.1 | 2500 |
| 0.0001% | 1 | 100 000 |
The left column refers to the mixture of active versus low activity clones that was compared with the clones recovered after flow cytometric sorting that showed a positive plate screening assay (right column). Cells per droplet gives the average droplet occupancy for each sample. Note that droplet shrinking (see section below on osmotic droplet volume changes) was employed to maintain the throughput at the higher occupancy used in the third experiment. Enrichment was determined by dividing the percentage of positives after sorting by that before sorting.
Figure 3.

Enzymatic assays in double emulsions. Model enrichment experiments of E. coli-expressing active wild-type arylsulfatase (PAS) or its inactive mutant AZ0 (see Table S-1), shown here with a sample in which 1 in 1000 compartmentalized cells expresses the active wild-type enzyme. (A) Overlay of fluorescent and visual microscope images showing one droplet exhibiting enzymatic activity (the full-scale images are shown in Figure S-5). The surrounding droplets lack enzymatic activity, because they are either unoccupied (∼90% of the droplets) or contain the low activity enzyme variant (∼10%). (B) In a plot of fluorescence versus forward scatter (derived from gated FSC/SSC data, Figure S-8) two droplet populations are clearly distinguishable. The highly fluorescent population represents droplets with enzymatic activity. The fluorescent droplet displayed in A corresponds to the highly fluorescent population displayed in B.
The plasmid DNA recovered from the sorted double emulsions was transformed into E. coli cells, which were grown on agar plates overnight. The number of colonies obtained per sorted droplet reflected the efficiency of DNA recovery.20 Typically one to five transformants were obtained per sorted droplet (using the high copy plasmid pASK-IBA63b-plus with ∼1000 plasmids per cell), thus ensuring that DNA from the majority of the sorted droplets was recovered. Our results confirm the previously described finding that the transformation of one cell requires on average 400 plasmid molecules with our experimental setup.20 To determine enrichment as a quantitative measure of successful sorting, the clones obtained after sorting were rescreened on agar plates for sulfatase activity using an indolyl sulfate substrate, which forms a blue precipitate product in active colonies (Figure S-3). The enrichment was calculated as the percentage of positive colonies after sorting divided by the percentage of active cells before sorting. For example, the sample with an initial content of 0.1% active cells showed 80% active, blue variants after sorting, giving an enrichment of 800-fold (= 80/0.1) (see Table 1), whereas a sample with 0.01% active cells in the starting population was enriched 2500-fold.
Our enrichment compares favorably with previously published work in which sorting of model libraries in polydisperse double emulsions gave enrichment values of 40- to 290-fold.37,41 Although the details of the experimental protocol differ between the different reports, it is clear that the approach we present here surpasses previous efforts, with our sorted samples approaching purity. This success prompted us to test our system with a challenging sample containing just one positive hit per million cells.
Osmotic Droplet Volume Changes Enable Production of High Occupancy Droplets for Sorting of Extremely Rare Events
For enrichment of very rare events (less frequent than 1 in 100 000) in large libraries (>107 members), droplet occupancy must be increased to avoid the need to sort an overwhelming number of droplets. Increasing the cell occupancy is, however, challenging due to cell deposition at channel walls (and subsequent channel blockage) and because high density cell suspensions decrease the stability of single emulsion water-in-oil droplets such that widespread coalescence is observed within 1 h. These problems can be counteracted to some degree by producing larger droplets, which decreases the required density of the cell suspension and makes use of wider microfluidic channels that are less likely to get blocked during droplet formation. However, to ensure stable droplet break-off during FACS sorting, the particle size should not exceed one-third of the nozzle diameter. This means that a common flow cytometer setup with a 70 μm nozzle can only sort droplets with a diameter of less than 23 μm. We address this practical problem with a method that makes use of osmosis to shrink large droplets to a size suitable for FACS sorting (Figure S-6). For example, exposing double emulsion droplets to an external solution with an ionic strength 10-fold higher than that of the buffer inside the droplets resulted in a 10-fold decrease of the volume of the inner aqueous droplet (Figure S-6, Table S-2). This represents a 2.2-fold decrease in inner droplet diameter, with the diameter of the whole double emulsion droplet being decreased by 23%. The overall double emulsion shrinkage is less dramatic than that of the inner droplet as the volume of encapsulating oil remains constant, and so it forms a thicker layer as the droplet shrinks. Thus, while the size change of the inner droplet is directly dependent on the molarity of the outer solution, the overall size change depends on the thickness of the oil layer surrounding the inner droplet, with a thinner oil layer enabling a greater degree of shrinkage.
Applying this approach to decrease droplet size, enrichment of very rare variants was attempted. A sample that initially contained only 1 hit in 1 000 000 cells (0.0001% cells expressing active protein) was successfully enriched to yield 10% active variants after only one sorting round of droplets with an average occupancy of one cell per droplet, corresponding to an enrichment of 100 000-fold.
Control of Assay Duration
The ability to control the duration of an enzymatic assay is key to controlling the stringency and hence selection pressure of the assay, and it is an important issue to consider in any directed evolution experiment. To demonstrate that reaction times can be controlled at will in our screening system, we performed a discontinuous assay by compartmentalizing PAS enzyme solution (crude lysate of cells expressing wild-type PAS) along with substrate in droplets and heat inactivating the enzyme after chosen assay times. The assay development in these droplets was compared to a progress curve obtained using the same lysate in a plate reader. The use of cell lysate simplified the analysis by excluding the Poisson distribution that would complicate cell-based experiments. Lysate sample droplets were mixed with reference droplets (negative) containing substrate only. Inclusion of “negative” droplets provided a reference for each reading and also allowed monitoring of leakage of product from the assay droplets.54,55 The mixture of lysate-containing and reference droplets was heat-inactivated at the indicated time points, and after all samples were collected, they were independently transformed into double emulsions and analyzed by flow cytometry (Figure 4A).
Figure 4.
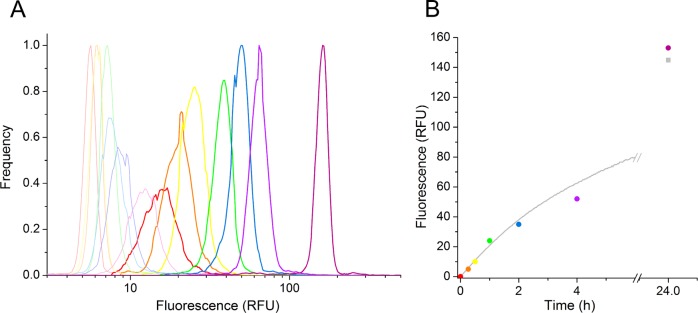
Introduction of time control by stopping the reaction at different time points. Diluted PAS-containing cell lysate was mixed with substrate on a microfluidic chip (Figure 2B) upon droplet formation. (A) FACS analysis of droplets with inactivated cell lysate. Heat inactivation was performed immediately after collection (t = 0, red), after 15 min (orange), 30 min (yellow), 1 h (green), 2 h (blue), 4 h (light violet), and 24 h (dark violet; end point measurement). The fluorescence distribution diagrams of heat-inactivated enzymatic reaction in droplets (left), measured 30 h after the reaction was started, show the background control droplets (with substrate only) in pale and droplets containing cell lysate in dark colors. (B) Overlay of normalized relative fluorescence versus time data obtained from FACS analysis (colored points corresponding to peaks in (A) and kinetics measurement in 96-well format (gray curve).
The FACS histogram verifies that clearly distinguishable positive and negative populations were still present after heat inactivation. In this lysate assay, the average coefficient of variation (standard deviation/mean fluorescence) of the positive peaks was 0.13, highlighting the monodispersity of the double emulsion generated using the two-chip method described here. A small amount of leakage from positive to reference droplets containing substrate alone (Figure 4A, pale curves) during heat inactivation at 95 °C for 5 min is reflected in the slightly increased fluorescence of negative peaks at the later time points. This leakage resulted in a 2-fold shift of the reference droplets over the course of the assay, whereas the positive droplets show more than a 10-fold increase in fluorescence.
In parallel with the droplet-based assay, a progress curve for the reaction carried out under the same conditions, but without encapsulation, was recorded in a microplate. The overlay of the normalized progress curve with normalized mean fluorescence values from FACS analysis shows identical reaction progress in 96-well plates and droplets (Figure 4B).
Until now, all screening efforts carried out on chip or in polydisperse emulsions have depended on the screening being carried out before the end point of the assay to allow valid comparison of samples, leading to considerable constraints in terms of user-friendliness of the system. The ability to introduce time control for stringent screening in a directed evolution experiment is an outstanding feature of the two-chip system. Its technical implementation by heat inactivation permits reactions to be stopped at any desired time point, permitting variation of assay duration, and hence stringency of the subsequent selection, to be altered at will. Furthermore, the ability to stop the assay allows the subsequent sample screening to be carried out when convenient for the experimenter, greatly improving the usability of this screening system.
Stopping Reactions in Discontinuous Workflow
The high stability of double emulsion droplets is the basis for their storage in frozen form at low temperatures so that they can be later analyzed or used in subsequent steps of more complex workflows. After being shock frozen in 20% glycerol, double emulsion droplets can be stored at −20 °C or −80 °C for at least 1 month without change. During freezing, the glycerol in the outer aqueous solution causes shrinking of double emulsions through osmosis. However, after sample thawing and rehydration by buffer exchange to a buffer isotonic with the buffer inside the droplets, the original size of the double emulsion is readily restored (Figure 5A). Flow cytometric analysis of a thawed and rehydrated sample (a mixture of high and low fluorescence droplets) showed that there was no significant change in fluorescence compared to an aliquot that was not frozen (Figure 5B). Although a small decrease in fluorescence of both high and low fluorescence droplets in the frozen sample is seen, the relative position of the populations does not change significantly, nor does the ratio of their mean fluorescence values. Thus, these data (Figure 5B) do not indicate significant small molecule transfer during the freezing–thawing procedure and demonstrate that sample identity is maintained after storage in a frozen state.
Figure 5.

Double emulsion droplets can be stored long-term after freezing (A) Shock freezing of droplets in 20% glycerol solution leads to shrinking of the inner aqueous droplet due to osmosis; however, rehydration in a solution of low molarity (150 mM) is readily achieved. Full-scale source images are shown in Figure S-7. (B) FACS analysis confirms that the relative fluorescence difference of droplets before (black) and after freezing (gray) does not change significantly. Peak centers are 4.5, 12.1, 1480, and 1750 RFU, giving positive/negative fluorescence ratios of 145 before freezing and 330 after.
This procedure contributes to the convenience of double emulsions for screening and also enables standardization of FACS measurements obtained at different times. The ability to store samples allows production of multiple samples over several days to weeks followed by their simultaneous analysis, saving time and enabling workflows that suit the experimenter. The creation of standard samples that can be used for adjustment of FACS parameters, such as the gain on each detection channel, facilitates the comparison of data collected during different FACS sessions.
Conclusions
We have presented here a simple, versatile, and user-friendly procedure for sorting of monodisperse double emulsion droplets in which the activity of an intracellularly expressed enzyme is assayed in cell lysate. The use of two chips for double emulsion generation (at 6–12 kHz) simplifies the monodisperse emulsion generation procedure, and offers flexibility in controlling droplet sizes and oil shell thickness as well as enabling manipulation of the sample, for example by thermal inactivation to stop the enzyme assay at chosen time point(s). A library of 107 double emulsion droplets is produced in 90 min. The sorting step (at a rate of 10–15 kHz) takes advantage of fluorescence-activated cell sorting (FACS), a well-established method enabling a throughput of >108 droplets per day.5 FACS sorters are widespread and readily used due to their ability to record numerous parameters simultaneously, such as relative volume, internal granularity, and fluorescence in multiple channels.
The method we describe here is broadly applicable, although the usual limits of droplet-based approaches still apply: enzymes that are to be evolved must yield a fluorescent readout (either directly as the product or via a coupled reaction) to be amenable to FACS. There are, however, a variety of fluorogenic probes that are readily available commercially. Furthermore, substrate and, particularly, the product, must not leak from the droplets within the assay time frame (i.e., for a period required to produce detectable fluorophore readout).
We also present a method for long-term storage of frozen double emulsions that can be reliably and reproducibly thawed and analyzed when convenient. Finally, the semipermeable nature of the oil shell used here allows double emulsions to be shrunk (or expanded) to a size convenient for sorting. This feature was exploited to allow the single-step enrichment by 100 000-fold of a sample containing just one positive cell per 1 000 000 negative cells. This is the greatest enrichment measured to date in a model selection and indicates that very rare events can be reliably retrieved using our experimental setup.
Hitherto, single water-in-oil emulsion droplets handled on-chip had been the only well-established format that combined high-precision assays in monodisperse compartments with ultrahigh throughput (>107) multistep processes.20 The ready access to monodisperse double emulsions, the degrees of freedom in manipulating droplet contents offline, and the extraordinary enrichment ratios achieved collectively suggest that our format for sorting of double emulsions can usefully complement the toolkit of in vitro compartmentalization. Further improvements to throughput will come through increasing the rate-limiting step of droplet production, possibly by either multiplexing55 or developing new and improved oils and/or surfactant combinations that allow higher flow rates. However, the current throughput already exceeds that of currently used screening systems (e.g., based on robotic liquid handling) at a fraction of their cost. For those embarking on compartmentalized experiments for the first time, the procedures outlined here may be the simplest entry point to harness the power of droplet microfluidics.
Acknowledgments
We thank Nigel Miller for constant help and advice with flow cytometric measurements. This research was funded by the Engineering and Physical Sciences Research Council (EPSRC), the Biotechnology and Biological Sciences Research Council (BBSRC), and the European Research Council (ERC). F.H. is an ERC Starting Investigator. A.Z. was supported by the BBSRC, the Cambridge Home and EU Scholarship Scheme (CHESS) and the EU Marie-Curie networks PhosChemRec and ENEFP. S.R.A.D., B.K., and M.F. were supported by postdoctoral EU Marie-Curie fellowships.
Supporting Information Available
Experimental details, practical notes, chip designs, images illustrating droplet manipulation, and movies showing formation of single and double emulsions are available as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- Lin H.; Cornish V. W. Angew. Chem., Int. Ed. 2002, 41, 4402–4425. [DOI] [PubMed] [Google Scholar]
- Turner N. J. Nat. Chem. Biol. 2009, 5, 567–573. [DOI] [PubMed] [Google Scholar]
- Agresti J. J.; Antipov E.; Abate A. R.; Ahn K.; Rowat A. C.; Baret J.-C.; Marquez M.; Klibanov A. M.; Griffiths A. D.; Weitz D. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Withers S. G. ChemBioChem 2009, 10, 2704–2715. [DOI] [PubMed] [Google Scholar]
- Tawfik D. S.; Griffiths A. D. Nat. Biotechnol. 1998, 16, 652–656. [DOI] [PubMed] [Google Scholar]
- Kiss M. M.; Ortoleva-Donnelly L.; Beer N. R.; Warner J.; Bailey C. G.; Colston B. W.; Rothberg J. M.; Link D. R.; Leamon J. H. Anal. Chem. 2008, 80, 8975–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaerli Y.; Hollfelder F. Mol. Biosyst. 2009, 5, 1392–1404. [DOI] [PubMed] [Google Scholar]
- Kintses B.; van Vliet L. D.; Devenish S. R. A.; Hollfelder F. Curr. Opin. Chem. Biol. 2010, 14, 548–555. [DOI] [PubMed] [Google Scholar]
- Theberge A. B.; Courtois F.; Schaerli Y.; Fischlechner M.; Abell C.; Hollfelder F.; Huck W. T. S. Angew. Chem., Int. Ed. 2010, 49, 5846–5868. [DOI] [PubMed] [Google Scholar]
- Guo M. T.; Rotem A.; Heyman J. A.; Weitz D. A. Lab. Chip 2012, 12, 2146. [DOI] [PubMed] [Google Scholar]
- Schaerli Y.; Kintses B.; Hollfelder F. In Protein Engineering Handbook; Lutz S.; Bornscheuer U. T., Eds.; Wiley VCH: Weinheim, 2012; Vol. 3, pp 73–89. [Google Scholar]
- Garti N.; Aserin A. Adv. Colloid Interface Sci. 1996, 65, 37–69. [Google Scholar]
- Seifriz W. J. Phys. Chem. 1925, 29, 587–600. [Google Scholar]
- Griffiths A. D.; Tawfik D. S. Trends Biotechnol. 2006, 24, 395–402. [DOI] [PubMed] [Google Scholar]
- Kaltenbach M.; Devenish S. R. A.; Hollfelder F. Lab Chip 2012, 12, 4185–4192. [DOI] [PubMed] [Google Scholar]
- Umbanhowar P. B.; Prasad V.; Weitz D. A. Langmuir 2000, 16, 347–351. [Google Scholar]
- Utada A. S.; Lorenceau E.; Link D. R.; Kaplan P. D.; Stone H. A.; Weitz D. A. Science 2005, 308, 537–541. [DOI] [PubMed] [Google Scholar]
- Huebner A.; Olguin L. F.; Bratton D.; Whyte G.; Huck W. T.; de Mello A. J.; Edel J. B.; Abell C.; Hollfelder F. Anal. Chem. 2008, 80, 3890–3896. [DOI] [PubMed] [Google Scholar]
- Kintses B.; Hein C.; Mohamed M. F.; Fischlechner M.; Courtois F.; Lainé C.; Hollfelder F. Chem. Biol. 2012, 19, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Cohen H. M.; Tawfik D. S.; Griffiths A. D. Protein Eng. Des. Sel. 2004, 17, 3–11. [DOI] [PubMed] [Google Scholar]
- Fallah-Araghi A.; Baret J.-C.; Ryckelynck M.; Griffiths A. D. Lab Chip 2012, 12, 882–891. [DOI] [PubMed] [Google Scholar]
- Mazutis L.; Baret J. C.; Treacy P.; Skhiri Y.; Araghi A. F.; Ryckelynck M.; Taly V.; Griffiths A. D. Lab Chip 2009, 9, 2902–2908. [DOI] [PubMed] [Google Scholar]
- Mazutis L.; Araghi A. F.; Miller O. J.; Baret J.-C.; Frenz L.; Janoshazi A.; Taly V.; Miller B. J.; Hutchison J. B.; Link D.; Griffiths A. D.; Ryckelynck M. Anal. Chem. 2009, 81, 4813–4821. [DOI] [PubMed] [Google Scholar]
- Shim J.; Ranasinghe R. T.; Smith C. A.; Ibrahim S. M.; Hollfelder F.; Huck W. T. S.; Klenerman D.; Abell C. ACS Nano 2013, 7, 5955–5964. [DOI] [PubMed] [Google Scholar]
- Shim J.; Olguin L. F.; Whyte G.; Scott D.; Babtie A.; Abell C.; Huck W. T. S.; Hollfelder F. J. Am. Chem. Soc. 2009, 131, 15251–15256. [DOI] [PubMed] [Google Scholar]
- Gadd J. C.; Kuyper C. L.; Fujimoto B. S.; Allen R. W.; Chiu D. T. Anal. Chem. 2008, 80, 3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen F.; van Vliet L.; Koprowski B. T.; Devenish S. R. A.; Fischlechner M.; Edel J. B.; Niu X.; deMello A. J.; Hollfelder F. Anal. Chem. 2013, 85, 4761–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner A.; Sharma S.; Srisa-Art M.; Hollfelder F.; Edel J. B. Lab Chip 2008, 8, 1244–1254. [DOI] [PubMed] [Google Scholar]
- Ahn K.; Kerbage C.; Hunt T. P.; Westervelt R. M.; Link D. R.; Weitz D. A. Appl. Phys. Lett. 2006, 88, 024104–024104. [Google Scholar]
- Baret J. C.; Miller O. J.; Taly V.; Ryckelynck M.; El-Harrak A.; Frenz L.; Rick C.; Samuels M. L.; Hutchison J. B.; Agresti J. J.; Link D. R.; Weitz D. A.; Griffiths A. D. Lab Chip 2009, 9, 1850–1858. [DOI] [PubMed] [Google Scholar]
- Shapiro H. M.Practical Flow Cytometry; 4th ed.; John Wiley & Sons Inc.: Hoboken, NJ, 2003. [Google Scholar]
- Aharoni A.; Thieme K.; Chiu C. P.; Buchini S.; Lairson L. L.; Chen H.; Strynadka N. C.; Wakarchuk W. W.; Withers S. G. Nat. Methods 2006, 3, 609–614. [DOI] [PubMed] [Google Scholar]
- Bernath K.; Hai M.; Mastrobattista E.; Griffiths A. D.; Magdassi S.; Tawfik D. S. Anal. Biochem. 2004, 325, 151–157. [DOI] [PubMed] [Google Scholar]
- Gupta R. D.; Goldsmith M.; Ashani Y.; Simo Y.; Mullokandov G.; Bar H.; Ben-David M.; Leader H.; Margalit R.; Silman I.; Sussman J. L.; Tawfik D. S. Nat. Chem. Biol. 2011, 7, 120–125. [DOI] [PubMed] [Google Scholar]
- Hai M.; Magdassi S. J. Controlled Release 2004, 96, 393–402. [DOI] [PubMed] [Google Scholar]
- Mastrobattista E.; Taly V.; Chanudet E.; Treacy P.; Kelly B. T.; Griffiths A. D. Chem. Biol. 2005, 12, 1291–1300. [DOI] [PubMed] [Google Scholar]
- Miller O. J.; Bernath K.; Agresti J. J.; Amitai G.; Kelly B. T.; Mastrobattista E.; Taly V.; Magdassi S.; Tawfik D. S.; Griffiths A. D. Nat. Methods 2006, 3, 561–570. [DOI] [PubMed] [Google Scholar]
- Griffiths A. D.; Tawfik D. S. EMBO J. 2003, 22, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodanovic R.; Ostafe R.; Blanusa M.; Schwaneberg U. Anal. Bioanal. Chem. 2012, 404, 1439–1447. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Amitai G.; Bernath K.; Magdassi S.; Tawfik D. S. Chem. Biol. 2005, 12, 1281–1289. [DOI] [PubMed] [Google Scholar]
- Levin I.; Aharoni A. Chem. Biol. 2012, 19, 929–931. [DOI] [PubMed] [Google Scholar]
- Bauer W.-A. C.; Fischlechner M.; Abell C.; Huck W. T. S. Lab. Chip 2010, 10, 1814–1819. [DOI] [PubMed] [Google Scholar]
- Nisisako T.; Okushima S.; Torii T. Soft Matter 2005, 1, 23–27. [DOI] [PubMed] [Google Scholar]
- Romanowsky M. B.; Abate A. R.; Rotem A.; Holtze C.; Weitz D. A. Lab Chip 2012, 12, 802–807. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Ho Y.-P.; Chiu Y.-L.; Chan H. F.; Chlebina B.; Schuhmann T.; You L.; Leong K. W. Biomaterials 2013, 34, 4564–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. W.; Abate A. R. Lab Chip 2013, 13, 4563–4572. [DOI] [PubMed] [Google Scholar]
- Olguin L. F.; Askew S. E.; O’Donoghue A. C.; Hollfelder F. J. Am. Chem. Soc. 2008, 130, 16547–16555. [DOI] [PubMed] [Google Scholar]
- Jonas S.; Hollfelder F. Pure Appl. Chem. 2009, 81, 731–742. [Google Scholar]
- Mohamed M. F.; Hollfelder F. Biochim. Biophys. Acta 2013, 1834, 417–420. [DOI] [PubMed] [Google Scholar]
- Boltes I.; Czapinska H.; Kahnert A.; von Bülow R.; Dierks T.; Schmidt B.; von Figura K.; Kertesz M. A.; Usón I. Structure 2001, 9, 483–491. [DOI] [PubMed] [Google Scholar]
- Koster S.; Angile F. E.; Duan H.; Agresti J. J.; Wintner A.; Schmitz C.; Rowat A. C.; Merten C. A.; Pisignano D.; Griffiths A. D.; Weitz D. A. Lab Chip 2008, 8, 1110–1115. [DOI] [PubMed] [Google Scholar]
- Courtois F.; Olguin L. F.; Whyte G.; Theberge A. B.; Huck W. T. S.; Hollfelder F.; Abell C. Anal. Chem. 2009, 81, 3008–3016. [DOI] [PubMed] [Google Scholar]
- Skhiri Y.; Gruner P.; Semin B.; Brosseau Q.; Pekin D.; Mazutis L.; Goust V.; Kleinschmidt F.; El Harrak A.; Hutchison J. B.; Mayot E.; Bartolo J.-F.; Griffiths A. D.; Taly V.; Baret J.-C. Soft Matter 2012, 8, 10618–10627. [Google Scholar]
- Paegel B. M.; Joyce G. F. Chem. Biol. 2010, 17, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.