

Abstract
Congenital disorders of glycosylation (CDGs) result from mutations in various N-glycosylation genes. The most common type, phosphomannomutase-2 (PMM2)-CDG (CDG-Ia), is due to deficient PMM2 (Man-6-P → Man-1-P). Many patients die from recurrent infections, but the mechanism is unknown. We found that glycosylation-deficient patient fibroblasts have less intercellular adhesion molecule-1 (ICAM-1), and because of its role in innate immune response, we hypothesized that its reduction might help explain recurrent infections in CDG patients. We, therefore, studied mice with mutations in Mpi encoding phosphomannose isomerase (Fru-6-P → Man-6-P), the cause of human MPI-CDG. We challenged MPI-deficient mice with an intraperitoneal injection of zymosan to induce an inflammatory response and found decreased neutrophil extravasation compared with control mice. Immunohistochemistry of mesenteries showed attenuated neutrophil egress, presumably due to poor ICAM-1 response to acute peritonitis. Since phosphomannose isomerase (MPI)-CDG patients and their cells improve glycosylation when given mannose, we provided MPI-deficient mice with mannose-supplemented water for 7 days. This restored ICAM-1 expression on mesenteric endothelial cells and enhanced transendothelial migration of neutrophils during acute inflammation. Attenuated inflammatory response in glycosylation-deficient mice may result from a failure to increase ICAM-1 on the vascular endothelial surface and may help explain recurrent infections in patients.
Keywords: CDG, ICAM-1, inflammatory response, mannose supplementation
Introduction
Congenital disorders of glycosylation (CDG) are caused by genetic defects in N-linked Glycosylation (Freeze and Sharma 2010). CDG affects most organ systems, and the patients exhibit highly variable motor and intellectual disabilities, seizures, developmental delay, hypoglycemia, clotting and digestion abnormalities (Freeze et al. 2012). Phosphomannomutase-2 (PMM2)-CDG, due to deficient PMM2, is the most common type of CDG and accounts for nearly 80% of all diagnosed cases (Haeuptle and Hennet 2009). Nearly 20% of patients die in infancy, often due to recurrent infection (Blank et al. 2006). Blank et al. (2006) proposed impaired innate immunity in those patients by analyzing neutrophil extravasation. They found diminished chemotaxis in patient neutrophils, but they did not observe aberrant expression of adhesion molecules such as Mac-1a, L-selectin, P-selectin glycoprotein ligand-1 (PSGL-1) and platelet endothelial cell adhesion molecule-1 (PECAM-1) in neutrophils from two cases. The importance of cell-surface glycoproteins in inflammatory response has been demonstrated in CDG-IIc, which is also known as leukocyte adhesion deficiency II (LAD II) (Etzioni et al. 1992). This disorder results from a rare genetic defect in terminal fucosylation of selectin ligands that are required for leukocyte rolling prior to their firm adhesion and extravasation. Incomplete ligand glycosylation leads to recurrent infections and leukocytosis (von Andrian et al. 1993; Hanna and Etzioni 2012). And in a few cases, providing dietary fucose supplements restores the ligand and normalizes rolling and extravasation.
These studies did not examine endothelial ligands such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) that lack the critical fucosylated ligands. Using quantitative proteomics, we previously found reduced ICAM-1 in N-glycosylation-deficient Chinese hamster ovary cells and in various subtypes of CDG type I patient fibroblasts (He et al. 2012). ICAM-1 is an inducible cell-surface glycoprotein and is a member of the immunoglobulin supergene family which is normally expressed at low levels on vascular endothelium, lymphocytes and macrophages (Otto et al. 2006). In response to infection, increased ICAM-1 cell-surface expression enhances cellular adhesive interactions by binding to its receptors, leukocyte function-associated antigen-1 and macrophage-1 (Mac-1), on leukocytes (Diamond et al. 1991). This facilitates transendothelial migration of leukocytes to the affected region and elimination of pathogens (Langer and Chavakis 2009). ICAM-1 is heavily glycosylated, and owing to its key role in innate immune response, we hypothesized that its reduced expression might explain recurrent infections in CDG patients. To test this hypothesis, we first analyzed ICAM-1 expression and functions in endothelial cells under glycosylation deficiency and inflammation conditions. Then, we investigated whether ICAM-1 expression could be induced in response to an inflammatory challenge in mice carrying mutations analogous to those in MPI-CDG patients.
Results
Tunicamycin blocked TNFα-induced ICAM-1 and VCAM-1 production
During acute inflammation, induction of adhesion molecules on the vascular endothelia facilitates their binding to the counter-receptors on the circulating leukocytes. Enhanced leukocyte adhesion is one of the key steps in molecular cascade events during innate immune response (Granger and Senchenkova 2010). Adhesion molecules, like ICAM-1 and VCAM-1 (Figure 1A), can be induced by TNF in vivo (Rothlein et al. 1988; Min et al. 2005), in which pronounced increases of fully glycosylated (top band) and partially glycosylated (multiple bands below the top one) ICAM-1 and VCAM-1 were observed. Tunicamycin blocks the synthesis of N-glycans by inhibiting enzyme GlcNAc phosphotransferase, which catalyzes the transfer of N-actelyglucosamine-1-phosphate from UDP-N-acetylglucosamine to dolichol phosphate in the first step of N-glycosylation. We treated human umbilical vein endothelial cells (HUVECs) with 0.1 μg/mL tunicamycin for 24 h followed by TNFα treatment for 6 h. We found that tunicamycin treatment led to drastic reduction in both fully glycosylated ICAM-1 and VCAM-1, and observed poorly glycosylated ICAM-1 and VCAM-1 in TNFα-treated HUVECs, comparable with TNFα treated samples with Peptide -N-Glycosidase F digestion (Figure 1A). This indicates that N-glycosylation deficiency can prevent ICAM-1 and VCAM-1 induction following TNFα-induced inflammation in HUVECs. We further tested whether the failure to induce adhesion molecule production in these cells diminished attachment to human monocyte cell line, THP-1. As shown in Figure 1B, tunicamycin completely abolished TNFα's enhancement of monocyte adhesion, indicating that glycosylation insufficiency impaired leukocytes adhesion to the endothelial cells with TNFα challenge.
Fig. 1.

Tunicamycin prevented TNF-triggered ICAM-1 increase and monocyte attachment. (A) HUVECs were treated with dimethyl sulfoxide (DMSO) or 0.1 μg/mL tunicamycin for 24 h, and then treated with or w/o 2 ng/mL TNFα for 6 h. Total proteins were harvested and TNF-treated lysates were also treated with Peptide -N-Glycosidase F to remove the N-glycans. Tubulin is used as protein loading marker. (B) HUVECs grow into 100% confluence in 96-well plate followed by treating with DMSO or 0.2 μg/mL tunicamycin for 16 h, and then were treated with or w/o TNF (2 ng/mL) for 6 h. The Calcein labeled THP-1 cells were mounted to the HUVEC monolayer. The adhesion percentage is calculated as the fluorescence of adherent cells divided by the total fluorescence of cells added to well. Each error bar in the histogram represents standard deviation (SD) of three independent assays.
Knockdown of PMM2 impeded TNFα-induced ICAM-1 up-regulation
Next, we tested whether the effects of tunicamycin on ICAM-1 and VCAM-1 expression could be mimicked by knocking down PMM2 which converts mannose-6-phosphate (Man-6-P) into mannose-1-phosphate (Man-1-P) (Supplementary data, Figure S1). In HUVECs, a PMM2 knockdown of 70% decreased TNFα-stimulated ICAM-1 induction by 25% (P = 0.05) (Figure 2A), but it had no effect on VCAM-1 expression (Figure 2B), indicating a preferential impairment of ICAM-1 response to inflammation.
Fig. 2.

siRNA knockdown of PMM2 impeded TNF-induced ICAM-1 (A) not vascular cell adhesion molecule 1 (VCAM-1) (B) up-regulation in HUVECs. HUVECs were transfected with scrambled or PMM2 siRNA for 72 h, and then treated with or w/o 2 ng/mL TNF for 6 h. Protein Coomassie blue stain serves as loading control. The lower graphs are plotted based on the calculation of western blot data. Gray intensity of ICAM-1, VCAM-1 and PMM2 bands was divided by that of the loading control. Each assay was repeated at least twice, and the values are the average of 3 experimental data. Each error bar in the histogram represents SD of three independent assays.
MPI-deficient mice show decreased leukocyte extravasation in response to acute peritonitis
CDG-Ib (MPI-CDG) is caused by mutations in MPI, encoding phosphomannose isomerase, which converts fructose-6-phosphate (Fru-6-P) into Man-6-P (Supplementary data, Figure S1). Like other type I CDGs, we found dramatic reduction of ICAM-1 in CDG-Ib patient fibroblasts (He et al. 2012). Since Mpi knockout (KO) causes embryonic lethality in mice, we used a line carrying a hypomorphic allele of Mpi to examine their response to an inflammatory challenge. This line carries a homozygous Y255C point mutation in Mpi, resulting in ∼12% enzyme activity in MPI, comparable with patients carrying this mutation (Sharma et al. 2014). We adopted a well-established acute peritonitis mouse model (Srikrishna et al. 2001) and included an ICAM1 KO line as a positive control. Four hours after challenge with zymosan injection, we calculated the number of neutrophils in peritoneal cavity as described in the Materials and methods section. As shown in Figure 3A, in phosphate buffered saline (PBS) treatment, nearly 5 × 104 neutrophils on average were found in peritoneal cavity in different groups of mice. Zymosan induced a surge of neutrophil exudation in all mice, but Mpi-KI mice exhibited more than a 2-fold decrease of neutrophil extravasation compared with wild-type (WT) mice (1.5 vs. 3.3 × 106, P = 0.007) comparable with ICAM1 KO mice (1.3 × 106). At 16 h, zymosan also induced monocyte extravasation and Mpi-mutant mice showed less monocyte exudation as well (Supplementary data, Figure S2A).
Fig. 3.
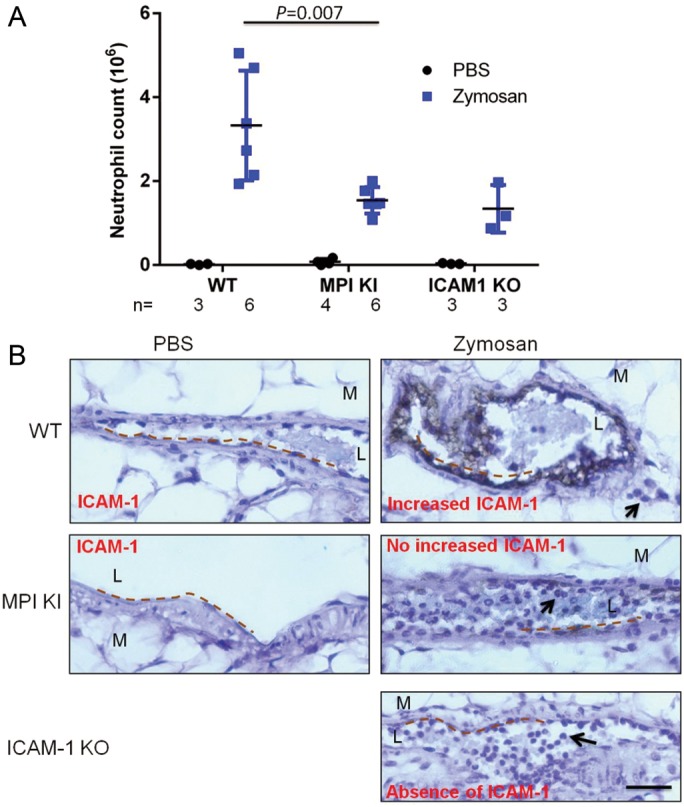
MPI deficient mice exhibited decrease of leukocytes extravasation in response to acute peritonitis. (A) Mice aged 6–10 weeks were i.p injected with zymosan or PBS, and the peritoneal lavage fluid was collected 4 h later. The Gr-1 positive cells by fluorescence-activated cell sorting analysis were defined as neutrophils. The total cell counts were calculated by TC20 automated cell counter. The absolute neutrophil number was calculated as total cell count × neutrophil%. (B) After zymosan injection for 4 h, the mesenteries were collected and fixed. The paraffinized slides were immunohistochemically stained by anti-ICAM-1 antibody, and ABC kit and DAB substrate were used to detect positive staining followed by hematoxylin counterstain. The images were captured with a Nikon Eclipse E800 microscope (Tokyo, Japan), using a Nikon ×40/0.75 air objective lens and a Nikon Digital Camera DXM2000. Images were processed using the Nikon ACT-1 (version 2.7) software and ImageJ 1.41 (NIH). The dotted lines indicate the vascular endothelial surface. The black arrows show the transendothelial leukocytes in wild-type and accumulated leukocytes within the vascular lumen in Mpi knockin (KI) and ICAM1 KO mice in acute inflammation. L, lumen; M, mesentery. The black bar represents 50 µm. Images were obtained from 1 of 3–6 individual mice, which gave representative results.
To determine whether the reduction of neutrophil exudation in Mpi-KI mice correlates with an ICAM-1 deficiency, we immunohistochemically stained mesentery tissues with anti-ICAM-1 antibody. We found that ICAM-1 expression on the endothelial surface in venules was markedly induced by zymosan in WT mice, whereas the ICAM-1 induction was prevented in Mpi-KI mice and totally absent in ICAM1 KO mice (Figure 3B, dotted line). However, we did not observe clear induction of VCAM-1 by inflammation in all mice (Supplementary data, Figure S3). We also found more of ICAM-1 staining in mice with zymosan treatment after 16 h (Supplementary data, Figure S2B). Of note, owing to leukocytes exudation (Figure 3B, black arrow in right upper panel), fewer cells were observed in inflamed vessels in WT mice. In contrast, considerably more leukocytes accumulated in venular lumen in Mpi and ICAM1-mutant mice following zymosan challenge (Figure 3B, black arrow in right middle and lower panels). These data indicate that impaired inflammatory response in MPI hypomorphic mice may be partially due to the failure of ICAM-1 induction caused by glycosylation defect.
Mannose supplementation enhanced neutrophils exudation in MPI-KI mice
Mannose supplementation improves glycosylation in MPI-deficient patients and their cells; it also restores ICAM-1 expression (He et al. 2012). In pilot experiments, we tried providing mice with 1–5 mM mannose in PBS by intravenous or intraperitoneal injection. Even though we could detect increase of serum mannose by 30–50% 3–8 h after injection, we did not detect ICAM-1 induction on endothelial surface and increase of neutrophil extravasation with zymosan challenge (data not shown). We then provided mice with 5% mannose in their drinking water for 7 days, providing ∼200 mg/day. Mannose supplementation increased serum mannose level by 20–40% (Supplementary data, Figure S7). After 7 days, mice were treated with zymosan for 4 h. This increased ICAM-1 expression on mesenteric endothelial cells of zymosan-challenged mice (Figure 4A) and enhanced transendothelial migration of neutrophils compared with untreated controls (P = 0.01) (Figure 4B). This result suggests that mannose supplementation may partially “fix” impaired inflammatory response in MPI-deficient mice presumably by restoration of ICAM-1 expression in vasculature.
Fig. 4.
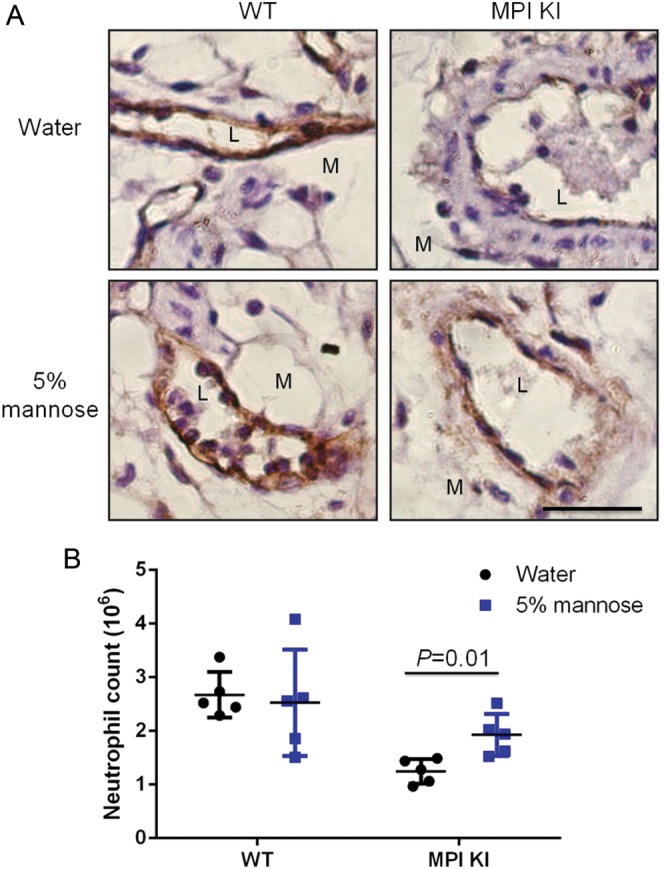
Mannose supplementation enhanced neutrophils exudation in Mpi-KI mice. Mice were fed 5% mannose water or water for 7 days prior to zymosan challenge. Mesenteries and lavaged cells were collected posterior to zymosan injection for 4 h. ICAM-1 protein level (A) and exudated neutrophils (B) were analyzed in a manner similar to that shown in Figure 3. Each treatment group included five mice.
Discussion
Leukocyte transmigration to the site of infection or injury is the first line of host defense against harmful pathogens. This innate immune response is characterized by increased interactions between circulating leukocytes and the endothelium, which consists of a well-defined and regulated multistep cascade involving the sequential steps of rolling, firm adhesion and ultimately transmigration from blood vessel to the affected region (Langer and Chavakis 2009; Granger and Senchenkova 2010). An impaired host response at any of these steps may result in increased susceptibility to infections and severe host tissue damage and can eventually lead to life-threatening complications such as multiple organ failure (Munoz et al. 1997; Cohen 2002; Wang et al. 2013). In CDG patients, especially PMM2-CDG, recurrent infection is the major cause of infant death (Blank et al. 2006), which may be caused by immunological dysfunction, but this is unproven.
We found reduction of ICAM-1 in fibroblasts from many types of CDG patients (He et al. 2012). ICAM-1 is one of the key adhesion molecules on the endothelium and is required for firm adhesion of leukocytes by binding to its receptor, CD18. Mice lacking this receptor have significant morbidity and mortality in response to bacterial infections (Tan et al. 1995; Scharffetter-Kochanek et al. 1998). Based on these facts, we reasoned that ICAM-1 reduction in CDG patients or glycosylation-deficient mice might impair innate immune response and consequently lead to recurrent infections.
Proinflammatory cytokines, such as TNFα, can stimulate inflammatory response by enhancing leukocyte adhesion to the endothelium. A widely accepted mechanism is induction of endothelial adhesion molecule expression, such as ICAM-1 (Bevilacqua et al. 1985; Sawa et al. 2007). We consistently observed dramatic induction of ICAM-1 expression by TNFα in HUVECs (Figure 1). Tunicamycin inhibits N-glycosylation at the initial step of the dolichol-linked precursor assembly. Chen et al. (2012) demonstrated that tunicamycin prevents TNFα-induced ICAM-1 expression in human retinal pigment epithelial cells. We also confirmed this in HUVECs (Figure 1). Induction of ICAM-1 may contribute to, but does not fully account for, TNFα-induced vascular leakage (Clark et al. 2007). ICAM-1 deficiency reduced neutrophil transmigration by 70% (Schnoor et al. 2011). Endothelial N-glycosylated VCAM-1 is recognized by β-1 integrin receptors on leukocytes that assists ICAM-1 and leukocyte function-associated antigen-1 in the process of rolling and firm adhesion (Elices et al. 1990). We observed similar TNFα and tunicamycin effects on VCAM-1 as ICAM-1 (Figure 1) indicating that glycosylation deficiency may impact on the response of multiple adhesion glycoproteins to inflammation in HUVECs. PMM2 knockdown prevented ICAM-1, but not VCAM-1, induction by TNF (Figure 2). Unexpectedly, we observed elevated levels of ICAM-1 protein using various transfection conditions with either lipid on nonlipid transfection reagents (data not shown). This likely explains why there is still a large amount of ICAM-1 present when HUVECs transfected (Figure 2A, Lanes 1 and 3), even without TNFα treatment, and this is not present in VCAM-1 (Figure 2B, Lanes 1 and 3). This reagent-specific effect on ICAM-1 may partially mask or dampen the effects of PMM2 knockdown on ICAM-1, and may explain why ICAM-1 reduction was less than that seen with tunicamycin and TNFα treatment. These cell model-derived data indicate that glycosylation can specifically cause failure of ICAM-1 response to inflammation.
Inflammatory and immune responses are impaired in mice deficient in ICAM-1 (Sligh et al. 1993; Tan et al. 1995). We consistently found much less leukocyte exudation in ICAM1 KO mice with acute peritonitis comparable with MPI-deficient mice (Figure 3A). We ascribed this reduction to the failure of ICAM-1 response to zymosan as demonstrated in Figure 3B, since we did not find VCAM-1 alterations with this treatment (Supplementary data, Figure S3). CD11b is one of the key components in ICAM-1's receptor Mac-1 and is also N-glycosylated, but we did not observe its expression change on neutrophils from peripheral blood in Mpi-mutant mice (Supplementary data, Figure S4A). These data indicate that the impaired innate immunity in Mpi-mutant mice may be primarily caused by ICAM-1 deficiency. Winning et al. (2010) reported that monocytic ICAM-1 may also play an important role in the adhesion of monocytes to the endothelium under hypoxia. And ICAM–ICAM interaction has been proposed in T cell–T cell synaptic interactions during the differentiation of protective CD8+ T cells (Gerard et al. 2013) and for the cross-talk of dendritic cells and B lymphocytes during the formation of germinal centers (Springer 1990). We did observe ICAM-1 reduction with vehicle treatment, yet found normal induction of ICAM-1 on neutrophils in Mpi-mutant mice (Supplementary data, Figure S4B) indicating that ICAM-1 on leukocytes in our model may not play important roles in extravasation. Considering the potential role of adhesion molecules in neutrophil mobilization from the bone marrow, we flushed out bone marrow cells (BMCs) after inflammation challenge and clearly detected a decreased proportion of neutrophils. However, we did not find more accumulation of neutrophils in Mpi-mutant mice compared with controls (Supplementary data, Figure S5). Neither did we observe any abnormalities of peripheral WBC proportions and numbers in Mpi-mutant mice under physiological and inflammatory conditions (Supplementary data, Figure S6). Taken together, the major immunological dysfunction in Mpi-mutant mice is diminished inflammatory response caused by ICAM-1 deficiency in vascular endothelia.
Mannose supplementation could improve glycosylation and is used to treat MPI-CDG (CDG-Ib) patients (Freeze and Sharma 2010; He et al. 2012). We found a partial restoration of ICAM-1 and inflammatory response when Mpi-mutant mice are given mannose, suggesting that ICAM-1 may serve as potential therapeutic target in management of recurrent infections in CDG patients. Besides VCAM-1 and CD11b, other adhesion molecules, like selectins and integrins, are also involved in inflammation response. These proteins are N-glycosylated and might be deficient in vivo when glycosylation is inhibited (Larsen et al. 1992; Pahlsson et al. 1995) and this could contribute to the impaired inflammatory response as well (Tan et al. 1995; Munoz et al. 1997; Scharffetter-Kochanek et al. 1998). The fact that ICAM1 KO mice phenocopy the quantitative responses of leukocyte egress strongly suggests that ICAM-1 plays a predominant role in MPI-deficient mice. Systematic investigation of the involvement of alternative adhesion molecules would be one of our future directions. Further, it will be competent to assess more patient-relevant inflammatory challenges, like bacterial infection, in CDG mice and evaluate mannose therapeutic effects in those models.
To summarize, glycosylation deficiency caused ICAM-1 reduction contributes to diminished inflammatory response both in HUVECs and in glycosylation-deficient mice, which improves on mannose treatment.
Materials and methods
Most of the reagents were purchased from Sigma-Aldrich (St. Louis, MO). Endothelial cell growth medium was purchased from Cell Applications, Inc. (San Diego, CA). RPMI 1640 medium was obtained from Mediatech, Inc. (Manassas, VA). Fetal bovine serum (FBS) was obtained from Hyclone Laboratories (Logan, UT). Recombinant human TNFα was purchased from Cell Sciences (Canton, MA). Vybrant™ Cell Adhesion Assay Kit, Lipofectamine™ RNAiMAX and NuPAGE Novex Bis-Tris Mini Gels were purchased from Life Technologies (Carlsbad, CA). PMM2 siRNA Smartpool (siGENOME) was designed and synthesized by Dharmacon (Thermo Scientific, Pittsburgh, PA). Scrambled siRNA (NC1) was purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Goat Anti-mouse ICAM-1 antibody was purchased from R&D Systems (Minneapolis, MN). Fluorescein isothiocyanate (FITC) anti-mouse CD54 (ICAM-1) antibody was purchased from Biolegend (San Diego, CA). PE rat anti-mouse Gr-1, PerCP Cy5.5-conjugated anti-mouse CD11b (macrophage 1) antibody and eight chamber tissue culture treated glass slide was purchased from Becton Dickinson Labware (Franklin Lakes, NJ). Anti-VCAM-1 antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Anti-alpha-tubulin antibody was obtained from Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). Horseradish peroxidase-conjugated second antibodies were purchased from Jackson Immunoresearch Lab (West Grove, PA). Immun-Star WesternC Kit was purchased from Bio-Rad (Hercules, CA). Polyvinylidene fluoride membrane was purchased from Whatman (Dassel, Germany). Vectastain ABC kit and 3,3′-diaminobenzidine (DAB) Peroxidase Substrate Kit were purchased from Vector labs (Burlingame, CA). ACK Lysing buffer was purchased from KD Medical Inc (Columbia, MD).
Cell culture
THP-1 monocyte cell line was maintained in RPMI 1640 medium supplemented with 25 mM HEPES, 50 µM beta-mercaptoethanol and 10% FBS. HUVECs were cultured in Endothelial cell growth medium. All cells were maintained in recommended media at 37°C and 5% CO2.
Mice- Institutional Animal Care and Use Committee approved all the animal studies. MpiY255C/Y255C (Mpi-KI) mice were created in C57BL/6 strain (Sharma et al. 2014). OT-II Thy ICAM1 KO mice on a C57BL/6 background were kind gifts from Dr. Bas Baaten in the Institute. All mutations in mice were confirmed by PCR genotyping.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot
As described by Jones et al. (2012). Immun-Star WesternC Kit was used as enhanced chemiluminescence substrate. The images were captured by Bio-Rad CCD imaging system. The gray intensity of bands was calculated by the Image Lab 3.0.1 (Beta 2) software designed by Bio-Rad.
Cell adhesion assay
For the procedures and calculation, refer to the Vybrant™ Cell Adhesion Assay Kit manufacturer protocol. The fluorescence was read out by Flx800 Microplate Fluorescence Reader (Bio-Tek Instruments, Inc., Winooski, VT)
siRNA knockdown of PMM2 and TNFα treatment
Lipofectamine™ RNAiMAX reagents were used to deliver 20 nM scrambled and PMM2 siRNA Smartpool into the HUVECs according to the manufacturer's protocol. Seventy-two hours after transfection, cells were treated with 2 ng/mL TNFα for 6 h.
Zymosan-induced acute peritonitis
Mice aged 6–10 weeks were intraperitoneally injected with 1 mg zymosan in 500 µL PBS. Four or sixteen hours later, 200 µL of blood was used to measure 18-parameter blood count (CBC) by VetScan HMII Hematology System (Abaxis, Union City, CA). After euthanasia by CO2, peritoneal lavage was collected as described by Srikrishna et al. (2001). BMCs were collected by flushing tibia and femur using Hank's Balanced Salt Solution buffer. Whole blood, lavaged cells and BMCs were treated with ACK lysis buffer to remove red blood cells. The lavaged cells were counted with TC20™ Automated Cell Counter (Bio-Rad,). Mice were given 5% (w/v) mannose or regular drinking water for 7 days followed by zymosan treatment.
Flow cytometry analysis
The cells were immunostained with 1:500 diluted PE-conjugated anti-mouse Gr-1, PerCP Cy5.5-conjugated anti-mouse CD11b antibody and 1:100 diluted FITC anti-mouse CD54 (ICAM-1) antibody. Flow cytometry was performed with a BD Biosciences FACSCalibur analyzer (San Jose, CA) and analyzed with the Cell Quest Pro software. Gr-1 positive and Gr-1low/CD11bhigh cells were defined as neutrophils and monocytes, respectively. The number of neutrophils/monocyte showing transendothelial migration was calculated as lavaged cell count by TC20 multiplied by the percentage of neutrophil/monocyte.
Immunohistochemical staining
The intestine mesenteries were sectioned from these mice and were fixed in 10% zinc formalin followed by embedded in paraffin. The slides were deparaffinized and blocked with 3% hydrogen peroxide/methanol 5 min. Vectastain ABC kit was used for the following immunohistochemical (IHC) staining. The sections were incubated for 16 h at 4°C with the anti-ICAM-1 (1:200 diluted) and VCAM-1 (1:100 diluted) primary antibodies. The peroxidase activity was produced by immersion in DAB.
Measurement of serum mannose
Peripheral blood was sampled after feeding mice with 5% mannose in water for 7 days. Ten micro litres of serum and mannose standards were added to 50 µL 50 mg/mL hydroxylamine hydrochloride in 1-methyl-imidazole for 30 min at 65°C. Then the mixture was derivatized by first adding 100 µL acetic anhydride, and after vortexing, add 100 µL chloroform and then 200 µL water. Most of the top aqueous layer was removed after vortexing and short spin. Water extraction was repeated, and 100 µL chloroform extract containing sugars was isolated and dried by speed vacuum, followed by resuspension in 100 µL chloroform for gas chromatography/mass spectrometry analysis (Sharma et al. 2011). A fragmentation series of m/z 217, 242 and 314 represents hexoses. The quantitation of mannose based on the intensity count of each fragment.
Statistics
Unpaired t-test was used to compare means between two groups, and P < 0.05 was considered statistically significant. Microsoft Excel 2007 and GraphPad Prism 6 software programs were used to plot the data.
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Funding
This work was supported by the National Institutes of Health (grant number R01-DK55615) and the Rocket Fund.
Conflict of interest
None declared.
Abbreviations
BMCs, bone marrow cells; CDGs, congenital disorders of glycosylation; DAB, 3,3′-diaminobenzidine; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; Fru-6-P, fructose-6-phosphate; HUVECs, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molecule-1; IHC, immunohistochemical; LAD II, leukocyte adhesion deficiency; Man-1-P, mannose-1-phosphate; Man-6-P, mannose-6-phosphate; MPI, phosphomannose isomerase; PBS, phosphate buffered saline; PMM2, phosphomannomutase-2; PSGL, P-selectin glycoprotein ligand; SD, standard deviation; TNFα, tumor necrosis factor alpha; VCAM-1, vascular cell adhesion molecule-1.
Supplementary Material
Acknowledgements
We acknowledge the technical support provide by Buddy Charbono in our animal facility and Guillermina Garcia and Robbin Newlin in histology core. We appreciate Dr. Bas Baaten of this Institute for providing ICAM1 KO mice. We also acknowledge the technical support provided by Jamie Smolin, Neal Nathan, Larry Nguyen and Joseph Russell in our laboratory.
References
- Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA., Jr Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. 1985;76:2003–2011. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank C, Smith LA, Hammer DA, Fehrenbach M, Delisser HM, Perez E, Sullivan KE. Recurrent infections and immunological dysfunction in congenital disorder of glycosylation Ia (CDG Ia) J Inherit Metab Dis. 2006;29:592. doi: 10.1007/s10545-006-0275-2. [DOI] [PubMed] [Google Scholar]
- Chen CL, Liang CM, Chen YH, Tai MC, Lu DW, Chen JT. Glucosamine modulates TNF-alpha-induced ICAM-1 expression and function through O-linked and N-linked glycosylation in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2012;53:2281–2291. doi: 10.1167/iovs.11-9291. [DOI] [PubMed] [Google Scholar]
- Clark PR, Manes TD, Pober JS, Kluger MS. Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J Invest Dermatol. 2007;127:762–774. doi: 10.1038/sj.jid.5700670. [DOI] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 1990;60:577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, Gershoni-Baruch R. Brief report: Recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. 1992;327:1789–1792. doi: 10.1056/NEJM199212173272505. [DOI] [PubMed] [Google Scholar]
- Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11:453–466. doi: 10.1016/S1474-4422(12)70040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH, Sharma V. Metabolic manipulation of glycosylation disorders in humans and animal models. Semin Cell Dev Biol. 2010;21:655–662. doi: 10.1016/j.semcdb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard A, Khan O, Beemiller P, Oswald E, Hu J, Matloubian M, Krummel MF. Secondary T cell-T cell synaptic interactions drive the differentiation of protective CD8+ T cells. Nat Immunol. 2013;14:356–363. doi: 10.1038/ni.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger DN, Senchenkova E. Leukocyte–Endothelial Cell Adhesion. Inflammation and the Microcirculation. San Rafael Morgan, CA: Claypool Life Sciences; 2010. [PubMed] [Google Scholar]
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. 2012;1250:50–55. doi: 10.1111/j.1749-6632.2011.06389.x. [DOI] [PubMed] [Google Scholar]
- He P, Ng BG, Losfeld ME, Zhu W, Freeze HH. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. J Biol Chem. 2012;287:18210–18217. doi: 10.1074/jbc.M112.355677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MA, Ng BG, Bhide S, Chin E, Rhodenizer D, He P, Losfeld ME, He M, Raymond K, Berry G, et al. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet. 2012;90:363–368. doi: 10.1016/j.ajhg.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–1220. doi: 10.1111/j.1582-4934.2009.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen GR, Sako D, Ahern TJ, Shaffer M, Erban J, Sajer SA, Gibson RM, Wagner DD, Furie BC, Furie B. P-selectin and E-selectin. Distinct but overlapping leukocyte ligand specificities. J Biol Chem. 1992;267:11104–11110. [PubMed] [Google Scholar]
- Min JK, Kim YM, Kim SW, Kwon MC, Kong YY, Hwang IK, Won MH, Rho J, Kwon YG. TNF-related activation-induced cytokine enhances leukocyte adhesiveness: Induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor and protein kinase C-dependent NF-kappaB activation in endothelial cells. J Immunol. 2005;175:531–540. doi: 10.4049/jimmunol.175.1.531. [DOI] [PubMed] [Google Scholar]
- Munoz FM, Hawkins EP, Bullard DC, Beaudet AL, Kaplan SL. Host defense against systemic infection with Streptococcus pneumoniae is impaired in E-, P-, and E-/P-selectin-deficient mice. J Clin Invest. 1997;100:2099–2106. doi: 10.1172/JCI119744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto VI, Damoc E, Cueni LN, Schurpf T, Frei R, Ali S, Callewaert N, Moise A, Leary JA, Folkers G, et al. N-glycan structures and N-glycosylation sites of mouse soluble intercellular adhesion molecule-1 revealed by MALDI-TOF and FTICR mass spectrometry. Glycobiology. 2006;16:1033–1044. doi: 10.1093/glycob/cwl032. [DOI] [PubMed] [Google Scholar]
- Pahlsson P, Strindhall J, Srinivas U, Lundblad A. Role of N-linked glycosylation in expression of E-selectin on human endothelial cells. Eur J Immunol. 1995;25:2452–2459. doi: 10.1002/eji.1830250907. [DOI] [PubMed] [Google Scholar]
- Rothlein R, Czajkowski M, O'Neill MM, Marlin SD, Mainolfi E, Merluzzi VJ. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines. Regulation by pharmacologic agents and neutralizing antibodies. J Immunol. 1988;141:1665–1669. [PubMed] [Google Scholar]
- Sawa Y, Sugimoto Y, Ueki T, Ishikawa H, Sato A, Nagato T, Yoshida S. Effects of TNF-alpha on leukocyte adhesion molecule expressions in cultured human lymphatic endothelium. J Histochem Cytochem. 2007;55:721–733. doi: 10.1369/jhc.6A7171.2007. [DOI] [PubMed] [Google Scholar]
- Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoor M, Lai FP, Zarbock A, Klaver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Ichikawa M, He P, Scott DA, Bravo Y, Dahl R, Ng BG, Cosford ND, Freeze HH. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J Biol Chem. 2011;286:39431–39438. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Nayak J, Derossi C, Charbono A, Ichikawa M, Ng BG, Grajales-Esquivel E, Srivastava A, Wang L, He P, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J. 2014 doi: 10.1096/fj.13-245514. doi:10.1096/fj.13-245514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sligh JE, Jr, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Toomre DK, Manzi A, Panneerselvam K, Freeze HH, Varki A, Varki NM. A novel anionic modification of N-glycans on mammalian endothelial cells is recognized by activated neutrophils and modulates acute inflammatory responses. J Immunol. 2001;166:624–632. doi: 10.4049/jimmunol.166.1.624. [DOI] [PubMed] [Google Scholar]
- Tan TQ, Smith CW, Hawkins EP, Mason EO, Jr, Kaplan SL. Hematogenous bacterial meningitis in an intercellular adhesion molecule-1-deficient infant mouse model. J Infect Dis. 1995;171:342–349. doi: 10.1093/infdis/171.2.342. [DOI] [PubMed] [Google Scholar]
- von Andrian UH, Berger EM, Ramezani L, Chambers JD, Ochs HD, Harlan JM, Paulson JC, Etzioni A, Arfors KE. In vivo behavior of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J Clin Invest. 1993;91:2893–2897. doi: 10.1172/JCI116535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shiratori I, Uehori J, Ikawa M, Arase H. Neutrophil infiltration during inflammation is regulated by PILRalpha via modulation of integrin activation. Nat Immunol. 2013;14:34–40. doi: 10.1038/ni.2456. [DOI] [PubMed] [Google Scholar]
- Winning S, Splettstoesser F, Fandrey J, Frede S. Acute hypoxia induces HIF-independent monocyte adhesion to endothelial cells through increased intercellular adhesion molecule-1 expression: The role of hypoxic inhibition of prolyl hydroxylase activity for the induction of NF-kappa B. J Immunol. 2010;185:1786–1793. doi: 10.4049/jimmunol.0903244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.