

Abstract
Background and PurposeSince monoacylglycerol lipase (MAGL) has been firmly established as the predominant catabolic enzyme of the endocannabinoid 2-arachidonoylglycerol (2-AG), a great need has emerged for the development of highly selective MAGL inhibitors. Here, we tested the in vivo effects of one such compound, KML29 (1,1,1,3,3,3-hexafluoropropan-2-yl 4-(bis(benzo[d][1,3]dioxol-5-yl)(hydroxy)methyl)piperidine-1-carboxylate).
Experimental ApproachIn the present study, we tested KML29 in murine inflammatory (i.e. carrageenan) and sciatic nerve injury pain models, as well as the diclofenac-induced gastric haemorrhage model. KML29 was also evaluated for cannabimimetic effects, including measurements of locomotor activity, body temperature, catalepsy, and cannabinoid interoceptive effects in the drug discrimination paradigm.
Key ResultsKML29 attenuated carrageenan-induced paw oedema and completely reversed carrageenan-induced mechanical allodynia. These effects underwent tolerance after repeated administration of high-dose KML29, which were accompanied by cannabinoid receptor 1 (CB1) receptor desensitization. Acute or repeated KML29 administration increased 2-AG levels and concomitantly reduced arachidonic acid levels, but without elevating anandamide (AEA) levels in the whole brain. Furthermore, KML29 partially reversed allodynia in the sciatic nerve injury model and completely prevented diclofenac-induced gastric haemorrhages. CB1 and CB2 receptors played differential roles in these pharmacological effects of KML29. In contrast, KML29 did not elicit cannabimimetic effects, including catalepsy, hypothermia and hypomotility. Although KML29 did not substitute for Δ9-tetrahydrocannabinol (THC) in C57BL/6J mice, it fully and dose-dependantly substituted for AEA in fatty acid amide hydrolase (FAAH) (−/−) mice, consistent with previous work showing that dual FAAH and MAGL inhibition produces THC-like subjective effects.
Conclusions and ImplicationsThese results indicate that KML29, a highly selective MAGL inhibitor, reduces inflammatory and neuropathic nociceptive behaviour without occurrence of cannabimimetic side effects.
Linked ArticlesThis article is part of a themed section on Cannabinoids 2013. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-6
Keywords: 2-arachidonoylglycerol, allodynia, cannabinoid, CB1, CB2, gastric haemorrhage, inflammatory pain, monoacylglycerol lipase, mouse, neuropathic pain
Introduction
The antinociceptive effects of Cannabis sativa have been known for millennia (Russo, 2007; Russo et al., 2008); however, current therapeutic use of cannabinoids remains limited because of psychomimetic side effects that include sensorimotor, affective and cognitive disturbances (Di Marzo, 2008). Recent research efforts have focused on development of drugs targeting components of the endogenous cannabinoid system, including CB1 and CB2 cannabinoid receptors, and enzymes regulating the biosynthesis and degradation of the endogenous cannabinoids N-arachidonoylethanolamine (anandamide; AEA) and 2-arachidonoylglycerol (2-AG; Ahn et al., 2008; Lichtman et al., 2010; Petrosino and Di Marzo, 2010). The major respective catabolic enzymes of AEA and 2-AG, fatty acid amide hydrolase (FAAH; Cravatt et al., 1996; 2001) and monoacylglycerol lipase (MAGL; Di Marzo et al., 1999; Goparaju et al., 1999; Blankman et al., 2007), are particularly promising targets for pharmacological modulation of the endocannabinoid system.
Inhibitors of endocannabinoid catabolic enzymes elevate endocannabinoid levels in the brain, allowing prolonged activation of their receptor targets. It is well established that FAAH inhibitors produce antinociceptive effects in multiple preclinical nociceptive assays, including neuropathic, (Lichtman et al., 2004a; Chang et al., 2006; Jhaveri et al., 2006; Russo et al., 2007; Kinsey et al., 2009; 2010; Starowicz and Przewlocka, 2012), inflammatory (Jayamanne et al., 2006; Ahn et al., 2009a, 2009b; 2011; Clapper et al., 2010; Naidu et al., 2010; Booker et al., 2012), and acute thermal nociception (Kathuria et al., 2003; Lichtman et al., 2004b). Moreover, FAAH-compromised mice show sustained antinociceptive phenotypes, with no evidence of CB1 receptor functional tolerance (Cravatt et al., 2001; Falenski et al., 2010; Schlosburg et al., 2010; Busquets-Garcia et al., 2011), although the antinociceptive effects of the FAAH inhibitor (3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl)-cyclohexylcarbamate (URB597) in the rat carrageenan model were lost following repeated administration (Okine et al., 2012). While the impact of selective FAAH inhibition has been investigated for approximately a decade, selective inhibitors of MAGL have only recently been developed to examine systematically the in vivo consequences of elevating 2-AG levels.
KML29 (1,1,1,3,3,3-hexafluoropropan-2-yl 4-(bis(benzo[d][1,3]dioxol-5-yl)(hydroxy)methyl)piperidine-1-carboxylate) is one of the most selective MAGL inhibitors developed to date and the first that increases 2-AG levels, but does not possess cross-activity with FAAH or other serine hydrolases, as described by Chang et al., 2012. Previous MAGL inhibitors [e.g. [1,1′-biphenyl]-3-yl-carbamic acid, cyclohexyl ester (URB602) or N-arachidonyl maleamide] lack potency in vivo (Hohmann et al., 2005; Guindon and Hohmann, 2009) and show insufficient selectivity (Burston et al., 2008; Long et al., 2009a). URB602 increases AEA levels, without altering 2-AG levels, following local administration. However, insufficient selectivity of this compound for MAGL over FAAH following systemic administration limits its use (Hohmann et al., 2005, Guindon and Hohmann, 2009). 4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) is the first long-lasting MAGL inhibitor that was effective in vivo (Long et al., 2009b). However, JZL184 has cross-activity with FAAH and is considerably less potent in inhibiting MAGL in rats than in mice and nonhuman primates (Long et al., 2009b). Although acute administration of JZL184 elevates 2-AG, and is insufficient to elevate brain AEA levels, repeated administration of JZL184 results in elevation of both 2-AG and AEA brain levels (Schlosburg et al., 2010). In contrast, KML29 does not show any detectable activity with FAAH even at high doses or following repeated administration, and unlike JZL184, KML29 does not inactivate carboxylesterase enzymes in peripheral tissues (Chang et al., 2012). Moreover, KML29 is considerably more potent than JZL184 in inhibiting MAGL in rats (Chang et al., 2012). These attributes make KML29 a potentially very useful tool to explore the consequences of inhibiting MAGL in the whole animal and in multiple species, and provides greater selectivity than JZL184 in inhibiting MAGL. Although a single injection of KML29 does not elevate AEA brain levels, the consequences of repeated administration have yet to be examined.
Systemic administration of JZL184 (Long et al., 2009a; 2009b; Schlosburg et al., 2010; Chang et al., 2012) elevates brain 2-AG levels, reduces nociceptive behaviour in models of neuropathic (Kinsey et al., 2009; 2010), inflammatory (Ghosh et al., 2013), cisplatin (Guindon et al., 2013), and bone cancer (Khasabova et al., 2011) pain, as well as in tail withdrawal, acetic acid abdominal stretching, and formalin tests in mice (Long et al., 2009a; 2009b; Busquets-Garcia et al., 2011). Local intraplantar injection of JZL184 reduces nociceptive behaviour in the formalin test (Guindon et al., 2011) and inhibits capsaicin-evoked nocifensive behaviour and thermal hypersensivity (Spradley et al., 2010). JZL184 also inhibits antinociceptive processing at the spinal level in the rat carrageenan-induced inflammatory pain model (Woodhams et al., 2012). Consistent with the observation that JZL184 is far less potent in inhibiting MAGL in rats than in mice (Long et al., 2009b), no changes in spinal levels of 2-AG or 2-oleoylglycerol hydrolysis were detected, although the CB1 receptor antagonist AM251 inhibited this effect in rats (Woodhams et al., 2012). In addition to its antinociceptive effects in neuropathic and inflammatory pain models and its gastro-protective actions, JZL184 produces a subset of cannabimimetic side effects, including hypomotility and hyperreflexia (Long et al., 2009a; 2009b).
In the present study, we investigated whether KML29 produces antinociception in well-established models of inflammatory and neuropathic pain and in a model of nonsteroidal anti-inflammatory drug (NSAID)-induced gastropathy. Additionally, we examined whether the antinociceptive effects of KML29 undergo tolerance following repeated administration and if functional CB1 receptor tolerance occurred, using the CP-55 940-stimulated guanosine 5′-O-(3-[35S]thio)triphosphate binding assay. Brain levels of 2-AG, AEA, palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) were quantified following acute or repeated KML29 administration. Because MAGL participates in the formation of arachidonic acid (AA) from 2-AG in the brain (Nomura et al., 2011), we also quantified this lipid modulator. We also determined whether KML29 produces cannabimimetic side effects, consisting of measurements of locomotor activity, body temperature, nociceptive behaviour and catalepsy (Martin et al., 1991). Finally, we assessed whether KML29 substitutes for Δ9-tetrahydrocannabinol (THC) or AEA in mice trained in the drug discrimination paradigm. The involvement of CB1 and CB2 receptors was assessed through the use of selective receptor antagonists or in CB1 (−/−) and CB2 (−/−) mice.
Here we report that KML29, a novel selective MAGL lipase inhibitor, produces antinociceptive effects in models of inflammatory and neuropathic pain, as well as gastro-protective actions, in the absence of cannabimimetic side effects.
Materials and methods
Animals
Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA) were utilized in models of inflammatory and neuropathic pain as well as in the tetrad assay (i.e. consisting of hypomotility, antinociception, catalepsy and hypothermia) and drug discrimination paradigm. Male Institute of Cancer Research (ICR) mice (Harlan Laboratories, Indianapolis, IN, USA) were used in a model of NSAID-induced gastric damage. Male FAAH (−/−) mice were used in some of the drug discrimination experiments. Male and female CB1 (−/−) and CB2 (−/−) mice and their respective littermate controls [CB1 (+/+) and CB2 (+/+) mice] were used in the carrageenan-induced inflammatory pain model. CB1 (−/−) and CB2 (−/−) mice were backcrossed onto a C57BL/6J background for more than 13 and 7 generations respectively. FAAH (−/−) mice were backcrossed onto a C57BL/6J background for at least 13 generations. All genetically manipulated mice were obtained from the Center Transgenic Colony at Virginia Commonwealth University. The subjects weighed between 18 and 40 g. Four to five mice were housed per cage in all experiments, except in the neuropathic pain model and drug discrimination experiments in which subjects were singly housed.
Animals were maintained in a 12/12 hour light/dark cycle (0600 h on/1800 h off) in a temperature (20–22°C) and humidity (55 ± 10%) controlled AAALAC-approved facility. Animals had ad libitum access to water and food, with the exception of animals used in drug discrimination experiments that were food restricted to 85–90% of free feeding body weight. In addition, mice were food deprived for 24 hours prior to induction of NSAID-induced gastric damage. All tests were conducted during the light phase. The sample sizes selected for each treatment group in each experiment were based on previous studies from our laboratory.
All animal protocols were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University or West Virginia University and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). After testing was completed, all mice were humanely euthanized via CO2 asphyxia, followed by rapid cervical dislocation. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 1991).
Experimental procedures
Carrageenan model of inflammatory pain
Oedema was induced via intraplantar injection of 0.3% carrageenan (Sigma, St Louis, MO, USA) in a 20 μL volume using a 30 G needle into the hind left paw. Paw thickness was measured with an electronic digital micrometer (Traceable Calipers, Friendswood, TX, USA), prior to and 5 h following carrageenan administration, which corresponds to peak oedema (Wise et al., 2008). Paw oedema data are expressed as the difference in paw thickness between the 5 h and pre-injection measures. Mechanical allodynia was assessed using von Frey filaments at the same peak time point (see the full description in supplementary methods).
KML29 or vehicle was given 2 h before testing. In experiments assessing cannabinoid receptor mechanism of action, the CB1 receptor antagonist rimonabant (1 mg·kg−1) or the CB2 receptor antagonist SR144528 (3 mg·kg−1) was administered 30 min prior to KML29 or vehicle (Lichtman et al., 2004a; La Rana et al., 2006; Russo et al., 2007; Kinsey et al., 2009; 2011; Ghosh et al., 2013). To assess further the involvement of cannabinoid receptors, the anti-oedematous and anti-allodynic effects of KML29 were evaluated in CB1 (−/−) and CB2 (−/−) mice and their wild-type littermates.
In order to assess the effects of repeated treatment of high-dose KML29 on paw oedema and allodynia, mice were divided into the following three experimental groups (i) Vehicle Control (six days of vehicle treatment); (ii) Acute KML29 (five days of vehicle treatment and injected with 40 mg·kg−1 KML29 on day 6); and (iii) Repeated KML29 (six days of injections of 40 mg·kg−1 KML29). On day 6, 3 h after injection of carrageenan, all groups received their respective treatments. Oedema and mechanical allodynia were then assessed 2 h later (i.e. 5 h after carrageenan).
Neuropathic pain model – chronic constriction nerve injury (CCI)
CCI was induced according to surgical procedures described previously (Kinsey et al., 2009), as detailed in the supplementary methods. Mice were randomly assigned to a drug treatment and tested in a counterbalanced Latin square within subject design with at least 5 day wash out period. Mechanical and cold allodynia were assessed as described previously (Kinsey et al., 2009, Ghosh et al., 2013). See Supporting Information Methods for details.
Model of NSAID-induced gastric lesions
Gastric haemorrhages were induced as described by Kinsey et al. (2011). Male ICR mice were weighed, then placed on a wire mesh barrier (Thoren Caging Systems, Inc, Hazleton, PN, USA) to prevent coprophagia, and food deprived for 24 h, with ad libitum access to water. To induce the gastric haemorrhages, the mice were weighed, then administered the NSAID, diclofenac sodium (100 mg·kg−1, p.o.) or vehicle, and returned to their home cage. Mice were pretreated 2.5 h prior to haemorrhage induction with the CB1 antagonist rimonabant (3 mg·kg−1, i.p.) or vehicle, then administered KML29 (40 mg·kg−1) 2 h prior to haemorrhage induction. A second group of animals received diclofenac sodium (100 mg·kg−1, p.o.) or vehicle following pretreatment with KML29 at 1, 5 or 20 mg·kg−1. The mice were humanely euthanized via CO2 asphyxiation 6 h after administration of diclofenac, and their stomachs were harvested and photographed. The procedure of harvesting stomachs and gastric haemorrhage scoring was performed as described previously (Liu et al., 1998; Kinsey et al., 2011) and detailed in the supplementary methods.
Tetrad assay
Mice (counterbalanced Latin square within subject design) were housed individually overnight. The behavioural testing was conducted in the following order: bar test (catalepsy), tail withdrawal test, rectal temperature and locomotor activity. Testing was performed according to previously described procedures (Long et al., 2009c; Schlosburg et al., 2010), as detailed in supplementary methods.
Drug discrimination
Male FAAH (−/−) and C57BL/6J mice (20–25 g) were tested according to procedures described previously (Long et al., 2009c; Walentiny et al., 2013) with minor modifications. See supplementary methods for details.
Extraction and quantification of endocannabinoids by liquid chromatography-tandem mass spectrometry
2-AG, AEA, PEA, OEA and AA levels were quantified in the whole brain of C57BL/6J mice after acute or repeated treatment with KML29 or the vehicle. Animals were killed 2, 4 or 24 h following KML29 treatment or 2 h following acute administration of vehicle and in experiments examining effects of repeated KML29 treatment. Immediately after individual mice were euthanized via rapid decapitation (at 1100–1300 h), their brains were harvested, snap-frozen in dry ice, and stored at −80°C until the time of processing. Tissues were further processed according to methods described previously (Ramesh et al., 2011). See supplementary methods for details.
[35S]GTPγS binding assay
CP-55 940-stimulated [35S]GTPγS binding was performed in brains of mice subjected to repeated KML29 (40 mg·kg−1) or vehicle administration and tested in the carrageenan assay, as described previously (Schlosburg et al., 2010). See supplementary methods for details. Percent stimulation = [(agonist-stimulated − basal)/basal × 100%]. Curves were fit using nonlinear regression in GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
Drugs
KML29 was synthesized at the Scripps Research Institute (Chang et al., 2012) and JZL184 was synthesized by Organix Inc. (Woburn, MA, USA), as described previously (Long et al., 2009a). The CB1 receptor antagonist rimonabant [N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide-HCl] (SR141716A), the CB2 antagonist SR144528 [5-(4-chloro-3-methylphenyl)-1-[ (4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-rimethylbicyclo [2.2.1] hept-2-yl]-1H-pyrazole-3-carboxamide] and THC were obtained from the National Institute on Drug Abuse (Bethesda, MD, USA). AEA was provided by Organix Inc. The NSAID diclofenac sodium was purchased from Sigma-Aldrich. Drugs were dissolved in a vehicle consisting of a mixture of ethanol, alkamuls-620 (Sanofi-Aventis, Bridgewater, NJ, USA), and saline (0.9% NaCl) in a ratio of 1:1:18. Each drug was given via the intraperitoneal (i.p.) route of administration, with two exceptions. In the drug discrimination studies, injections were given via subcutaneous (s.c.) route of administration, and diclofenac was administered orally (p.o.). THC, AEA, rimonabant, SR144528 and diclofenac were administered in a volume of 10 μL·g−1 body mass; JZL184 and KML29, in a volume of 20 μL·g−1 body mass. The doses of KML29 selected were based on results reported by Chang et al. (2012) indicating that acute administration of 20 mg·kg−1 of KML29 produces maximal inhibition of MAGL lipase in the mouse brain. Nomenclature used throughout the paper conforms to BJP's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
Data analysis
All data are presented as mean ± SEM. Data were analysed using t-tests, one-way or two-way anova. Dunnett's test was used for post hoc analysis in the dose–response experiments and Tukey's test was used for post hoc analyses comparing different treatment groups. Multiple comparisons following two-way anova were conducted with Bonferroni post hoc comparison. Differences were considered significant at the level of P < 0.05. Statistical analysis was performed with IBM Statistical Package for the Social Sciences (SPSS) Statistics, version 20.0 (IBM Corp., Armonk, NY, USA).
Results
Evaluation of brain endocannabinoid levels following KML29 treatment
Acute administration of KML29 (40 mg·kg−1) significantly elevated 2-AG brain levels at 2 and 4 h post administration, but by 24 h, 2-AG brain levels were similar to those from vehicle-treated mice [F(3,12) = 22.2, P < 0.001, Figure 1A]. KML29 significantly reduced free AA brain levels at 2, 4 and 24 h after treatment [F(3,12) = 38.6, P < 0.001; Figure 1B]. In contrast, KML29 did not elicit any significant changes in brain levels of AEA [P = 0.34; Figure 1C] or OEA [P = 0.94; Figure 1E] across the 24 h time course. A small, but statistically significant increase of PEA [P = 0.01; Figure 1D], was observed, but none of the time points differed from the vehicle-treated control group.
Figure 1.
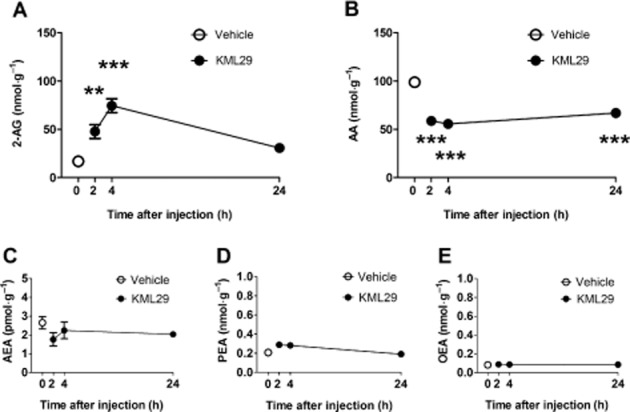
Time course of 2-AG and other lipid signaling molecules following KML29 (40 mg·kg−1). (A) 2-AG brain levels were significantly increased at 2 and 4 h following drug administration. (B) Brain AA concentrations were reduced throughout the entire 24 h time course. According to post hoc analyses, no significant changes were found in brain levels of (C) AEA, (D) PEA, or (E) OEA. Data presented as mean ± SEM, n = 4 mice per group; **P < 0.01, ***P < 0.001 vs. vehicle-treated control group.
KML29 reduces carrageenan-induced paw oedema and allodynia
Administration of KML29 significantly prevented the occurrence of carrageenan-induced paw oedema [Figure 2A; F(3,22) = 53.4, P < 0.0001] and mechanical allodynia [F(3,22) = 5.15, P < 0.01; Figure 2B]. For each dependent variable, 20 and 40 mg·kg−1 KML29 significantly differed from the vehicle condition. Use of pharmacological and genetic approaches revealed differential involvement of CB1 and CB2 receptors underlying these two effects. As shown in Figure 3, the CB2 receptor antagonist, SR144528, completely blocked the anti-oedematous effects of KML29 [F(1,18) = 28.2, P < 0.0001; Figure 3A]. However, the CB1 receptor antagonist, rimonabant had no effect on oedema (P = 0.66). Similarly, the anti-oedematous effects of KML29 were retained in CB1 (−/−) mice (Figure 3B), but were absent in CB2 (−/−) mice [interaction between genotype and drug: F(1,20) = 20.1, P < 0.001; Figure 3C]. These results indicate that the anti-oedematous effects of KML29 require CB2 receptors, while CB1 receptors are expendable. The data depicted in Figure 4A show that the anti-allodynic effects of KML29 were completely blocked by pretreatment with either rimonabant [interaction: F(1,18) = 6.91, P < 0.05] or SR144528 [interaction F(1,18) = 4.9, P < 0.05]. Similarly, CB1 (−/−) mice [interaction: F(1,22) = 10.4, P < 0.01] and CB2 (−/−) mice [interaction: F(1,20) = 7.1, P < 0.05] were completely resistant to the anti-allodynic effects of KML29 (Figure 4B and 4C). Carrageenan induced a similar magnitude of paw oedema and allodynia in C57BL/6J mice treated with rimonabant or SR144528 and in CB1 (−/−) and CB2 (−/−) mice as compared with the appropriate littermate controls.
Figure 2.
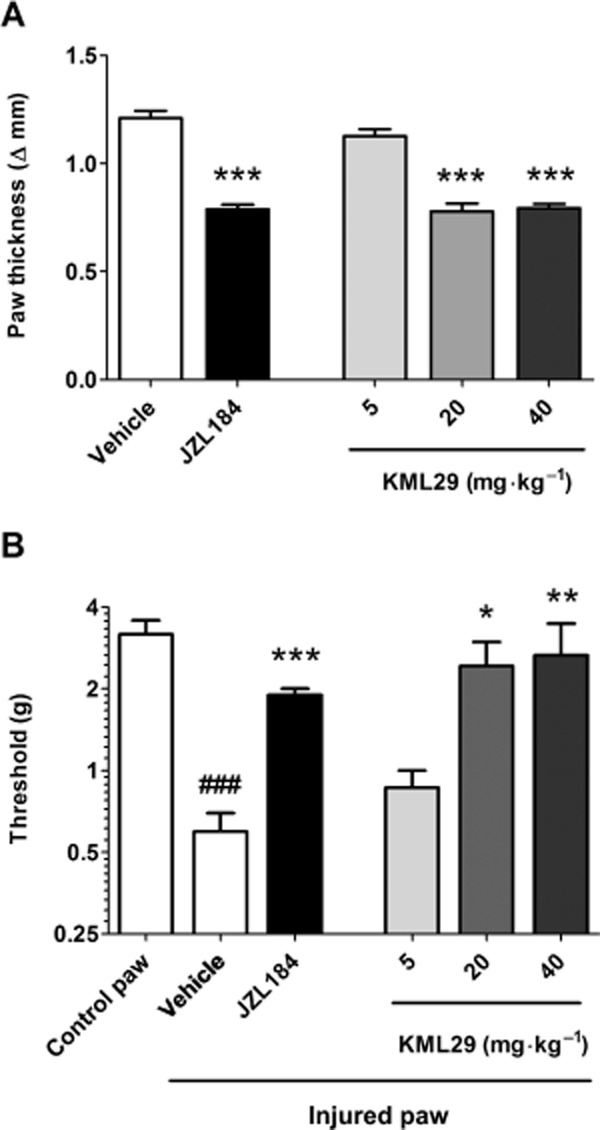
The selective MAGL inhibitor KML29 significantly reduces paw oedema (A) and mechanical allodynia (B) induced by intraplantar injection of carrageenan. Mice received i.p. injection of vehicle, KML29 or JZL184 (40 mg·kg−1) 2 h before testing. Data presented as mean ± SEM, n = 6–8 mice per group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle; ###P < 0.001 vs. control paw.
Figure 3.
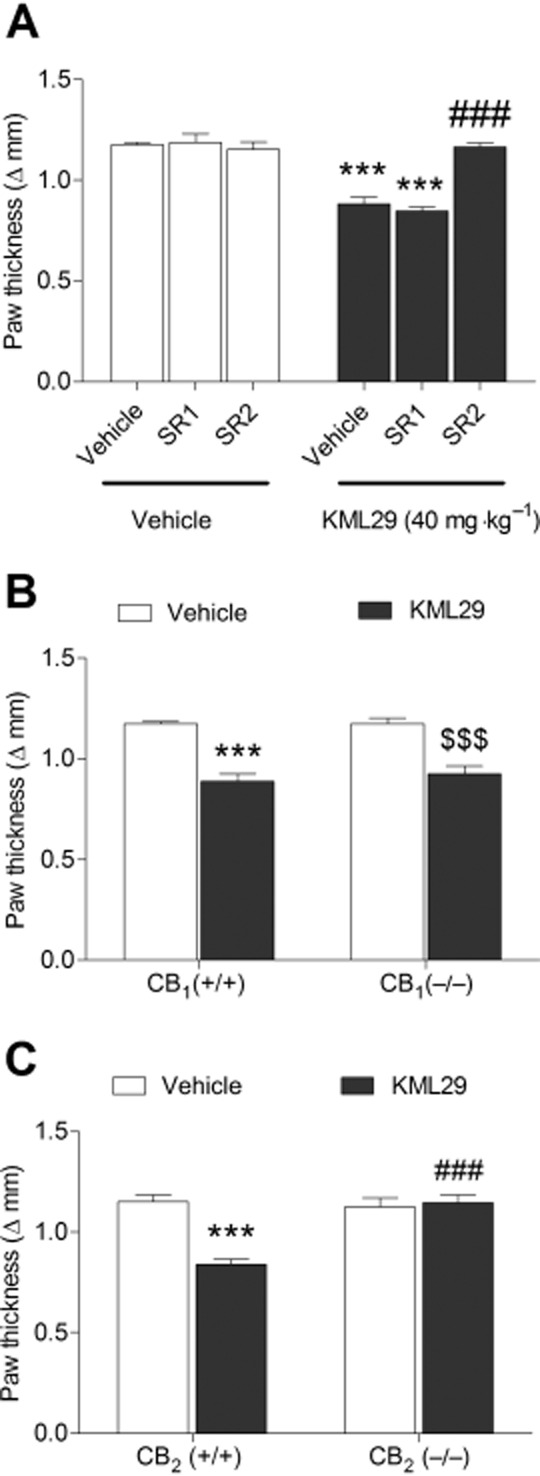
The anti-oedematous effects of KML29 (40 mg·kg−1) in carrageenan-induced model of inflammatory pain require CB2, but not CB1 receptors. (A) The anti-oedematous effects of KML29 were not abated in wild-type mice treated with the CB1 receptor antagonist, rimonabant (SR1, 3 mg·kg−1), but were completely blocked by the CB2 receptor antagonist SR144528 (SR2, 1 mg·kg−1). Similarly, the anti-oedematous effects of KML29 were present in CB1 (−/−) mice (B), but were absent in CB2 (−/−) mice (C). Data presented as mean ± SEM, n = 5–8 mice per group; ***P < 0.001 vs. vehicle; ###P < 0.001 compared with other KML29-treated groups; $$$P < 0.001 compared with vehicle-treated CB1 (−/−) mice.
Figure 4.
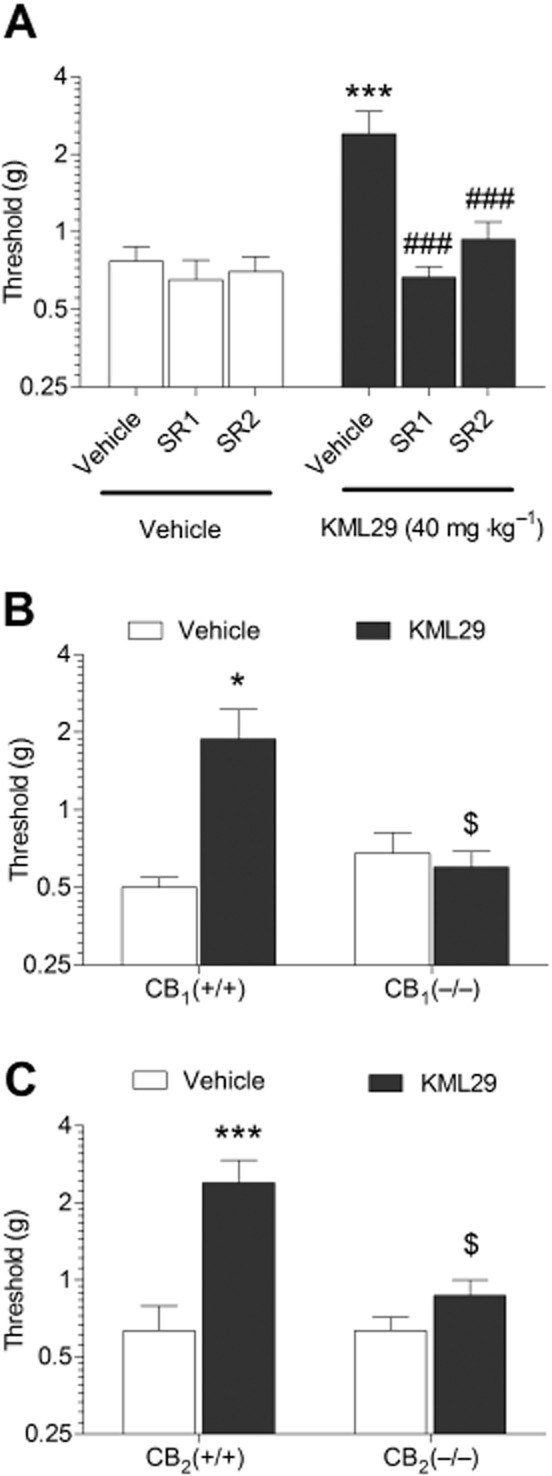
The anti-allodynic effects of KML29 (40 mg·kg−1) in carrageenan-induced model of inflammatory pain require both CB1 and CB2 receptors. (A) The anti-allodynic effects of KML29 were completely blocked in wild-type mice treated with either CB1 antagonist, rimonabant (SR1, 1 mg·kg−1) or CB2 receptor antagonist SR144528 (SR2, 3 mg·kg−1). Similarly, anti-allodynic effects of KML29 were absent in both CB1 (−/−) mice (B) and in CB2 (−/−) mice (C). Data presented as mean ± SEM, n = 5–8 mice per group; *P < 0.05 vs. vehicle; ###P < 0.001 compared with KML29-treated mice pretreated with vehicle; $P < 0.05 compared with CB1 (+/+) or CB2 (+/+) mice treated with KML29.
In the next experiment, mice were treated repeatedly with vehicle or high-dose KML29 (40 mg·kg−1) and on the test day were injected with the appropriate treatments 3 h after carrageenan (i.e. 2 h before assessing allodynia and paw thickness). The anti-oedematous effects of KML29 were attenuated in mice subjected to repeated KML29 administration, suggesting partial tolerance [F(2,15) = 15.0, P < 0.001; Figure 5A]. As shown in Figure 5B, carrageenan-induced mechanical allodynia was significantly reversed following acute administration of KML29, but this effect was absent in mice subjected to repeated treatment with KML29 [F(2,15) = 6.71, P < 0.01].
Figure 5.
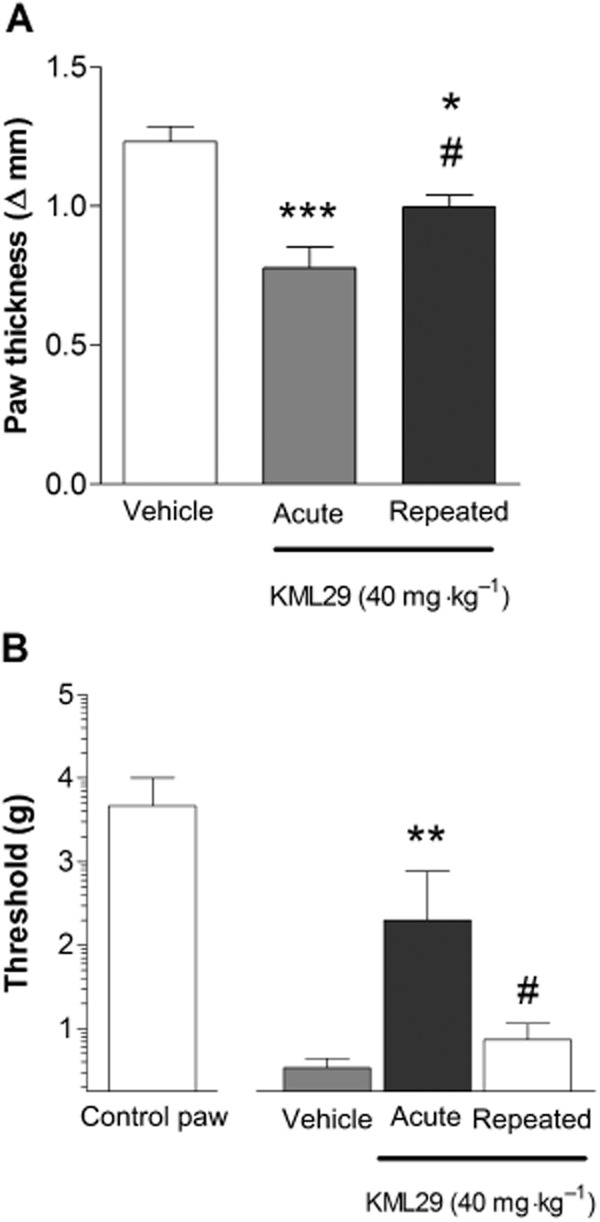
Comparison of acute and repeated administration of high-dose KML29 (40 mg·kg−1) in the carrageenan assay. (A) The anti-oedematous effects of KML29 underwent partial tolerance following repeated treatment with 40 mg·kg−1. (B) The anti-allodynic effects of KML29 underwent tolerance and were absent in animals receiving repeated KML29 treatment. Data presented as mean ± SEM, n = 6 mice per group; **P < 0.01, ***P < 0.001 vs. vehicle; #P < 0.05 vs. acute KML29.
Immediately after assessing allodynia and paw thickness (approximately 2 h after injection) brains were harvested for determination of brain levels of endocannabinoids and other lipids, as well as assessment of CB1 receptor function utilizing CP-55 940-stimulated [35S]GTPγS binding assay. As depicted in Figure 6, repeated KML29 treatment significantly reduced CP-55 940-stimulated [35S]GTPγS binding [KML29 main effect: F(1,60) = 159, P < 0.001; KML29 by CP-55 940 interaction: F(5,60) = 2.65, P < 0.05]. The Emax, as calculated based on nonlinear regression best-fit values of specific binding was 77.3% of vehicle-treated control levels, indicating that prolonged MAGL inhibition reduces CB1 receptor function.
Figure 6.
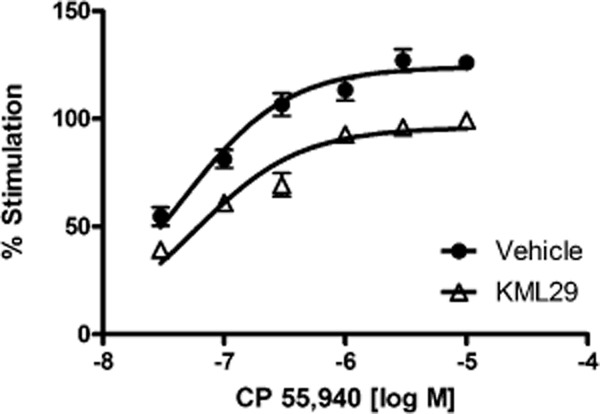
Repeated KML29 (40 mg·kg−1) treatment reduced activity of CB1 receptor as indicated by a significant decrease in CP-55 940-stimulated [35S]GTPγS binding in the whole brain. Data presented as mean ± SEM, n = 6 mice per group (each sample in triplicate).
Repeated KML29 (40 mg·kg−1) treatment significantly increased 2-AG brain levels approximately threefold, as compared with mice treated acutely with KML29 [F(2,17) = 241.3, P < 0.001 Figure 7A], which was accompanied by significantly reduced levels of free AA [F(2,17) = 40.53, P < 0.001; Figure 7B]. Unexpectedly, a small, but significant reduction of AEA levels was detected [F(2,17) = 16.29, P < 0.001; Figure 7C]. No significant changes in levels of PEA (P = 0.66) or OEA (P = 0.39) were found following repeated or single treatment with KML29 (Figure 7 D and E).
Figure 7.
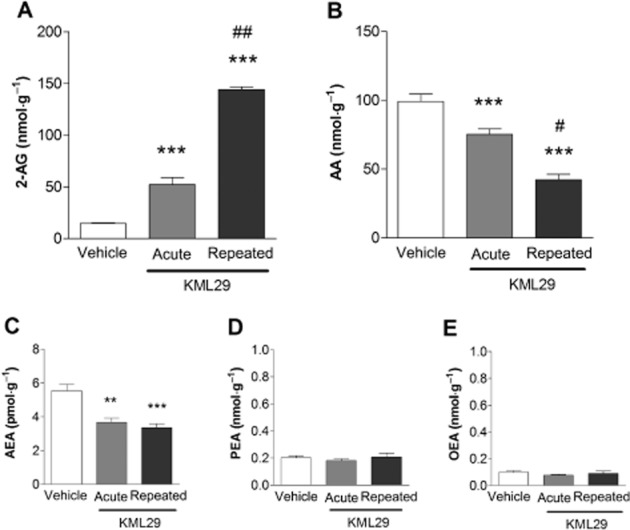
Repeated administration with KML29 (40 mg·kg−1) induced significant increases in brain 2-AG concentration (A) compared with vehicle as well as acute KML29 treatment. Increase in 2-AG levels was accompanied by decreases in AA concentration (B). Acute and repeated administration of KML29 reduced brain level of AEA (C), but did not produce any changes in levels of PEA (D) or OEA (E). Data presented as mean ± SEM, n = 4 mice per group; **P < 0.01, ***P < 0.001 vs. vehicle-treated control group #P < 0.05; ##P < 0.01 vs. acute KML29.
KML29 partially reverses mechanical and cold allodynia in the CCI model
As shown in Figure 8A, KML29 (40 mg·kg−1) significantly reversed allodynic response to cold stimulation [F(2,198) = 6.15, P < 0.01], with the most pronounced effects observed from 3 to 6 hour following treatment (Figure 8A). KML29 also significantly decreased mechanical allodynia [F(2,198) = 7.31, P < 0.001] that achieved peak effects 3 h following treatment (Figure 8B). In both nociceptive assays, the anti-allodynic effects were of a similar magnitude to those produced by JZL184 (40 mg·kg−1). No changes in latency to respond to cold simulation (P = 0.12) or paw withdrawal threshold (P = 0.47) were observed in animals treated with vehicle.
Figure 8.
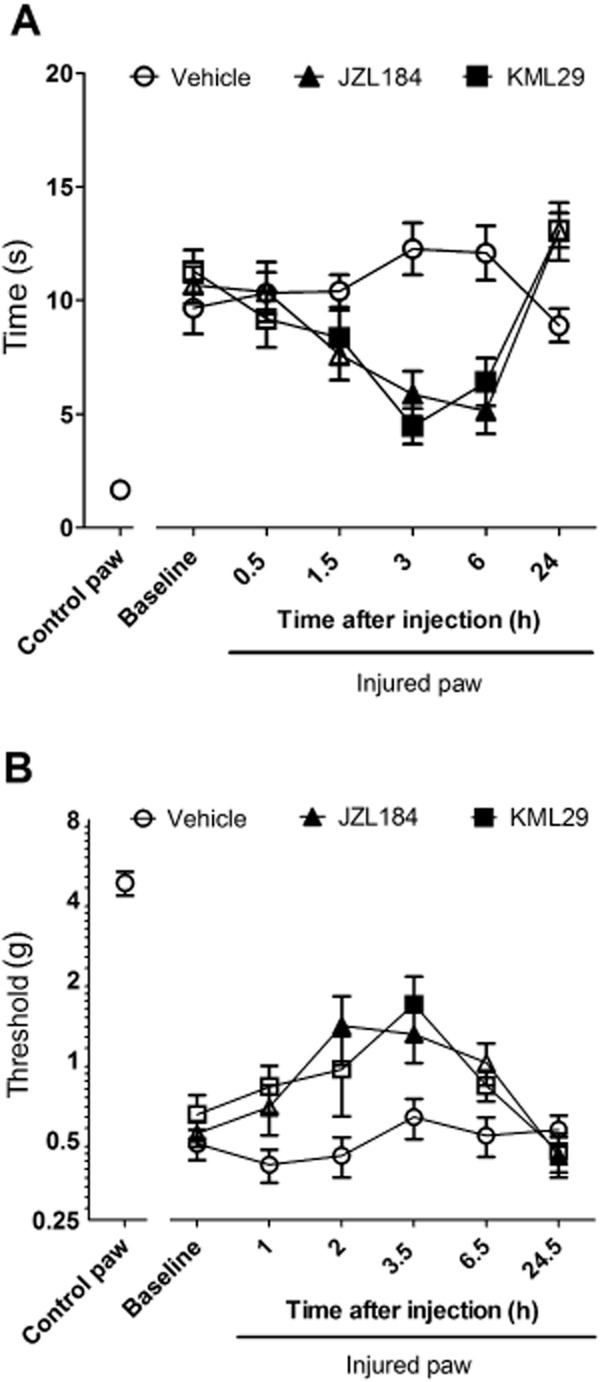
KML29 (40 mg kg−1) reduced cold (A) and mechanical allodynia (B) in the CCI model of neuropathic pain, with a similar magnitude of effects and time courses as JZL184 (40 mg kg−1). Data presented as mean ± SEM, n = 12 mice per group. Significant difference (P < 0.05) as compared with baseline measurements indicated as filled symbols.
The anti-allodynic effects of KML29 (40 mg·kg−1) in response to cold stimulation were blocked by rimonabant [Figure 9A; KML29 main effect: F(1,44) = 20.9, P < 0.001; rimonabant main effect: F(1,44) = 10.3, P < 0.05; KML29 by rimonabant interaction: P = 0.06]. In contrast, SR144528 did not prevent the decrease in time of response to cold stimulation produced by KML29 (main effect of SR144528: P = 0.76; KML29 by SR144528 interaction: P = 0.39). Rimonabant also blocked the anti-allodynic effects of KML29 in response to mechanical stimulation [Figure 9B; interaction F(1,44) = 10.4, P < 0.05], but they were unaffected by SR144528 (main effect of SR144528: P = 0.34; interaction: P = 0.82). None of the treatments affected responses to acetone (P = 0.8) or von Frey (P = 0.35) in the control paw. Neither rimonabant nor SR144528, in the absence of KML29, significantly affected responses in von Frey or acetone responses in either CCI or control paws.
Figure 9.
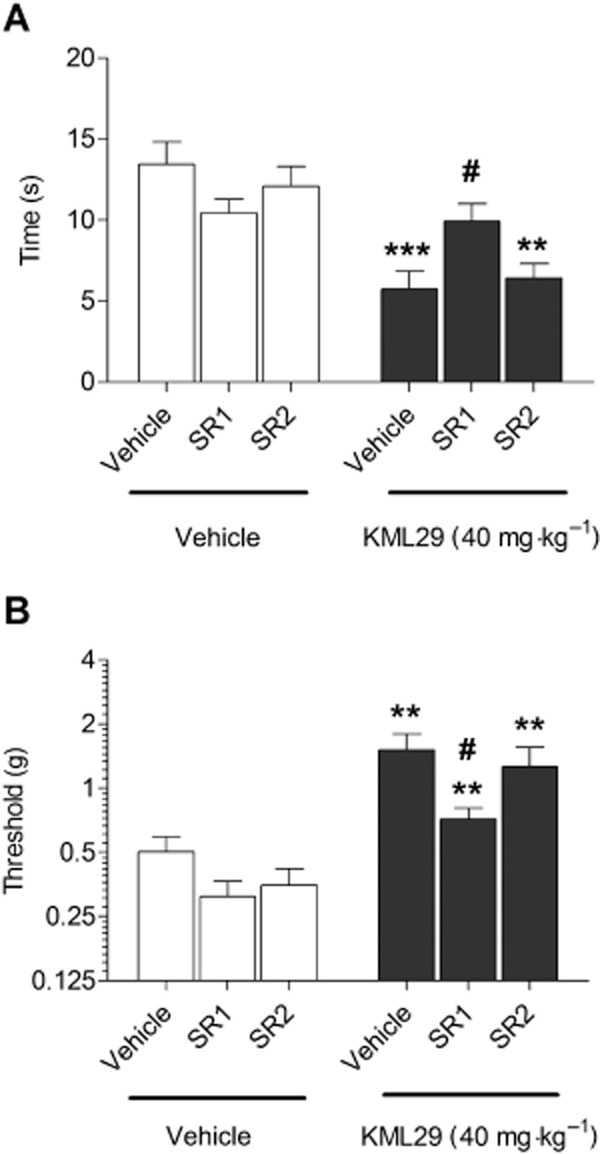
The anti-allodynic effects of KML29 (40 mg kg−1) in the CCI model of neuropathic pain are mediated by CB1, but not CB2 receptors. The CB1 receptor antagonist rimonabant (SR1, 3 mg·kg−1, 10 min pretreatment) prevented the anti-allodynic effects of KML29 (40 mg·kg−1, pretreatment time 2 h) in tests of cold (A) and mechanical allodynia (B), while the CB2 receptor antagonist SR144528 (SR2, 3 mg·kg−1, 10 min pretreatment) did not affect the anti-allodynic properties of KML29 in either assay (A, B). Data presented as mean ± SEM, n = 12 mice per group; **P < 0.01; ***P < 0.001 vs. vehicle-treated controls; #P < 0.05 vs. KML29 treated group pretreated with vehicle.
KLM29 blocked NSAID-induced gastric damage
Mice were fasted for 24 h and then the NSAID diclofenac was administered to induce gastric haemorrhages. Administration of KML29 dose-dependently blocked the development of gastric haemorrhages [F(4,34) = 11.5, P < 0.0001; Figure 10A]. Pretreatment with rimonabant (3 mg·kg−1) prevented the gastroprotective effects of KML29 [40 mg·kg−1; F(2,21) = 18.9, P < 0.0001; Figure 10B, left panel], while the CB2 receptor antagonist SR144528 was without effect (Figure 10B, right panel).
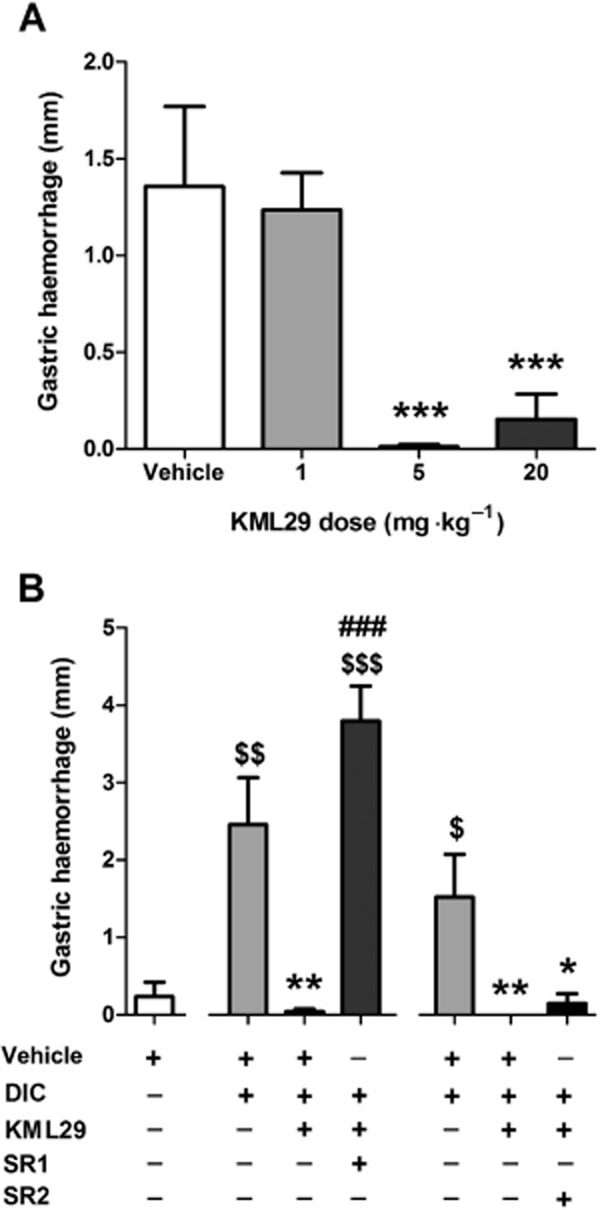
Protective effects of KML29 in the NSAID-induced gastric haemorrhage model, (A) KML29 dose-dependently prevented the development of gastric haemorrhages induced by oral administration of diclofenac (DIC, 100 mg·kg−1). (B) The gastroprotective effects of KML29 (40 mg·kg−1) were blocked by CB1 antagonist rimonabant (SR1, 3 mg·kg−1, 30 min pretreatment, left side of the panel), but not CB2 antagonist SR144528 (SR2, 3 mg·kg−1, 30 min pretreatment, right side of the panel). Data presented as mean ± SEM, n = 8–10 mice per group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle + DIC; ###P < 0.001 vs. KML29 + DIC; $P < 0.05, $$P < 0.01; $$$P < 0.001 vs. vehicle alone.
KML29 does not elicit cannabimimetic side effects
KML29 (40 mg·kg−1) produced significant antinociception in the tail withdrawal test [F(1,70) = 13.2, P < 0.001], which was most pronounced 2 and 4 h following KML29 administration (Figure 11A). However, KML29 did not elicit catalepsy (Figure 11B), hypothermia (Figure 11C) or decreases in locomotor activity (Figure 11D).
Figure 11.
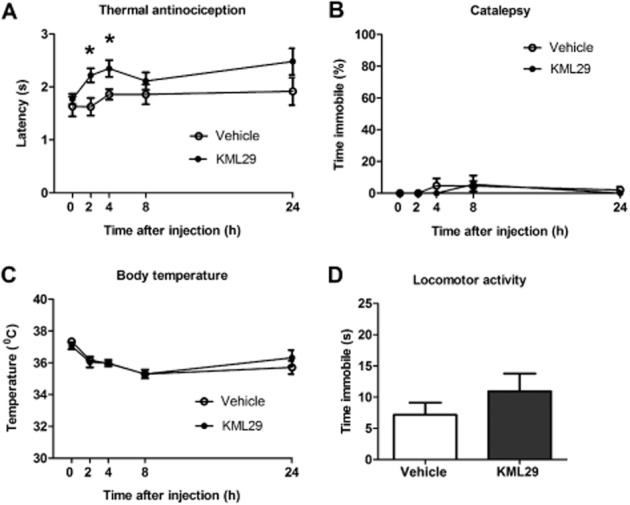
KML29 (40 mg·kg−1) produced significant antinociceptive effects in the tail withdrawal test 2 and 4 h (A) without the occurrence of catalepsy (B), hypothermia (C) or changes in locomotor activity (2 h following treatment) (D). Data presented as mean ± SEM; n = 8 mice per group; *P < 0.05 vs. vehicle.
Evaluation of KML29 in the drug discrimination paradigm
KML29 (40 mg·kg−1) failed to substitute for THC (Figure 12A) in C57BL/6J mice trained to discriminate THC (5.6 mg·kg−1) from vehicle and did not affect response rates at doses assessed (Figure 12B). These findings suggest that selective blockade of MAGL does not produce THC-like discriminative stimulus effects. However, KML29 fully and dose-dependently substituted for AEA in FAAH (–/–) mice [F(3,20) = 20.25, P < 0.001; Figure 13A]. KML29 produced full substitution at 40 mg·kg−1and partial AEA substitution at 20 mg·kg−1, but 5 mg·kg−1 failed to elicit any substitution for AEA. KML29 (40 mg·kg−1)-induced substitution for AEA was fully blocked by rimonabant [1 mg·kg−1; interaction F(1,20) = 66.83, P < 0.001; Figure 13C]. Rate of responding was not affected by KML29 administration in FAAH (–/–) mice (Figure 13B and D).
Figure 12.
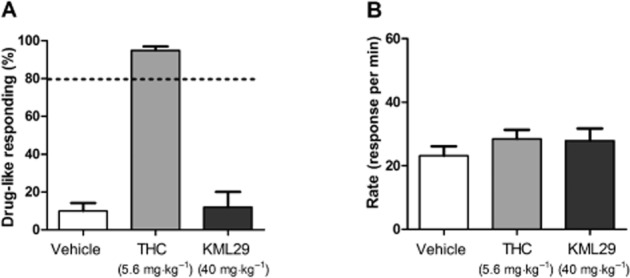
KML29 (40 mg·kg−1) did not substitute for THC (5.6 mg·kg−1) in C57BL/6J mice trained to discriminate THC (5.6 mg·kg−1) from vehicle (A). KML29 did not affect response rates (B) in the same animals. Data presented as mean ± SEM, n = 7 mice per group.
Figure 13.
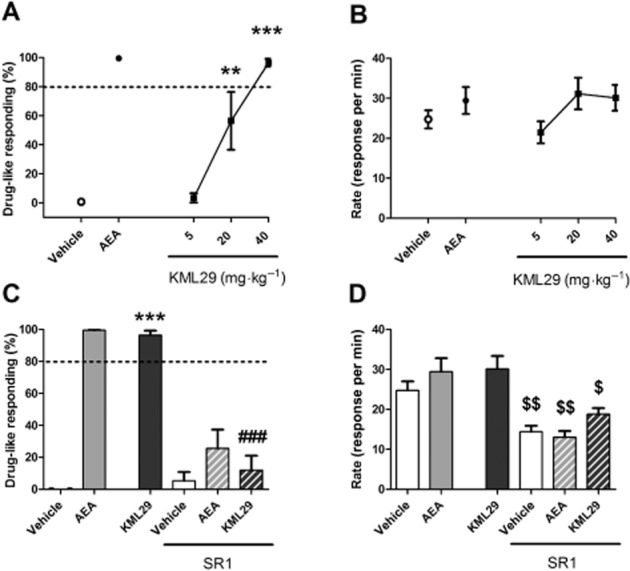
KML29 dose-dependently substituted for AEA (6 mg·kg−1) in FAAH (−/−) mice trained to discriminate AEA (6 mg·kg−1) from vehicle (A). KML29 (40 mg·kg−1) substitution for AEA was blocked by pretreatment with CB1 antagonist rimonabant (SR1, 1 mg·kg−1, 10 min pretreatment) (C). KML29 did not affect response rates under any condition (B, D). Rimonabant on its own produced a decrease in responding (D). Data presented as mean ± SEM, n = 6 mice per group; **P < 0.01, ***P < 0.001 vs. vehicle; ###P < 0.001 vs. KML29; $P < 0.05, $$P < 0.01 vs. appropriate control.
Discussion
Here, we report the results of an extensive evaluation of the selective MAGL inhibitor, KML29, in mice. Acute or repeated administration of KML29 significantly increased brain levels of 2-AG, but did not elevate brain AEA levels. This compound also produced significant anti-allodynic and anti-oedematous effects in the carrageenan model of inflammatory pain and partially reversed mechanical and cold allodynia in the CCI model of neuropathic pain. Moreover, KML29 protected against gastric damage induced by the NSAID diclofenac. Importantly, KML29 did not elicit THC-like discriminative stimulus effects in the drug discrimination paradigm and did not produce cannabimimetic effects in a battery of tests sensitive to the pharmacological actions of THC and other direct-acting CB1 receptor agonists.
Significant antinociceptive effects of KML29 in the tail withdrawal test were observed 2 and 4 h following KML29 administration, during which time levels of 2-AG in the brain significantly increased. The most pronounced anti-allodynic effects of KML29 occurred in the CCI model 3–6 h following KML29 treatment. It is important to note that peak 2-AG levels occurred at 4 h following acute drug administration. By 24 h after KML29 administration, no antinociceptive effects were observed in the tail withdrawal test or in the CCI model and 2-AG levels in the brain returned to baseline levels. No increases in whole brain levels of AEA or another FAAH substrate, OEA were observed at any time point following KML29 administration. A small increase in PEA levels was found; however, this finding was not replicated in the second experiment. KML29 significantly decreased whole brain levels of AA at 2, 4 and 24 h, which is consistent with the finding that MAGL represents a major biosynthetic enzyme of this eicosanoid in the brain (Nomura et al., 2011).
Using complementary pharmacological and genetic approaches, we found that the antinociceptive effects of KML29 were mediated through distinct cannabinoid receptor mechanisms of action. Its anti-oedematous actions in the carrageenan model did not occur in CB2 (−/−) mice or wild-type mice treated with SR144528. However, both CB1 and CB2 receptors were required for the anti-allodynic effects of KML29 in this assay. This differential involvement of cannabinoid receptor subtypes is in agreement with the effects of JZL184 in the carrageenan assay (Ghosh et al., 2013). In the CCI model of neuropathic pain, KML29 partially reversed mechanical allodynia and strongly reversed cold allodynia through a CB1 receptor mechanism, while CB2 receptors were expendable. These anti-allodynic effects were similar in magnitude as those produced by JZL184. Moreover, the present results replicate a previous report in which CB1 receptors, not CB2 receptors, mediated the antinociceptive properties of JZL184 in the CCI assay (Kinsey et al., 2009). It is noteworthy that both JZL184, which also inhibits FAAH, and KML29 yield a similar pattern of effects in the carrageenan and CCI models. Thus, both CB1 and CB2 receptors play necessary roles in the anti-allodynic effects of MAGL inhibitors in the carrageenan model of inflammatory pain, but only CB1 receptors are required for the anti-allodynic effects in the CCI neuropathic pain model.
Similarly, a differential involvement of cannabinoid receptor subtypes has also been observed in other models of nociception. In particular, both CB1 and CB2 receptors mediate the antinociceptive effects of JZL184 in formalin or capsaicin-induced thermal hyperalgesia (Spradley et al., 2010; Guindon et al., 2011), whereas only CB1 was found to mediate the antinociceptive effects in warm water tail withdrawal or acetic acid stretching tests (Long et al., 2009a; 2009b; Busquets-Garcia et al., 2011). Although differences observed in the various pain models could be related to the different end points being measured, this is not the case for the carrageenan and CCI models, as both employed von Frey filaments to assess tactile allodynia (Kinsey et al., 2009; Ghosh et al., 2013). Alternatively, this differential involvement of cannabinoid receptor subtypes in various models of pain could be due to multiple nociceptive mechanisms and different proportions of neural and inflammatory components of each nociceptive response. Accordingly, it is plausible that the CB2 receptor component in the anti-allodynic effects of MAGL inhibition in the various inflammatory pain models is associated with infiltration of immune cells that are known to have high CB2 expression (Galiegue et al., 1995). Likewise, activation of CB2 receptors associated with infiltration of immune cells during ongoing inflammatory process in the swollen paw likely mediated the anti-oedematous effects of MAGL inhibition. Conversely, the CB1 receptor component may be mediated by activation of these receptors located supraspinally, spinally or in peripheral nociceptors (Herkenham et al., 1991; Guindon and Hohmann, 2009; Starowicz and Przewlocka, 2012). The precise mechanism of involvement of cannabinoid receptor subtypes and their site of action in these nociceptive models remain to be determined.
Cannabinoid-induced antinociception involves supraspinal structures (e.g. periaqueductal gray, thalamus, rostral ventromedial medulla and amygdala), as well as the dorsal horn of the spinal cord (Guindon and Hohmann, 2009). In the CNS, MAGL is localized mainly presynaptically and is also expressed in microglia. Endocannabinoids are produced in dorsal root ganglion cells, a primary afferent input to spinal cord. However, anatomical localization of MAGL in the periphery is not known. Thus, the antinociceptive effects of MAGL inhibitors in pain models involving a strong inflammatory component, may involve both neuronal and non-neuronal sites of action, within the CNS and periphery.
There also appears to be differential involvement of the CB2 receptor in the CCI mouse model between FAAH and MAGL inhibitors (Kinsey et al., 2010). Whereas the anti-allodynic effects of FAAH inhibitors require both CB1 and CB2 receptors (Russo et al., 2007; Kinsey et al., 2009), the anti-allodynic effects of MAGL inhibitors require only CB1 receptors (Kinsey et al., 2009; 2010). As both AEA and 2-AG bind both cannabinoid receptors, a plausible explanation for the disparate involvement of CB2 receptors between the anti-allodynic effects of FAAH and MAGL inhibitors in the CCI mouse model may be related to differential production and release of these endogenous cannabinoids at key CB2 receptors. On the other hand, the antinociceptive effects of both FAAH and MAGL inhibitors in models of inflammatory pain require both CB1 and CB2 cannabinoid receptors (Guindon and Hohmann, 2009; Ghosh et al., 2013). To date, few studies have investigated the anatomical sites of action mediating the antinociceptive effects of MAGL and FAAH inhibitors (Jhaveri et al., 2006; Woodhams et al., 2012) Thus, further research in this area may reveal the mechanisms underlying differences between FAAH and MAGL inhibitors in various pain models.
Hydrolysis of 2-AG by MAGL represents an important pathway of AA formation in the brain (Nomura et al., 2011). Accordingly, the decreased pool of AA decreases brain levels of prostaglandins and pro-inflammatory cytokines following inflammatory insults (Nomura et al., 2011). Therefore, we cannot exclude the possibility that decreased prostaglandin synthesis, as a consequence of decreased availability of substrate for their production (i.e. AA), may at least partially contribute to the antinociceptive effects of KML29.
Repeated administration of high-dose, but not low-dose, JZL184 leads to functional tolerance in the carrageenan assay (Ghosh et al., 2013). Because repeated JZL184 significantly increased both 2-AG and AEA in the brain, it is difficult to discern whether the tolerance was driven primarily by the elevated levels of 2-AG or the elevated levels of AEA played a necessary role. Therefore, we used a high dose of KML29 (40 mg·kg−1) and administered daily over the course of 6 days to produce prolonged elevations of 2-AG, but not AEA, in the brain. Repeated KML29 administration increased 2-AG levels and concomitantly decreased free AA levels in the brain. Both effects were more pronounced following repeated KML29 treatment as compared with mice receiving KML29 acutely, suggesting an accumulation of 2-AG and decreased AA over time. Interestingly, a modest (less than 50%), but significant, decrease in AEA was found. However, no changes in other fatty acid amides were detected following KML29 administration. Thus, repeated administration of high-dose KML29 leads to tolerance to its anti-allodynic and anti-oedematous effects. These effects are both associated with prolonged and profound increased levels of 2-AG, but not AEA, in the brain. Repeated administration of high-dose KML29 decreased CP-55 940-stimulated [35S]GTPγS binding in the whole brain, which is consistent with the notion that prolonged brain 2-AG elevation contributes to the loss of anti-allodynic effects of KML29 upon repeated administration in the carrageenan model of inflammatory pain. However, further studies are needed to determine whether the CB1 receptor down-regulation/desensitization occurs differentially within discrete regions of the brain and spinal cord. Although the present study did not evaluate whether functional tolerance of the CB2 receptor occurs following repeated administration of a MAGL inhibitor, no change in CB2 receptor mRNA in the spleen was observed in MAGL (−/−) mice (Chanda et al., 2010). Accordingly, the impact of prolonged MAGL inhibition on CB2 receptor expression and function in the CNS as well as in the periphery remains to be evaluated.
It is important to note that the anti-allodynic and antioedematous effects, as well as gastroprotective and anxiolytic actions of JZL184 did not undergo tolerance and no impairment in CB1 receptor function was observed following repeated administration of low-dose JZL184 (Sciolino et al., 2011; Ghosh et al., 2013; Kinsey et al., 2013). These results suggest that partial MAGL inhibition may represent a therapeutic strategy to treat inflammatory and neuropathic pain disorders.
KML29 also produced a dose-dependent CB1 receptor mediated gastroprotective effect in the NSAID-induced gastric haemorrhage model. This observation is in agreement with previous studies showing that MAGL blockade with JZL184 protects against gastric haemorrhages induced by NSAIDs (Kinsey et al., 2011; 2013). Thus, MAGL inhibition may be an effective strategy for the prevention of NSAID-induced gastric ulcers. Although the CB2 receptor is traditionally viewed as having a strong anti-inflammatory function, the antihaemorrhagic effects of JZL184, as well as the phytocannabinoid THC, are prevented by genetic deletion or pharmacological antagonism of the CB1 receptor (Kinsey et al., 2011). In contrast, genetic deletion or pharmacological antagonism of the CB2 receptor had no effect. In addition, the FAAH inhibitor URB597 did not produce gastroprotective effects in CB1 (–/–) mice, but remained effective in CB2 (–/–) mice (Naidu et al., 2009). Thus, CB1 receptors are required for the gastroprotective effects of endocannabinoid catabolic enzyme inhibitors in the diclofenac-induced gastric ulcer model.
Importantly, high-dose KML29 (40 mg·kg−1) did not elicit catalepsy, hypothermia, or hypomotility. The lack of effects on catalepsy and hypothermia are in agreement with previous reports on actions of JZL184 (Long et al., 2009a; 2009b), although JZL184 has been reported to elicit hypomotility. Similar to JZL184 (Long et al., 2009a; 2009b; 2009c), KML29 elicited a small, but significant antinociceptive effect in the tail withdrawal test. Differences in the tetrad effects between JZL184 and KML29 could be due to the high selectivity of KML29 for MAGL i.e. JZL184 inhibits FAAH as well as peripheral enzymes (Chang et al., 2012), indicating that structural differences and slight differences in selectivity may impact the effects of these compounds.
In the drug discrimination assay, KML29 did not substitute for THC (5.6 mg·kg−1), which suggests that 10-fold increases of 2-AG in whole brain are not sufficient to produce THC-like discriminative stimulus effects. However, it is unclear whether the increased levels of 2-AG are signalling at CB1 receptors, which mediate the discriminative stimulus effects of THC. 2-AG is hypothesized to act as retrograde messengers in the brain and to be synthesized ‘on demand’ from phospholipid precursors (Di Marzo, 2009). Therefore, blockade of its degradation may not result in sufficient levels of 2-AG to stimulate CB1 receptors in circuits mediating the discriminative stimulus effects of THC. Whereas KML29 (40 mg·kg−1) failed to substitute for THC in C57BL/6J mice, it fully and dose-dependently substituted for AEA in FAAH (−/−) mice. Likewise, dual blockade of MAGL and FAAH using both pharmacological and genetic approaches produces THC-like effects (Long et al., 2009c). Accordingly, inhibition of a single endocannabinoid catabolic enzyme is not sufficient to produce THC-like cannabimimetic effects at doses that maximally inhibit MAGL. However, elevating both AEA and 2-AG fully substitutes for exogenously administered cannabinoid receptor agonists. AEA-like effects of KML29 in FAAH (−/−) mice were fully blocked by rimonabant, consistent with a CB1 receptor mechanism of action. KML29 did not affect rates of responding which is consistent with its lack of effects on spontaneous locomotor behavior, though other research has revealed decreases in locomotor behavior produced by MAGL inhibitor JZL184 (Long et al., 2009a; 2009b; 2009c).
In conclusion, the results of the present study indicate that selective MAGL inhibition produces beneficial antinociceptive and gastroprotective effects, without unwanted cannabimimetic side effects similar to those produced by Cannabis or THC. The observations that KML29 and JZL184 produce antinociceptive effects in a wide variety of animal models of pain, suggest that inhibition of MAGL represents a promising strategy to treat inflammatory and neuropathic pain, with no or minimal cannabimimetic side effects. Given its selectivity for MAGL and effectiveness in multiple species, KML29 offers a useful tool to investigate the consequences of blocking this enzyme in vivo.
Acknowledgments
Research was supported by US National Institutes of Health grants: DA009789, DA017259, DA026449 and DA005274 from the National Institute on Drug Abuse and Toni Rosenberg Memorial Fellowship. We would like to thank Khristina Bankhead for her technical assistance.
Glossary
- 2-AG
2-arachidonoylglycerol
- AEA
anandamide
- AA
arachidonic acid
- CCI
chronic constriction injury
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- THC
Δ9-tetrahydrocannabinol
Conflict of interest
None.
References
- Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov. 2009a;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009b;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C, et al. The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 acts in the nervous system to reverse LPS-induced tactile allodynia in mice. Br J Pharmacol. 2012;165:2485–2496. doi: 10.1111/j.1476-5381.2011.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burston JJ, Sim-Selley LJ, Harloe JP, Mahadevan A, Razdan RK, Selley DE, et al. N-arachidonyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonylglycerol through inhibition of monoacylglycerol lipase. J Pharmacol Exp Ther. 2008;327:546–553. doi: 10.1124/jpet.108.141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol Psychiatry. 2011;70:479–486. doi: 10.1016/j.biopsych.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Chang JW, Niphakis MJ, Lum KM, Cognetta AB, 3rd, Wang C, Matthews ML, et al. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol. 2012;19:579–582. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or to reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Orlando P, Wagner JA, et al. Biosynthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in circulating and tumoral macrophages. Eur J Biochem. 1999;264:258–267. doi: 10.1046/j.1432-1327.1999.00631.x. [DOI] [PubMed] [Google Scholar]
- Falenski KW, Thorpe AJ, Schlosburg JE, Cravatt BF, Abdullah RA, Smith TH, et al. FAAH–/– mice display differential tolerance, dependence, and cannabinoid receptor adaptation after delta 9-tetrahydrocannabinol and anandamide administration. Neuropsychopharmacology. 2010;35:1775–1787. doi: 10.1038/npp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF, et al. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci. 2013;92:498–505. doi: 10.1016/j.lfs.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju SK, Ueda N, Taniguchi K, Yamamoto S. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol. 1999;57:417–423. doi: 10.1016/s0006-2952(98)00314-1. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res. 2013;67:94–109. doi: 10.1016/j.phrs.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valinoc F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacol Res. 2011;64:60–67. doi: 10.1016/j.phrs.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, O'Neal ST, Long JZ, Mahadevan A, Cravatt BF, et al. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, et al. Repeated low dose administration of the monoacylglycerol lipase Inhibitor JZL184 retains CB1 receptor functional effects. J Pharmacol Exp Ther. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rana G, Russo R, Campolongo P, Bortolato M, Mangieri RA, Cuomo V, et al. Modulation of neuropathic and inflammatory pain by the endocannabinoid transport inhibitor AM404 [N-(4-hydroxyphenyl)-eicosa-5,8,11,14-tetraenamide. J Pharmacol Exp Ther. 2006;317:1365–1371. doi: 10.1124/jpet.105.100792. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004a;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004b;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Blankman JL, Cravatt BF. Endocannabinoid overload. Mol Pharmacol. 2010;78:993–995. doi: 10.1124/mol.110.069427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Okajima K, Murakami K, Harada N, Isobe H, Irie T. Role of neutrophil elastase in stress-induced gastric mucosal injury in rats. J Lab Clin Med. 1998;132:432–439. doi: 10.1016/s0022-2143(98)90114-7. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009b;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009c;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, et al. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav. 40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 1991;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Kinsey SG, Guo TL, Cravatt BF, Lichtman AH. Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J Pharmacol Exp Ther. 2010;334:182–190. doi: 10.1124/jpet.109.164806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies Press; 2011. [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okine BN, Norris LM, Woodhams S, Burston J, Patel A, Alexander SP, et al. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br J Pharmacol. 2012;167:627–640. doi: 10.1111/j.1476-5381.2012.02028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V. FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs. 2010;11:51–62. [PubMed] [Google Scholar]
- Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, et al. Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther. 2011;339:173–185. doi: 10.1124/jpet.111.181370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo EB. History of cannabis and its preparations in saga, science, and sobriquet. Chem Biodivers. 2007;4:1614–1648. doi: 10.1002/cbdv.200790144. [DOI] [PubMed] [Google Scholar]
- Russo EB, Jiang HE, Li X, Sutton A, Carboni A, del Bianco F, et al. Phytochemical and genetic analyses of ancient cannabis from Central Asia. J Exp Bot. 2008;59:4171–4182. doi: 10.1093/jxb/ern260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciolino NR, Zhou W, Hohmann AG. Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradley JM, Guindon J, Hohmann AG. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res. 2010;62:249–258. doi: 10.1016/j.phrs.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K, Przewlocka B. Modulation of neuropathic-pain-related behaviour by the spinal endocannabinoid/endovanilloid system. Philos Trans R Soc Lond B Biol Sci. 2012;367:3286–3299. doi: 10.1098/rstb.2011.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentiny DM, Vann RE, Mahadevan A, Kottani R, Gujjar R, Wiley JL. Novel 3-substituted rimonabant analogs lack Δ(9)-tetrahydrocannabinol-like abuse-related behavioral effects in mice. Br J Pharmacol. 2013;169:10–20. doi: 10.1111/bph.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise LE, Cannavacciulo R, Cravatt BF, Martin BF, Lichtman AH. Evaluation of fatty acid amides in the carrageenan-induced paw edema model. Neuropharmacology. 2008;54:181–188. doi: 10.1016/j.neuropharm.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams SG, Wong A, Barrett DA, Bennett AJ, Chapman V, Alexander SP. Spinal administration of the monoacylglycerol lipase inhibitor JZL184 produces robust inhibitory effects on nociceptive processing and the development of central sensitization in the rat. Br J Pharmacol. 2012;167:1609–1619. doi: 10.1111/j.1476-5381.2012.02179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]