

Abstract
Background and Purpose
A conserved amino acid within a protein family indicates a significance of the residue. In the centre of transmembrane helix (TM)-5, position V:13/5.47, an aromatic amino acid is conserved among class A 7TM receptors. However, in 37% of chemokine receptors – a subgroup of 7TM receptors – it is a leucine indicating an altered function. Here, we describe the significance of this position and its possible interaction with TM-3 for CCR5 activity.
Experimental Approach
The effects of [L203F]-CCR5 in TM-5 (position V:13/5.47), [I116A]-CCR5 in TM-3 (III:16/3.40) and [L203F;G286F]-CCR5 (V:13/5.47;VII:09/7.42) were determined in G-protein-and β-arrestin-coupled signalling. Computational modelling monitored changes in amino acid conformation.
Key Results
[L203F]-CCR5 increased the basal level of G-protein coupling (20–70% of Emax) and β-arrestin recruitment (50% of Emax) with a threefold increase in agonist potency. In silico, [I116A]-CCR5 switched χ1-angle in [L203F]-CCR5. Furthermore, [I116A]-CCR5 was constitutively active to a similar degree as [L203F]-CCR5. Tyr244 in TM-6 (VI:09/6.44) moved towards TM-5 in silico, consistent with its previously shown function for CCR5 activation. On [L203F;G286F]-CCR5 the antagonist aplaviroc was converted to a superagonist.
Conclusions and Implications
The results imply that an aromatic amino acid in the centre of TM-5 controls the level of receptor activity. Furthermore, Ile116 acts as a gate for the movement of Tyr244 towards TM-5 in the active state, a mechanism proposed previously for the β2-adrenoceptor. The results provide an understanding of chemokine receptor function and thereby information for the development of biased and non-biased antagonists and inverse agonists.
Keywords: CCR5, 7TM, GPCR, arrestin, chemokines, computer modelling, constitutive activity, superagonist, US28, biased signalling
Introduction
The superfamily of seven-transmembrane domain (7TM) receptors (Alexander et al., 2013) is conserved in diverse organisms such as mammals, yeast and viruses. The 7TM receptors regulate a variety of physiological mechanisms including sensory control, hormone production, cardiovascular balance and the immune system. The latter is highly dependent on signalling mediated by chemokine binding – a system responsible for leukocyte migration to sites of inflammation and during homeostasis. Malfunction of this system can result in autoimmune diseases as well as angiogenesis, cancer progression and metastasis (Viola and Luster, 2008). Furthermore, viruses have developed mechanisms to exploit the chemokine system. For example, HIV uses the chemokine receptors CCR5 and CXCR4 as co-receptors for cell entry (Deng et al., 1996; Feng et al., 1996), whereas several herpes viruses have acquired ligands for various human chemokine receptors (Kledal et al., 1997; Lüttichau et al., 2007). An essential component of the viral 7TM signalling and link to pathology is the ligand-independent activity. For instance, it has been suspected that the high constitutive activity, rapid recycling and chemokine-binding promiscuity of US28 (a human cytomegalovirus-encoded chemokine receptor) enables it to scavenge chemokines and thereby limit the inflammatory immune response (Bodaghi et al., 1998) in addition to its tumourigenic properties (Maussang et al., 2006).
Most human 7TM receptors are constrained towards inactivation with almost no constitutive activity and with the majority of small organic, drug-like ligands acting as antagonists (Schwartz et al., 2006). However, despite this inactive property, it has been shown that single amino acid substitutions may increase the basal receptor activity, for example, mutation of Glu113 [III:04/3.28, retinal Schiff base counter-ion in dark state rhodopsin (the generic numbering system proposed by Baldwin (Baldwin, 1993) and modified by Schwartz (Schwartz, 1994) followed by the Ballesteros/Weinstein numbering system (Ballesteros and Weinstein, 1995) are used in this paper)] or Lys296 (VII:10/7.43, retinal attachment site) in opsin (Robinson et al., 1992) and Asp130 (III:25/3.49, part of the ionic lock) in the β2-adrenoceptor (Rasmussen et al., 1999). It is conceivable that shifting the equilibrium between the inactive and active conformations in this way also will make the receptor ‘agonist-prone’, that is, more susceptible to ligand-induced activation. Indeed, some constitutively activating mutations (CAMs) cause a ligand ‘gain-of-function’, for example, antagonists/inverse agonists are converted into agonists or the potency of a given agonist enhances concomitantly with the degree of constitutive activity as seen in the dopamine D1 receptor and the bradykinin B2 receptor (Cho et al., 1996; Marie et al., 1999). The importance of naturally occurring CAMs has been illustrated in various pathologies, for example, in rhodopsin causing retinitis pigmentosa (Robinson et al., 1992) and in the thyrotropin receptor resulting in hyperthyroidism (Parma et al., 1993).
We have previously shown that a steric hindrance mutation in the centre of TM-7 of CCR5 [i.e. [G286F]-CCR5 (VII:09/7.42)] induced an efficacy switch conversion of an antagonist to an agonist and an increase in the basal level of activation through Gi (Steen et al., 2013). In contrast, the same amino acid substitution completely eliminated β-arrestin recruitment and thus induced functional selectivity or biased signalling. In line with our results, it has been shown that TM-7 is involved in β-arrestin binding as evident from the crystal structure of the β1-adrenoceptor, where the β-arrestin-biased agonists carvedilol and bucindolol interacted with additional residues in TM-7 compared with unbiased ligands (Warne et al., 2012). In addition, two papers simultaneously reported that β-arrestin-biased ligands of the β2-adrenoceptor and the arginine-vasopressin receptor type 2 provoked changes primarily in TM-7 (and not TM-6 as G-protein agonists did) measured in [19F]-NMR and a fluorescence-based assay respectively (Liu et al., 2012; Rahmeh et al., 2012). Biased ligands have great potential as drug candidates since they theoretically can minimize unwanted effects caused by activation of non-specific signalling pathways. For example, a ligand for the nicotinic GPR109A receptor (MK-0354) mediates beneficial anti-lipolytic effects through Gi but does not affect the β-arrestin pathway and thus does not cause the unwanted side effect of flushing that unbiased GPR109A agonists do (Richman et al., 2007; Semple et al., 2008).
We show here that an aromatic amino acid in position V:13/5.47 controls the level of basal activity in both CCR5 ([L203F-CCR5]) and the viral highly constitutive active counterpart US28 ([F197A]-US28). In silico analyses of CCR5 receptors show a rotamer change of Ile116 (III:16/3.40), and a slight movement of Tyr244 (VI:09/6.44) towards [L203F]-CCR5, which could explain the constitutive activity. An overview of the relevant positions for this paper is given in Figure 1A.
Figure 1.

Ligand-independent activation of CCR5 WT and [L203F]-CCR5. Helical wheel diagram of CCR5 indicating central residues (white on black) either mutated or of general importance for CCR5 (A). The most conserved amino acid in each TM is indicated (black on grey). (B) Alignment of class A 7TM receptors discussed in the text or similar to CCR5. Position V:13/5.47 is indicated. In addition, the level of constitutive activity in CCR5 WT and [L203F]-CCR5 in four different signalling pathways is depicted (C and D). The β-arrestin recruitment was assessed in U20S cells whereas COS-7 cells were used for the remaining. The data were normalized to CCL3 Emax on the respective receptor (C) or forskolin-induced cAMP activation in untransfected cells (shown as inhibition) (D). (E) Surface expression measured with elisa in COS-7 cells using N-terminal FLAG-tagged receptors. Data were normalized to WT. Statistical significance was calculated using Student's unpaired t-test. **P < 0.05, ***P < 0.001; ns, not significant; n = 3–25.
Methods
Materials
Human CCL3 and CCL5 were purchased from Peprotech. Human CCR5 cDNA was cloned from a spleen-derived cDNA library. TAK-779 and aplaviroc were kindly provided by Gary Bridger (AnorMED, Langley, Canada). [125I]-CCL3 was purchased from PerkinElmer (Boston, MA, USA). The chimeric G-protein GαΔ6qi4myr [Gqi4myr, converts Gi-related signalling into a Gq readout (Kostenis et al., 1998)] was kindly provided by Evi Kostenis (University of Bonn, Germany).
Molecular biology
FLAG-tagged receptor cDNA was cloned into expression vectors pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA; PI turnover, cAMP, [35S]-GTPγS binding, and competition binding) or pCMV-ProLink™1 (DiscoveRx, Birmingham, UK; β-arrestin recruitment). Mutations were constructed using QuikChange™ Site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions. All mutations were verified by DNA sequence analysis.
Transfections and tissue culture
COS-7 cells were grown at 10% CO2 and 37°C in DMEM 1885 supplemented with 10% FBS, 2 mM glutamine, 180 U·mL−1 penicillin and 45 μg·mL−1 streptomycin. PathHunter U2OS β-arrestin 2 cell line (DiscoveRx) were grown at 5% CO2 and 37°C in MEMα Glutamax medium (Invitrogen) supplemented with 10% FBS, 180 U·mL−1 penicillin, 45 μg·mL−1 streptomycin and 0.25 mg·mL−1 Hygromycin B (Invitrogen). Transfection of COS-7 cells for PI turnover, [35S]-GTPγS binding and competition binding was performed using the calcium phosphate precipitation method as previously described (Kissow et al., 2012) or by using Lipofectamine™ 2000 (Invitrogen; cAMP and elisa). U2OS cells were transfected using FuGENE® 6 Transfection reagent (Roche, Mannheim, Germany).
PI turnover
COS-7 cells were co-transfected with receptor cDNA and Gqi4myr. One day after transfection, the cells were seeded in 24-well plates (1.5 × 105 cells per well) and incubated with 2 μCi of [3H]-myo-inositol in 0.3 mL growth medium for 24 h. Cells were washed twice with HBSS supplemented with CaCl2 and MgCl2 and afterwards incubated for 15 min in buffer with 10 mM LiCl before ligand addition followed by 90 min incubation. The [3H]-inositol phosphate generated was purified on AG 1X8 anion exchange resin. Determinations were made in duplicate.
Membrane preparation
Membranes were prepared from transfected COS-7 cells. The cells were harvested with a rubber policeman in ice-cold PBS and homogenized with a Dounce homogenizer. The homogenate was centrifuged and the supernatants were collected and centrifuged at 21 000 × g at 4°C. The resulting membrane pellets were resuspended in 20 mM HEPES buffer containing 2 mM MgCl2 and complete protease inhibitor (Roche). The protein concentration was determined using the BCA protein assay kit (Pierce, Rockford, IL, USA).
[35S]-GTPγS binding
The membrane preparation (20 μg protein per well, 96-well plates) was diluted in assay buffer (50 mM HEPES, 2 mM MgCl2, 50 mM NaCl, 1 mM EGTA, 1 μM GDP, 0.1% BSA and complete inhibitor). CCL3 was added followed by [35S]-GTPγS (1250 Ci·mmol−1; 12.5 mCi·mL−1; PerkinElmer) diluted in assay buffer (1 nM). The membranes were incubated for 1 h at room temperature. Wheat germ agglutinin-coupled scintillation proximity assay beads (GE Healthcare, Buckinghamshire, UK) were added followed by 30 min incubation at room temperature. The radioactivity was measured on a TopCount scintillation counter (PerkinElmer). Non-specific binding was determined by adding unlabelled GTPγS (40 μM).
cAMP accumulation
COS-7 cells (35 000 cells per well) were seeded in 96-well plates one day before transfection. Two days after transfection, the cells were washed twice with HEPES-buffered saline (HBS) buffer and incubated with HBS and 1 mM IBMX for 30 min at 37°C. Forskolin (Sigma-Aldrich, St. Louis, MO, USA) was added and the cells were incubated for 30 min at 37°C. The HitHunter™ cAMP XS+ assay (DiscoveRx) was carried out according to the manufacturer's instructions. Determinations were made in triplicate.
β-Arrestin recruitment
Recruitment of β-arrestin was measured using the PathHunter™ β-arrestin assay (DiscoveRx). WT (wild type) CCR5 and mutants were fused with the ProLink™ pk1-tag (a small fragment of the enzyme β-galactosidase) and cloned into pCMV. Assays were performed in U20S cells stably expressing β-arrestin2 coupled to the large β-galactosidase fragment. Cells were seeded in 96-well plates, 20 000 cells per well and transfected the following day with 50 ng DNA using FuGENE® 6 (0.15 μL per well); 24 h after transfection, the medium was removed and 100 uL Opti-MEM® I (Gibco®, Carlsbad, CA, USA) was added. The following day, cells were stimulated with varying concentrations of agonist for 90 min at 37°C. The Detection Reagent Solution® (DiscoveRx) was added and incubated at room temperature for 60 min. β-arrestin recruitment was measured as chemiluminescence using Perkin Elmer EnVision 2104 Multilable Reader.
[125I]-CCL3 competition binding
COS-7 cells were seeded in wells 1 day after transfection with the number of cells seeded per well aimed at obtaining 5–10% specific binding of the added radioactive ligand (3–15 × 105 cells per well). Two days after transfection, cells were assayed by competition binding for 3 h at 4°C using 20–70 pM [125I]-CCL3 as well as unlabelled ligand in 50 mM HEPES buffer, pH 7.4, supplemented with 1 mM CaCl2, 5 mM MgCl2 and 0.5% (w v-1) BSA. After incubation, cells were washed in ice-cold binding buffer with 0.5 M NaCl. Non-specific binding was determined as the binding in the presence of 0.1 μM unlabelled CCL3. Determinations were made in duplicates.
elisa
COS-7 cells were transfected with FLAG-tagged (M1) receptor DNA in 96-well plates (3.5 × 104 cells per well). Two days after transfection, cells were washed in Tris-buffered saline (TBS), fixed in 3.7% formaldehyde for 15 min at room temperature, washed and incubated in blocking solution (TBS with 2% BSA). Cells were incubated for 2 h with anti-FLAG antibody (Sigma-Aldrich) at 2 μg·mL−1 in TBS with 1 mM CaCl2 and 1% BSA. After three washes with TBS/CaCl2/BSA, the cells were incubated with goat anti-mouse HRP-conjugated antibody (Abcam, Cambridge, UK). After extensive washing, the immunoreactivity was revealed by addition of TMB Plus substrate (Kem-En-Tec, Taastrup, Denmark), and the reaction was stopped with 0.2 M H2SO4. Absorbance was measured at 450 nm on a Wallac VICTOR2 plate reader (PerkinElmer).
Calculations
IC50/EC50 and Kd/Ki values and statistical significance were determined by non-linear regression and Bmax values were calculated using the GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA).
CCR5 comparative models and docking of aplaviroc
Computational modelling was performed as described previously (Steen et al., 2013). Briefly, a pair-wise sequence alignment, and the construction of a comparative homology model of the human CCR5 receptor was produced in the Internal Coordinate Mechanics (ICM) software package (Molsoft LLC, La Jolla, CA, USA) using human CXCR4 (PDB entry 3ODU; Wu et al., 2010) as template. The CCR5 models were subjected to full-atom structure relaxation using the ROSETTA membrane force field (Barth et al., 2007) in Rosetta 3.2.1 (Leaver-Fay et al., 2011). Full flexible ligand docking was performed using the biased probability Monte Carlo docking routine in ICM (Totrov and Abagyan, 2008; Bottegoni et al., 2009). Individual best scored docking poses were optimized using a combined Monte Carlo and minimization procedure (using the MMFF94 force field). A final stack of 50 conformations was generated, scored and manually analysed to identify the complexes between aplaviroc and CCR5.
Conformational sampling and statistics of rotamer states and atomic distances
The [L203F]-CCR5 receptor variant was constructed from the initial CCR5 WT model using the residue substitution function in the Rosetta 3.2.1 (Leaver-Fay et al., 2011). CCR5 WT as well as [L203F]-CCR5 were subjected to full-atom structure relaxation using the ROSETTA membrane force field (Barth et al., 2007) in Rosetta 3.2.1 (Leaver-Fay et al., 2011) to optimize the structures and repack the side chain. A total of 1000 models were generated for both receptors. Statistics on side chain rotamer states and atomic distances were analysed using customized scripts and function in the CCP4 software package (Winn et al., 2011).
Phylogenetic tree
Amino acid sequence alignments of gene sequences were generated using the MAFFT multiple-aligner plug-in of Geneious Pro 5.1.4 software (Biomatters Ltd, Auckland, New Zealand; Katoh et al., 2002) and phylogenetic trees were determined (all alignments are available on request). Phylogenetic relationships were investigated with trees built by the maximum likelihood (ML) method, using the PhyML 2.0.1 plug-in (Guindon et al., 2010).
Results
L203F increases the activity of CCR5
Position V:13/5.47 is an aromatic residue among 81% of class A receptors (Mirzadegan et al., 2003). Of the chemokine receptors, 37% contain a Leu, as is the case for CCR5 (Leu203), together with CCR1-4, CCR8 and CXCR4. This residue is important in several 7TM receptors, for example, the ghrelin receptor, GPR39, GPR119, the β2-adrenoceptor and the NK1 receptor (all of which are Gs-or Gq-coupled) where removal of the Phe in this position reduced the level of constitutive activity and agonist-dependent activity (Holst et al., 2010; see alignment in Figure 1B). This prompted us to investigate the importance of this position in a Gi-coupled receptor, CCR5. [L203F]-CCR5 was constitutively active in all four signalling pathways examined (Figure 1C and D). Thus, the mutant bound ∼65% more GTP than WT in [35S]-GTPγS binding and induced PI turnover to ∼20% of Emax in the absence of an agonist. CCR5 WT displayed ∼30% constitutive activity in β-arrestin recruitment, as we have shown previously (Steen et al., 2013); however, [L203F]-CCR5 displayed a basal activity to ∼50% relative to Emax of CCL3 (Figure 1C). Furthermore, [L203F]-CCR5 ligand independently inhibited 60% of the level of forskolin-induced cAMP accumulation in untransfected cells, whereas no inhibition was observed for CCR5 WT (Figure 1D). In G-protein signalling, the potencies of both CCL3 and CCL5 were similar to WT, whereas in β-arrestin recruitment, the potencies were about threefold increased (Table 1). Homologous competition binding against [125I]-CCL3 showed that the affinity to [L203F]-CCR5 was twofold higher than CCR5 WT (Table 2). Figure 1E shows that the expression level of WT and the mutant receptor measured in elisa were similar implying that the increase in basal activity is not a result of receptor overexpression. In contrast, a nearly threefold increased Bmax value was observed for [L203F]-CCR5, suggesting an increase in the maximum CCL3-binding capacity (Rosenkilde and Schwartz, 2000).
Table 1.
Expression level and functional analyses of agonists on CCR5 WT and mutants as measured in PI turnover and β-arrestin recruitment
| PI turnover | β-Arrestin recruitment | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression level | Constitutive activity (%) | Emax | CCL3 | CCL5 | Constitutive activity (%) | Emax | CCL3 | CCL5 | ||||||||||
| Residue | % of WT ± SEM | (basal/Emax) × 100 | % of WT ± SEM | log EC50 ± SEM | Fold EC50 | (n) | log EC50 ± SEM | Fold EC50 | (n) | (basal/Emax) × 100 | % of WT ± SEM | log EC50 ± SEM | Fold EC50 | (n) | log EC50 ± SEM | Fold EC50 | (n) | |
| CCR5 WT | 100 ± 0.00 | 1.6 ± 1.8 | 100 ± 0.00 | −8.1 ± 0.07 | 1.0 | (41) | −9.1 ± 0.07 | 1.0 | (37) | 29 ± 2.2 | 100 ± 0.00 | −8.1 ± 0.08 | 1.0 | (40) | −8.4 ± 0.24 | 1.0 | (8) | |
| I116A | 102 ± 6.0 | 27 ± 3.3*** | 97 ± 16 | −8.1 ± 0.28 | 1.1 | (6) | −9.1 ± 0.34 | 0.93 | (4) | 52 ± 4.6** | 91 ± 13 | −7.9 ± 0.16 | 1.8 | (9) | −7.4 ± 0.41 | 9.5** | (4) | |
| L203F | 108 ± 10 | 21 ± 1.7*** | 81 ± 17 | −8.0 ± 0.09 | 1.3 | (7) | −9.2 ± 0.14 | 0.84 | (4) | 50 ± 3.0*** | 88 ± 8.5 | −8.5 ± 0.16 | 0.35** | (15) | −8.8 ± 0.22 | 0.36 | (4) | |
| L203F+G286F | 13 ± 1.7*** | 38 ± 3.1*** | 21 ± 1.5*** | −7.8 ± 0.14 | 2.0 | (4) | −9.0 ± 0.78 | 1.2 | (4) | NA | NA | NA | (9) | NA | (3) | |||
Significant change from WT calculated by Student's unpaired t-test.
P < 0.05,
P < 0.001.
NA, no activity.
Table 2.
Binding affinities of CCL3 and small-molecule antagonists on CCR5 WT and mutants
| Bmax ± SEM | CCL3 | TAK-779 | Aplaviroc | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Residue | % of WT | log Kd ± SEM | Fold Kd | (n) | log Ki ± SEM | Fold Ki | (n) | log Ki ± SEM | Fold Ki | (n) |
| CCR5 WT | 100 ± 0.00 | −8.4 ± 0.06 | 1.0 | (19) | −7.6 ± 0.05 | 1.0 | (15) | −7.3 ± 0.03 | 1.0 | (15) |
| I116A | 63 ± 16* | −7.9 ± 0.09 | 2.7** | (4) | NT | −6.9 ± 0.02 | 2.3** | (2) | ||
| L203F | 82 ± 14 | −8.6 ± 0.10 | 0.49** | (4) | −7.7 ± 0.05 | 0.94 | (3) | −7.1 ± 0.02 | 1.4 | (3) |
| L203F+G286F | 38 ± 11** | −9.0 ± 0.34 | 0.21*** | (3) | −7.0 ± 0.21 | 4.5** | (3) | −6.8 ± 0.26 | 2.8** | (3) |
Bmax values were calculated from the homologous binding curves.
Significant change from WT calculated by Student's unpaired t-test,
P < 0.1,
P < 0.05,
P < 0.001.
NT, not tested.
We recently described an ‘efficacy switch’ of aplaviroc, a well-characterized CCR5 small-molecule antagonist; upon mutations, (both CAMs and non-CAMs, in the centre of TM-6 and −7) aplaviroc switched from being antagonist to agonist (Steen et al., 2013). On CCR5 WT, aplaviroc acts as a full antagonist with no intrinsic activity (Figure 2A and B) and with a potency and an affinity similar to previously published data (Maeda et al., 2004). However, when testing aplaviroc on [L203F]-CCR5, it was converted to a partial agonist for PI turnover, that is, it only partially inhibited CCL3-induced activation and activated the receptor on its own (Figure 2C). The antagonistic potency (i.e. inhibition of CCL3-induced activation) was decreased approximately fivefold, whereas the agonistic potency was increased approximately threefold compared with the antagonistic WT potency (Table 3). In contrast, aplaviroc fully inhibited CCL3-induced β-arrestin recruitment on [L203F]-CCR5, with an approximately ninefold decrease in potency (Table 3, Figure 2D). The affinity of aplaviroc on [L203F]-CCR5 was in the same range as on WT (Table 2).
Figure 2.
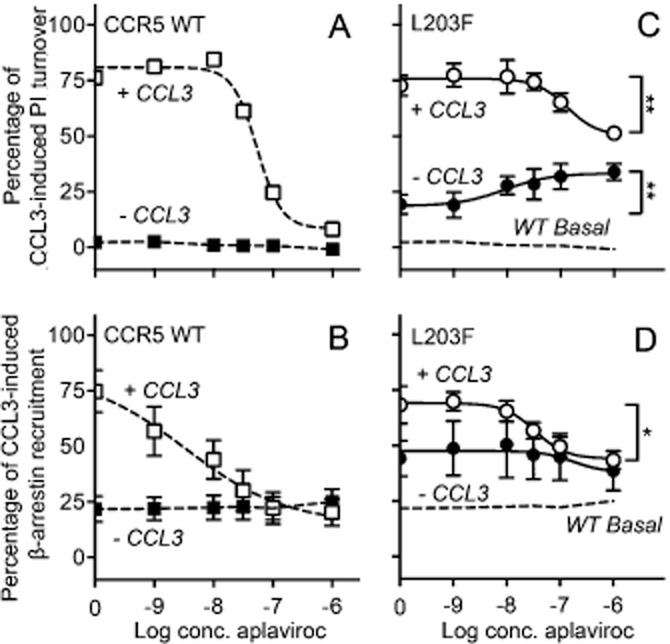
Effect of aplaviroc on PI turnover and β-arrestin recruitment when acting on [L203F]-CCR5 and WT. The effect of aplaviroc was measured as G-protein signalling (PI turnover in COS-7 cells, A and C) and β-arrestin recruitment in U20S cells (B and D). The graphs represent CCR5 WT (A and B) and [L203F]-CCR5 (C and D) and are shown for aplaviroc with CCL3 and without CCL3. The data were normalized to Emax of CCL3 on the respective receptor. The basal level of activation for CCR5 WT is indicated with dashed lines on the graphs for the mutant (C and D). Statistical significance between untreated cells and cells treated with maximum concentration of aplaviroc was calculated using Student's unpaired t-test. *P < 0. 1, **P < 0.05, ***P < 0.001; ns, not significant; n = 4–13.
Table 3.
PI turnover and β-arrestin recruitment in response to antagonists on CCR5 WT and mutants
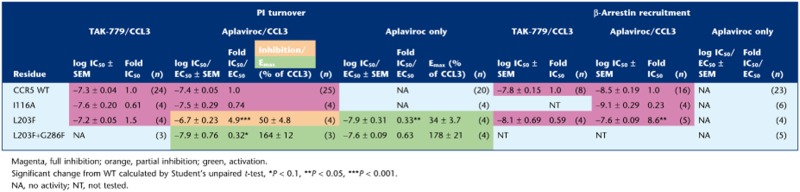 |
To investigate whether this was a phenomenon observed for aplaviroc specifically and not for CCR5 antagonists in general, we examined TAK-779, another well-described CCR5 small-molecule antagonist (Baba et al., 1999). As with aplaviroc, TAK-779 inhibited CCL3-induced PI turnover and β-arrestin recruitment and displaced the chemokine on CCR5 WT with a potency and affinity that resembled previously published data (Shiraishi et al., 2000; Tables 1998 and 1999 respectively). However, in contrast to aplaviroc, TAK-779 acted as a full antagonist for both PI turnover and β-arrestin recruitment on [L203F]-CCR5 with potencies in the same range as WT (Table 3). Similarly, the affinity was similar to the WT (Table 2).
L203F induces rotation of Ile116 and sliding of Tyr244 in silico
To further investigate the molecular mechanism behind the increased activation of [L203F]-CCR5, a computational model of WT and [L203F]-CCR5 in complex with aplaviroc was constructed. Residues possibly interacting with Leu/Phe203 were examined for changes in side chain movement and rotation in 1000 models constructed for both WT and [L203F]-CCR5. Measurement of χ1-angles in CCR5 WT showed that the distribution of the torsion angle of Ile116 (III:16/3.40) was gauche− (−60°) in 90% and trans (180°) in 10%. Upon mutation of Leu203 to Phe, the distribution of the χ1-angle of Ile-116 changed to trans in 98% (Figure 3A and B). Importantly, no change was observed in the distribution of χ1-angles in the neighbouring residues: Phe112 (III:12/3.36), Tyr244 (VI:09/6.44), Trp248 (VI:13/6.48) and Tyr251 (VI:16/6.51). However, a tendency to ‘sliding’ of the side chain of Tyr244 towards TM-5 was observed: from being on average 8.94 ± 0.01 Å (mean ± SEM, calculated for all 1000 models) away from Leu203 in the WT models, it moved to only 8.25 ± 0.01 Å away from Phe203 in the mutant as measured from C4 in the phenol ring of Tyr244 to the Cα of Leu/Phe203 (Figure 3C and D). Furthermore, the distance between the Cγ1 of Ile116 and C4 in Tyr244 was increased in [L203F]-CCR5 compared with WT (5.1 ± 0.01 Å vs. 4.0 ± 0.01 Å), indicating disruption of the van der Waals interaction. Consistent with these data, we have previously demonstrated that Tyr244 plays an essential role in the activation of CCR5 (Steen et al., 2013).
Figure 3.
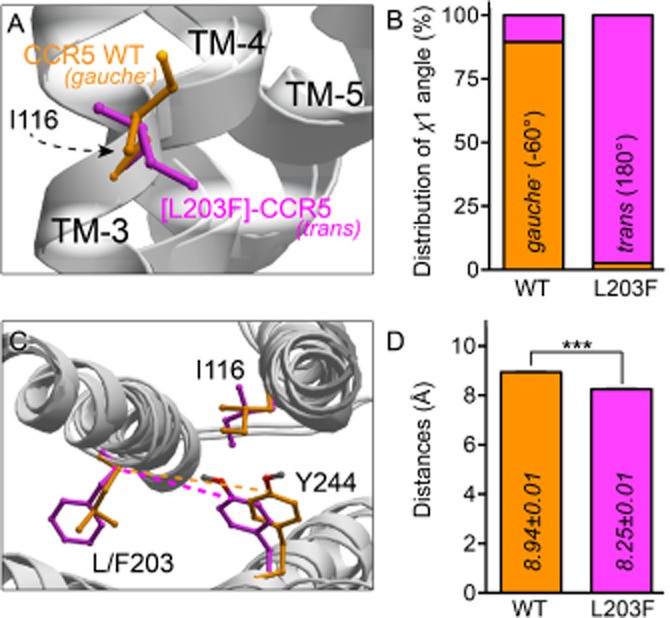
Computational modelling of CCR5 WT and [L203F]-CCR5. In silico models of CCR5 WT (orange) and [L203F]-CCR5 (magenta). (A) Side view of the orientation of the χ1-angle of Ile116 and (B) the percentage distribution between gauche− (−60°) and trans (180°) measured in 1000 models of CCR5 WT and [L203F]-CCR5 respectively. (C) Top view of TM-3, TM-5 and TM-6 showing an example of the distance between Tyr244 and Leu/Phe203 (dashed lines emphasize the difference) for both WT and mutant receptor. (D) Mean distances between Tyr244 and Leu/Phe203 calculated for all 1000 models for each receptor. Statistical significance was calculated using Student's unpaired t-test. ***P < 0.001; n = 1000.
Elimination of Ile116 provokes constitutive activity similar to [L203F]-CCR5
To confirm that Ile116 is involved in the activation mechanism, [I116A]-CCR5 was constructed. Interestingly, for both PI turnover and β-arrestin recruitment, this mutation induced constitutive activity similar to [L203F]-CCR5 (Figure 4A compared with Figure 1C). As for [L203F]-CCR5, the increase in basal activity was not caused by an increase in overall receptor expression (Figure 4B), but rather in high-affinity CCL3 conformations indicated by an increased Bmax value (Table 2). The potency of CCL3 and CCL5 on [I116A]-CCR5 was similar to WT for PI turnover but 2–10-fold lower for β-arrestin recruitment (Table 1), whereas the affinity of CCL3 was slightly decreased (2.7-fold) compared with WT (Table 2).
Figure 4.
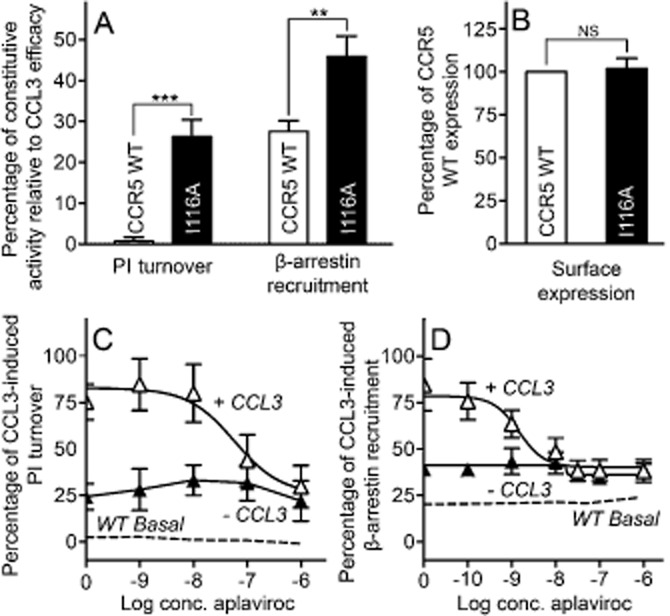
Ligand-independent and aplaviroc-induced activation of [I116A]-CCR5. Constitutive activity normalized to Emax of the respective receptor in PI turnover and β-arrestin recruitment (A) and surface expression normalized to WT (B) of CCR5 WT (white columns) and [I11A]-CCR5 (black columns). (C and D) effect on PI turnover (C) and β-arrestin recruitment (D) of aplaviroc with CCL3 (white triangles) and without (black triangles) on [I116A]-CCR5. CCR5 WT basal level is indicated with dashed lines. Data is normalized to CCL3 Emax. COS-7 cells were used for PI turnover and N-terminally FLAG-tagged-based elisa, whereas U20S cells were used in β-arrestin recruitment. Statistical significance was calculated using Student's unpaired t-test. **P < 0.05, ***P < 0.001; ns, not significant; n = 3–25.
Despite increased activity, no efficacy switch of aplaviroc was observed [aplaviroc inhibited CCL3-induced activation in both PI turnover and β-arrestin recruitment to the basal level of [I116A]-CCR5 (Figure 4C and D)]. The potency of aplaviroc in PI turnover was similar to WT, but increased approximately fivefold in β-arrestin recruitment (Table 3). The affinity of aplaviroc at [I116A]-CCR5 was decreased 2.3-fold (Table 2). As with [L203F]-CCR5, TAK-779 acted as a full antagonist of this mutant with WT-like potency (Table 3).
Additive effect of constitutively activating mutations in TM-5 and TM-7
We have previously demonstrated how a steric hindrance mutation in the centre of TM-7 (G286F) shifts the conformational equilibrium towards the active state, that is, [G286F]-CCR5 was constitutively active, and furthermore, induced efficacy switch of aplaviroc in Gi-coupled signalling pathways (Steen et al., 2013). Here, we show how another steric hindrance mutation in the centre of TM-5, L203F, induces the same activity state. To investigate whether these effects were cumulative, a double mutant was constructed combining L203F and G286F. Indeed, [L203F;G286F]-CCR5 increased the constitutive activity in PI turnover above the level of both [G286F]-CCR5 and [L203F]-CCR5, that is, 1.5-and twofold increase respectively (Figure 5A). The potencies of the chemokines were similar (CCL5) or slightly decreased (CCL3) (Table 1); however, consistent with the higher activity, the affinity of CCL3 on the double mutant was increased fivefold compared with WT (Table 2). The level of constitutive activity was also assessed in cAMP inhibition and here a slight increase in basal activity was observed for [L203F;G286F]-CCR5 as compared with [L203F]-CCR5, but no difference from the level observed in [G286]-CCR5 (Figure 5B). The double mutant was unable to recruit β-arrestin – both ligand dependently and independently – as was the case for [G286F]-CCR5 (Table 1; Steen et al., 2013).
Figure 5.
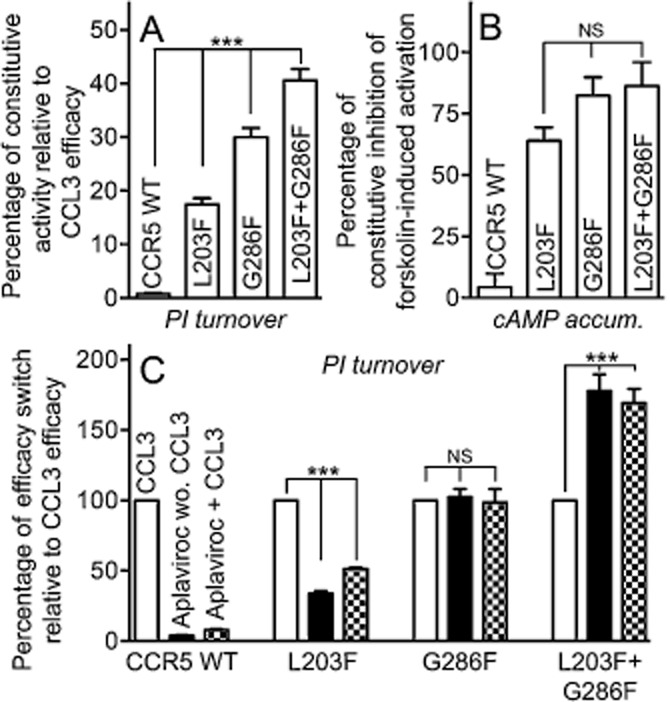
Effect of combining L203F and G286F on constitutive activity and aplaviroc efficacy. Shown as bar graphs is the ligand-independent activation of PI turnover (A) and inhibition of forskolin-induced cAMP accumulation (B) for CCR5 WT, [L203F]-, [G286F]-and [L203F;G286F]-CCR5 normalized to CCL3 Emax (A) or maximum level of forskolin-stimulated cAMP in untransfected cells (B). (C) CCL3 activation as well as the intrinsic effect of aplaviroc and aplaviroc-induced inhibition of CCL3 activation, normalized to CCL3 Emax. Data from both assays were obtained in COS-7 cells. Statistical significance was calculated using one-way anova, ***P < 0.001; ns, not significant; n = 3–25.
Interestingly, the efficacy increase (shift) for aplaviroc was also cumulative on the double mutant [L203F;G286F]-CCR5. Figure 5C shows an overview of the efficacy profile of aplaviroc compared with CCL3 on CCR5 WT, the two single mutants and the double mutant. In CCR5 WT, aplaviroc had no intrinsic effect and acted as a full antagonist of CCL3-induced activity. In contrast, it was able to activate [L203F]-CCR5 and [G286F]-CCR5 to ∼30% (partial agonist) and ∼100% (full agonist) of Emax of CCL3 respectively. Consistent with this, it did not inhibit CCL3-induced activation on [G286F]-CCR5 and only partially on [L203F]-CCR5 (to ∼50%). Intriguingly, in [L203F;G286F]-CCR5, aplaviroc was able to induce activation to ∼170% of CCL3 Emax both in the absence and presence of CCL3 and thus acts as a superagonist on the double mutant. TAK-779 was also tested on the double mutant, and was unable to inhibit CCL3-induced PI turnover despite maintained binding (Tables 1999 and 1998).
Elimination of Phe197 (V:13/5.47) in US28 supports the importance of this residue for constitutive activity
US28 is a viral chemokine receptor, which displays a high level of ligand-independent activity (Kledal et al., 1998; Casarosa et al., 2001). Among the human chemokine receptors, US28 clusters with the group of receptors that CCR5 belongs to (Figure 6A). This is not surprising as it binds the same chemokines as CCR5 (e.g. CCL3) in addition to other chemokines (Kledal et al., 1998). Interestingly, all CC-chemokine receptors in this cluster carry a Leu in position V:13/5.47, whereas US28 carries a Phe thereby providing an opportunity to examine if this naturally occurring Phe in a receptor homologous to CCR5 is indeed involved in the ligand-independent activation. Hence, Phe197 was mutated to Ala ([F197A]-US28) and subjected to PI turnover (Figure 6B), where it reduced the level of constitutive activity by ∼50%. It did not affect the affinity of CCL3, [0.71 ± 0.19 nM (WT) vs. 0.64 ± 0.22 nM ([F197A]-US28) (Ki ± SEM)] and there was no significant decrease in expression measured in elisa (Figure 6C) or Bmax (Figure 6D). Thus, a Phe in position V:13 is also very important for the activity level of US28.
Figure 6.
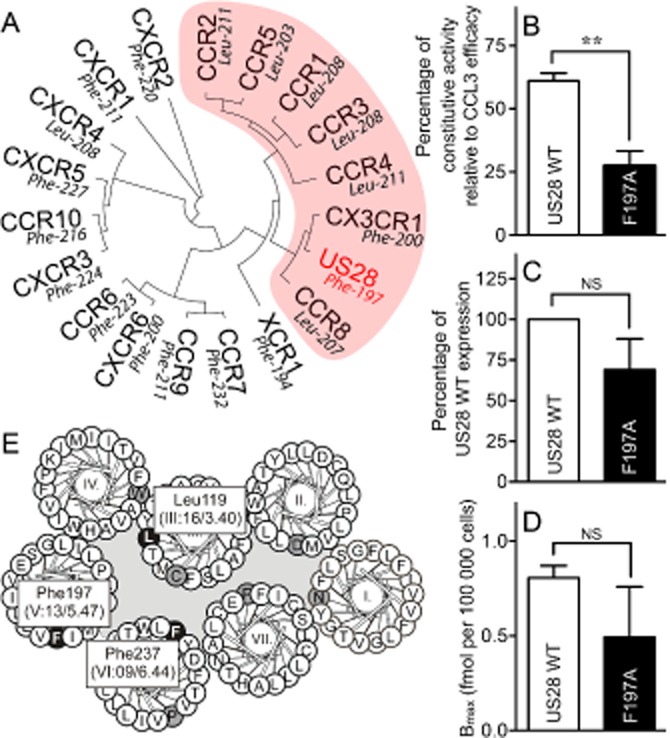
Relationship between the viral chemokine receptor US28 and the human chemokine family. (A) Phylogenetic tree of the human chemokine receptors together with the virus-encoded chemokine receptor US28. Indicated in red is the subgroup of receptors in which both CCR5 and US28 belong. The amino acid in position V:13/5.47 is given for each receptor. Effect on agonist-independent PI turnover (B), surface expression measured in elisa (C) or Bmax calculated from homologous competition binding with [125I]-CCL3 (D) upon mutation of Phe197 in US28 ([F197A]-US28) is shown compared with US28 WT. (E) Helical wheel diagram of US28 showing the most conserved residues (black on grey) and the residues discussed in current paper (white on black). Data is normalized to maximum CCL3 efficacy on the respective receptor (B) or to US28 WT expression (C) or not normalized (D). Statistical significance was calculated using Student's unpaired t-test. **P < 0.05; ns, not significant; n = 3–4.
Discussion and conclusions
Here, we demonstrated how insertion of Phe in V:13/5.47 induces an increase in the basal activity of CCR5. Moreover, removing the naturally occurring Phe in US28 markedly reduced constitutive activity. In addition, conformational changes in surrounding residues indicated a possible mechanism for the occurrence of constitutive activity in [L203F]-CCR5.
Areas in CCR5 of importance for constitutive activity – implications for disease?
The example of CAMs in CCR5 described here and numerous other examples of CAMs in the 7TM receptor family show us that while WT receptors are mostly found in an inactive conformation, subtle changes can convert them to active receptors. In accordance with the increased basal activity of [I116A]-and [L203F]-CCR5, efficacy switch of the antagonist aplaviroc to agonist is observed, indicating an overall more agonist-prone nature. As we and others have demonstrated previously, the binding site of aplaviroc differs from TAK-779 and other CCR5 antagonists in that it also involves extracellular loop 2 residues (Maeda et al., 2006; Thiele et al., 2011, Steen et al., 2013) and this could explain why it alone acquires agonistic properties.
There are numerous examples of diseases caused by altered activity of receptors. For example, it has been shown that CCR5 is constitutively expressed in Hodgkin's lymphoma cells and might play a role in tumour cell growth (Aldinucci et al., 2008). Not only US28, but in fact the majority of viral receptors are constitutively active. Thus, constitutive activity is a common trait for the ORF74 and the BILF1 receptors encoded by oncogenic γ2-and γ1-herpes viruses respectively (Bais et al., 1998; McLean et al., 2004; Rosenkilde et al., 2004; 2005,; Beisser et al., 2005; Paulsen et al., 2005; Rosenkilde, 2005; Lyngaa et al., 2010). Because of the close homology to human chemokine receptors, it is conceivable that a few naturally occurring mutations could be tumourigenic. In fact, we show here that exchanging Leu203 with Phe increases the basal activity of CCR5. This mutation corresponds to the sequence of the cancer-causing viral CCR5 homologue US28 and as shown here is important for its constitutive activity. If this CAM – or an alternative – occurs in vivo, it could possibly lead to uncontrollable cell growth.
The position and nature of CAMs provide clues to the difference between inactive and active receptor conformations. Many articles have reported areas important for constitutive activity, foe example, CAMs in TM-6 in the muscarinic receptors (Spalding et al., 1998), TM-3 in the angiotensin II type 1A receptor (Parnot et al., 2000) and TM-3 and TM-6 interaction in the α1B-adrenoceptor (Greasley et al., 2001). Here, we show that the centres of TM-3 and TM-5, and in particular, the interplay between these two regions and possibly TM-6, are involved in the activation of CCR5. This information is useful in the search for compounds for the treatment of diseases where altered signalling of CCR5 either is part of the underlying cause or could be beneficial for recovery.
Gating function of Ile116 (III:16/3.40) – a possible mechanism for the increased activity state
Computational modelling of WT and [L203F]-CCR5 indicates that the CAM L203F provokes a conformational change of the side chain of Ile116. Although this change does not seem to affect the overall position of TM-3, the movement of Ile116 indicates increased flexibility of the residue, presumably caused by a disruption of interactions between TM-3 and TM-5. This could also explain why elimination of the larger side chain in [I116A]-CCR5 increases the basal level to a similar degree to [L203F]-CCR5 in vitro. This interaction is consistent with the crystal structures of inactive 7TM receptors, which all show many interactions between TM-3 and TM-5 (Venkatakrishnan et al., 2013).
Another difference between the two models was the decreased distance from Leu/Phe203 to Tyr244 in TM-6 due to a tendency of Tyr244 to slide towards TM-5 in [L203F]-CCR5. In the recently published inactive crystal structure of CCR5, Ile116 and Tyr244 interact directly [PDB: 4MBS (Tan et al., 2013)]. The close proximity in the WT structure presented here (4.0 Å) also indicate that the two residues form a van der Waals interaction; however, in [L203F]-CCR5, the distance is increased to 5.1 Å suggesting that this bond is broken. It is therefore conceivable that mutating these residues stabilizes active – or weakens ground state – conformations by diminishing interactions, which stabilize Tyr244 in its inactive state, one of which is the interaction between Ile116 and Tyr244. We have previously shown that Tyr244 is central for the activation of CCR5 and therefore necessary for stabilization of the active state (Steen et al., 2013). The two active structures of the β2-adrenoceptor (Rasmussen et al., 2011a; 2011b,) showed that a small movement of the extracellular end of TM-5 resulted in a rotamer switch of Ile121 (III:16/3.40) and rotation of TM-6, which in turn was coupled to a 4 Å movement of Phe282 (VI:09/6.44). Recently, further evidence for this mechanism of the β2-adrenoceptor was published (Valentin-Hansen et al., 2012). Here, it was shown that Phe282 is locked between the backbone and two hydrophobic residues in TM-3 in the inactive state, and upon activation slides towards TM-5 into a hydrophobic pocket between TM-3 and TM-5. Meanwhile, Ile121 acts as a gate for the transition. As we observed the same rotamer change of Ile116 and movement of Tyr244 upon disruption of the TM-3/5 interface (although to a lesser degree), this theory could very well be adopted for CCR5. Moreover, the decrease in constitutive activity upon removal of PheV:13 in US28 – which has amino acids with the same chemical properties in III:16/3.40 and VI:09/6.44 (Figure 6E) as CCR5 – further supports this theory. However, CX3CR1 groups with CCR5 and US28 (Figure 6A) and has a Phe in V:13/5.47 but is not constitutively active to our knowledge. However, in TM-3, there is cluster of aromatic amino acids surrounding IleIII:16/3.40 and this could influence the gating function of Ile and explain the lack of constitutive activity. Interestingly, CCR5 is coupled to Gi and US28 is Gq-coupled, whereas the β2-adrenoceptor interacts with Gs, and thus, this could indicate a common 7TM receptor mechanism for activation.
An overview of the orientation of Ile in III:16/3.40 in all published crystal structures is given in Figure 7 (only those carrying an Ile are included, i.e. 46 structures out of 75 in total). The only receptor where an inactive and a truly active structure (i.e. in complex with agonist and G-protein) can be compared is the β2-adrenoceptor, and here Ile121 has a different conformation in the inactive compared with the two active structures (trans vs. gauche−), indicating a conserved function of this residue across the class A 7TM receptors. However, the χ1-angle in the inactive structures is not consistently trans in the different receptors and indeed the ‘active’ conformation of Ile116 we observed in CCR5 is the opposite of what was seen in the β2-adrenoceptor (trans in CCR5 WT and gauche− in the β2-adrenoceptor). Moreover, in the crystal structure of CCR5 (Tan et al., 2013), the ‘inactive’ conformation of Ile116 is in trans, which is contrary to our model (which is build with CXCR4 as structural template). This indicates that while this residue changes rotamer state between inactive and active receptor conformations, there is no consensus of the active conformation of IleIII:16/3.40 between the different receptors, suggesting that the conformation of the residue is of regulatory significance rather than positional, that is, it serves a gating function rather than being an actual switch. However, the change in rotamer state could also be related to the major ‘rigid body’ movement of the entire TM-3 upon activation, sliding along its axis towards the extracellular side by ∼2 Å, relative to the TM ‘core’ of TM-1 to TM-4, which with varying degrees are seen in both opsin (Standfuss et al., 2011), the β2-adrenoceptor (Rasmussen et al., 2011a) and the A2A receptor (Lebon et al., 2011). In this context, the change in conformation of the Ile side chain (which lack proper electron densities in some crystal structures) seems small when the residue together with others are relocated several Å.
Figure 7.

Overview of the orientation of the χ1-angle of Ile in position III:16/3.40 in all relevant crystal structures (46 of the available 75 structures contain Ile in this position). The middle panels depict a side and top view the orientation of Ile in gauche− (−60°, magenta) and trans (180°, orange) position, represented by the β2-adrenoceptor structures [PDB #2RH1 (inactive) and #3SN6 (active)]. Right and left panels list the position of Ile side chain in each structure for gauche− and trans respectively. Different PDB files with identical complexes (identical ligand and receptor) are only listed once. The structures were taken from the Protein Data Bank (http://www.rcsb.org/pdb) and visualized in Molsoft Browser Pro (Molsoft LLC).
Molecular mechanisms for constitutive activity of CCR5
Recently, we showed that insertion of a steric hindrance mutation in the centre of TM-7 ([G286F]-CCR5, VII:09/7.42) induced constitutive activity and efficacy switch of aplaviroc (Steen et al., 2013). Through computational modelling, we observed a rotation of the χ1-angle of Trp-248 (VI:13/6.48) in [G286F]-CCR5 and proposed that the molecular mechanism for the increased activity was an alteration in the conformation of Trp248. Differences in side chain conformations were not observed in any other residues analysed, including the ones described in the current paper. Conversely, there was no influence of the L203F mutation on the χ1-angle of Trp248. Thus, the increased activity of [L203F]-and [G286F]-CCR5 seems to occur via different molecular interactions. A double mutation combining L203F and G286F showed an additional increase in both constitutive activity and agonist efficacy of aplaviroc. This could indicate that it is two different molecular mechanisms that are responsible for the increased activation state of CCR5. On the contrary, it could also be two interlinked interactions, as movement in Trp248 is likely to alter the position of Tyr244.
TM-3 and TM-5 are central for G-protein-dependent and-independent activation
[G286F]-CCR5 exhibits biased signalling as β-arrestin recruitment is eliminated concomitantly with increased G-protein signalling (Steen et al., 2013). This indicates that TM-7 has alternating roles for the two pathways, as also supported by several other studies showing that TM-7 is especially important for β-arrestin recruitment (Liu et al., 2012; Rahmeh et al., 2012; Warne et al., 2012). In the present study, we showed that mutations in TM-3 and TM-5 produce similar responses in G-protein-dependent and β-arrestin signalling, that is, they enhance the signalling in both cases. This suggests a general importance of these two receptor regions for the activity states of CCR5, independently of the nature of the downstream signalling. These structure-function observations are important for the development of novel biased and unbiased drugs for manipulating the activity states of CCR5, and putatively of class A 7TM receptors in general.
Acknowledgments
The authors would like to thank Katja Spiess for construction of the phylogenetic tree and Olav Larsen for excellent technical assistance. This work was carried out with financial support from The Danish Council for Independent Research | Medical Sciences, the Novo Nordisk Foundation, the Lundbeck Foundation, the A. P. Møller Foundation for the Advancement of Medical Science and the Aase and Einar Danielsen Foundation.
Glossary
- 7TM
seven-transmembrane domain
- CAM
constitutively activating mutation
- HBS
HEPES-buffered saline
- TM
transmembrane domain
Conflicts of interest
The authors state that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12553
Representative data sets depicting the constitutive activity of [L203F]-CCR5. Dose response curves for GTPγS binding, PI turnover, and β-arrestin recruitment are shown. The β-arrestin recruitment was assessed in U20S cells whereas COS-7 cells were used for the remaining. [L203F]-CCR5 is shown in black, while CCR5 WT is shown in white. The level of activation in untransfected cells is shown in dashed lines. Statistical significance was calculated using unpaired t-test. **P < 0.05, ***P < 0.001. cpm, counts per minute, rlu, relative luminescence units.
Representative dose response curves for [I116A]-CCR5 in PI turnover and β-arrestin recruitment. The β-arrestin recruitment was assessed in U20S cells whereas COS-7 cells were used PI turnover. [I116A]-CCR5 is shown in black, while CCR5 WT is shown in white. The level of activation in untransfected cells is shown in dashed lines. Statistical significance was calculated using unpaired t-test. *P < 0.1, **P < 0.05. cpm, counts per minute, rlu, relative luminescence units.
Representative dose response curve for [L203F;G286F]-CCR5 (white squares) in PI turnover (first panel). CCR5 WT is shown in white squares and the level of activation in untransfected cells is shown in dashed lines. Emax, Bmax, and the expression level are shown in columns diagrams representing values given in Tables 1 and 2. Statistical significance was calculated using unpaired t-test. **P < 0.05. cpm, counts per minute.
Representative PI turnover dose response curve of US28 WT (white squares) and [F197A]-US28 (black squares) as well as untransfected cells (dashed line). Statistical significance was calculated using unpaired t-test. ***P < 0.001. cpm, counts per minute.
References
- Aldinucci D, Lorenzon D, Cattaruzza L, Pinto A, Gloghini A, Carbone A, et al. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int J Cancer. 2008;122:769–776. doi: 10.1002/ijc.23119. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/2014. G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 1999;96:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, et al. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391:86–89. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104:15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisser PS, Verzijl D, Gruijthuijsen YK, Beuken E, Smit MJ, Leurs R, et al. The Epstein–Barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J Virol. 2005;79:441–449. doi: 10.1128/JVI.79.1.441-449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodaghi B, Jones TR, Zipeto D, Vita C, Sun L, Laurent L, et al. Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J Exp Med. 1998;188:855–866. doi: 10.1084/jem.188.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009;52:397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarosa P, Bakker RA, Verzijl D, Navis M, Timmerman H, Leurs R, et al. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem. 2001;276:1133–1137. doi: 10.1074/jbc.M008965200. [DOI] [PubMed] [Google Scholar]
- Cho W, Taylor LP, Akil H. Mutagenesis of residues adjacent to transmembrane prolines alters D1 dopamine receptor binding and signal transduction. Mol Pharmacol. 1996;50:1338–1345. [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Greasley PJ, Fanelli F, Scheer A, Abuin L, Nenniger-Tosato M, DeBenedetti PG, et al. Mutational and computational analysis of the alpha(1b)-adrenergic receptor. Involvement of basic and hydrophobic residues in receptor activation and G protein coupling. J Biol Chem. 2001;276:46485–46494. doi: 10.1074/jbc.M105791200. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissow H, Hartmann B, Holst JJ, Viby N-E, Hansen LS, Rosenkilde MM, et al. Glucagon-like peptide-1 (GLP-1) receptor agonism or DPP-4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul Pept. 2012;179:91–100. doi: 10.1016/j.regpep.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Kledal TN, Rosenkilde MM, Coulin F, Simmons G, Johnsen AH, Alouani S, et al. A broad-spectrum chemokine antagonist encoded by Kaposi's sarcoma-associated herpesvirus. Science. 1997;277:1656–1659. doi: 10.1126/science.277.5332.1656. [DOI] [PubMed] [Google Scholar]
- Kledal TN, Rosenkilde MM, Schwartz TW. Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Lett. 1998;441:209–214. doi: 10.1016/s0014-5793(98)01551-8. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Zeng FY, Wess J. Functional characterization of a series of mutant G protein alphaq subunits displaying promiscuous receptor coupling properties. J Biol Chem. 1998;273:17886–17892. doi: 10.1074/jbc.273.28.17886. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttichau HR, Johnsen AH, Jurlander J, Rosenkilde MM, Schwartz TW. Kaposi sarcoma-associated herpes virus targets the lymphotactin receptor with both a broad spectrum antagonist vCCL2 and a highly selective and potent agonist vCCL3. J Biol Chem. 2007;282:17794–17805. doi: 10.1074/jbc.M702001200. [DOI] [PubMed] [Google Scholar]
- Lyngaa R, Nørregaard K, Kristensen M, Kubale V, Rosenkilde MM, Kledal TN. Cell transformation mediated by the Epstein–Barr virus G protein-coupled receptor BILF1 is dependent on constitutive signaling. Oncogene. 2010;29:4388–4398. doi: 10.1038/onc.2010.173. [DOI] [PubMed] [Google Scholar]
- McLean KA, Holst PJ, Martini L, Schwartz TW, Rosenkilde MM. Similar activation of signal transduction pathways by the herpesvirus-encoded chemokine receptors US28 and ORF74. Virology. 2004;325:241–251. doi: 10.1016/j.virol.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, et al. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol. 2004;78:8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Das D, Ogata-Aoki H, Nakata H, Miyakawa T, Tojo Y, et al. Structural and molecular interactions of CCR5 inhibitors with CCR5. J Biol Chem. 2006;281:12688–12698. doi: 10.1074/jbc.M512688200. [DOI] [PubMed] [Google Scholar]
- Marie J, Koch C, Pruneau D, Paquet JL, Groblewski T, Larguier R, et al. Constitutive activation of the human bradykinin B2 receptor induced by mutations in transmembrane helices III and VI. Mol Pharmacol. 1999;55:92–101. doi: 10.1124/mol.55.1.92. [DOI] [PubMed] [Google Scholar]
- Maussang D, Verzijl D, van Walsum M, Leurs R, Holl J, Pleskoff O, et al. Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:13068–13073. doi: 10.1073/pnas.0604433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadegan T, Benkö G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parma J, Duprez L, Van Sande J, Cochaux P, Gervy C, Mockel J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- Parnot C, Bardin S, Miserey-Lenkei S, Guedin D, Corvol P, Clauser E. Systematic identification of mutations that constitutively activate the angiotensin II type 1A receptor by screening a randomly mutated cDNA library with an original pharmacological bioassay. Proc Natl Acad Sci U S A. 2000;97:7615–7620. doi: 10.1073/pnas.110142297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen SJ, Rosenkilde MM, Eugen-Olsen J, Kledal TN. Epstein-Barr virus-encoded BILF1 is a constitutively active G protein-coupled receptor. J Virol. 2005;79:536–546. doi: 10.1128/JVI.79.1.536-546.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A. 2012;109:6733–6738. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U. Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol. 1999;56:175–184. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- Rasmussen SGF, Choi H-J, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman JG, Kanemitsu-Parks M, Gaidarov I, Cameron JS, Griffin P, Zheng H, et al. Nicotinic acid receptor agonists differentially activate downstream effectors. J Biol Chem. 2007;282:18028–18036. doi: 10.1074/jbc.M701866200. [DOI] [PubMed] [Google Scholar]
- Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM. Virus-encoded chemokine receptors-putative novel antiviral drug targets. Neuropharmacology. 2005;48:1–13. doi: 10.1016/j.neuropharm.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Schwartz TW. Potency of ligands correlates with affinity measured against agonist and inverse agonists but not against neutral ligand in constitutively active chemokine receptor. Mol Pharmacol. 2000;57:603–609. doi: 10.1124/mol.57.3.602. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, McLean KA, Holst PJ, Schwartz TW. The CXC chemokine receptor encoded by herpesvirus saimiri, ECRF3, shows ligand-regulated signaling through Gi, Gq, and G12/13 proteins but constitutive signaling only through Gi and G12/13 proteins. J Biol Chem. 2004;279:32524–32533. doi: 10.1074/jbc.M313392200. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Kledal TN, Schwartz TW. High constitutive activity of a virus-encoded seven transmembrane receptor in the absence of the conserved DRY motif (Asp-Arg-Tyr) in transmembrane helix 3. Mol Pharmacol. 2005;68:11–19. doi: 10.1124/mol.105.011239. [DOI] [PubMed] [Google Scholar]
- Schwartz TW. Locating ligand-binding sites in 7TM receptors by protein engineering. Curr Opin Biotechnol. 1994;5:434–444. doi: 10.1016/0958-1669(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation-a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Semple G, Skinner PJ, Gharbaoui T, Shin Y-J, Jung J-K, Cherrier MC, et al. 3-(1H-tetrazol-5-yl)-1,4,5,6-tetrahydro-cyclopentapyrazole (MK-0354): a partial agonist of the nicotinic acid receptor, G-protein coupled receptor 109a, with antilipolytic but no vasodilatory activity in mice. J Med Chem. 2008;51:5101–5108. doi: 10.1021/jm800258p. [DOI] [PubMed] [Google Scholar]
- Shiraishi M, Aramaki Y, Seto M, Imoto H, Nishikawa Y, Kanzaki N, et al. Discovery of novel, potent, and selective small-molecule CCR5 antagonists as anti-HIV-1 agents: synthesis and biological evaluation of anilide derivatives with a quaternary ammonium moiety. J Med Chem. 2000;43:2049–2063. doi: 10.1021/jm9906264. [DOI] [PubMed] [Google Scholar]
- Spalding TA, Burstein ES, Henderson SC, Ducote KR, Brann MR. Identification of a ligand-dependent switch within a muscarinic receptor. J Biol Chem. 1998;273:21563–21568. doi: 10.1074/jbc.273.34.21563. [DOI] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D'Antona A, Fransen M, Xie G, Oprian DD, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen A, Thiele S, Guo D, Hansen LS, Frimurer TM, Rosenkilde MM. Biased and constitutive signaling in the CC-chemokine receptor CCR5 by manipulating the interface between transmembrane helices 6 and 7. J Biol Chem. 2013;288:12511–12521. doi: 10.1074/jbc.M112.449587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele S, Steen A, Jensen PC, Mokrosinski J, Frimurer TM, Rosenkilde MM. Allosteric and orthosteric sites in CC chemokine receptor (CCR5), a chimeric receptor approach. J Biol Chem. 2011;286:37543–37554. doi: 10.1074/jbc.M111.243808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Hansen L, Holst B, Frimurer TM, Schwartz TW. PheVI:09 (Phe6.44) as a sliding micro-switch in 7TM G protein-coupled receptor activation. J Biol Chem. 2012;287:43516–43526. doi: 10.1074/jbc.M112.395137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- Warne T, Edwards PC, Leslie AGW, Tate CG. Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative data sets depicting the constitutive activity of [L203F]-CCR5. Dose response curves for GTPγS binding, PI turnover, and β-arrestin recruitment are shown. The β-arrestin recruitment was assessed in U20S cells whereas COS-7 cells were used for the remaining. [L203F]-CCR5 is shown in black, while CCR5 WT is shown in white. The level of activation in untransfected cells is shown in dashed lines. Statistical significance was calculated using unpaired t-test. **P < 0.05, ***P < 0.001. cpm, counts per minute, rlu, relative luminescence units.
Representative dose response curves for [I116A]-CCR5 in PI turnover and β-arrestin recruitment. The β-arrestin recruitment was assessed in U20S cells whereas COS-7 cells were used PI turnover. [I116A]-CCR5 is shown in black, while CCR5 WT is shown in white. The level of activation in untransfected cells is shown in dashed lines. Statistical significance was calculated using unpaired t-test. *P < 0.1, **P < 0.05. cpm, counts per minute, rlu, relative luminescence units.
Representative dose response curve for [L203F;G286F]-CCR5 (white squares) in PI turnover (first panel). CCR5 WT is shown in white squares and the level of activation in untransfected cells is shown in dashed lines. Emax, Bmax, and the expression level are shown in columns diagrams representing values given in Tables 1 and 2. Statistical significance was calculated using unpaired t-test. **P < 0.05. cpm, counts per minute.
Representative PI turnover dose response curve of US28 WT (white squares) and [F197A]-US28 (black squares) as well as untransfected cells (dashed line). Statistical significance was calculated using unpaired t-test. ***P < 0.001. cpm, counts per minute.