

Abstract
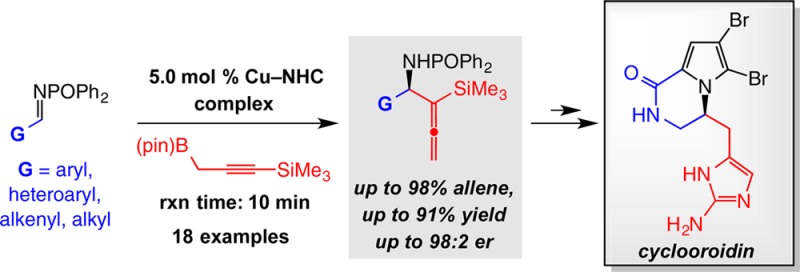
A catalytic method for the efficient and enantioselective addition of a 1-trimethylsilyl-substituted allene moiety to phosphinoylimines is presented. Transformations are promoted by 5.0 mol % of a copper complex of an N-heterocyclic carbene in the presence of a propargylboron reagent that can be readily prepared in multigram quantities. Within 10 min of reaction, products are obtained in up to 91% yield, 98% allene (vs propargyl) selectivity, and 98:2 enantiomeric ratio. An assortment of aldimines serve as suitable substrates. The phosphinoyl and silyl groups can be removed efficiently and orthogonally. The silylallene moiety may be transformed to versatile derivatives that are difficult to access via nonsilylated allenes. The special features and utility of the approach are highlighted through a succinct enantioselective synthesis of marine alkaloid S-(−)-cyclooroidin.
Development of catalytic protocols for enantioselective synthesis of N-containing organic molecules is a compelling research objective in organic chemistry.1 An array of strategies has been introduced for the preparation of homoallylic amines;2 catalytic reactions that furnish homopropargyl amines have been recently developed.3 Homoallenylamines are another set of compounds of substantial potential utility; such entities present unique functionalization possibilities4 that generate enantiomerically enriched derivatives not easily obtainable through manipulation of an allyl, an alkyne, or a propargyl group. Nonetheless, catalytic enantioselective methods that deliver allene-substituted amines are scarce, and the small number of existing protocols offers limited scope. In one investigation propargylstannanes were added to especially activated α-imino esters;3a in another, highly electrophilic allenoates were employed.5 These pioneering studies were focused on additions to aryl-substituted imines almost exclusively; heteroaryl- or alkenyl-containing variants were not included, and one alkyl-substituted case was shown to react with lower efficiency and enantioselectivity.5 A later report, centered on allyl additions promoted by an In-based salt and a Ag-based chiral complex, included only two examples of homoallenylamides generated as the major component of a 3–4:1 mixture with the homopropargyl addition products.6
Herein, we present an efficient and broadly applicable method for the synthesis of silyl-substituted homoallenylamides formed through catalytic transformations involving aryl-, heteroaryl-, alkyl-, and alkenyl-substituted imines. Transformations, promoted by a catalyst generated in situ from CuCl and an imidazolinium salt, deliver the desired products in up to 98:2 enantiomeric ratio (er). Phosphinoylimines, the chiral ligand, and the propargylboron reagent can be synthesized in gram quantities. Unfunctionalized allenes or unprotected amines are accessed selectively under mild conditions, and the silyl group can be manipulated to reveal versatile and otherwise difficult-to-access functional groups. The utility of the approach is demonstrated through application to a concise enantioselective synthesis of naturally occurring S-(−)-cyclooroidin.7
Selective addition of an allene group via a propargyl–metal intermediate (e.g., B, R = H; Scheme 1) requires identification of a reaction pathway that entails mainly a γ-addition process. One challenge stems from the lower stability of entities represented by B, which are generally less favored than the allenyl isomer (A; R = H) and exist in minimal quantities. We have previously exploited this stability difference, calculated to be ∼10 kcal/mol in favor of A (vs B, R = H), toward the development of a catalytic protocol for enantioselective synthesis of homopropargylamides.3c In considering a route that involves B as the predominant isomer and leads to homoallenylamides selectively, we surmised that an exclusive preference for B might not be necessary. We reasoned that the Cu–propargyl species might well be more nucleophilic by virtue of the higher donor ability of a Cu–Csp3 (vs Cu–Csp2 in A)8 and that, if available in only sufficient amounts, the transformation could proceed predominantly through the more nucleophilic homopropargyl–Cu complex (Curtin–Hammett kinetics; cf. Scheme 1). We were heartened by the fact that DFT calculations reinforced the above contention, indicating that the activation barrier for reaction via B would be lower than that involving allenyl–Cu complex A.10
Scheme 1. Selective Allene Addition through Curtin–Hammett Kinetics.
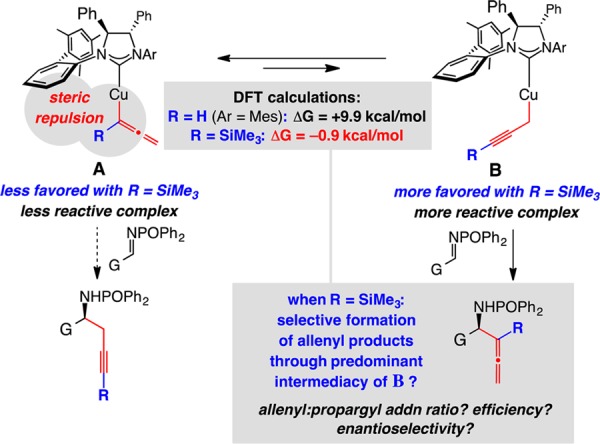
To induce the preferred shift in equilibrium favoring B, we opted to incorporate a trimethylsilyl (TMS) group with the organocopper complex, as illustrated in Scheme 1 (R = SiMe3); in the presence of the sizable N-heterocyclic carbene (NHC) ligand, the allenyl–Cu species A would then become less favored due to steric repulsion.9 Indeed, DFT calculations pointed out that when R = SiMe3 (Scheme 1), the preference for Cu–allene complex A is reduced substantially such that ΔG = −0.9 kcal/mol (vs ∼10 kcal/mol with R = H).10 Nonetheless, we remained concerned that the presence of a silyl unit at the carbon directly involved in C–C bond formation might diminish reaction rates due to steric factors.
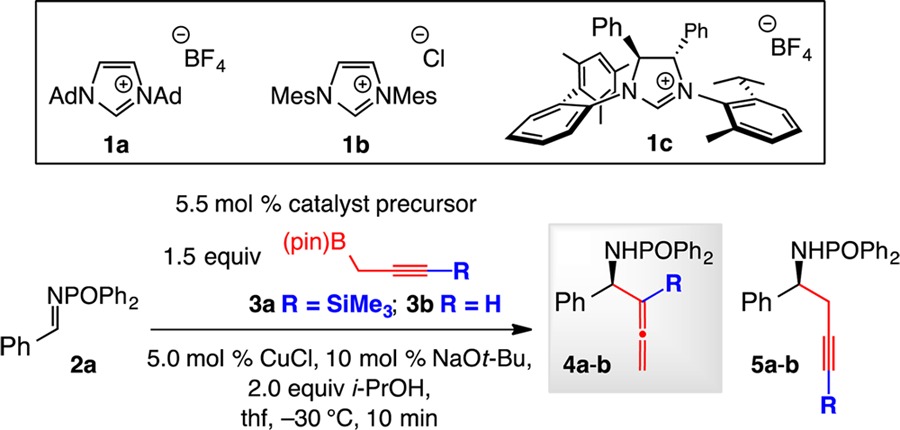
Based on the above considerations, we examined the NHC–Cu-catalyzed transformations between phosphinoylimine 2a and silyl-substituted propargyl–B(pin) reagent 3a, which is accessible in multigram quantities through a one-step operation (Table 1).11 With the complex derived from bis-adamantyl imidazolium salt 1a and CuCl, 83% conv was observed within 10 min, but it was homopropargyl amide 5a that was obtained with 92% selectivity (entry 1). With the catalyst derived from 1b (entry 2), carrying two wider spanning mesityl units, reaction with 0.5 g of 2a afforded 4a as the major product (83:17 allene:propargyl, 96% allene after purification; 76% yield). Use of the larger chiral imidazolinium salt 1c (entry 3) in a transformation performed with 0.3 g of 2a resulted in >98% conv in 10 min, delivering 4a in 98:2 allene:propargyl selectivity, 80% yield, and 95:5 er after purification. The mechanistic scenario depicted in Scheme 1 was further substantiated by the strong preference for the formation of homopropargylamide 5a with the Cu catalyst lacking an NHC ligand (98% 5a, entry 4), or with 3b, which does not carry a TMS group, serving as the reagent (R = H; entry 5; 98% 5b).
Table 1. Initial Examination of Catalysts and Reagentsa.
| entry | reagent (R); catalyst precursor | conv (%);b allene: propargylb | yield (%);c allene: propargyld | er (of allenyl isomer)e |
|---|---|---|---|---|
| 1 | 3a; 1a | 83; 8:92 | 75; 8:92 | na |
| 2f | 3a; 1b | >98; 83:17 | 76; 96:4 | na |
| 3g | 3a; 1c | >98; 95:5 | 80; 98:2 | 95:5 |
| 4 | 3a; none | 95; 2:98 | 81; 2:98 | na |
| 5 | 3b; 1c | 70; 2:98 | 44; 2:98 | 92:8 |
Reactions performed under N2 atmosphere.
By analysis of 1H NMR spectra of unpurified mixtures (±2%).
Yields of purified products (±5%).
Allenyl/propargyl ratios after purification.
By HPLC analysis.
Performed on 0.5 g of 2a.
Reaction performed on 0.3 g of 2a; 95% allenylamide before purification. See the Supporting Information for details. Ad = adamantyl; Mes = 2,4,6-Me3-C6H2; B(pin) = (pinacolato)boron; na = not applicable.
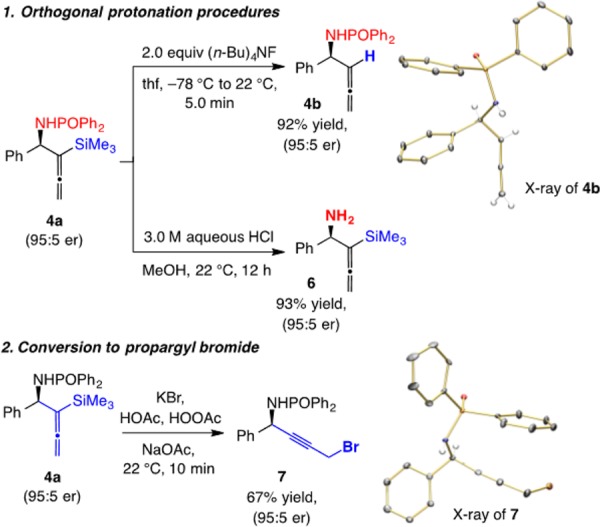
The transformations in Scheme 2, resulting in the efficient and selective formation of desilylated allene 4b (92% yield)12 or silyl-substituted amine 6 (93% yield), underscore a notable feature of the present strategy: The TMS group may be deleted, and the phosphinoyl unit, orthogonally removed. Another attribute of the approach is that the silylallene moiety can be manipulated to allow access to enantiomerically enriched products that cannot be prepared through homoallenylamides or synthesized easily by alternative pathways. A noteworthy example is illustrated in Scheme 2: Treatment of homoallenylamide 4a with KBr in acetic acid and peracetic acid at ambient temperature for 10 min generated propargyl bromide 7.13 In all the cases shown in Scheme 2, there was neither loss of enantiomeric purity nor formation of the homopropargyl isomer. The X-ray structures for 4b and 7 allowed us to establish the stereochemical identity of the products.
Scheme 2. Representative Functionalization of Homoallenyamides.
Yields are for purified products. See the Supporting Information for details.
Syntheses of the enantiomerically enriched aryl-substituted homoallenylamides delineate the scope of the method. Additions to o-substituted arylimines are facile (Scheme 3). The preference for allene transfer is diminished as the size of the substituent is increased: 4c–4e were formed with 95:5, 75:25, and 57:43 allene:propargyl addition selectivity, respectively. Product mixtures were further enriched by routine silica gel chromatography, as revealed by the ratios indicated in the parentheses (Scheme 3), to deliver samples of 96–98% allene in 46–84% yield and 92:8–95:5 er. Control experiments show that the diminution in allene addition selectivity with a larger ortho substituent in a sluggish NHC–Cu-catalyzed addition originates from competitive non-Cu-catalyzed processes that engender exclusive transfer of a propargyl unit.14 When the aryl substituent is at the meta (4f) or para position (4g–h), the preference for allene addition remains strong (≥90:10); after chromatography, 4f–h were isolated in 67–87% yield and 91:9–98:2 er along with ≤3% of the homopropargyl derivative. The lower conversion observed with 4h suggests that an electron-donating substituent can diminish the rate of addition. Heteroaryl-substituted products were prepared efficiently and with excellent group selectivity and enantioselectivity: pyridyl 4i, furyl 4j, and thienyl 4k, containing 84–98% of the allene product, in 76–85% yield and 96:4–98:2 er. The X-ray structure for 4f was obtained, supporting the absolute stereochemical assignment that is consistent with the stereochemical model formerly proposed.15,16
Scheme 3. Enantioselective Synthesis to Aryl and Heteroaryl Homoallenylamides.

For conditions, see Table 1 (entry 3). Yields refer to purified products. See the Supporting Information for details.
A significant feature of the Cu-catalyzed protocol is that reactions of alkyl-substituted phosphinoylimines proceed with excellent group transfer and enantioselectivity; amides 8a–b were isolated in 72–90% yield, 95–97% allene:propargyl selectivity, and 97:3–98:2 er (Scheme 4). Formation of the sterically encumbered cyclohexyl-substituted 8c proceeded less readily (85% conv), delivering the desired amide in 55% yield and 96:4 er (98% allene). The applicability of the approach can be extended to reactions involving imines derived from α,β-unsaturated aldehydes that possess different electronic attributes; homoallenylamides 9a–c (Scheme 4) were isolated in 86–91% yield and 96:4 er (93–96% allene). The transformation leading to sterically hindered 10 proceeded to 62% conv, affording the desired product in 56% yield and 91:9 er (93% allene).
Scheme 4. Enantioselective Additions to Alkyl and Alkenyl Imines.

For conditions, see Table 1 (entry 3). Yields refer to purified products. See the Supporting Information for details.
Enantioselective synthesis of cyclooroidin,17 outlined in Scheme 5, highlights some of the attractive features of the NHC–Cu-catalyzed approach. Treatment of 11, prepared in three steps from readily available materials (52% overall yield),18 with NaHCO3 generated the requisite phosphinoylimine; direct subjection of the latter compound to the enantioselective addition conditions led to the formation of homoallenylamide 12 in 62% overall yield and 97:3 er (98:2 allene:propargyl); the sequence (11→12) was complete within a total of 70 min. The phosphinoyl group was then selectively removed under the mild acidic conditions mentioned previously (Scheme 2) without unmasking of the silyl unit, which is needed at a later key step, or deprotection to afford the primary amine, the exposure of which would also have to be implemented at a later stage. Subsequent conversion of the enantiomerically enriched amine to 2-carboxylic ester substituted pyrrole 14 was effected through reaction with ethyl 2,5-dioxopentanoate (13);19 the silyl-substituted allene thus remained intact after two processes carried out in acidic media. Next, simultaneous with the transformation of silyl-substituted allene to the derived bromo-propargyl derivative, the heterocyclic moiety was outfitted with two halogen atoms to afford tribromide 15 in 63% yield. Subsequent treatment with bis-Boc-guanidine (16) and K2CO3 in dimethylformamide (22 °C, 8.0 h) furnished 17 in 62% yield as a result of three operations: alkylation of the propargyl bromide,20 removal of the Fmoc unit, and cyclization to the six-membered ring amide. Intramolecular Ag-catalyzed hydroamination of the alkyne21 proceeded with >98% preference for the five-membered ring heterocycle. The ensuing removal of the remaining Boc group21b allowed for the generation of the heterocyclic aromatic ring, delivering the target molecule in 90% overall yield (for the last two-step sequence; 97:3 er). The route depicted in Scheme 5 (11 steps, including a three-step synthesis of 11) is shorter than the two previously disclosed enantioselective syntheses (17–18 steps).17
Scheme 5. Application to Enantioselective Synthesis of Cyclooroidin.
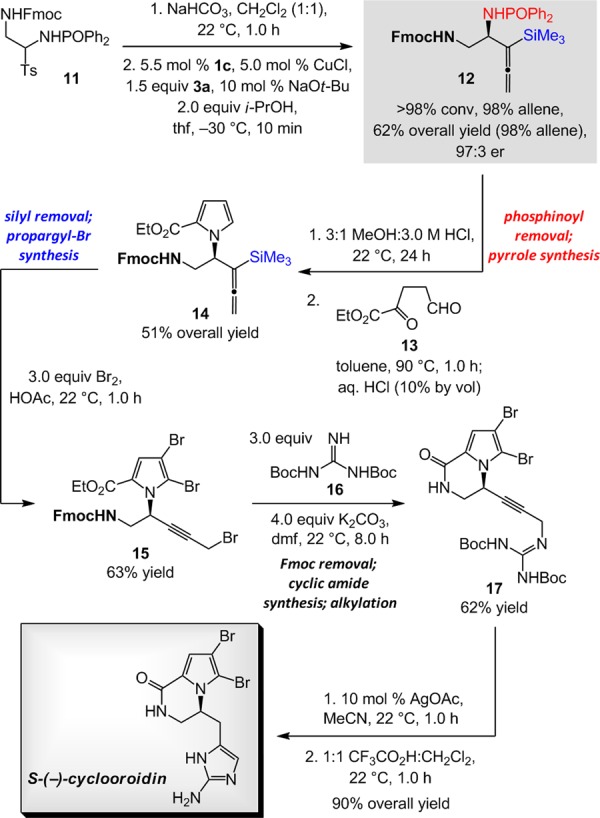
See the Supporting Information for details. Ts = p-toluenesulfonyl; Fmoc = fluorenylmethyloxycarbonyl.
We present the first general catalytic protocol for the enantioselective synthesis of silyl-substituted homoallenylamides and their various derivatives (including amines; cf. 14). The approach is applicable to readily accessible aldimines. The versatility of the approach is underscored by efficient removal of the phosphinoyl unit while the C–Si bond is retained, and selective reaction of the silyl-substituted allene with Br2/HOAc without adversely impacting an Fmoc-protected amine. Another key aspect is the possibility of converting a silyl-substituted homoallenylamide or amine to a propargyl bromide, a versatile electrophilic moiety that offers the possibility of a variety of functionalization options.
Acknowledgments
Financial support was provided by the NIH (GM-57212). We are grateful to Dr. Bo Li for his assistance with obtaining X-ray data as well as to D. L. Silverio, E. M. Vieira, and H. Wu for helpful discussions. We thank Boston College Research Services for access to computational facilities.
Supporting Information Available
Experimental procedures, spectral and analytical data for all products, and crystallographic (CIF) and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Nugent T. C.; El-Shazly M. Adv. Synth. Catal. 2010, 352, 753. [Google Scholar]; b Chiral Amine Synthesis; Nugent T. C., Ed.; Wiley–VCH: Weinheim, 2010. [Google Scholar]
- For a recent comprehensive review, see:Yus M.; González-Gómez J. C.; Foubelo F. Chem. Rev. 2011, 111, 7774. [DOI] [PubMed] [Google Scholar]
- a Kagoshima H.; Uzawa T.; Akiyama T. Chem. Lett. 2002, 31, 298. [Google Scholar]; b Wisniewska H. M.; Jarvo E. R. Chem. Sci. 2011, 2, 807. [Google Scholar]; c Vieira E. M.; Haeffner F.; Snapper M. L.; Hoveyda A. H. Angew. Chem., Int. Ed. 2012, 51, 6618. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent overview, see:; d Wisniewska H. M.; Jarvo E. R. J. Org. Chem. 2014, 78, 11629. [DOI] [PubMed] [Google Scholar]
- For representative examples, see:; a Modern Allene Chemistry, Krause N., Hashmi A. S. K., Eds; Wiley–VCH: Weinheim, Germany, 2004. [Google Scholar]; b Burks H. E.; Liu S.; Morken J. P. J. Am. Chem. Soc. 2007, 129, 8766. [DOI] [PubMed] [Google Scholar]; c Yu S.; Ma S. Angew. Chem., Int. Ed. 2012, 51, 3074. [DOI] [PubMed] [Google Scholar]; d Zbieg J. R.; McInturff E. L.; Leung J. C.; Krische M. J. J. Am. Chem. Soc. 2011, 133, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cooke M. L.; Xu K.; Breit B. Angew. Chem., Int. Ed. 2012, 51, 10876. [DOI] [PubMed] [Google Scholar]; f Yuan W.; Ma S. Adv. Synth. Catal. 2012, 354, 1867. [Google Scholar]; g Semba K.; Shinomiya M.; Fujihara T.; Terao J.; Tsuji Y. Chem.—Eur. J. 2013, 19, 7125. [DOI] [PubMed] [Google Scholar]; h Meng F.; Jung B.; Haeffner F.; Hoveyda A. H. Org. Lett. 2013, 15, 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cowen B. J.; Saunders L. B.; Miller S. J. J. Am. Chem. Soc. 2009, 131, 6105. [DOI] [PubMed] [Google Scholar]; For enantioselective additions of trisubstituted allenoates to N-tosylimines, see:; b Hashimoto T.; Sakata K.; Tamakuni F.; Dutton M. J.; Maruoka K. Nat. Chem. 2013, 5, 240. [DOI] [PubMed] [Google Scholar]
- Huang Y.-Y.; Chakrabarti A.; Morita N.; Schneider U.; Kobayashi S. Angew. Chem., Int. Ed. 2011, 50, 11121. [DOI] [PubMed] [Google Scholar]
- a Ferretti C.; Marengo B.; De Ciucis C.; Nitti M.; Pronzato M. A.; Marinari U. M.; Pronzato R.; Manconi R.; Domenicotti C. Int. J. Oncol. 2007, 30, 161. [PubMed] [Google Scholar]; b Movassaghi M.; Siegel D. S.; Han S. Chem. Sci. 2010, 1, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent studies that address the issue of propargyl- to allenyl-metal complex isomerization and its attendant consequences, see:; a Lambert C.; Schleyer P. v. R.; Würthwein E. U. J. Org. Chem. 1993, 58, 6377. [Google Scholar]; b Alcaide B.; Almendros P.; Aragoncillo C.; Rodríguez-Acebes R. J. Org. Chem. 2001, 66, 5208. [DOI] [PubMed] [Google Scholar]; c Reich H. J.; Holladay J. E.; Walker T. G.; Thompson J. L. J. Am. Chem. Soc. 1999, 121, 9769. [Google Scholar]; d Lin M.-J.; Loh T.-P. J. Am. Chem. Soc. 2003, 125, 13042. [DOI] [PubMed] [Google Scholar]; e Guo L.-N.; Gao H.; Mayer P.; Knochel P. Chem.—Eur. J. 2010, 16, 9829. [DOI] [PubMed] [Google Scholar]
- Similar trends in allene vs propargyl addition to ketones (nonenantioselective) have been recently reported to be the result of the size of Cu–bisphosphine complexes involved. See:; a Fandrick K. R.; Ogikubo J.; Fandrick D. R.; Patel N. D.; Saha J.; Lee H.; Ma S.; Grinberg N.; Busacca C. A.; Senanayake C. H. Org. Lett. 2013, 15, 1214. [DOI] [PubMed] [Google Scholar]; For a Cr-catalyzed approach to the formation of the corresponding silyl-substituted alcohols, see:; b Inoue M.; Nakada M. Angew. Chem., Int. Ed. 2006, 45, 252. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details of the DFT calculations.
- Fandrick D. R.; Roschangar F.; Kim C.; Hahm B. J.; Cha M. H.; Kim H. Y.; Yoo G.; Kim T.; Reeves J. T.; Song J. J.; Tan Z.; Qu B.; Haddad N.; Shen S.; Grinberg N.; Lee H.; Yee N.; Senanayake C. H. Org. Process Res. Dev. 2012, 16, 1131. [Google Scholar]
- Reddy L. R. Chem. Commun. 2012, 48, 9189. [DOI] [PubMed] [Google Scholar]
- Hernandez E.; Burgos C. H.; Alicea E.; Soderquist J. A. Org. Lett. 2006, 8, 4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- As an example, subjection of 2a to 1.5 equiv of 3a and 2.0 equiv of i-PrOH in thf at 22 °C leads to 66% conv to 5a in 1 h (4a:5a = 2:98).
- Vieira E. M.; Snapper M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For DFT calculations in connection with reactions involving imidazolinium salt 1c, see the Supporting Information.
- a Patel J.; Pelloux-Léon N.; Minassian F.; Vallée Y. Tetrahedron Lett. 2006, 47, 5561. [Google Scholar]; b Mukherjee S.; Sivappa R.; Yousufuddin M.; Lovely C. J. Org. Lett. 2010, 12, 4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for experimental details.
- Hughes C. C.; Kauffman C. A.; Jensen P. R.; Fenical W. J. Org. Chem. 2010, 75, 3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi J.; Seiple I. B.; Young I. S.; O’Malley D. P.; Maue M.; Baran P. S. Angew. Chem., Int. Ed. 2008, 47, 3578. [DOI] [PubMed] [Google Scholar]
- a Ermolat’ev D. S.; Bariwal J. B.; Steenackers H. P. L.; De Keersmaecker S. C. J.; Van der Eycken E. V. Angew. Chem., Int. Ed. 2010, 49, 9465. [DOI] [PubMed] [Google Scholar]; b Gainer M. J.; Bennett N. R.; Takahashi Y.; Looper R. E. Angew. Chem., Int. Ed. 2011, 50, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.