

Abstract
A streamlined approach to the tertiary amine-containing core of the calyciphylline A and daphnicyclidin A-type Daphniphyllum alkaloids is presented. A known carvone derivative is converted into the core structure in only four synthetic operations, and it is well poised for further elaboration. The key enabling methodology is a radical cyclization cascade beginning with addition of a secondary, neutral aminyl radical to the β-position of an enone, followed by trapping with a pendant alkyne.
The Daphniphyllum alkaloids are a structurally alluring family of plant alkaloids isolated from trees of the genus Daphniphyllum.1 Biosynthetic proposals from Kobayashi suggest that they are derived from rearrangements of secodaphnane,1a which in turn, arises via cyclization and condensation of squalene.2 Members of this family of alkaloids exhibit cytotoxicity against murine leukemia (L1210, P-388, and A-549 cell lines), demonstrate antioxidant activity against PC12 cells, show vasorelaxant effects in the rat aorta, and increase mRNA expression of nerve growth factor.1a Despite these promising bioactivities, the biological role of the vast majority of Daphniphyllum alkaloids has not been determined. The range of known Daphniphyllum alkaloid bioactivities, their significant structural diversity, and the sheer number of compounds in this family of alkaloids demand further investigation of the therapeutic potential of these compounds and related structures. Accordingly, the synthetic community has taken an increased interest in these targets in recent years.1a,3
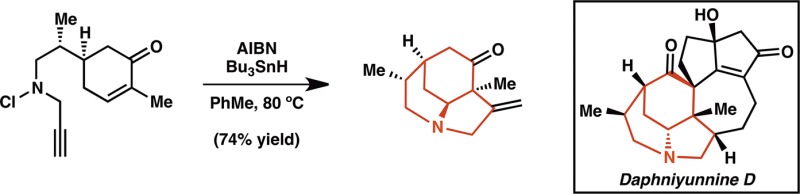
The combination of fused and bridging ring systems in the daphnicyclidin A-type (1) and calyciphylline A-type (2) alkaloids piqued our interest (Scheme 1). Additionally, these alkaloids pose several other synthetic challenges. The calyciphylline A subfamily boasts a pair of vicinal all-carbon quaternary stereocenters, each of which is positioned at the junction of three rings. Meanwhile, the daphnicyclidin A subfamily is comprised of two 7-membered rings fused by a unique fulvene moiety. Both subtypes contain a tertiary aliphatic amine at the junction of two rings. Importantly, we felt either subclass of alkaloids could be accessed from a single core structure such as tricyclic β-amino cyclohexanone 3, which could be used directly to access the calyciphylline A-type alkaloids or could furnish the daphnicyclidin A alkaloids upon ring expansion. We envisioned generation of tricyclic core 3 via tandem radical cyclization of an N-centered radical, which could be produced from chloroamine 4. Enone 4 would, in turn, be generated from known lactone 5, a derivative of (+)-(S)-carvone (6).
Scheme 1. Targeted Daphniphyllum Alkaloids.
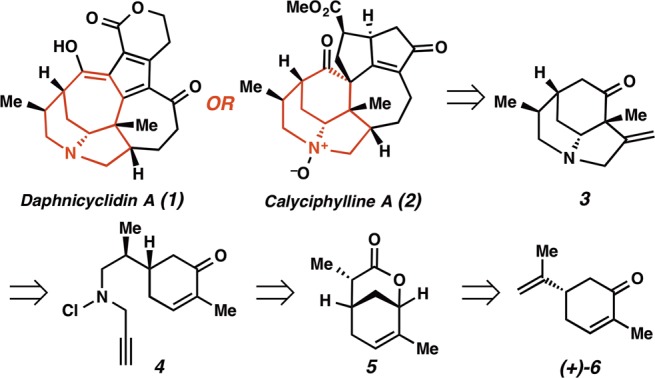
We began our synthetic efforts with bicyclic lactone 5, which is accessed in three steps from (R)-carvone (Scheme 2).4 This lactone could be appropriately functionalized to generate the cyclization substrate in just three synthetic operations. Thus, DIBAL-H reduction to the lactol was followed by reductive amination with propargylamine to afford amino alcohol 7 in 86% yield over two steps. Propargylamine derivative 7 was reacted with NCS in methylene chloride at −78 °C, the reaction mixture was warmed to 0 °C, and Dess–Martin periodinane was added. Finally, the reaction was allowed to come to ambient temperature. This protocol afforded the cyclization substrate (ent-4) in 96% yield.
Scheme 2. Synthesis of Radical Cyclization Substrate.

We envisioned that propargyl chloroamine derivative ent-4 would be a viable substrate in a tandem radical cyclization initiated at nitrogen, directly providing desired core structure ent-3. As illustrated in Scheme 3, homolytic cleavage of the N–Cl bond (4) would provide N-centered radical (NCR) 8. Intramolecular 6-exo addition to the β-position of cyclic enone would result in the formation of radical 9. We anticipated that the polarization of the enone would encourage cyclization of NCR 8 in preference to nonselective hydrogen atom abstraction.5 Tertiary radical 9 would then be trapped by the pendant alkyne in a C–C bond-forming event to form vinyl radical 10. Finally, hydrogen atom abstraction would afford tricyclic core ent-3.
Scheme 3. Strategy for Facilitating Tandem Cyclization of Aminyl Radical To Access Core Structure.

At the outset of our work, there was little known about the viability of a secondary neutral N-centered radical cyclizing with a highly polarized olefin such as an enone. Although amidyl, iminyl, and aminium radicals have been fairly well studied,6 fewer reports have described the cyclization of neutral aminyl radicals.6b,7,8 Curiously, only one prior example of a 6-exo cyclizations of a neutral aminyl radical has been reported.9 In primary aminyl radicals, competitive cyclization between 5-exo and 6-endo pathways is observed and can be tuned by adjusting the substituents on the olefin.7b Cyclizations of secondary neutral aminyl radicals are less common,7a,7b,8b−8d and this might be attributable to an increase in their nucleophilicity relative to primary NCRs. The more nucleophilic NCR would have a higher SOMO energy, resulting in less favorable overlap with the HOMO of a neutral or electron-rich olefin. Similar effects have been observed with alkoxyl radicals, which have intermediate SOMO energies and react as electrophilic radicals with electron rich olefins, but as nucleophilic radicals with electron poor olefins.10 On the basis of this reactivity, it is reasonable to expect that N-centered radicals might exhibit a similar dichotomy, and the secondary aminyl radicals would react well with electron poor olefins. However, there are limited examples of cyclization of any type of NCR with Michael acceptors, such as enones.8 Guindon and co-workers reported cyclization of a neutral aminyl radical (derived from 11, Bth = benzothiazole) onto the β-position of an α,β-unsaturated carbonyl system to generate pyrrolidine 12 (Scheme 4).8b Efficient cyclization was observed for benzyl derivatives and primary tributylstannyl NCRs, with only a small amount of reduction product 13. However, no cyclization occurred for dialkyl N-centered radicals.8b In the synthesis of (+)-clividine by Banwell and co-workers, an arylogous unsaturated lactone (14) was employed in a radical cyclization, initiated at nitrogen, to afford pyrrolidine 15 in 83% yield.8c A major byproduct in the reaction of related structures was the uncyclized N–Cl reduction product.8c,8d In general, neutral aminyl radicals are particularly prone to nonselective hydrogen atom abstraction reactions,5 and reduction of the NCR by reaction with tributyltin hydride often out-competes cyclization.11
Scheme 4. Known Cyclizations of Neutral Secondary Aminyl Radicals onto Polarized Olefins.

With these precedents in mind, we were poised to evaluate the key cyclization reaction. Attempts to induce cyclization of ent-4 with BEt3 and O2 led to a 52% yield of N–Cl reduction products.12 Use of neutral CuCl conditions13 provided a low yield of the vinyl chloride. We were excited to find that reaction with AIBN and tributyltin hydride at 80 °C in toluene afforded 40–50% yields of tandem cyclization product ent-3 (Table 1). In an effort to avoid use of tin reagents, a photolytic reaction was conducted using a sunlamp as the source.14 Unfortunately, only a 17% yield was observed. The major side products in the thermally induced cyclization were N–Cl reduction plus alkenyl radical trapping products, including the alkenyl chloride, the alkenylstannane, and the benzyl-substituted derivative arising from •CH2Ph trapping. To avoid the tolyl radical-derived byproduct, the solvent was changed to trifluorotoluene.8a Surprisingly, this lowered the yield to 27%. Addition of the AIBN and Bu3SnH over 1 h in toluene resulted in a 61% yield of the desired product, with the major byproduct being the alkenyl chloride. In benzene, this protocol afforded only a 21% yield of the tricyclic product. The final hydrogen atom abstraction seemed to be the limiting factor in improving the yield, and so the reaction was heated to reflux to increase the rate of the final radical trapping.15 Gratifyingly, this adjustment led to a 74% yield16 of the cyclization product as a single diastereomer, with no N–Cl reduction byproducts. Interestingly, we did not observe evidence of [1,5]-hydrogen atom transfer (to form the amine and an allylic radical) in these reactions.
Table 1. Optimization of the Tandem NCR Cyclizationa.
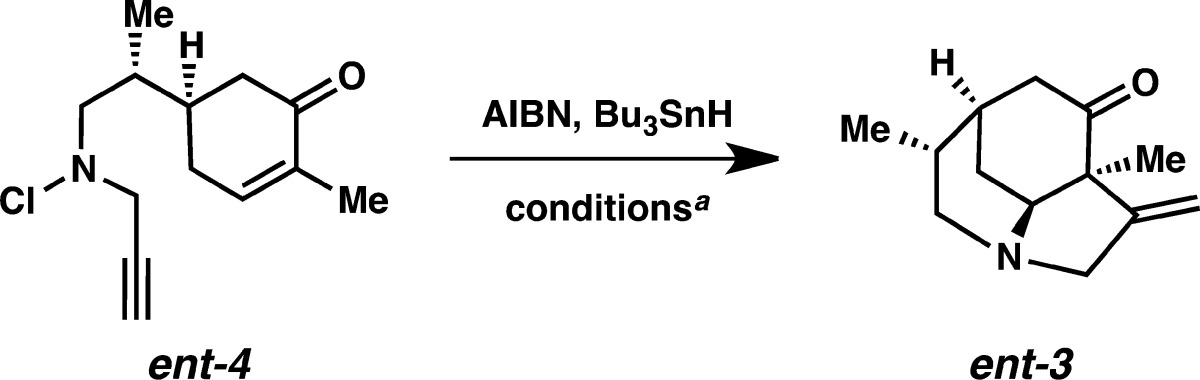
| temp (°C) | solvent | addition time (h) | % yield (isolated) |
|---|---|---|---|
| 80 | Ph-Me | 0 | 40–50 |
| 21 (sunlamp) | Ph-Me | 0 | 17 |
| 80 | Ph-CF3 | 0 | 27 |
| 80 | Ph-Me | 1 | 61 (+Cl· transfer pdt.) |
| 80 | Ph-H | 1 | 21 |
| 110 | Ph–Me | 1 | 74 |
All reactions were performed using 0.5 equiv of AIBN, 2 equiv of Bu3SnH, at 0.01 M. Addition over 1 h indicates a solution of AIBN and Bu3SnH was added dropwise over 1 h to a heated solution of the substrate in the indicated solvent.
As mentioned previously, we believed that the polarization of the olefin by the enone carbonyl was important to the observed reactivity. To validate this hypothesis, we synthesized allylic alcohol substrate 16 (Scheme 5). Treatment of this substrate with AIBN and Bu3SnH in toluene at 80 °C led exclusively to N–Cl reduction, and no products of tandem cyclization were observed. This experiment supports our assertion that the enone is crucial to the observed reactivity.
Scheme 5. Failed Cyclization of Allylic Alcohol Substrate.

In summary, the tertiary amine-containing core of the calyciphylline A and daphnicyclidin A alkaloids was generated in enantiopure fashion in four steps from known lactone ent-5. The key reaction (ent-4 → ent-3) represents the first addition of an N-centered radical to the β-position8a of an enone. It is among the highest yielding reported cyclizations of secondary aminyl radicals under neutral conditions,8c and it is the second report of a neutral aminyl radical undergoing a 6-exo cyclization. Reaction of the neutral secondary aminyl radical with an electrophilic system, such as an enone, seems to be essential to harnessing the reactivity of these species. Efforts to elaborate this core structure to the calyciphylline A and daphnicyclidin A-type alkaloids are ongoing.
Acknowledgments
We thank the National Institutes of Health (R00-GM097095 to J.L.S., R25-GM058905 IMSD Fellowship to A.M.L.) and Wayne State University for generous financial support. We also gratefully acknowledge the staff of the WSU Lumigen Instrument Center for spectroscopic support, in particular Dr. Yuriy Danylyuk and Dr. Bashar Ksebati.
Supporting Information Available
Experimental procedures and spectroscopic data (1H NMR, 13C NMR, IR, HRMS, and optical rotation) are provided for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
This manuscript is dedicated to Prof. David Y. Gin, in memoriam, with gratitude for his mentorship and support during the inception of this strategy.
Schemes 1, 2, 4, and the Supporting Information contained errors in the version published ASAP on February 7, 2014; the correct version reposted on February 11, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Kobayashi J.; Kubota T. Nat. Prod. Rep. 2009, 26, 936–962and references therein. [DOI] [PubMed] [Google Scholar]; b Zhang Y.; Di Y.-T.; Zhang Q.; Mu S.-Z.; Tan C.-J.; Fang X.; He H.-P.; Li S.-L.; Hao X.-J. Org. Lett. 2009, 11, 5414–5417. [DOI] [PubMed] [Google Scholar]; c He T.; Zhou Y.; Wang Y.-H.; Mu S.-Z.; Hao X.-J. Helv. Chim. Acta 2011, 94, 1019–1023. [Google Scholar]; d Yang T.-Q.; Di Y.-T.; He H.-P.; Zhang Q.; Zhang Y.; Hao X.-J. Helv. Chim. Acta 2011, 94, 397–403. [Google Scholar]; e Zhang Y.; Di Y.; He H.; Li S.; Lu Y.; Gong N.; Hao X. Eur. J. Org. Chem. 2011, 4103–4107. [Google Scholar]; f Tan C.-J.; Wang Y.-H.; Di Y.-T.; He H.-P.; Mu S.-Z.; La S.-F.; Zhang Y.; Hao X. J. Tetrahedron Lett. 2012, 53, 2588–2591. [Google Scholar]; g Cao M.; Zhang Y.; He H.; Li S.; Huang S.; Chen D.; Tang G.; Li S.; Di Y.; Hao X. J. Nat. Prod. 2012, 75, 1076–1082. [DOI] [PubMed] [Google Scholar]; h Zhang Q.; Zhang Y.; Yang T.-Q.; Di Y.-T.; Hao X.-J. RSC Adv. 2013, 3, 9658–9661. [Google Scholar]; i Cao M.-M.; Wang L.; Zhang Y.; He H.-P.; Gu Y.-C.; Zhang Q.; Li Y.; Yuan C.-M.; Li S.-L.; Di Y.-T.; Hao X.-J. Fitoterapia 2013, 89, 205–209. [DOI] [PubMed] [Google Scholar]
- Heathcock C. H. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 14323–14327and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dunn T. B; Ellis J. M.; Kofink C. C.; Manning J. R.; Overman L. E. Org. Lett. 2009, 11, 5658–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weiss M. E.; Carreira E. M. Angew. Chem., Int. Ed. 2011, 48, 11501–11505. [DOI] [PubMed] [Google Scholar]; c Coldham I.; Burrell A. J. M.; Guerrand H. D. S.; Oram N. Org. Lett. 2011, 13, 1267–1269. [DOI] [PubMed] [Google Scholar]; d Xu C.; Liu Z.; Wang H.; Zhang B.; Xiang Z.; Hao X.; Wang D. Z. Org. Lett. 2011, 13, 1812–1815. [DOI] [PubMed] [Google Scholar]; e Coldham I.; Watson L.; Adams H.; Martin N. G. J. Org. Chem. 2011, 76, 2360–2366. [DOI] [PubMed] [Google Scholar]; f Sladojevich F.; Michaelides I. N.; Darses B.; Ward J. W.; Dixon D. J. Org. Lett. 2011, 13, 5132–5135. [DOI] [PubMed] [Google Scholar]; g Bélanger G.; Boudreault J.; Lévesque F. Org. Lett. 2011, 13, 6204–6207. [DOI] [PubMed] [Google Scholar]; h Darses B.; Michaelides I. N.; Sladojevich F.; Ward J. W.; Rzepa P. R.; Dixon D. J. Org. Lett. 2012, 14, 1684–1687. [DOI] [PubMed] [Google Scholar]; i Xu C.; Wang L.; Hao X.; Wang D. Z. J. Org. Chem. 2012, 77, 6307–6313. [DOI] [PubMed] [Google Scholar]; j Fang B.; Zheng H.; Zhao C.; Jing P.; Li H.; Xie X.; She X. J. Org. Chem. 2012, 77, 8367–8373. [DOI] [PubMed] [Google Scholar]; k Yang M.; Wang L.; He Z.-H.; Wang S.-H.; Tu Y.-Q.; Zhang F.-M. Org. Lett. 2012, 14, 5114–5117. [DOI] [PubMed] [Google Scholar]; l Li H.; Zheng J.; Xu S.; Ma D.; Zhao C.; Fang B.; Xie X.; She X. Chem.—Asian J. 2012, 7, 2519–2522. [DOI] [PubMed] [Google Scholar]; m Yao Y.; Liang G. Org. Lett. 2012, 14, 5499–5501. [DOI] [PubMed] [Google Scholar]; n Lu Z.; Li Y.; Deng J.; Li A. Nature Chem. 2013, 5, 679–684. [DOI] [PubMed] [Google Scholar]; o Hanessian S.; Dorich S.; Menz H. Org. Lett. 2013, 15, 4134–4137. [DOI] [PubMed] [Google Scholar]; p Zheng C.; Wang L.; Li J.; Wang L.; Wang D. Z. Org. Lett. 2013, 15, 4046–4049. [DOI] [PubMed] [Google Scholar]
- a Corminboeuf O.; Overman L. E.; Pennington L. D. J. Am. Chem. Soc. 2003, 125, 6650–6652. [DOI] [PubMed] [Google Scholar]; b This protocol was optimized to reduce purification steps and improve the yields. See the Supporting Information for details.; c The (R)-enantiomer is less expensive than the (S)-enantiomer, which would lead to the correct enantiomer of the Daphniphyllum alkaloids.
- Stella L. Angew. Chem., Int. Ed. Engl. 1983, 22, 337–350and references cited therein. [Google Scholar]
- a Mackiewicz P.; Furstoss R. Tetrahedron 1978, 34, 3241–3260. [Google Scholar]; b Zard S. Z. Chem. Soc. Rev. 2008, 37, 1603–1618and references cited therein. [DOI] [PubMed] [Google Scholar]; c Quiclet-Sire B.; Zard S. Z. Beilstein J. Org. Chem 2013, 9, 557–576. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Quiclet-Sire B.; Zard S. Z. Pure Appl. Chem. 2011, 83, 519–551. [Google Scholar]; e Li Z.; Song L.; Li C. J. Am. Chem. Soc. 2013, 135, 4640–4643. [DOI] [PubMed] [Google Scholar]; f Zhuang S.; Liu K.; Li C. J. Org. Chem. 2011, 76, 8100–8106. [DOI] [PubMed] [Google Scholar]; g Wang Y.-F.; Zhang F.-L.; Chiba S. Org. Lett. 2013, 15, 2842–2845. [DOI] [PubMed] [Google Scholar]; h Clark A. J.; Filik R. P.; Thomas G. H.; Sherringham J. Tetrahedron Lett. 2013, 54, 4094–4097. [Google Scholar]
- a Prévost N.; Shipman M. Tetrahedron 2002, 58, 7165–7175. [Google Scholar]; b Banwell M. G.; Lupton D. W. Heterocycles 2006, 68, 71–92. [Google Scholar]; c Liu F.; Liu K.; Yuan X.; Li C. J. Org. Chem. 2007, 72, 10231–10234. [DOI] [PubMed] [Google Scholar]; d Zhai H.; Zlotorzynskya M.; Sammis G. Chem. Commun. 2009, 5716–5718. [DOI] [PubMed] [Google Scholar]; e Minozzi M.; Nanni D.; Spagnola P. Chem.—Eur. J. 2009, 15, 7830–7840and references cited therein. [DOI] [PubMed] [Google Scholar]; f Zhai H.; Wickenden J. G.; Sammis G. M. Synlett 2010, 3035–3038. [Google Scholar]; g Zlotorzynska M.; Zhai H.; Sammis G. M. J. Org. Chem. 2010, 75, 864–872. [DOI] [PubMed] [Google Scholar]
- a Cassayre J.; Gagosz F.; Zard S. Z. Angew. Chem., Int. Ed. 2002, 41, 1783–1785. [DOI] [PubMed] [Google Scholar]; b Guindon Y.; Guérin B.; Landry S. R. Org. Lett. 2001, 3, 2293–2296. [DOI] [PubMed] [Google Scholar]; c White L. V.; Schwartz B. D.; Banwell M. G.; Willis A. C. J. Org. Chem. 2011, 76, 6250–6257. [DOI] [PubMed] [Google Scholar]; d Schwartz B. D.; Jones M. T.; Banwell M. G.; Cade I. A. Org. Lett. 2010, 12, 5210–5213. [DOI] [PubMed] [Google Scholar]
- a Musa O. M.; Horner J. H.; Shahin H.; Newcomb M. J. Am. Chem. Soc. 1996, 118, 3862–3868. [Google Scholar]; b Horner J. H.; Martinez F. N.; Musa O. M.; Newcomb M.; Shahin H. E. J. Am. Chem. Soc. 1995, 117, 11124–11133. [Google Scholar]
- Kempter I.; Groß A.; Hartung J. Tetrahedron 2012, 68, 10378–10390. [Google Scholar]
- a Le Tadic-Biadatti M.-H.; Callier-Dublanchet A.-C.; Horner J. H.; Quiclet-Sire B.; Zard S. Z.; Newcomb M. J. Org. Chem. 1997, 62, 559–563. [DOI] [PubMed] [Google Scholar]; b Horner J. H.; Musa O. M.; Bouvier A.; Newcomb M. J. Am. Chem. Soc. 1998, 120, 7738–7748. [Google Scholar]
- The initial product of N–Cl reduction partially cyclizes in a conjugate addition fashion to afford a 1:1 ratio of cyclized and uncyclized reduction products.
- a Schulte-Wülwer I. A.; Helaja J.; Göttlich R. Synthesis 2003, 1886–1890. [Google Scholar]; See also:; b Boate D.; Fontaine C.; Guittet E.; Stella L. Tetrahedron 1993, 49, 8397–8406. [Google Scholar]; c Miura T.; Morimoto M.; Murakami M. Org. Lett. 2012, 14, 5214–5217. [DOI] [PubMed] [Google Scholar]
- All other hydrogen atom sources investigated (Ph3SnH, TMS3SiH, PhSH) led to N–Cl reduction or side reactions such as conjugate addition.
- Toluene could serve as a terminal hydrogen atom source in this reaction.
- Average of five runs.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.