

Abstract

The regulatory component (MMOB) of soluble methane monooxygenase (sMMO) has a unique N-terminal tail not found in regulatory proteins of other bacterial multicomponent monooxygenases. This N-terminal tail is indispensable for proper function, yet its solution structure and role in catalysis remain elusive. Here, by using double electron–electron resonance (DEER) spectroscopy, we show that the oxidation state of the hydroxylase component, MMOH, modulates the conformation of the N-terminal tail in the MMOH–2MMOB complex, which in turn facilitates catalysis. The results reveal that the N-terminal tail switches from a relaxed, flexible conformational state to an ordered state upon MMOH reduction from the diiron(III) to the diiron(II) state. This observation suggests that some of the crystallographically observed allosteric effects that result in the connection of substrate ingress cavities in the MMOH–2MMOB complex may not occur in solution in the diiron(III) state. Thus, O2 may not have easy access to the active site until after reduction of the diiron center. The observed conformational change is also consistent with a higher binding affinity of MMOB to MMOH in the diiron(II) state, which may allow MMOB to displace more readily the reductase component (MMOR) from MMOH following reduction.
Soluble methane monooxygenase (sMMO) is a multicomponent enzyme that hydroxylates a wide range of hydrocarbons.1,2 To accomplish this task, the enzyme finely orchestrates reactions involving four substrates, including protons, electrons, dioxygen, and the hydrocarbon (methane, for example):
The chemistry is effected by the interplay among three protein components.1−3 For sMMO from Methylococcus capsulatus (Bath), a 251-kilodalton (kDa) dimeric (α2β2γ2) hydroxylase designated MMOH houses a non-heme diiron active site in the α-subunit of each monomer.4 A 38.6-kDa reductase known as MMOR is responsible for reduction of the diiron center, transferring two electrons from NADH through its flavin adenine dinucleotide cofactor and [Fe2S2] cluster.5 The third component is a 15.9-kDa cofactorless regulatory protein, MMOB, which has a unique N-terminal sequence composed of approximately 35 residues.6−8 MMOB induces many effects upon binding to MMOH, including decreasing the redox potential of MMOH,9 changing the coordination of Fe2 as well as the global conformation of the reduced enzyme,10 and increasing the yield and rate of hydrocarbon hydroxylation.11,12
How MMOB exerts these regulatory effects is enigmatic. The structure and function of its N-terminal sequence are of particular interest. In the bacterial multicomponent monooxygenase (BMM) superfamily, the presence of an N-terminal tail in the regulatory protein is unique to sMMO,6 the only enzyme capable of oxidizing methane.1,2 The tail is required for steady-state catalytic activity8,13 as well as for the generation of reaction intermediates Hperoxo and Q.13 Solution-state nuclear magnetic resonance (NMR) spectroscopic studies revealed that free MMOB has a well-folded core, but that the N-terminal sequence is largely unstructured.7,14 As discovered in a recent crystal structure determination (Protein Data Bank (PDB) ID 4GAM) of two MMOB proteins complexed to one MMOH (H–2B complex), the MMOB core docks into the canyon region on each side of MMOH, while the N-terminal tail, strikingly, forms an unusual ring-like conformation on the surface of MMOH (Figure 1a), contacting helices H and 4.8 The MMOB core together with the N-terminal tail exert allosteric effects within MMOH, connecting cavities in MMOH for the ingress of gaseous substrates, and closing a pore proposed as the proton transfer pathway from MMOH surface to the diiron center.8 The oxidation state of the iron atoms in the H–2B complex could not be determined unambiguously from the crystal structure; coordination of residue E243 is similar to that of MMOH in the diiron(II) state (Hred), but the Fe···Fe and Fe–O distances are closer to those in the diiron(III) state (Hox).8
Figure 1.
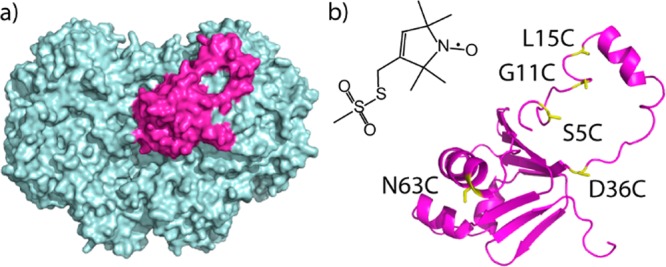
X-ray crystal structure of Hox–2B complex (Protein Data Bank (PDB) ID 4GAM). (a) View of the structure, with MMOH in cyan and MMOB in magenta. There is a second MMOB bound on the other side of MMOH. (b) Structure of the spin label, MTSL. Also depicted is MMOB in the Hox–2B complex, indicating two positions (N63C and D36C) within the core and three (S5C, G11C, and L15C) on the N-terminal tail, labeled for MMOB core-to-tail distance determinations.
To gain a more dynamic, solution-state view of the complex, and to study how MMOB responds to MMOH reduction priming the complex for catalysis, we used double electron–electron resonance (DEER) spectroscopy. DEER is a pulsed electron paramagnetic resonance technique that accurately determines distances between paramagnetic centers typically in the 1.8–6 nm range in frozen solution.15 In the present study, pairs of 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl methanethiosulfonate spin labels (MTSL, Figure 1b inset) were attached to pairs of amino acids strategically positioned in the MMOB core and its N-terminal tail, through the introduction of site-directed double cysteine mutations (Figure 1b). Five labeling positions, two on the core region (N63C and D36C) and three on the N-terminal tail (L15C, G11C, and S5C), were selected for measuring core-to-tail distances, which define the conformation of MMOB. The labeling positions are all solvent-exposed and not on the hydroxylase-binding surface, based on the NMR study7 and the X-ray crystal structure.8 Mutations at these positions cause minimal structural perturbation, as evidenced by the high catalytic activity retained by the spin-labeled MMOB mutants (Figure S1, Supporting Information [SI]).
We first investigated the solution conformation of MMOB in the oxidized complex, Hox–2B, and compared the DEER results with the crystal structure. With the spin-labeled L15C/N63C mutant in the absence of MMOH, the DEER-derived core-to-tail distance measurement featured a very broad distribution with multiple peaks/shoulders, ranging from less than 3 nm to more than 6 nm (Figure S2a, SI), consistent with disorder in the N-terminal tail as indicated by NMR spectroscopy.7,14 On forming the Hox–2B complex, the distance distribution narrowed, and a major peak emerged, centered at 3.6 nm (Figure 2a), a value in good agreement with that derived from a rotamer analysis of spin-labeled side chains of the L15C/N63C mutant based on the H–2B crystal structure (Figure 2a, right panel, red dashed line). This result suggests that, in solution, the N-terminal tail of MMOB adopts the crystallographically observed ring-like conformation upon binding to MMOH, at least for residues from L15 to the MMOB core. A minor peak centered at approximately 5 nm indicated the presence of other conformational states in solution.
Figure 2.

X-band DEER of spin-labeled MMOB double cysteine mutants in Hox–2B complex and the corresponding distance distributions. Left panels, background subtracted DEER data (black) and simulated fits (red) for spin-labeled MMOB double mutants L15C/N63C, L15C/D36C, G11C/N63C, and S5C/N63C in Hox–2B complex. Right panels, the corresponding distance distributions derived from the DEER data (black) and from the crystal structure of Hox–2B complex (red dashed lines). DEER data were processed by using DeerAnalysis 2011.16 The distance distributions derived from the crystal structure were modeled by using MMM program package 2011.2.17
We next placed the spin label on the core at another position, D36, by preparing the L15C/D36C mutant. In the absence of MMOH, this mutant again exhibited a broad distance distribution ranging from less than 2 to 5 nm, with a major peak centered at 2.8 nm (Figure S2b, SI). Upon binding to MMOH in the diiron(III) state, the major peak shifted to 2.3 – 2.4 nm (Figures 2b), in good agreement with the 2.5 nm distance derived from the crystal structure (Figure 2b, right panel, red dashed line). The width of the distance distribution, however, was broader than that compatible with the crystal structure, suggesting a more flexible N-terminal tail in solution.
As the labeling position on the N-terminal tail was moved closer to the N-terminus, wide distance distributions were observed for the Hox–2B complex. With the spin-labeled G11C/N63C mutant, a broad distance distribution was observed with three major peaks ranging from 2 to 5.5 nm (Figure 2c), whereas a single peak at 2.9 nm is expected for this mutant based on the crystal structure (Figure 2c, red dashed line). The distance distribution obtained with spin-labeled S5C/N63C (Figure 2d) was again much wider than that expected based on the crystal structure (Figures 2d, right panel, red dashed line).
These results indicate that the structure of the first several residues, at least from the N-terminus to G11, are highly flexible in the Hox–2B complex in solution, in contrast to the well-ordered structure observed crystallographically. Therefore, some of the key interactions between the N-terminal tail of MMOB and MMOH observed in the crystal structure are unlikely to be maintained stably in the Hox–2B complex in solution. In particular, in the crystalline state, Y8 of MMOB forms hydrogen bonds with R307 and E299 in MMOH, reorienting W308. These interactions together with those between the MMOB core and MMOH connect the cavities for gaseous substrates ingress.8 The importance of Y8 for catalysis was evidenced by the approximately 70% decrease in catalytic activity exhibited by the MMOB mutant with the first eight residues truncated,8 whereas full activity was achieved when only the first five residues were truncated (Figure S1, SI). In the solution state, however, Y8 is unlikely to form stable hydrogen bonding interactions with MMOH residues in the Hox–2B complex because S5 and G11 of MMOB are both highly flexible. This argument raises the possibility that the substrate ingress cavities are largely disconnected in the Hox–2B complex in solution, blocking O2 and possibly CH4 access to the diiron(III) center.
Upon reduction of MMOH from the diiron(III) to the diiron(II) state, however, the MMOB core-to-tail distance distributions in the Hred–2B complex narrowed significantly. With spin-labeled L15C/N63C and L15C/D36C mutants, single peaks at 3.6 and 2.1 nm, respectively, were observed (Figures 3a, b), in general agreement with the 3.6 and 2.5 nm distances derived from the crystal structure. More dramatic changes occurred when labeling positions were moved closer to the N-terminus. With spin-labeled G11C/N63C and S5C/N63C mutants, single dominant peaks at 3.7 and 3.0 nm, respectively, appeared (Figures 3c, d), slightly longer than the 2.9 and 2.5 nm distances derived from the crystal structure, but in stark contrast to the broad, multipeak distance distributions in the diiron(III) state.
Figure 3.
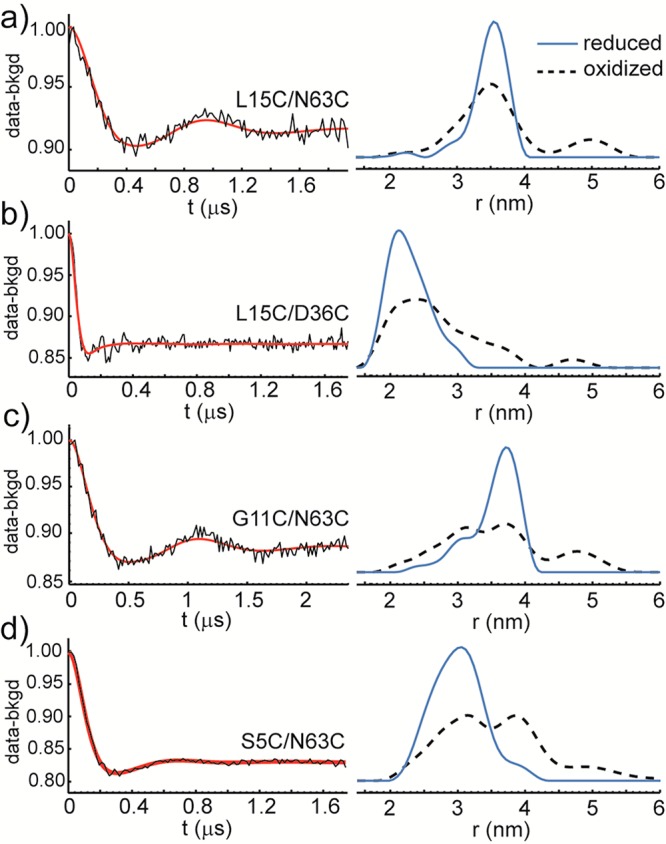
X-band DEER of spin-labeled MMOB double cysteine mutants in the Hred–2B complex and the corresponding core-to-tail distance distributions. Left panels, background subtracted DEER data (black) and simulated fits (red) for spin-labeled MMOB double mutants L15C/N63C, L15C/D36C, G11C/N63C, and S5C/N63C in the Hred–2B complex. Right panels, the corresponding distance distributions derived from the DEER data (blue); the DEER derived distance distributions of the corresponding Hox–2B complex are also shown (black dashed lines). DEER data were processed by using DeerAnalysis 2011.16 The distance distributions derived from the crystal structure were modeled by using MMM program package 2011.2.17
The narrower distance distributions observed with Hred–2B complex indicate that the N-terminal tail of MMOB adopts a significantly more ordered structure when MMOH is reduced. This finding has two important implications. First, the allosteric effects exerted by the MMOB N-terminal tail on the MMOH internal structure observed crystallographically are most likely relevant to the reduced form of the complex in the solution state. Such an effect was discussed above for the interaction between Y8 of the N-terminal tail and MMOH. It is at this stage, when the hydroxylase is reduced, that formation of stable interactions with MMOH, triggered by Y8, connect the substrate ingress cavities, allowing O2 and hydrocarbon access to the diiron(II) site. Second, the N-terminal tail and/or the entire MMOB molecule may bind with higher affinity to MMOH in the reduced state than in the oxidized state. To determine quantitatively how MMOB binding affinity might change with iron oxidation state, we determined the Kd values of H–2B complexes by fluorescence anisotropy titrations using fluorescently labeled MMOB. The results, shown in Figure 4, reveal stronger binding of MMOB to reduced MMOH. The titration curves were well fit, assuming that each MMOH has two noninteracting binding sites for MMOB. A Kd value of 0.55 ± 0.03 μM was observed for the oxidized complex, comparable to that previously determined by isothermal titration calorimetry.3 The Kd value dropped to 0.17 ± 0.02 μM when the complex was reduced, indicating tighter binding affinity of MMOB for the reduced MMOH. This change may allow (i) MMOB more readily to displace the reductase MMOR from MMOH after MMOH reduction, if they both bind to the canyon region of MMOH, situated just above the diiron active site, and (ii) the N-terminal tail of MMOB to form stable contacts with MMOH that allosterically lead to the connection of substrate ingress cavities and facilitate catalysis.
Figure 4.
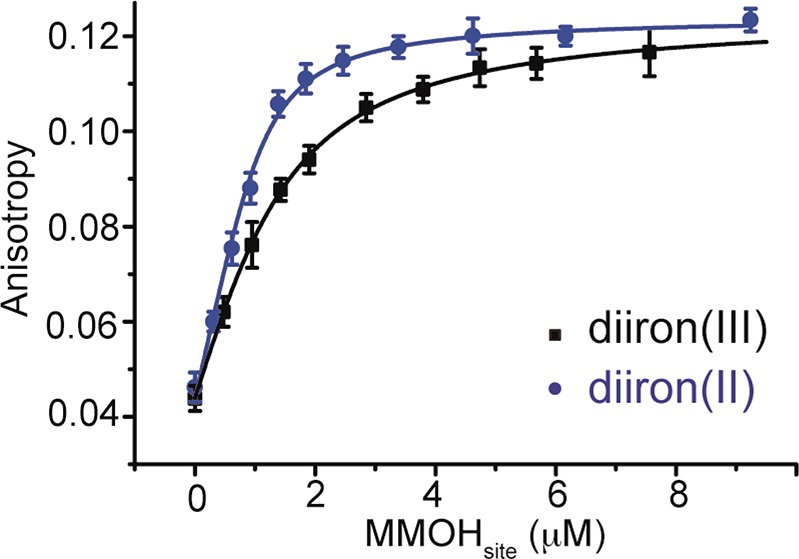
MMOB binding to MMOH in the diiron(III) state and the diiron(II) state. The fluorescence anisotropy of 1 μM 1,5-IAEDANS-labeled MMOB D36C mutant was recorded as MMOH was titrated in. Titration curves were fit, assuming each MMOH has two noninteracting MMOB binding sites (MMOHsite).
The modulation of protein conformation by the redox state of the active-site metal center, as observed with the H–2B complex, is also used by other enzyme systems as a mechanism to control catalysis. The heme-containing monooxygenase, cytochrome P450,18 provides one such example. Cytochrome P450 enzymes activate O2 at a heme iron center coordinated by a cysteine thiolate, which increases the pKa of the iron(IV) hydroxide intermediate.19 The two electrons required for O2 activation18 are delivered from a redox partner, which also serves as an effector protein.20 A recent solution-state study using DEER spectroscopy revealed that the conformation of the cytochrome P450cam in complex with its reductase/effector protein putidaredoxin depends on the oxidation state of the heme center.21
In summary, our results shed light on how the oxidation state of the iron atoms in MMOH modulates the conformation of MMOB, which in turn facilitates catalysis. The N-terminal tail of MMOB in the H–2B complex adopts ring-like conformations in solution, similar to that observed in the crystal structure. A more relaxed, flexible structure of the N-terminal tail was observed for the Hox–2B complex in solution, however. This result suggests that the crystallographically determined allosteric conformational changes triggered by interaction of Y8 in the MMOB N-terminal tail with MMOH are disfavored in the Hox–2B complex in solution, and that the substrate ingress cavities may not be connected until after reduction of the diiron center. The N-terminal tail switches to an ordered conformation in response to MMOH reduction to the diiron(II) state, producing more stable interactions with MMOH and allowing MMOB to exert its allosteric effects on the reduced MMOH, connecting cavities in MMOH for substrate ingress. We also demonstrate that, consistent with the observed conformational change, MMOB binds more tightly to reduced MMOH. This increased binding affinity may allow MMOB more readily to displace the reductase MMOR from the reduced MMOH.
Acknowledgments
We thank Prof. Robert G. Griffin for access to the pulsed EPR spectrometer in his laboratory. We thank Rebecca P. Luoh for assistance in the preparation and activity assay of the MMOB Δ2-5 mutant and Ali D. Liang for helpful discussions. This work was supported by NIH Grant GM032134 from the National Institute of General Medical Sciences.
Supporting Information Available
Experimental details, Supporting Information Figures S1–4, Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Merkx M.; Kopp D. A.; Sazinsky M. H.; Blazyk J. L.; Müller J.; Lippard S. J. Angew. Chem., Int. Ed. 2001, 40, 2782. [DOI] [PubMed] [Google Scholar]
- Wallar B. J.; Lipscomb J. D. Chem. Rev. 1996, 96, 2625. [DOI] [PubMed] [Google Scholar]
- Gassner G. T.; Lippard S. J. Biochemistry 1999, 38, 12768. [DOI] [PubMed] [Google Scholar]
- Rosenzweig A. C.; Frederick C. A.; Lippard S. J.; Nordlund P. Nature 1993, 366, 537. [DOI] [PubMed] [Google Scholar]
- Kopp D. A.; Gassner G. T.; Blazyk J. L.; Lippard S. J. Biochemistry 2001, 40, 14932. [DOI] [PubMed] [Google Scholar]
- Brandstetter H.; Whittington D. A.; Lippard S. J.; Frederick C. A. Chem. Biol. 1999, 6, 441. [DOI] [PubMed] [Google Scholar]
- Walters K. J.; Gassner G. T.; Lippard S. J.; Wagner G. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J.; McCormick M. S.; Lippard S. J.; Cho U. S. Nature 2013, 494, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen K. E.; Liu Y.; Fox B. G.; Lipscomb J. D.; Munck E.; Stankovich M. T. Biochemistry 1994, 33, 713. [DOI] [PubMed] [Google Scholar]
- Mitić N. a.; Schwartz J. K.; Brazeau B. J.; Lipscomb J. D.; Solomon E. I. Biochemistry 2008, 47, 8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. E.; Valentine A. M.; Wang D.; Huynh B. H.; Edmondson D. E.; Salifoglou A.; Lippard S. J. J. Am. Chem. Soc. 1995, 117, 10174. [Google Scholar]
- Liu Y.; Nesheim J. C.; Lee S.-K.; Lipscomb J. D. J. Biol. Chem. 1995, 270, 24662. [DOI] [PubMed] [Google Scholar]
- Chang S. L.; Wallar B. J.; Lipscomb J. D.; Mayo K. H. Biochemistry 2001, 40, 9539. [DOI] [PubMed] [Google Scholar]
- Chang S.-L.; Wallar B. J.; Lipscomb J. D.; Mayo K. H. Biochemistry 1999, 38, 5799. [DOI] [PubMed] [Google Scholar]
- Jeschke G. Annu. Rev. Phys. Chem. 2012, 63, 419. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Chechik V.; Ionita P.; Godt A.; Zimmermann H.; Banham J.; Timmel C. R.; Hilger D.; Jung H. Appl. Magn. Reson. 2006, 30, 473. [Google Scholar]
- Polyhach Y.; Bordignon E.; Jeschke G. Phys. Chem. Chem. Phys. 2011, 13, 2356. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Chem. Rev. 2005, 105, 2253. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Science 2013, 342, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson C. A.; Lipscomb J. D.; Gunsalus I. C. J. Biol. Chem. 1972, 247, 5777. [PubMed] [Google Scholar]
- Myers W. K.; Lee Y.-T.; Britt R. D.; Goodin D. B. J. Am. Chem. Soc. 2013, 135, 11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.