

Abstract
Recent advances in our understanding of the underlying genetic architecture of psychiatric disorders, has blown away “old” diagnostic boundaries defined by currently used diagnostic manuals. The DISC1 gene was originally discovered at the breakpoint of an inherited chromosomal translocation, which segregates with major mental illnesses. Many biological studies have indicated the role of DISC1 in early neurodevelopment and synaptic regulation. Given the additional insight that DISC1 is thought to drive a range of endophenotypes, which underlie major mental conditions, the biology of DISC1 may provide a hint on constructing new diagnostic categories for mental illnesses with more meaningful biological foundation.
Introduction
Major mental illnesses, such as schizophrenia and mood disorders, remain medical and financial burdens in our society. This is despite revolutions in treatment in the past fifty years with antipsychotic and antidepressant use and the current availability of generic forms of many of these drugs. Statistics from the website of the US research institute, the National Institute for Mental Health (NIMH) highlight this and show that in 2006 alone there was expenditure of nearly $58 billion on mental health in the US (http://www.nimh.nih.gov/statistics/4COST_AM2006.shtml). The often quoted, but unfortunately correct, reason for this predicament is the serendipitous identification of these medications, with no appreciation for the complex underlying pathophysiology. Thus the drugs treat a subset of symptoms but not the core pathology. The product of this approach has been that many disease domains, such as cognitive deficits and negative symptoms in schizophrenia, remain poorly treated1–5. In addition to the severe limitations in efficacy, most of these compounds have substantial side effects6–8 and improved medications are clearly needed. We predict that breakthrough medications for mental illnesses will be developed through an integrated understanding of the etiopathology, genetic underpinnings and altered brain circuitry of these illnesses. Fortunately with the advent of new genetic approaches, such as next generation sequencing9, and a growing toolbox for dissecting brain circuitry10, we are well positioned to deliver on this.
What is abundantly clear is that the major mental illnesses are multi-factorial brain disorders driven by a combination of genetic and environmental susceptibility factors11–13. Recent advances in the understanding of the underlying genetic architecture suggest that “old” diagnostic boundaries defined by currently used diagnostic manuals, such as Diagnostic and Statistical Manual (DSM) and International Classification of Diseases (ICD) may be difficult to reconcile with more biology-based assessments of mental illnesses. For example, shared risk variants between schizophrenia, bipolar disorder, and autism have been reported14–16. The common genetic underpinnings of schizophrenia with diseases which manifest in childhood, such as autism, has further strengthened the view that schizophrenia is likely to include neurodevelopmental etiologies and indicated the importance of looking at early life events for causative factors16,17. A number of key literatures support this notion: epidemiological studies have indicated that adverse events during the prenatal and perinatal periods are important18; premorbid behavioral and neurological signs are prominent in many patients with schizophrenia, whereas brain imaging approaches have identified abnormalities at disease onset17,19. In addition, there are a number of preclinical animal models of schizophrenia that are based on lesions early in development. They only manifest behavioral symptoms post-adolesence, thus mimicking the developmental trajectory of schizophrenia17,20 This delayed onset after puberty also indicates important roles for dynamic changes in adolescent brain maturation, such as loss of glutamatergic synapses (synaptic pruning), maturation of interneurons, dopaminergic neurons, and full development of myelination21. Amongst these possibilities, synaptic changes in adolescence could be very important because synaptic changes have been frequently reported in the pyramidal neurons of the cortex and hippocampus in brains from patients with schizophrenia, and are thought to reflect disease alterations in critical brain circuitry22–24.
Therefore, in studying molecular mechanisms underlying the pathophysiology of schizophrenia and related disorders, a sharp focus on the specific disease neurodevelopmental trajectory, especially in early development and adolescent brain maturation, is vitally important. This significance has been recently summarized in several review articles21,25. As we seek to identify ways and means to understand these complex disorders, the discovery of ‘Disrupted-in-Schizophrenia 1’ (DISC1), a molecule that has crucial roles both early in development and during adolescence, including synapse regulation, has provided an invaluable window onto the disease landscape. In this review, we will describe current thinking on the roles of DISC1 and its network of interacting proteins (interactome) in brain development and the mature brain, and the corresponding influences on phenotypic manifestations relevant to major mental illnesses. We will also critically discuss the therapeutic implications of these findings and the priorities for further study and rationalization across DISC1 biology.
Discovery of the Scottish pedigree, identification of the DISC1 gene, and functional studies: the 40-year history
The DISC1 gene was originally identified in a unique Scottish pedigree in which an inherited balanced chromosomal translocation disrupts this gene and segregates with major mental disorders, such as major depression, schizophrenia, and bipolar disorder (The details of the discovery of DISC1 can be seen in BOX 1)26–30. Following the identification of the Scottish pedigree, many groups have examined the genetic association with schizophrenia, bipolar disorder, major depression, autism, and developmental mental disorders31–36. Substantial numbers of studies have reported a modest, but significant, association of the DISC1 locus with these disorders. Indeed, recent reports have identified several ultra-rare DISC1 variants that might be associated with schizophrenia and/or bipolar disorder37,38. However, it should be noted that recent genome-wide association studies (GWAS) have not identified DISC1 as a prominent hit for either schizophrenia or bipolar disorder, and a meta-analysis failed to observe significant association between DISC1 and schizophrenia39–43.
Box 1. Discovery of the DISC1 t(1;11) pedigree and the DISC1 gene.
The discovery of the DISC1 pedigree has to be thankful to the UK Medical Research Council and its thorough collection of chromosomal abnormalities in the 1960s and 1970s, and their location in Scotland which enabled multiple generations of the pedigree to be traced27. The pedigree was originally identified and published over forty years ago by Patricia Jacobs and colleagues at the University of Edinburgh as part of a population survey of chromosomal abnormalities28. The original observation of a balanced translocation between chromosomes 1 and 11 [(1;11)(q42.1;q14.3)] was made in an 18-year old boy with adolescent conduct disorder who had been placed in a young offenders institute28. Analysis of his family identified additional members with the translocation and critically, over the next 20 years members of the family were interviewed by psychiatrists Professors David St Clair, Walter Muir and Douglas Blackwood using a standard diagnostic instrument26. Their work confirmed a diagnosis of schizophrenia or other major mental disorder in several family members. In fact all members with a major mental illness carried the translocation. The last detailed clinical report on the family was published in 200130. In 2000, another major breakthrough with the pedigree occurred with Millar and Porteous cloning the gene impacted on chromosome 1. They named the 13 exon gene, with the breakpoint between exons 8 and 9 as ‘Disrupted in Schizophrenia 1’ (DISC1)29. AlthoughDISC1 is the only disrupted transcript within an open reading frame, a transcript occurring on the antisense strand, DISC2, is also disrupted29. The biological consequences of this chromosomal disruption are under debate68–70,173,174 (see Box 2). The scheme is adapted from the previous publication by Blackwood et al30.
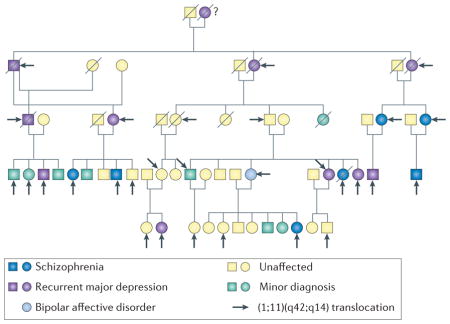
How can these apparent genetic contradictions be reconciled? Accumulating evidence has been reported in association studies of a genetic link between DISC1 and specific endophenotypes commonly associated with major mental illnesses, including behavioral (anhedonia, frontal lobe-associated memory) electrophysiological (auditory event-related potential) and anatomical (cortical thickness, hippocampal gray matter volume, white matter integrity) characteristics34,44–56. Furthermore, a unique condition of a clinical mixture of schizophrenia and mood disorders, such as schizoaffective disorder, is associated more significantly with the DISC1 locus32,35. Thus, DISC1 may not be a key risk factor for schizophrenia nor other DSM-defined disorders, but does confer a genetic risk at the level of endophenotypes or brain circuitry that underlies many major mental disorders. We can also note that GWAS may require more samples to detect entire repertoires of disease-associated common variants and that most of the genome-wide studies for copy number variants may not fully detect small chromosomal deletions/duplications57,58. Such technical issues may also underlie the failure in identifying DISC1 as a prominent hit in association with schizophrenia.
In parallel with such clinical-genetic studies, efforts to elucidate the biological function of DISC1 have made significant progress in the past decade. DISC1 is highly expressed in the developing brain, and is also expressed in various brain areas including the olfactory bulb, cortex, hippocampus, hypothalamus, cerebellum, and brain stem after birth59. In adulthood, the highest expression of DISC1 is observed in the dentate gyrus of the hippocampus60. DISC1 is expressed not only in neurons (both pyramidal neurons and interneurons)59, but also in glial cells61.
Much of the understanding on DISC1’s biological function comes through an understanding of the biology of DISC1-interacting proteins and the functions of these protein complexes62,63. Though there have been multiple yeast-two hybrid studies reported with DISC164–67, the most comprehensive catalogue of interactions to date was published as the ‘DISC1 interactome’ by Camargo and colleagues63 in 2007 (See Fig. 1 for a modified interactome highlighting relevant proteins discussed in this review). This study comprised iterative yeast-two hybrid screens with DISC1 as the starting protein, followed by screens with a diverse set of DISC1 interactors, representing the most prevalent functional protein classes from the original bioinformatic analysis. The data from this study, together with the early biochemical studies of DISC1, pioneered the concept that DISC1 is likely to be an intracellular scaffold protein with many protein interactors68–70. Since this study, individual interactions have been confirmed in other experimental systems21,71–74. Identification of the specific binding sites for individual interactions has played a major role in deciphering the function of the various DISC1 partnerships, the most important of which are shown in Fig. 2. Studies of the protein interactors of DISC1 have shown that it has at least two major roles in the brain: regulation of key processes in early brain development and synapse formation/maintenance. Such roles are consistent with current hypotheses for the etiopathology of major mental illness and thus provide us a ‘DISC1-shaped’ window to view these diseases.
Figure 1. The ‘DISC1 interactome’ points towards the multiple functions of DISC1 during neuronal development.

The important DISC1 interactions relevant for this review are highlighted in this figure. They are a small subset of the original dataset63, which have all been confirmed in other biological systems66,82,90,94,95,110,111,140,200. The functional relevance of the different interactions is highlighted on the perimeter of the cartoon, with arrows representing the chronological order of events during development. DISC1, Disrupted-in-schizophrenia-1; GSK3β, Glycogen synthase kinase 3β; Dixdc1, DIX domain containing 1; NDEL1, Nuclear distribution protein nudE-like 1; LIS1, Lissencephaly protein 1; PCM1, Pericentriolar material 1; BBS, Bardet-Biedl Syndrome protein; PDE4; Phosphodiesterase type 4; Kal-7, Kalirin-7; TNIK, TRAF2 and NCK-interacting protein kinase; and PSD95; Postsynaptic density protein 95.
Figure 2. DISC1 protein interaction domains and relationship to the location of rare human variants associated with mental disorders.

Diagram of protein-protein interaction domains mapped onto DISC1 and key DISC1 genetic variants (top) possibly associated with mental disorders34–38,78,82,87,90,95,110,111,141,185,200. PCM1, Pericentriolar material 1; BBS, Bardet-Biedl Syndrome; Dixdc1, DIX domain containing 1; GSK3β Glycogen synthase kinase 3β; LIS1, Lissencephaly protein 1; NDE1, Nuclear Distribution protein NudE homolog 1; NDEL1, Nuclear Distribution Protein NudE-Like 1; PDE4, Phosphodiesterase type 4; TNIK, TRAF2 and NCK-interacting protein kinase; and Kal-7, Kalirin-7. Genetic variants at Q264R, L607F, and S704C (common variants) are associated with schizophrenia (SZ). Furthermore, associations with L607F (*) and S704C (#) are reported for schizoaffective (SA) and major depression (MD), respectively. BP, bipolar disorder. Please note that the binding sites have been mapped using a range of different approaches so the resolution achieved in individual interactions is different (dotted lines are sites awaiting fine mapping).
Role for DISC1 in early neurodevelopment
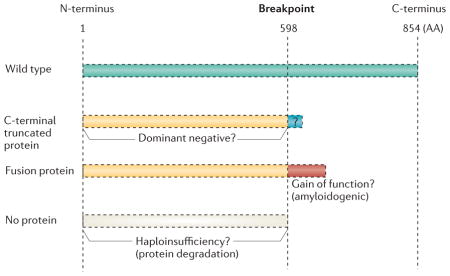
Using yeast two-hybrid screening, multiple labs independently determined that DISC1 interacts with a class of proteins that interact with microtubules and associated complexes (Fig. 1). These proteins include NDEL1, NDE1, LIS1, 14-3-3, dynactin, MIPT3, and MAP1A64,66,75–77. The interactions of DISC1 with these microtubule-related proteins have also been confirmed biochemically64,66,75,76. Immunofluorescent cell staining and biochemical studies have indicated that DISC1 is located to the centrosome, which is the microtubule-organizing center, and also associates with microtubules themselves64,76,78. A C-terminal truncated form of DISC1 (amino acids 1-598), that could be generated by the chromosomal abnormality identified in the original Scottish pedigree functions as a dominant-negative, blocking proper accumulation of DISC1 (A discussion on possible pathological mechanisms due to this chromosomal abnormality is introduced in BOX 2), NDEL1, and their dynein associated proteins to the centrosome and disturbing microtubular dynamics78. It is now appreciated that regulation of microtubules is critical for key neurodevelopmental processes such as neuronal migration. What is the major role for DISC1 in association with the microtuble-centrosomal network in the brain? Studies of Miller-Dieker lissencephaly syndrome, a human developmental brain disorder caused by neuronal migration defects resulting in abnormal layering of the cerebral cortex, have shown that LIS1, a DISC-1 interacting protein, is causal for the disease79. Furthermore, RNAi knockdown of LIS1 and NDEL1 in developing mouse brains impairs centrosome-associated microtubular dynamics and leads to defects of neuronal migration in the developing cortex80. These results together with the finding that DISC1 expression is higher in the developing cortex59,66 led to an intriguing hypothesis that DISC1 may be implicated in neurodevelopmental processes, in particular neuronal migration.
Box 2. What are the possible pathological mechanism of the DISC1 t(1;11) chromosomal abnormality?
There are three major hypotheses at present which address this question. Sawa and Snyder68 initially proposed that this chromosomal abnormality in the Scottish pedigree could lead to a C-terminal truncated DISC1 protein and/or degradation of the protein. Data from cell models indicated that the putative C-terminal truncated protein could act as a dominant-negative78, but the existence of this protein product has not been examined in the brain yet. Furthermore, at least in the lymphoblastoid cell limes from a few subjects with this chromosomal abnormality, the truncated protein was not identified by Western blotting140, Nonetheless, the complexity of DISC1 isoforms and their differences between lymphoblasts and brain, as well as between developing and adult brain, precludes making a strong conclusion here. Most importantly, either a dominant-negative protein or a partial loss of protein leads to an overall loss-of DISC1 function. In addition, the notion that a fusion transcript could form across the breakpoints of both chromosomes 1 and 11 and lead to a fusion protein, which has been shown to be amyloidogenic when overexpressed in cultured cells, has been proposed166.
Roles for DISC1 in corticogenesis in vivo
Radial migration of cortical neurons is an important stage of cortical development and is known to involve microtuble-centrosomal machinery81. A key role for DISC1 in neuronal migration was illustrated by DISC1 knockdown using in utero introduction of short-hairpin RNA (shRNA) at embryonic day 14.5 (E14.5). This elicited robust defects in radial migration in the cortex78 similar to those produced by knockdown of NDEL1 or LIS180,81. Consistent with this finding, expression of the C-terminal truncated dominant-negative DISC1 (described DN-DISC1 in the following sections) results in a similar delay in neuronal migration78. The similarity of phenotypes elicited in these experiments suggests a possible disturbance of the motor-microtubule-centrosome machinery. Indeed, a number of molecular components of this machinery have since been shown to be novel protein interactors of DISC1. These proteins include Bardet-Biedl Syndrome (BBS) proteins, such as BBS1 and BBS4, and Pericentriolar Material 1(PCM1) (Fig. 1). In the developing cortex, cooperative action of DISC1 and BBS4 is required for accumulation of PCM1 to the centrosome and proper neuronal migration82. It should also be noted that the gene coding for PCM1 by itself is a promising candidate for schizophrenia82–84. Furthermore, two nonsynonymous genetic variants, i.e. mutation leads to a change in amino-acid sequence, in the DISC1 locus (S704C, L607F) affect centrosomal PCM1 localization85,86. Taken together, these findings indicate that DISC1 is likely to be an intrinsic factor that regulates migration in the developing cortex (Fig. 3).
Figure 3. DISC1 function during early development.

DISC1 regulates both progenitor cell proliferation and postmitotic neuronal migration. In proliferating progenitors, DISC1 inhibits premature cell cycle exit and differentiation of proliferating progenitors87: In these cells, DISC1 inhibits GSK3β through direct physical interaction, which in turn reduces β-catenin phosphorylation and stabilizes β-catenin. Green circles with the black and white stripe indicate the rapid degradation of this protein. In postmitotic neurons DISC1 preferentially interacts with proteins in the dynein motor complexes associated with microtubules (pink tubes) and the centrosome (two grey connected ovals), including BBS proteins, which mediates neuronal migration78,82. As corticogenesis progresses, the affinity of DISC1 for GSK3β is reduced, likely through phosphorylation at S713; in contrast, the interaction with BBS proteins is increased. Furthermore, DISC1 regulates the transition from neuronal progenitor proliferation to neuronal migration through an increase in phosphorylation of DISC1 at S710, which occurs at embryonic stage E13-15: DISC1/BBS binding recruits DISC1 away from a GSK3β-regulated Wnt/β-catenin activity and thus contributes to the switch from neuronal progenitor proliferation to neuronal migration91. DISC1, Disrupted-in-Schizophrenia-1; BBS, Bardet-Biedl Syndrome protein; and GSK3β, Glycogen synthase kinase 3β.
In cortical development, proper control of progenitor cell proliferation is another key element that operates in parallel to postmitotic neural migration. Following the initial discovery of a role for DISC1 in neural migration, the Tsai lab87 explored a role for DISC1 in progenitor cell proliferation in the developing cortex (Fig. 3). Reduction of DISC1 protein levels by shRNA at E13 results in substantial impairment of cortical progenitor proliferation and reduction of cells in the ventricular and subventricular zones. A key underlying mechanism was shown to be the loss the ability of DISC1 to interact with and inhibit glycogen synthase kinase-3β (GSK3β) (Fig. 1), which in turn phosphorylates β-catenin and mediates its degradation87. β-catenin is an important component of canonical Wnt signaling, which controls progenitor cell proliferation, and implicates DISC1 as a regulator of this pathway. Consistent with this observation, a transgenic mouse that exogenously expressed a portion of the DISC1 genomic locus within a ‘Bacterial Artificial Chromosome (BAC)’, that mimics the chromosomal 1 truncation found in the Scottish pedigree (BAC-DN), exhibits reduced neural proliferation during cortical neurogenesis88 (Table 1).
Table 1.
DISC1 animal models.
| Mutants | Dominant negative transgenic | Knockdown | ||||||
|---|---|---|---|---|---|---|---|---|
| Constitutive | Inducible | |||||||
| Name | Q31L | L100P | Δ25 bp/stop codon | BAC-DN | CaMK-DN | CaMK-DN | CaMK-cc | In utero RNAi |
| Neurogenesis | ↓ (CTX) | ↓ (DG) | ↓ (CTX) | |||||
|
Pyramidal neurons (CTX, HP) Granule neurons (DG) |
Misposition, ↓Dendrites, ↓ Spine | Alteration | ↓ Spine | ↓Dendrite | ↓Dendrite | |||
| Interneurons (mPFC) | → PV, CB | ↓ PV | ↓ PV | ↓ PV | ↓ PV | |||
| Dopamine | ↑ D2High (STR) | ↓ DA, DOPAC (CTX) | ↓ TH, DA (CTX) | |||||
| Behavioral abnormalities | ||||||||
| Amphetamine hypersensitivity | yes | yes | yes | |||||
| MK-801 hypersensitivity | yes | |||||||
| Sensorimotor gating (PPI) | yes | yes | no | yes |
|
yes | ||
| Latent inhibition | yes | yes | yes | |||||
| Short term memory (Y maze) |
|
|
||||||
| Working memory (DNMTP) | yes | yes | yes | yes | yes | |||
| Spatial learning/memory (MWM) | no | no | no | no |
|
|||
| Fear conditioning | no | yes | ||||||
| Antidepressant-related (FST) | yes | no | yes | yes |
|
yes | no | |
| Sociability | yes | no | yes |
|
yes | |||
| Anxiety (EPM) | no | no | no |
|
no | |||
| References | Clapcote 2007, Lipina 2010, Lee 2011 |
Koike 2006 Kvajo 2008 |
Shen 2008 | Hikida 2007, Ibi 2010 | Pletnikov 2008, Ayhan 2010, Abazyan 2010 | Li 2008 | Niwa 2010 | |
The upper part describes histological and neurochemical abnormalities, studied in either the cortex or hippocampus. The lower part summarizes whether the mice display abnormalities in the indicated behavioral tests. All DISC1 animal models show substantially overlapping behavioral profiles. Two studies have combined an environmental stressor (poly I:C) with genetic risk (the putative dominant-negative C-terminal truncated human DISC1 under regulation by the CaMKII promoter)156,157. The behavioral outcome when this environmental stressor is added to the genetic model is depicted in red. CTX, cortex; DG, dentate gyrus; HP, hippocampus; mPFC, medial prefrontal cortex; STR, striatum; PV, parvalbumin; CB, calbindin; DA, dopamine; DOPAC, 3,4-Dihydroxyphenylacetic acid; TH, tyrosine hydroxylase; PPI, prepulse inhibition; DNMTP, delayed non-matching to place; MWM, Morris water maze; FST, forced swim test; and EPM, elevated plus maze. ↓, reduction; →, no change; and ↑, augmentation.
The DISC1 ‘interactome’ has identified other DISC1-interacting proteins that are involved in progenitor proliferation and neural migration. For example, ‘Coiled-coil protein Associated with Myosin II and DISC1’ (CAMDI) plays a role in neuronal migration and interacts preferentially with phospho-myosin II and induces an accumulation of phosphomyosin II at the centrosome in a DISC1-dependent manner89. Indeed, a single nucleotide polymorphism in the Camdi gene (R828W) leads to a reduction of protein binding between CAMDI and phosphomyosin II, and mice overexpressing R828W in neurons exhibit impaired radial migration. Furthermore, another recently identified DISC1 regulator and interacting protein is DIX domain containing-1 (Dixdc1), the third mammalian gene discovered to contain a Disheveled-Axin (DIX) domain90 (Fig. 1). Dixdc1 regulates dual roles for DISC1 in embryonic neural progenitor proliferation and neuronal migration. Dixdc1 functionally interacts with DISC1 to regulate neural progenitor proliferation by co-modulating Wnt-GSK3β-β-catenin signaling. In contrast, phosphorylation of Dixdc1 by cyclin-dependent kinase 5 (Cdk5) facilitates DISC1-NDEL1 interaction and in turn neuronal migration.
Roles for DISC1 in proliferation-migration transition in vivo
Although many molecules have been shown to regulate the processes of progenitor cell proliferation and neuronal migration, the molecular mechanisms responsible for the transition from neuronal proliferation to neuronal migration are largely unknown. A mass spectrometry effort to identify posttranslational modifications which might be critical for this transition identified a specific phosphorylation event at serine-710 in mouse DISC1 which could be a critical marker for this process91. GSK3β preferentially binds to DISC1 when it is non-phosphorylated at this site (S710-DISC1), whereas BBS1 and BBS4 interact preferentially with DISC1 when phosphorylated at S710. Thus, through an increase in phosphorylation of DISC1 at S710, which occurs at embryonic stage E13-15, DISC1/BBS binding recruits DISC1 away from a GSK3β-regulated Wnt/β-catenin activity, and contributes to the switch from neuronal progenitor proliferation to neuronal migration. In summary, accumulating evidence consistently uncovers dual roles for DISC1 in corticogenesis, from regulation of progenitor proliferation to mediation of postmitotic neural migration91. The phosphorylation of residue S710 on DISC1 is a key regulator to coordinate these two roles91 (Fig. 3).
It is very important to note that the data discussed above, which suggests a key role for DISC1 in neuronal proliferation and migration, has used principally RNAi-based approaches which reduce levels of a particular protein in order to study its biological effect. Looking at alternative approaches, the systematic histological analysis of mice with point mutations in the DISC1 gene (Q31L and L100P)92 has shown a reduction in neuron number and altered neuron distribution in the cerebral cortex, compared to wild-type littermates. Although these phenotypes are suggestive of a role for DISC1 in neurogenesis and neuron migration (Table 1), the phenotypes in the mouse models are more modest than those in the models mediated by RNAi. This would suggest the existence of parallel mechanism(s) that may compensate for the DISC1-mediated disturbance and mask the resultant phenotypes in the mutant mice.
Implications from studies of the hippocampus: commonality and differences in roles for DISC1 between cortical and hippocampal development
DISC1 is highly expressed in the hippocampus, especially in the dentate gyrus where neurogenesis takes place, not only in early development but also in adulthood60. To address roles for DISC1 in the hippocampus, most studies have also utilized RNAi methods. Downregulation of DISC1 in adult dentate gyrus leads to accelerated neuronal integration, resulting in aberrant morphological development and mispositioning of new dentate granule cells in a cell-autonomous fashion93. Newborn neurons with reduced DISC1 levels exhibit enhanced excitability and accelerated dendritic development and synapse formation93. Considering that the same group observed delayed neuronal migration in the developing cortex with the same shRNA to DISC193, this phenotype is likely to be unique to adult dentate gyrus. This unique phenotype seems to be mediated by AKT and the DISC1 interacting protein KIAA1212/Girdin94 (Fig. 1). Interestingly, knockdown of Girdin also leads to overextended migration and mispositioning of dentate granule neurons95. Knockdown of DISC1 in newborn granule cells in adulthood also leads to defects in axonal targeting and development of synaptic outputs96. Influence of DISC1 on newborn neurons seems to be developmental stage dependent: downregulation of DISC1 hinders the migration of dentate gyrus granule cells during early development, contrasting to its influences in adult dentate gyrus97. In CA1 of the developing hippocampus, knockdown of DISC1 at E15 leads to clearly disturbed migration at postnatal day 3 (P3)97, although more subtle alteration does appear at P298. Some of these results obtained with DISC1 RNAi have been consistently observed in other DISC1 mouse models. An example is data from a mouse model containing a 25-base deletion in exon 6 of DISC1, derived from the 129 mouse strain, and a premature polyadenylation site in intron 8 (described as the deletion model in the following sections), which results in loss of some DISC1 isoforms, dendritic misorientation, and impaired growth phenotypes in newborn neurons of the adult dentate gyrus99 (Table 1). An intraperitoneal injection of the NMDA-type glutamate receptor antagonist memantine into adult male mice also leads to a decrease in expression of DISC1 and overextended migration of newborn neurons in the dentate gyrus, which is rescued by exogenous expression of DISC1100. Decrease in DISC1 expression in the adult dentate gyrus is also reported in long-term kindled rats101. Together, these results indicate that DISC1 also plays roles in neurodevelopmental processes in the hippocampus, but its influences on phenotypes in adult dentate gyrus is unique compared to those in many other brain areas.
DISC1, Spines and Synapses
Deficits in dendritic spines and glutamatergic neurotransmission are shared across a number of mental illnesses to which DISC1 has been genetically associated102. Dendritic spines contain the postsynaptic compartment of the majority of excitatory synapses in the brain, and their morphology has been inextricably linked to neuronal activation and synaptic plasticity103–108. Recent reports make a compelling case for DISC1 localization and function at the postsynaptic density (PSD) and a role in the development and maintenance of synapses.
Role for DISC1 in synapses: from neuron culture to mouse models
There is now evidence from electron microscopy, immunocytochemistry, and biochemical analyses that DISC1 is found at synapses. An initial study with human post-mortem frontal and parietal cortex samples showed that a pool of DISC1 is localized in the synapse109. Approximately 40% of synapses, especially excitatory synapses, were labeled for DISC1. It is worthwhile noting that there was staining of inhibitory symmetrical synapses and that this is an area worth re-visiting in future studies. A similar PSD-localization was also recently shown in rat cortical and hippocampal cultures as well as mouse striatum110–112. Supporting the imaging studies, DISC1 has been shown by classical subcellular fractionation approaches to be enriched in the post-synaptic density fraction110,111,113.
In cortical neuron culture, knockdown of DISC1 with shRNA has been shown to regulate spine size, spine number, surface levels of the AMPA-type glutamate receptor subunit GluR1, and the frequency of miniature excitatory post-synaptic currents (mEPSCs)110. The delivery of GluR1 to the plasma membrane, probably to extra-synaptic domains, and then subsequent insertion into synaptic sites is crucial for increasing spine size, morphology and synapse strength114,115. Consistent with these observations in neuronal culture, many mouse models with DISC1 also display synaptic pathology. For example, the Q31L and L100P mutant mice show reduced spine density on pyramidal neurons in layers III and V of the frontal cortex and CA1 neurons in hippocampus92 (Table 1). Another mouse model (the so-called deletion model; see previous section for details) shows decreases in synaptic spines and associated abnormalties in the dentate gyrus99 (Table 1). A knockdown model of the NMDA-type glutamate receptor subunit NR1, which has been previously characterized to exhibit behavioral deficits relevant to mental disorders116, shows reduced levels of DISC1 and a key DISC1 interactor 14-3-3 epsilon at synapses and also shows spine shrinkage in the striatum suggesting that there is an intimate relationship between the activity of NMDA receptors and either DISC1 stability or perhaps subcellular targeting112. By contrast, knockdown of DISC1 in the dentate gyrus by shRNA in adult C57BL/6 mice reportedly leads to accelerated formation of dendritic spines in newborn neurons that have both glutamatergic and GABAergic synapses, as measured functionally by electrophysiology93. These studies provided the impetus to understand the function of DISC1 in regulating spine form and function.
Molecular mechanisms underlying the functions of DISC1 in the synapse
The ‘DISC1 interactome’ is a protein-protein interaction study based on iterative yeast-two hybrid data screens with DISC1 as the core ‘starter’ protein62,63. The findings from these studies predicted that DISC1 would be a key synaptic player, as the DISC1 protein environment was enriched in proteins found at the synapse and involved in NMDA receptor-dependent signaling63 (Fig. 4).
Figure 4. DISC1 function at the synapse.

DISC1 has been recently shown to regulate dendritic spines through regulation of a Kal-7-Rac1-PAK pathway110 and synapse maintenance through regulation of TNIK111. DISC1 enhances binding of Kal-7 and PSD95, blocking the access of Kal-7 to Rac1. Once the NMDA receptor is activated, these protein interactions are weakened, allowing the free access of Kal-7 to Rac1, leading to the activation of Rac1110. Regulation of Kal-7’s access to Rac1 by DISC1 in an activity-dependent fashion is crucial for the proper maintenance of the synaptic spine. It is currently unknown if TNIK is found in similar or different synapses as DISC1 and kalirin-7 complexes. Inhibition of TNIK function leads to the selective degradation of a number of key postsynaptic proteins including GluR1 and the scaffold protein PSD-95 and a loss of GluR1 from the membrane surface111. The degradation of protein is either lysosomal or proteasomal mediated. The E3 ubiquitin ligase, Neuronal precursor cell-expressed developmentally downregulated protein 4-1 (Nedd4-1)134,137, has been identified as a TNIK binding partner. Nedd4-1 has been shown to ubiquitinate GluR1 and drive its lysosomal degradation139. Hypothetically, inhibition of TNIK, could lead to an elevated activity of Nedd4-1, increased ubiquitination of GluR1 (or other proteins) and lead to their subsequent degradation. An integrated view of these two pathways is currently unavailable, though both pathways have common end-points related to actin cytoskeleton regulation and AMPA receptor trafficking. Independent studies suggest that TNIK may regulate GluR1 endocytosis through a complex with the E3 ligase Nedd4-1138. Complexes of DISC1 with PDE4, GSK3 and NDEL1 (not shown) are also found at the synapse but their function is not known. DISC1, Disrupted-in-Schizophrenia-1; TNIK, TRAF2 and NCK-interacting protein kinase; Nedd4-1, E3 ubiquitin ligase, Neuronal precursor cell-expressed developmentally downregulated protein 4-1; GSK3β, Glycogen synthase kinase 3β; NDEL1, Nuclear distribution protein nudE-like 1; LIS1, Lissencephaly protein 1; PDE4; Phosphodiesterase type 4; Kal-7, Kalirin-7; and PSD95; Postsynaptic density protein 95; NMDAR, N-methyl-D-aspartic acid type glutamate receptor; AMPAR, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionatetyoe glutamate receptor.
Amongst DISC1 interacting partners known to localize to the synapse, Kalirin-7 (Kal-7) was a attractive candidate to explain the effects of DISC1 knock down in cultured neurons (Fig. 1). In autopsied brain, Kal-7 mRNA (Duo in humans) is reduced in patients with schizophrenia117, and a GWAS in the Japanese population showed association at the KALRN locus (including Kal-7 and other isoforms) on 3q21.2 with schizophrenia118,119. In addition, re-sequencing of KALRN exons identified a number of rare mutations, two of which have been shown to be significantly associated with schizophrenia119. Furthermore, Kalrn knockout mice have deficits in working memory, social interaction, and prepulse inhibition, all of which are relevant as endophenotypes of schizophrenia and related disorders120. Kal-7, the most abundant isoform of the Kalrn genes found in the brain, is a Rac1-GTP exchange factor (GEF) that activates the small GTPase Rac1 and has been shown to be localized to the PSD121,122. A growing body of literature has shown that Kal-7 is crucial for spine form and function in a number of different contexts122. In immature hippocampal neurons ephrinB/EphB signaling regulates spine maturation through Kal-7 and activation of Rac1 and PAK1123. Activity-dependent spine morphogenesis in mature neurons has also implicated Kal-7, for example NMDA-receptor dependent activation of CaMKII results in phosphorylation of Kal-7 which increases its GEF activity and subsequent increases spine size and synaptic strength through increasing synaptic levels of GluR1124. Kal-7 has also been shown to be regulated in cortical interneurons by Neuregulin-1 (NRG1)/ErbB4 signaling to control dendritic arborization125,126. The interaction of Kal-7 with DISC1 was shown originally in three independent yeast two-hybrid screens63,67,110, and confirmed by standard biochemical techniques as part of a trimolecular complex with PSD-95110. At the baseline condition, DISC1 enhances binding of Kal-7 and PSD95, blocking the access of Kal-7 to Rac1. Once the NMDA receptor is activated, these protein interactions are weakened, allowing the free access of Kal-7 to Rac1, leading to the activation of Rac1110. Although Rac1 activity is crucial for synapse formation and maintenance, excess activation of Rac1 leads to synaptic shrinkage. Thus, regulation of Kal-7’s access to Rac1 by DISC1 in an activity-dependent (e.g., NMDA receptor activation-dependent) fashion is crucial for the proper maintenance of the synaptic spine110. Of note, a recent paper suggests that DISC1 can mediate axon guidance via regulating TRIO, another GEF for Rac1127.
Understanding the role of DISC1 in maintaining synapse form and function further has come via studies of a kinase known as Traf- and nck-interacting kinase (TNIK), a member of the Ste20 kinase family111. Specifically TNIK is a Germinal Center Kinase Type IV kinase (GCK type IV) and is characterized by a conserved N-terminal kinase domain and a C-terminal citron-homology domain (CNH), and a less well conserved intermediate domain128. Initial functions ascribed to TNIK are activation of the c-Jun N-terminal kinase (JNK) pathway, and regulation of the actin cytoskeleton by disruption of filamentous actin (F-actin), in a kinase activity-dependent fashion129. Until recently though, there was little known about the function of TNIK in the brain. Interest emerged from unbiased proteomic profiling studies of PSDs of the mouse and rat, where TNIK was identified by mass-spectrometry methods130,131. Initial links to schizophrenia have emerged from GWAS studies that implicated TNIK as a risk factor for schizophrenia and as a determinant of prefrontal cortical function in schizophrenics42,132. The DISC1 link to TNIK has origins in the ‘DISC1 interactome’ study, where TNIK was clearly one of the more heavily connected partners63 (Fig. 1). TNIK and DISC1 are found colocalized in dendritic spines in cultured primary neurons, which was confirmed by electron microscopy111. DISC1 binds specifically to the kinase domain of TNIK, which clearly implicated DISC1 as a regulator of TNIK kinase activity, which was subsequently experimentally confirmed111. Mapping of the binding site of TNIK on DISC1 identified a crucial thirteen amino-acid long sequence between residues 335–347. It is worth noting that a rare SNP, R338Q, linked to bipolar disorder is found within this binding site38 (Fig. 2). The binding domain peptide was serendipitously characterized as a potent inhibitor of TNIK kinase activity111. Inhibition of TNIK function, by either this peptide or a shRNA knockdown approach, leads to the selective degradation of a number of key postsynaptic proteins including GluR1 and the scaffold protein PSD-95, a loss of GluR1 from the membrane surface and decreases in the amplitude and frequency of mEPSCs111. A second report has shown similar cellular phenotypes after knockdown of the related kinase Misshapen like kinase (MINK)133. This implicates DISC1 and TNIK in the maintenance of synapse structure, probably through both proteasomal and lysosomal degradation pathways. Recently the role of protein degradation and the ‘ubiquitin-proteasome system’ (UPS) in particular, in regulating synaptic plasticity and behavior, has become more widely appreciated134. Co-administering the TNIK peptide inhibitor with either lysosome or proteasome inhibitors shows a clear blockade of protein degradation seen with the TNIK peptide inhibitor alone, in a manner consistent with our current knowledge about routes of degradation for the specific proteins involved134–136. To understand how TNIK regulates protein degradation the spot-light has fallen on Neuronal precursor cell-expressed developmentally downregulated protein 4-1 (Nedd4-1), a member of the Homologous to E6-associated protein C-terminus (HECT) class of E3 ubiquitin ligases134,137. Kawabe and co-workers138 recently showed that Nedd4-1 is critical for dendrite extension and arborization and for normal synapse formation, and identified TNIK as a specific Nedd4-1 binding partner. In addition, Nedd4-1 has been shown to directly interact with, and ubiquitinate GluR1 and drive its endocytosis and subsequent lysosomal degradation in mature hippocampal cultures139. In our own studies either knockdown of TNIK or inhibition of TNIK kinase activity, which both led to a decrease in GluR1 protein levels, could lead to the elevated activity of Nedd4-1, increased ubiquitination of GluR1 and subsequent degradation. It is possible that TNIK might regulate Nedd4-1 activity either through a scaffolding function or more directly through phosphorylation. Furthermore this suggests that changes in DISC1 function which impact TNIK activity, could regulate synapse form and function through regulation of protein degradative pathways.
In addition to Kal-7 and TNIK, there are other DISC1-interacting proteins that are likely to play a role in the synapse, including GSK3 and phosphodiesterase-4 (PDE4)87,140,141 (Fig. 1). The relationship between DISC1 and GSK3 has clearly been highlighted in terms of regulating neuronal precursor proliferation and differentiation. To date there have been no specific studies of this complex in the mature brain or at the synapse, but it is worth noting that GSK3 has been shown to play roles in both LTP and LTD142. PDE4 has been localized to synapses and specifically shown to co-localize with DISC1 at PSDs in the pre-frontal cortex (PFC) of non-human primates143. There are preliminary suggestions that DISC1-localized PDE4 is essential for regulating cAMP levels in the PFC and acts as a buffer against stress-induced PFC dysfunction143. Finally, a recent report indicates that synaptic modulators Nrxn1 and Nrxn3 are disregulated in L100P-DISC1 mice144. The challenge now is to understand how DISC1 regulates multiple pathways at the synapse over the course of nervous system development. Are the different functional modules independent or dependent on each other? Are they found in different cell types and synapses? And how does all of this change with ageing, key developmental milestones and of course disease?
Animal models for DISC1: behavioral outcome, link to environmental factors
There are eight distinct animal models for DISC1 already published (Table 1). Surprisingly there is not a traditional knockout mouse amongst them, which may be in part due to the complexities of DISC1 isoforms, which are still very poorly understood (see below). As described above, several studies have addressed whether anatomical and histological defects exist in these models and shown alterations in the cortex and hippocampus72,145–147. We have already discussed the significant but modest phenotypes in models of germ-line genomic lesions, which is in part because DISC1 pathways are likely to be regulated by a number of redundant mechanisms. Notably, all DISC1 animal models show a substantially overlapping behavioral profile. There are some differences seen across models, but at this stage it is too early to understand the significance of any differences until the models are compared alongside each other in the same laboratory. The different DISC1 mouse models have been recently comprehensively reviewed148.
As shown in Table 1, frontal deficits characterized by impairments in working memory [measured by a Delayed Non-Matching to Place (DNMTP) task] and latent inhibition are evident in all the DISC1 models thus far tested, whereas spatial learning and memory are not disturbed99,113,149–152. In addition to these frontal deficits, most DISC1 animal models show deficits in prepulse inhibition, a measure of sensory-motor gating and another representative endophenotype relevant to schizophrenia113,150,153. However, as expected from the mixed clinical phenotype of the DISC1 Scottish pedigree and other genetic association studies30,41, behavioral changes are not limited to endophenotypes of schizophrenia. For example, many DISC1 models show increased immobility time in the forced swim test, a traditional but well-accepted behavioral assay to test the efficacy of antidepressants154.
The presence of multiple different clinical diagnoses in a single pedigree, as seen with the Scottish DISC1 family, is not uncommon but typical of families with a high mental-illness ‘load’ 26,155. What could be the underlying mechanism(s) that account for family members with different psychiatric diagnoses elicited by the same genetic mutation? In the case of DISC1 it is possible to hypothesize that dysregulation of different DISC1 interacting partners may contribute to the different diagnoses seen. This is purely hypothetical at this moment but is a very attractive model to consider. Because major mental illnesses are believed to occur via a combination of genetic and environmental factors, one possible mechanism might be modification by environmental factor(s). To address this hypothesis from a DISC1-centric viewpoint, several publications have addressed how environmental factors affect behavioral phenotypes of DISC1 animal models. Ibi and colleagues156 administered polyriboinosinic-polyribocytidylic acid (polyI:C), which mimics innate immune responses elicited by viral infection early in development, to a transgenic model expressing the putative dominant-negative C-terminal truncated human DISC1 under regulation by the CaMKII promoter150 (constitutive CaMK-DN mice described in Table 1). They reported that this environmental stressor, which mimics an early environmental insult linked with schizophrenia, administered between P2 and P6, augments a range of behavioral and anatomical phenotypes, including a decreased immunoreactivity in parvalbumin-positive interneurons in the medial prefrontal cortex (Table 1). More recently, it has been reported that in another transgenic model expressing the dominant-negative human DISC1, prenatal immune activation with poly I:C to pregnant dams at gestational day 9 can also augment, and in some cases reveal, a particular phenotype157. In this experimental paradigm, anxiety and depression-like responses and social behavior deficits become prominent in the presence of this prenatal environmental stressor (Table 1). These results suggest that hereditary DISC1 animal models may be good templates to study gene--environment interactions for major mental illnesses, which can be used to address questions of whether and how environmental stressors augment intrinsic abnormalities due to genetic factors and modify final phenotypic manifestations.
In addition to behavioral abnormalities, neurochemical and signaling cascades that underlie such behaviors are consistently altered in DISC1 animal models (Table 1). Prominent changes in dopaminergic neurotransmission have been reported153,158,159. A model based on transient knockdown of DISC1 in cells of the pyramidal neuron lineage in the developing cortex via in utero shRNA transfer at E14, driving knockdown for 1–2 weeks, shows deficits in several behavioral domains only in adulthood. These include working memory, prepulse inhibition, and methamphetamine-induced locomotion153. Consistent with these behavioral changes, insufficient maturation of mesocortical dopaminergic projections and an augmented surge of dopamine release in the nucleus accumbens upon amphetamine challenge were reported. Interestingly, these effects were observed only after puberty153, which parallels the initial etiology of human schizophrenia, which tends to emerge in young adulthood. Therefore, this model mimics a developmental trajectory that is characteristic to schizophrenia and is distinct from autism and other neurodevelopmental disorders25. In a CaMK-DN model, a significant decrease of tissue dopamine (DA) and 3,4 dihydroxy-phenylacetic acid (DOPAC) in cortex is observed159. Of note, decreased immunoreactivity in parvalbumin-positive interneurons, a cell-type known to be decreased in the frontal cortex of schizophrenic patients and thought to contribute to the establishment of cognitive deficits, is observed in both these two models153,157,159. This might be significant, considering that a bi-directional relationship between dopaminergic defects and interneuron dysfunction underlying behavioral defects is a currently hotly debated topic in schizophrenia research160. Furthermore, in L100P-DISC1 mice, a two-fold increase in the proportion of striatal D2 dopamine receptors in the high affinity state is observed158. The effects on intracellular signaling pathways that are known to be downstream of dopamine receptors, such as cyclic AMP signaling and GSK-3, have also been addressed in DISC1 models161,162. In the Q31L-DISC1 mice, a decrease in activity of phosphodiesterase-4 (PDE4) and an increased sensitivity to a GSK-3 inhibitor, TDZD-8, are observed161. In L100P-DISC1 mice, the protein interaction of DISC1 with GSK-3 is reduced, and both pharmacological and genetic inactivation of GSK3 ameliorate prepulse inhibition, latent inhibition and locomotor activity deficits161. Furthermore, combined treatment of L100P-DISC1 mice with the PDE4 inhibitor rolipram and the GSK3 inhibitor TDZD-8 at sub-threshold doses can correct defects in prepulse inhibition and hyperactivity, possibly suggesting synergistic interactions between PDE4, GSK3, and DISC1161. Many reports with DISC1 animal models in the last several years have reinforced the notion that DISC1 is implicated in higher brain functions, such as cognition and emotion. Although care needs to be taken when extrapolating these preclinical observations to a role for DISC1 in human mental disorders, it is clear that disturbance of DISC1 in mouse models impacts endophenotypes that are relevant to schizophrenia and related disorders.
There are a number of major areas of particular importance for future studies of DISC1 animal models. As described above, there are a number of consistent behavioral changes observed across the different DISC1 mouse models but one of the biggest gaps in this area though is a neuronal circuit-based discussion to explain the different phenotypes. To date there have been no reports of optogenetic approaches applied to DISC1 biology to understand the specific cells which are relevant to its function. There are also no conditional knock-out mice available to understand the selective loss of DISC1 in distinct circuits. These scenarios are also compounded.by no publications showing the electrophysiological properties of cell or networks from these mice. It is possible though to make some suggestions from observations made at the cellular level in relation to the behavioral effects seen. For example, as described above, more than one DISC1 model shows a consistent decrease in parvalbumin immunoreactivity in the interneurons of the frontal cortex. This decrease is known to reflect hypofunction of specific types of interneurons that regulate cortical synchrony, being represented by gamma oscillations, and in turn underlie working memory163. Furthermore, working memory is also properly maintained by coherence of the hippocampus and prefrontal cortex164, both of which are particularly impaired in many DISC1 animal models (Table 1). It is now very important to address the functional connectivity of the prefrontal cortex and hippocampus experimentally. In the future, it will be critical to conduct behavioral studies alongside in vivo electrophysiology experiments.
Furthermore, as described, DISC1 is a multifunctional protein with expression not only in neuronal cells but also in glial cells61 and in cells of a neuronal lineage, roles for DISC1 are likely to differ in a developmental and brain-region dependent manner. To address the influences of DISC1 in each context on circuitry and behavior, animal models that allow precise control of DISC1 expression in a spatially and temporally specific manner, most likely through the generation of conditional knockout mice, are not available yet but are eagerly awaited by those working in this area. Second, animal models that can address epistatic effects of DISC1 and its interactors need to be studied in the context of mental disorders. Cross-breeding of DISC1 model mice with other transgenic lines, and introduction of viral vectors that modulate expression of DISC1 interactors in DISC1 models may be realistic approaches. Alternately, models with in utero gene transfer, which allow the introduction of multiple expression/knockdown constructs153,165, may also be useful. Finally, as described, the study of gene-environment interactions by using DISC1 animal models will be important.
The Interactome Suggests DISC1 Functions Beyond Neurodevelopment and Synapse Function
In addition to roles for DISC1 in early brain development and synapse formation and maintenance, there are several intriguing reports that shed light on other functions of DISC1. The characterization of these functions has to be treated as preliminary as in general it has been limited to in vitro assays.
An increasing number of studies are exploring possible links between schizophrenia susceptibility factors both at genetic and functional levels. Wood and colleagues166 demonstrated that the knockdown of disc1 by morpholino antisense methods in zebrafish disturbs oligodendrocyte development. It does so by promoting specification of olig2-positive cells in the hindbrain and other brain regions, which resembles the phenotypes seen with knockdown of neuregulin1 (nrg1), a major susceptibility gene for schizophrenia in humans. Consistent with this initial suggestion, induction of an isoform of DISC1 (a 130 kDa isoform) by NRG1 is observed in primary cortical neuron cultures from rodents61. Consistently, downregulation of DISC1 is observed in embryos of nrg1 knockout mice. Furthermore, in β-site amyloid precursor protein cleaving enzyme-1 (BACE1) knockout mice where the active form of NRG1 is no longer generated, similar downregulation of DISC1 is seen61. Induction of DISC1 by NRG1 is blocked by LY294002, a small molecule inhibitor of PI3K, indicating that this cascade is possibly mediated through AKT1. As described in a section above, DISC1 and AKT are functionally connected via KIAA1212/Girdin94,95. Demonstration of links amongst these schizophrenia susceptibility factors, such as DISC1, NRG1, AKT, and Olig2 may further support a notion of convergent molecular pathways underlying the pathology of the disease.
In addition, a possible protein interaction of DISC1 with Dysbindin (the protein encoded by DTNBP1) is intriguing167. Dysbindin is another promising risk factor for schizophrenia and linked to the cognitive deficits seen in the disease168. At the cellular level it has been recently linked to synapse function through both pre- and post-synaptic activities169,170. DISC1 and Dysbindin have been suggested to share a number of common binding partners from the results of the DISC1 interactome63,167. A recent report indicates that Dysbindin binds to DISC1 aggregates in recombinant systems and co-fractionates with DISC1 high-molecular weight aggregates from human postmortem tissue167. This study has two major implications: first, it further confirms a promising working hypothesis on the convergent pathways in mental illnesses; second, this is consistent with a previous study that DISC1 protein is found within insoluble fractions in brains from patients with schizophrenia and affective disorders, but not normal controls171. This observation supports an intriguing hypothesis that a mechanism associated with protein misfolding and aggregate formation may underlie the pathology of schizophrenia, like those for neurodegenerative disorders, such as Alzheimer’s disease and Huntington’s disease. Interestingly, a protein interaction of DISC1 and Huntingtin, the disease protein for Huntington’s disease, has been suggested by the existence of many common binding proteins and subsequent bioinformatic analyses63,66,67,78,172. Direct validation of this protein interaction at the experimental level is awaited. Also of interest is that in the original Scottish pedigree there are reports that a fusion transcript could form across the breakpoint on chromosome 1 (encodes N-terminal 1-598 amino acids of DISC1) and the breakpoint on chromosome 11. Critically this transcript can form a protein, which has been shown to be amyloidogenic when overexpressed in cultured cells173,174. This misfolded DISC1 fusion protein might play a pathological role in patients of the Scottish pedigree. Taken together, although it is still hypothetical, a role for misfolded DISC1 in the pathology of schizophrenia and related disorders may be an attractive concept, worthy of future investigation and clarification.
The story around DISC1 and mitochondria is growing. DISC1 has been found in mitochondria using immunocytochemical techniques in cultured primary neurons and also electron microscopy in rat hippocampus and mouse striatum66,75,175. The brain demands a large percentage of the body energy output due to the needs of synaptic transmission and action potential generation176,177. Mitochondria are localized to synapses to provide the required ATP for high fidelity synaptic neurotransmission and also to act as a calcium buffer. Synaptic activity, in a NMDA receptor-dependent manner, has been shown to regulate mitochondrial localization to dendritic spines through tethering to specific proteins176. Recently it was shown that knockdown or overexpression of DISC1 regulates the trafficking of mitochondria in cultured hippocampal neurons178. DISC1 has been shown to bind to a range of mitochondrial proteins that could possibly play a role in these events and perhaps in targeting mitochondria to the synapse. For example ‘Fasciculation and elongation factor 1’ (FEZ1) has been shown to bind DISC1 and together play a role in neuronal outgrowth65, while independently FEZ1 has a role in regulating mitochondrial motility179. The inner mitochondrial membrane protein Mitofillin has also been shown to bind DISC1180. Decreases in DISC1 or Mitofillin were both shown to decrease NADH dehydrogenase and mitochondrial monoamine activities and to cause abnormal mitochondrial calcium dynamics. These effects could explain the effects on the trafficking of mitochondria mentioned previously178. Intriguingly, DISC1 stabilizes Mitofillin and prevent its degradation by the proteasome, explaining the phenocopies of effects after knocking down either protein. It will of course be interesting to see what is responsible for the degradation here and whether Nedd4-1 has a role. Together these partnerships suggest that DISC1 could play an important role in mitochondrial activity and possibly its synaptic localization in neurons.
A pool of DISC1 is enriched in the nucleus, in particular within the promyelocytic leukemia (PML) nuclear body, and is likely to regulate gene transcription181. A yeast two-hybrid screen, followed by biochemical confirmation, indicates that DISC1 binds with activating transcription factor 4 (ATF4) [aka, cAMP-response element binding protein-2 (CREB2)] and ATF564. In cultured cells, such as HeLa cells, DISC1 interacts with ATF4/CREB2 and a co-repressor N-CoR, modulating CRE-mediated gene transcription181. DISC1-dependent transcriptional repression is also reported in zebrafish cranial neural crest for the foxd3 and sox10 genes182. Furthermore, DISC1 reportedly regulates expression of N-cadherin and β1-integrin in PC12 cells183. Functional roles for nuclear DISC1 in the mammalian brain remain to be elucidated. So far, a role of nuclear DISC1 in sleep homeostasis in drosophila melanogaster and in neural crest migration and differentiation in zebrafish has been suggested181,182. In a study with human autopsied brains (focus on orbitofrontal cortices), an altered distribution of a 75 kDa isoform of DISC1 to the nucleus, is observed in association with psychosis and substance and alcohol abuse184.
Analysis of a role for DISC1 in neurite outgrowth, mainly within PC12 cells, has suggested several novel functions for DISC1. PC12 cells stably overexpressing wild-type DISC1 show an increase in neurite outgrowth65, whereas knockdown of DISC1 via RNAi or expression of a dominant-negative C-terminal truncated DISC1 leads to a decrease in outgrowth66,78. The protein interaction of DISC1 with NDEL1 is required this effect185. However, the fact that an endo-oligopeptidase activity of NDEL1 is also required for neurite outgrowth can be inhibited by its interaction with DISC1186 suggests that this interaction is likely complex and has dual roles in this process. In addition to the DISC1-NDEL1 interaction, binding of DISC1 with FEZ1 also reportedly participates in this process65. Furthermore, a novel protein DISC1-Binding Zinc finger protein (DBZ) can modulate DISC1-dependent neurite outgrowth, which may be influenced by exogenous Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP)187. Although all of these proposed mechanisms are interesting, whether they occur in the brain and which specific circuits they regulate, requires validation.
DISC1: priorities for future neurobiological and translational studies
Over the past ten years, since the isolation and cloning of the DISC1 gene, many studies of its corresponding protein have been conducted. Almost all support the notion that this is an intracellular anchoring protein that has many protein interactors, and is found in multiple subcellular domains. Many of the key interacting proteins are also genetic risk factors for schizophrenia and other mental illnesses, suggesting that DISC1 is a hub for several pathways underlying mental disorders. Nonetheless, several key questions remain to be answered.
First, the molecular complexity of DISC1, especially its protein isoforms, is not well understood. Initial studies following the identification of the gene suggested that DISC1 has four isoforms (L, Lv, S, Es)188. However, the multiple specific protein bands seen by Western blotting experiments, with a multitude of anti-DISC1 antibodies, were not as predicted from the mRNA experiments. This enigma was further amplified by observed DISC1 immunoreactivity by Western blotting of brain from the 129 strain of the mouse, in which a 25-base deletion in coding exon 6 of the Disc1 gene exists. DISC1 immunoreactivity obtained from brain extracts of 129 and C57BL/6 strains (no 25-base deletion) are not distinguishable with many antibodies against DISC1189, including very popular commercial reagents that have been used in key studies of DISC1 suggesting careful characterization of antisera and mouse models, with a range of control experiments and antisera respectively is absolutely required (see below for details) 87,90,93,96,101,190–192. Accordingly, the Gogos/Karayiorgou lab99 has suggested caution in using antibodies against DISC1 and in characterizing RNAi experiments, which is strongly dependent on Western blotting with these same antibodies. As many findings on DISC1 have come from experiments with RNAi78,87,90,91,93,96,98,110,111,153,158,193,194, this experimental issue should not to be ignored. However, the following recently published studies are likely to provide a solution to move this debate along and indicate future strategies that should be taken in DISC1 research: the Weinberger lab195 has recently reported that DISC1 has many isoforms (many more than those classically reported). Consistent with this report, DISC1 RNAi-s that have been used for many studies have functional effects in both 129 and C57BL/6 strains, suggesting that 129 strain maintains functional DISC1 isoforms165. Although the publication from the Weinberger lab supports the concept that there are still many unknown isoforms of DISC1, this paper even does not fully account for all the mysterious patterns of DISC1 protein bands seen by Western blotting. Further studies of both mRNA and protein are needed. In addition, we need to pay attention to posttranslational modifications and their contribution to the complexity of DISC1 signals in Western blotting. Taking all this together we propose the following criteria for all future DISC1 research: (1) when we use animal models for DISC1, the use of the C57BL/6 strain without the 25-base deletion is strongly recommended; (2) to detect DISC1 protein in brain samples, reliable pairs of antibodies against DISC1 should be used in immunoprecipitation-Westerns (one antibody is used for immunoprecipitation and the other for Western blotting), leading to enhanced DISC1 signals in the immunoprecipitates; and (3) use of multiple RNAi targeting constructs to different regions of DISC1 is recommended. The design of these constructs should take into consideration the different DISC1 isoforms, and be combined with a rescue experiment by co-expressing a known DISC1 isoform, such as full-length isoform L. Through these experiments, we can be confident that functions of DISC1 revealed by RNAi experiments are at least associated with known isoforms.
Second, some unanswered questions on the molecular nature of DISC1 may be clarified with novel cell engineering technology, which generates induced pluripotent stem cells (iPS cells) and induced neural cells (iN cells). This technology allows us to generate neurons from peripheral tissues, such as skin fibroblasts, of patients and normal controls. This technology is being taken up rapidly and it is already being rewardrd across the psychiatric diseases, such as Rett syndrome and schizophrenia196. Although the implication of the genetic variant to psychiatric illnesses is unclear197,198, generation of iPS cells carrying a DISC1 frameshift mutation was recently reported199. In the future it will be interesting to establish iPS cells from the Scottish pedigree with the chromosomal translocation.
Third, although many possible functions for DISC1 have been suggested, functional validation in vivo has been sufficiently achieved only in the context of early development, such as corticogenesis and adult neurogenesis in the dentrate gyrus. Studies on synaptic DISC1 have involved functional validation in vivo, but assessment of real-time spine dynamics using multiphoton microscopy and impact on distinct circuitry by optogenetic approaches is awaited. In addition, DISC1 is expressed in both neurons and glial cells (astrocytes, oligodendrocytes, and microglia). Thus, the spatial and temporal specificity of DISC1 activity is a key outstanding point to address. So far, most of the studies have focused on DISC1 in progenitor cells and pyramidal neurons in the cortex and hippocampus. It is very important to address the significance of DISC1 in interneurons and glial cells. Minor isoforms that were reported in brain homogenates may reflect important isoforms in specific cell types. Thus, when we conduct functional assays, cell type-specific targeting is very important. Furthermore, brain development and function are determined by the interaction of genetic and environmental factors. Therefore, how environmental factors and stressors may influence DISC1 cascades during development and the overall brain function need to be further addressed.
Finally, although DISC1 has been established to be a key regulatory molecule in diverse processes of neurodevelopment and synaptic function, its translational potential may be different from the one originally expected under the name of “Disrupted-in-Schizophrenia.” As described above, the importance of DISC1 as a genetic risk factor is rather weak in schizophrenia, instead the molecular pathways involving DISC1 seems to be implicated in unique endophenotypes that underlie many distinct mental disorders labeled by current diagnostic manuals. Thus, DISC1 biology may possibly provide a hint on constructing new diagnostic criteria for mental disorders at a more biological (and likely relevant) basis. DISC1 animal models need to be further characterized, with a more thorough cross-lab, circuit-based approach. Then the phenotypes can be explained by the underlying circuit deficits, which should allow better correlation to the clinical data in patients. Lastly, we are optimistic that important drug targets will be identified from understanding the molecular pathways of DISC1, which will provide therapeutic advantages beyond current drugs across multiple mental illness diagnoses.
On-Line Summary.
DISC1 was identified in a Scottish family through characterization of a balanced chromosomal translocation found to associate with major mental illnesses (schizophrenia, bipolar disorder and depression). Additional studies have shown that DISC1 is associated with endophenotypes underlying these disorders.
Identification of the ‘DISC1 interactome’ has enabled discovery of roles for DISC1 in brain development and function. This has been complemented with extensive RNAi experimental approaches and the use of DISC1 animal models.
DISC1 has been shown to play a key role in neurodevelopmental processes in particular in regulating cortical development (progenitor proliferation and neuronal migration) and hippocampal neurogenesis.
DISC1 has been found to play a key role in synapse function. In particular interactions with the molecules Kalirin-7 and TNIK have suggested roles in synapse formation and maintenance.
DISC1 is considered an important tool for drug discovery approaches. Interactions between DISC1 and proteins, such as PDE4 and GSK3, previously suggested as therapeutic targets for mental illnesses, have implicated DISC1 as a possible interacting hub for drug targets. DISC1 animal models are also being considered for drug discovery screening.
Acknowledgments
We thank Ms. Yukiko L. Lema for preparing figures and organizing the manuscript and Prof. Douglas Blackwood for preparation of Box 1. We also thank Drs. Hanna Jaaro-Peled, Koko Ishizuka, and Minae Niwa for critical reading of the manuscript. Grant supports (A.S.): from MH-084018, MH-94268 Silvo O. Conte center, MH-069853, MH-085226, MH-088753, MH-092443, Stanley, RUSK, S-R foundations, NARSAD and Maryland Stem Cell Research Fund.
GLOSSARY
- DOMAIN
Domain refers to a specific cluster of symptoms that can be grouped together and described by a single descriptive neuropsychological or clinical construct, e.g., information processing, working memory, and attention
- BALANCED CHROMOSOMAL TRANSLOCATION
refers to an exchange of genetic material between non-homologous chromosomes with no overall loss or gain of genes
- NON-SYNONYMOUS MUTATION
mutation in a gene which leads to a change in amino-acid sequence
- INDUCED PLURIPOTENT STEM CELLS (IPS CELLS)
are created from differentiated cell-types e.g. fibroblasts which are reprogrammed by a cocktail of transcription factors (or other approaches) back to a pluripotent state. These cells can now be differentiated into cells of distinct lineages e.g. neurons
- INDUCED NEURAL CELLS (IN CELLS)
are created from differentiated cell-types e.g. fibroblasts which are reprogrammed directly to a neuronal lineage by a cocktail of transcription factors
Biographies
Nick Brandon leads research efforts into Psychiatry and Behavioral Disorders at Pfizer Neuroscience. Nick was educated at the University of Cambridge and University College London and then worked at Merck and Wyeth. His lab is focused on identifying therapeutic strategies to treat mental illness, including schizophrenia and autism.
Akira Sawa is Director of the Johns Hopkins Schizophrenia Center and Professor, Departments of Psychiatry and Neuroscience. Akira started his career as a psychiatrist and later conducted postdoctoral research training under Solomon Snyder. Since 2001, Akira has served as faculty at Johns Hopkins University.
References
- 1.Philip NS, Carpenter LL, Tyrka AR, Price LH. Pharmacologic approaches to treatment resistant depression: a re-examination for the modern era. Expert Opin Pharmacother. 2010;11:709–722. doi: 10.1517/14656561003614781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter CS, Barch DM. Cognitive neuroscience-based approaches to measuring and improving treatment effects on cognition in schizophrenia: the CNTRICS initiative. Schizophr Bull. 2007;33:1131–1137. doi: 10.1093/schbul/sbm081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green MF, et al. Approaching a consensus cognitive battery for clinical trials in schizophrenia: the NIMH-MATRICS conference to select cognitive domains and test criteria. Biol Psychiatry. 2004;56:301–307. doi: 10.1016/j.biopsych.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 4.Kern RS, Glynn SM, Horan WP, Marder SR. Psychosocial treatments to promote functional recovery in schizophrenia. Schizophr Bull. 2009;35:347–361. doi: 10.1093/schbul/sbn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkpatrick B, Fenton WS, Carpenter WT, Jr, Marder SR. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr Bull. 2006;32:214–219. doi: 10.1093/schbul/sbj053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer JM. Antipsychotic safety and efficacy concerns. J Clin Psychiatry. 2007;68 (Suppl 14):20–26. [PubMed] [Google Scholar]
- 7.Meyer JM. Antipsychotics and metabolics in the post-CATIE era. Curr Top Behav Neurosci. 2010;4:23–42. doi: 10.1007/7854_2010_45. [DOI] [PubMed] [Google Scholar]
- 8.Gartlehner G, et al. Comparative risk for harms of second-generation antidepressants: a systematic review and meta-analysis. Drug Saf. 2008;31:851–865. doi: 10.2165/00002018-200831100-00004. [DOI] [PubMed] [Google Scholar]
- 9.Williams HJ, Owen MJ, O’Donovan MC. Schizophrenia genetics: new insights from new approaches. Br Med Bull. 2009;91:61–74. doi: 10.1093/bmb/ldp017. [DOI] [PubMed] [Google Scholar]
- 10.Arenkiel BR, Ehlers MD. Molecular genetics and imaging technologies for circuit-based neuroanatomy. Nature. 2009;461:900–907. doi: 10.1038/nature08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuang M. Schizophrenia: genes and environment. Biol Psychiatry. 2000;47:210–220. doi: 10.1016/s0006-3223(99)00289-9. [DOI] [PubMed] [Google Scholar]
- 12.van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature. 2010;468:203–212. doi: 10.1038/nature09563. [DOI] [PubMed] [Google Scholar]
- 13.Moffitt TE, Caspi A, Rutter M. Strategy for investigating interactions between measured genes and measured environments. Arch Gen Psychiatry. 2005;62:473–481. doi: 10.1001/archpsyc.62.5.473. [DOI] [PubMed] [Google Scholar]
- 14.Owen MJ, Craddock N, Jablensky A. The genetic deconstruction of psychosis. Schizophr Bull. 2007;33:905–911. doi: 10.1093/schbul/sbm053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuthbert B, Insel T. The data of diagnosis: new approaches to psychiatric classification. Psychiatry. 2010;73:311–314. doi: 10.1521/psyc.2010.73.4.311. [DOI] [PubMed] [Google Scholar]
- 16.Owen MJ, O’Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry. 2011;198:173–175. doi: 10.1192/bjp.bp.110.084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marenco S, Weinberger DR. The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol. 2000;12:501–527. doi: 10.1017/s0954579400003138. [DOI] [PubMed] [Google Scholar]
- 18.Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- 19.Cannon M, Jones P. Schizophrenia. J Neurol Neurosurg Psychiatry. 1996;60:604–613. doi: 10.1136/jnnp.60.6.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powell SB. Models of neurodevelopmental abnormalities in schizophrenia. Curr Top Behav Neurosci. 2010;4:435–481. doi: 10.1007/7854_2010_57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaaro-Peled H, et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32:485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 23.Goldman-Rakic PS, Selemon LD. Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull. 1997;23:437–458. doi: 10.1093/schbul/23.3.437. [DOI] [PubMed] [Google Scholar]
- 24.Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- 25.Insel TR. Rethinking schizophrenia. Nature. 2010;468:187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- 26.St Clair D, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. Landmark paper in the DISC1 field presenting the DISC1 Scottish pedigree for the first time carrying the balanced translocation t(1;11)(q42.1 q14.3) [DOI] [PubMed] [Google Scholar]
- 27.Muir WJ, Pickard BS, Blackwood DH. Disrupted-in-Schizophrenia-1. Curr Psychiatry Rep. 2008;10:140–147. doi: 10.1007/s11920-008-0025-2. [DOI] [PubMed] [Google Scholar]
- 28.Jacobs P, Brunton M, Freackiewicz A, Newton M, Cook PJL, Robson EB. Studies on a family with three cytogenetic markers. Ann Hum Genet. 1970;33:325–336. [Google Scholar]
- 29.Millar JK, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. Ten years after the pedigree were published in the Lancet the genes at the site of the translocation on chromosome 1 were cloned and named Disrupted in Schizophrenia 1 and 2. [DOI] [PubMed] [Google Scholar]
- 30.Blackwood DH, et al. Schizophrenia and affective disorders--cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. This paper provides the last clinical update on the pedigree. The LOD score of the pedigree and major mental illness shown to be 7.1. Critically showed that the translocation results in a P300 electrophysiological deficit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hennah W, et al. Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet. 2003;12:3151–3159. doi: 10.1093/hmg/ddg341. [DOI] [PubMed] [Google Scholar]
- 32.Hamshere ML, et al. Genomewide linkage scan in schizoaffective disorder: significant evidence for linkage at 1q42 close to DISC1, and suggestive evidence at 22q11 and 19p13. Arch Gen Psychiatry. 2005;62:1081–1088. doi: 10.1001/archpsyc.62.10.1081. [DOI] [PubMed] [Google Scholar]
- 33.Kilpinen H, et al. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- 34.Callicott JH, et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci U S A. 2005;102:8627–8632. doi: 10.1073/pnas.0500515102. This paper is one of the first reports that successful link genetic variations of DISC1 to brain function and anatomy in human brains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodgkinson CA, et al. Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862–872. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto R, et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet. 2006;15:3024–3033. doi: 10.1093/hmg/ddl244. [DOI] [PubMed] [Google Scholar]
- 37.Song W, et al. Identification of high risk DISC1 structural variants with a 2% attributable risk for schizophrenia. Biochem Biophys Res Commun. 2008;367:700–706. doi: 10.1016/j.bbrc.2007.12.117. [DOI] [PubMed] [Google Scholar]
- 38.Song W, et al. Identification of high risk DISC1 protein structural variants in patients with bipolar spectrum disorder. Neurosci Lett. 2010;486:136–140. doi: 10.1016/j.neulet.2010.09.027. [DOI] [PubMed] [Google Scholar]
- 39.Mathieson I, Munafo MR, Flint J. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. doi: 10.1038/mp.2011.41. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Donovan MC, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 41.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sprooten E, et al. Association of white matter integrity with genetic variation in an exonic DISC1 SNP. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.15. [DOI] [PubMed] [Google Scholar]
- 45.Raznahan A, et al. Common functional polymorphisms of DISC1 and cortical maturation in typically developing children and adolescents. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blackwood DH, Muir WJ. Clinical phenotypes associated with DISC1, a candidate gene for schizophrenia. Neurotox Res. 2004;6:35–41. doi: 10.1007/BF03033294. [DOI] [PubMed] [Google Scholar]
- 47.Cannon TD, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. This paper is one of the first reports that successful link genetic variations of DISC1 to brain function and anatomy in human brains. [DOI] [PubMed] [Google Scholar]
- 48.Hennah W, et al. A haplotype within the DISC1 gene is associated with visual memory functions in families with a high density of schizophrenia. Mol Psychiatry. 2005;10:1097–1103. doi: 10.1038/sj.mp.4001731. [DOI] [PubMed] [Google Scholar]
- 49.Di Giorgio A, et al. Association of the SerCys DISC1 polymorphism with human hippocampal formation gray matter and function during memory encoding. Eur J Neurosci. 2008;28:2129–2136. doi: 10.1111/j.1460-9568.2008.06482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szeszko PR, et al. DISC1 is associated with prefrontal cortical gray matter and positive symptoms in schizophrenia. Biol Psychol. 2008;79:103–110. doi: 10.1016/j.biopsycho.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prata DP, et al. Effect of disrupted-in-schizophrenia-1 on pre-frontal cortical function. Mol Psychiatry. 2008;13:915–917. 909. doi: 10.1038/mp.2008.76. [DOI] [PubMed] [Google Scholar]
- 52.Carless MA, et al. Impact of DISC1 variation on neuroanatomical and neurocognitive phenotypes. Mol Psychiatry. doi: 10.1038/mp.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brauns S, et al. DISC1 is associated with cortical thickness and neural efficiency. Neuroimage. doi: 10.1016/j.neuroimage.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi T, et al. The Disrupted-in-Schizophrenia-1 Ser704Cys polymorphism and brain morphology in schizophrenia. Psychiatry Res. 2009;172:128–135. doi: 10.1016/j.pscychresns.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 55.Mata I, et al. Additive effect of NRG1 and DISC1 genes on lateral ventricle enlargement in first episode schizophrenia. Neuroimage. 2010;53:1016–1022. doi: 10.1016/j.neuroimage.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 56.Tomppo L, et al. Association of variants in DISC1 with psychosis-related traits in a large population cohort. Arch Gen Psychiatry. 2009;66:134–141. doi: 10.1001/archgenpsychiatry.2008.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merikangas AK, Corvin AP, Gallagher L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet. 2009;25:536–544. doi: 10.1016/j.tig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 58.Kim Y, Zerwas S, Trace SE, Sullivan PF. Schizophrenia genetics: where next? Schizophr Bull. 2011;37:456–463. doi: 10.1093/schbul/sbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schurov IL, Handford EJ, Brandon NJ, Whiting PJ. Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol Psychiatry. 2004;9:1100–1110. doi: 10.1038/sj.mp.4001574. [DOI] [PubMed] [Google Scholar]
- 60.Austin CP, Ky B, Ma L, Morris JA, Shughrue PJ. Expression of Disrupted-In-Schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience. 2004;124:3–10. doi: 10.1016/j.neuroscience.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 61.Seshadri S, et al. Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc Natl Acad Sci U S A. 2010;107:5622–5627. doi: 10.1073/pnas.0909284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brandon NJ. Dissecting DISC1 function through protein-protein interactions. Biochem Soc Trans. 2007;35:1283–1286. doi: 10.1042/BST0351283. [DOI] [PubMed] [Google Scholar]
- 63.Camargo LM, et al. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. Original disclosure of the ‘DISC1 interactome’, a complex protein-protein interaction network based off yeast-two hybrid screens using DISC1 and a set of DISC1 interactors as baits. Identified majority of key DISC1 related pathways. [DOI] [PubMed] [Google Scholar]
- 64.Morris JA, Kandpal G, Ma L, Austin CP. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum Mol Genet. 2003;12:1591–1608. doi: 10.1093/hmg/ddg162. [DOI] [PubMed] [Google Scholar]
- 65.Miyoshi K, et al. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry. 2003;8:685–694. doi: 10.1038/sj.mp.4001352. [DOI] [PubMed] [Google Scholar]
- 66.Ozeki Y, et al. Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci U S A. 2003;100:289–294. doi: 10.1073/pnas.0136913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Millar JK, Christie S, Porteous DJ. Yeast two-hybrid screens implicate DISC1 in brain development and function. Biochem Biophys Res Commun. 2003;311:1019–1025. doi: 10.1016/j.bbrc.2003.10.101. [DOI] [PubMed] [Google Scholar]
- 68.Sawa A, Snyder SH. Genetics. Two genes link two distinct psychoses. Science. 2005;310:1128–1129. doi: 10.1126/science.1121114. [DOI] [PubMed] [Google Scholar]
- 69.Ishizuka K, Paek M, Kamiya A, Sawa A. A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- 70.Porteous DJ, Millar JK. Disrupted in schizophrenia 1: building brains and memories. Trends Mol Med. 2006;12:255–261. doi: 10.1016/j.molmed.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 71.Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Jaaro-Peled H, Sawa A, Brandon NJ. How has DISC1 enabled drug discovery? Mol Cell Neurosci. 2008;37:187–195. doi: 10.1016/j.mcn.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 73.Brandon NJ, et al. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci. 2009;29:12768–12775. doi: 10.1523/JNEUROSCI.3355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sawamura N, Sawa A. Disrupted-in-schizophrenia-1 (DISC1): a key susceptibility factor for major mental illnesses. Ann N Y Acad Sci. 2006;1086:126–133. doi: 10.1196/annals.1377.018. [DOI] [PubMed] [Google Scholar]
- 75.Brandon NJ, et al. Subcellular targeting of DISC1 is dependent on a domain independent from the Nudel binding site. Mol Cell Neurosci. 2005;28:613–624. doi: 10.1016/j.mcn.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 76.Miyoshi K, et al. DISC1 localizes to the centrosome by binding to kendrin. Biochem Biophys Res Commun. 2004;317:1195–1199. doi: 10.1016/j.bbrc.2004.03.163. [DOI] [PubMed] [Google Scholar]
- 77.Wang Q, Brandon NJ. Regulation of the cytoskeleton by Disrupted-in-Schizophrenia 1 (DISC1) Mol Cell Neurosci. 2011 Jan 12; doi: 10.1016/j.mcn.2011.06.004. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 78.Kamiya A, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. The first paper elucidating a molecular mechanism of DISC1 in vivo: DISC1 plays a critical role in the early cortical development and in the regulation of centrosome function. Approach taken to understand DISC1 function in this paper has provided a platform for many recent studies to further elucidate DISC1s role. [DOI] [PubMed] [Google Scholar]
- 79.Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838–2842. doi: 10.1001/jama.270.23.2838. [DOI] [PubMed] [Google Scholar]
- 80.Shu T, et al. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 81.Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 82.Kamiya A, et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: a candidate for psychiatric illnesses. Arch Gen Psychiatry. 2008;65:996–1006. doi: 10.1001/archpsyc.65.9.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Datta SR, et al. A threonine to isoleucine missense mutation in the pericentriolar material 1 gene is strongly associated with schizophrenia. Mol Psychiatry. 2010;15:615–628. doi: 10.1038/mp.2008.128. [DOI] [PubMed] [Google Scholar]
- 84.Gurling HM, et al. Genetic association and brain morphology studies and the chromosome 8p22 pericentriolar material 1 (PCM1) gene in susceptibility to schizophrenia. Arch Gen Psychiatry. 2006;63:844–854. doi: 10.1001/archpsyc.63.8.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eastwood SL, Walker M, Hyde TM, Kleinman JE, Harrison PJ. The DISC1 Ser704Cys substitution affects centrosomal localization of its binding partner PCM1 in glia in human brain. Hum Mol Genet. 2010;19:2487–2496. doi: 10.1093/hmg/ddq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eastwood SL, Hodgkinson CA, Harrison PJ. DISC-1 Leu607Phe alleles differentially affect centrosomal PCM1 localization and neurotransmitter release. Mol Psychiatry. 2009;14:556–557. doi: 10.1038/mp.2009.13. [DOI] [PubMed] [Google Scholar]
- 87.Mao Y, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. Initial description of the relationship between DISC1 and canonical wnt signaling. The interaction was shown to be mediated by a direct interaction between DISC1 and GSK3. Inhibition of GSK3 shown to be able to compensate for the loss of DISC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shen S, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fukuda T, Sugita S, Inatome R, Yanagi S. CAMDI, a novel disrupted in schizophrenia 1 (DISC1)-binding protein, is required for radial migration. J Biol Chem. 2010;285:40554–40561. doi: 10.1074/jbc.M110.179481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh KK, et al. Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron. 2010;67:33–48. doi: 10.1016/j.neuron.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ishizuka K, et al. DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature. 2011;473:92–96. doi: 10.1038/nature09859. Delineated the importance of DISC1 phosphorylation at serine 710 (mouse) in corticogenesis. More specifically this post-tranlational modification event switches the preferred binding partners for DISC1 and promotes neuronal migration versus proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee FH, et al. Disc1 Point Mutations in Mice Affect Development of the Cerebral Cortex. J Neurosci. 2011;31:3197–3206. doi: 10.1523/JNEUROSCI.4219-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duan X, et al. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. Compelling paper showing the role of DISC1 in adult hippocampal neurogenesis. Knockdown of DISC1 regulates neuronal integration of new-born neurons with neurons exhibiting accelerated synapse formation and dendritic development and increased excitability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim JY, et al. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Enomoto A, et al. Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron. 2009;63:774–787. doi: 10.1016/j.neuron.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 96.Faulkner RL, et al. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci U S A. 2008;105:14157–14162. doi: 10.1073/pnas.0806658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tomita K, Kubo KI, Ishii K, Nakajima K. Disrupted-in-Schizophrenia-1 (Disc1) is necessary for migration of the pyramidal neurons during mouse hippocampal development. Hum Mol Genet. doi: 10.1093/hmg/ddr194. (in press) [DOI] [PubMed] [Google Scholar]
- 98.Meyer KD, Morris JA. Disc1 regulates granule cell migration in the developing hippocampus. Hum Mol Genet. 2009;18:3286–3297. doi: 10.1093/hmg/ddp266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kvajo M, et al. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Namba T, et al. NMDA receptor regulates migration of newly generated neurons in the adult hippocampus via Disrupted-In-Schizophrenia 1 (DISC1) J Neurochem. 2011;118:34–44. doi: 10.1111/j.1471-4159.2011.07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fournier NM, et al. The effect of amygdala kindling on hippocampal neurogenesis coincides with decreased reelin and DISC1 expression in the adult dentate gyrus. Hippocampus. 2010;20:659–671. doi: 10.1002/hipo.20653. [DOI] [PubMed] [Google Scholar]
- 102.Penzes P, Cahill ME, Jones KA, Vanleeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55:1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 104.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 105.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 106.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 107.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 108.Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- 109.Kirkpatrick B, et al. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol. 2006;497:436–450. doi: 10.1002/cne.21007. [DOI] [PubMed] [Google Scholar]
- 110.Hayashi-Takagi A, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. First paper to clearly show a functional role for DISC1 in regulating synapse function and spine morphology, using RNAi approaches. Also, showed the importance of the DISC1-kalirin7 complex in mediating these effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Q, et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.87. Characterized the interaction between DISC1 and the kinase TNIK. This was initially found by the DISC1 interactome study. Clearly showed that this complex is critical for regulating the stability of a range of post-synaptic proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ramsey AJ, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc Natl Acad Sci U S A. 2011;108:5795–5800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Clapcote SJ, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 114.Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang Y, Wang XB, Frerking M, Zhou Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci U S A. 2008;105:11388–11393. doi: 10.1073/pnas.0802978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 117.Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- 118.Ikeda M, et al. Genome-wide association study of schizophrenia in a Japanese population. Biol Psychiatry. 2011;69:472–478. doi: 10.1016/j.biopsych.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 119.Kushima I, et al. Resequencing and Association Analysis of the KALRN and EPHB1 Genes And Their Contribution to Schizophrenia Susceptibility. Schizophr Bull. 2010 doi: 10.1093/schbul/sbq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cahill ME, et al. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 2009;106:13058–13063. doi: 10.1073/pnas.0904636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Penzes P, Cahill ME, Jones KA, Srivastava DP. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008;18:405–413. doi: 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 122.Penzes P, Jones KA. Dendritic spine dynamics--a key role for kalirin-7. Trends Neurosci. 2008;31:419–427. doi: 10.1016/j.tins.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Penzes P, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- 124.Xie Z, et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cahill ME, et al. Control of interneuron dendritic growth through NRG1/erbB4-mediated kalirin-7 disinhibition. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006;60:132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 127.Chen SY, Huang PH, Cheng HJ. Disrupted-in-Schizophrenia 1-mediated axon guidance involves TRIO-RAC-PAK small GTPase pathway signaling. Proc Natl Acad Sci U S A. 2011;108:5861–5866. doi: 10.1073/pnas.1018128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11:220–230. doi: 10.1016/s0962-8924(01)01980-8. [DOI] [PubMed] [Google Scholar]
- 129.Fu CA, et al. TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J Biol Chem. 1999;274:30729–30737. doi: 10.1074/jbc.274.43.30729. [DOI] [PubMed] [Google Scholar]
- 130.Peng J, et al. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- 131.Collins MO, et al. Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem. 2005;280:5972–5982. doi: 10.1074/jbc.M411220200. [DOI] [PubMed] [Google Scholar]
- 132.Potkin SG, et al. A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr Bull. 2009;35:96–108. doi: 10.1093/schbul/sbn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hussain NK, Hsin H, Huganir RL, Sheng M. MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function. J Neurosci. 2010;30:14786–14794. doi: 10.1523/JNEUROSCI.4124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Colledge M, et al. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 137.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 138.Kawabe H, Brose N. The ubiquitin E3 ligase Nedd4-1 controls neurite development. Cell Cycle. 2010;9 doi: 10.4161/cc.9.13.12236. [DOI] [PubMed] [Google Scholar]
- 139.Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Millar JK, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. Original identification of the relationship between DISC1 and PDE4. DISC1 shown to bind and regulate PDE4. Critically, also identified 2 members of a family with mental illness with a translocation in PDE4B. Convergence of data re-ignited interest in PDE4 as a target for psychoses. [DOI] [PubMed] [Google Scholar]
- 141.Murdoch H, et al. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci. 2007;27:9513–9524. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peineau S, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 143.Gamo NJ, et al. Society for Neuroscience 2010. San Diego, USA: 2010. [Google Scholar]
- 144.Brown SM, et al. Synaptic modulators Nrxn1 and Nrxn3 are disregulated in a Disc1 mouse model of schizophrenia. Mol Psychiatry. 2011;16:585–587. doi: 10.1038/mp.2010.134. [DOI] [PubMed] [Google Scholar]
- 145.Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36:301–313. doi: 10.1093/schbul/sbp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kellendonk C, Simpson EH, Kandel ER. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci. 2009;32:347–358. doi: 10.1016/j.tins.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Johnstone M, et al. DISC1 in schizophrenia: genetic mouse models and human genomic imaging. Schizophr Bull. 2011;37:14–20. doi: 10.1093/schbul/sbq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kelly MP, Brandon NJ. Taking a bird’s eye view on a mouse model review: a comparison of findings from mouse models targeting DISC1 or DISC1-interacting proteins. Future Neurology. 2011;6:661–677. [Google Scholar]
- 149.Pletnikov MV, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- 150.Hikida T, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li W, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A. 2007;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Niwa M, et al. Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron. 2010;65:480–489. doi: 10.1016/j.neuron.2010.01.019. The first paper that address a context-dependent role for DISC1 (spatially and temporally) in overall brain function, such as behaviors. Critically a selected deficit of DISC1 in the developing cortex led to a range of neurochemical and behavioral phenotypes later after puberty, mirroring the time course of schizophrenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pollak DD, Rey CE, Monje FJ. Rodent models in depression research: classical strategies and new directions. Ann Med. 2010;42:252–264. doi: 10.3109/07853891003769957. [DOI] [PubMed] [Google Scholar]
- 155.Van Snellenberg JX, de Candia T. Meta-analytic evidence for familial coaggregation of schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2009;66:748–755. doi: 10.1001/archgenpsychiatry.2009.64. [DOI] [PubMed] [Google Scholar]
- 156.Ibi D, et al. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206:32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Abazyan B, et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lipina TV, et al. Enhanced dopamine function in DISC1-L100P mutant mice: implications for schizophrenia. Genes Brain Behav. 2010;9:777–789. doi: 10.1111/j.1601-183X.2010.00615.x. [DOI] [PubMed] [Google Scholar]
- 159.Ayhan Y, et al. Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry. 2011;16:293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.O’Donnell P. Adolescent maturation of cortical dopamine. Neurotox Res. 2010;18:306–312. doi: 10.1007/s12640-010-9157-3. [DOI] [PubMed] [Google Scholar]
- 161.Lipina TV, Wang M, Liu F, Roder JC. Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology. doi: 10.1016/j.neuropharm.2011.02.020. (in press) [DOI] [PubMed] [Google Scholar]
- 162.Carlyle BC, Mackie S, Christie S, Millar JK, Porteous DJ. Co-ordinated action of DISC1, PDE4B and GSK3beta in modulation of cAMP signalling. Mol Psychiatry. doi: 10.1038/mp.2011.17. (in press) [DOI] [PubMed] [Google Scholar]
- 163.Nakazawa K, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jones MW, Wilson MA. Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol. 2005;3:e402. doi: 10.1371/journal.pbio.0030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kubo K, et al. Migration defects by DISC1 knockdown in C57BL/6, 129X1/SvJ, and ICR strains via in utero gene transfer and virus-mediated RNAi. Biochem Biophys Res Commun. 2010;400:631–637. doi: 10.1016/j.bbrc.2010.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wood JD, Bonath F, Kumar S, Ross CA, Cunliffe VT. Disrupted-in-schizophrenia 1 and neuregulin 1 are required for the specification of oligodendrocytes and neurones in the zebrafish brain. Hum Mol Genet. 2009;18:391–404. doi: 10.1093/hmg/ddn361. [DOI] [PubMed] [Google Scholar]
- 167.Ottis P, et al. Convergence of Two Independent Mental Disease Genes on the Protein Level: Recruitment of Dysbindin to Cell-Invasive Disrupted-In-Schizophrenia 1 Aggresomes. Biol Psychiatry. doi: 10.1016/j.biopsych.2011.03.027. (in press) [DOI] [PubMed] [Google Scholar]
- 168.Guo AY, et al. The dystrobrevin-binding protein 1 gene: features and networks. Mol Psychiatry. 2009;14:18–29. doi: 10.1038/mp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ito H, et al. Dysbindin-1, WAVE2 and Abi-1 form a complex that regulates dendritic spine formation. Mol Psychiatry. 2010;15:976–986. doi: 10.1038/mp.2010.69. [DOI] [PubMed] [Google Scholar]
- 170.Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Leliveld SR, et al. Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci. 2008;28:3839–3845. doi: 10.1523/JNEUROSCI.5389-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Boxall R, Porteous DJ, Thomson PA. DISC1 and Huntington’s disease--overlapping pathways of vulnerability to neurological disorder? PLoS One. 2011;6:e16263. doi: 10.1371/journal.pone.0016263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhou X, Chen Q, Schaukowitch K, Kelsoe JR, Geyer MA. Insoluble DISC1-Boymaw fusion proteins generated by DISC1 translocation. Mol Psychiatry. 2010;15:669–672. doi: 10.1038/mp.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou X, Geyer MA, Kelsoe JR. Does disrupted-in-schizophrenia (DISC1) generate fusion transcripts? Mol Psychiatry. 2008;13:361–363. doi: 10.1038/sj.mp.4002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Millar JK, James R, Christie S, Porteous DJ. Disrupted in schizophrenia 1 (DISC1): subcellular targeting and induction of ring mitochondria. Mol Cell Neurosci. 2005;30:477–484. doi: 10.1016/j.mcn.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 176.MacAskill AF, Atkin TA, Kittler JT. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci. 2010;32:231–240. doi: 10.1111/j.1460-9568.2010.07345.x. [DOI] [PubMed] [Google Scholar]
- 177.MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 178.Atkin TA, Macaskill AF, Brandon NJ, Kittler JT. Disrupted in Schizophrenia-1 regulates intracellular trafficking of mitochondria in neurons. Mol Psychiatry. 2011;16:122–124. doi: 10.1038/mp.2010.110. [DOI] [PubMed] [Google Scholar]
- 179.Ikuta J, et al. Fasciculation and elongation protein zeta-1 (FEZ1) participates in the polarization of hippocampal neuron by controlling the mitochondrial motility. Biochem Biophys Res Commun. 2007;353:127–132. doi: 10.1016/j.bbrc.2006.11.142. [DOI] [PubMed] [Google Scholar]
- 180.Park YU, et al. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc Natl Acad Sci U S A. 2010;107:17785–17790. doi: 10.1073/pnas.1004361107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sawamura N, et al. Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol Psychiatry. 2008;13:1138–1148. 1069. doi: 10.1038/mp.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Drerup CM, Wiora HM, Topczewski J, Morris JA. Disc1 regulates foxd3 and sox10 expression, affecting neural crest migration and differentiation. Development. 2009;136:2623–2632. doi: 10.1242/dev.030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hattori T, et al. DISC1 regulates cell-cell adhesion, cell-matrix adhesion and neurite outgrowth. Mol Psychiatry. 2010;15:778, 798–809. doi: 10.1038/mp.2010.60. [DOI] [PubMed] [Google Scholar]
- 184.Sawamura N, Sawamura-Yamamoto T, Ozeki Y, Ross CA, Sawa A. A form of DISC1 enriched in nucleus: altered subcellular distribution in orbitofrontal cortex in psychosis and substance/alcohol abuse. Proc Natl Acad Sci U S A. 2005;102:1187–1192. doi: 10.1073/pnas.0406543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kamiya A, et al. DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum Mol Genet. 2006;15:3313–3323. doi: 10.1093/hmg/ddl407. [DOI] [PubMed] [Google Scholar]
- 186.Hayashi MA, et al. Assessing the role of endooligopeptidase activity of Ndel1 (nuclear-distribution gene E homolog like-1) in neurite outgrowth. Mol Cell Neurosci. 2010;44:353–361. doi: 10.1016/j.mcn.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 187.Hattori T, et al. A novel DISC1-interacting partner DISC1-Binding Zinc-finger protein: implication in the modulation of DISC1-dependent neurite outgrowth. Mol Psychiatry. 2007;12:398–407. doi: 10.1038/sj.mp.4001945. [DOI] [PubMed] [Google Scholar]
- 188.Taylor MS, Devon RS, Millar JK, Porteous DJ. Evolutionary constraints on the Disrupted in Schizophrenia locus. Genomics. 2003;81:67–77. doi: 10.1016/s0888-7543(02)00026-5. [DOI] [PubMed] [Google Scholar]
- 189.Ishizuka K, et al. Evidence that many of the DISC1 isoforms in C57BL/6J mice are also expressed in 129S6/SvEv mice. Mol Psychiatry. 2007;12:897–899. doi: 10.1038/sj.mp.4002024. [DOI] [PubMed] [Google Scholar]
- 190.Meyer KD, Morris JA. Immunohistochemical analysis of Disc1 expression in the developing and adult hippocampus. Gene Expr Patterns. 2008;8:494–501. doi: 10.1016/j.gep.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 191.Lipina TV, et al. Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse. 2011;65:234–248. doi: 10.1002/syn.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Young-Pearse TL, Suth S, Luth ES, Sawa A, Selkoe DJ. Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J Neurosci. 2010;30:10431–10440. doi: 10.1523/JNEUROSCI.1445-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shinoda T, et al. DISC1 regulates neurotrophin-induced axon elongation via interaction with Grb2. J Neurosci. 2007;27:4–14. doi: 10.1523/JNEUROSCI.3825-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Taya S, et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J Neurosci. 2007;27:15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nakata K, et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proc Natl Acad Sci U S A. 2009;106:15873–15878. doi: 10.1073/pnas.0903413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sachs NA, et al. A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol Psychiatry. 2005;10:758–764. doi: 10.1038/sj.mp.4001667. [DOI] [PubMed] [Google Scholar]
- 198.Green EK, et al. Evidence that a DISC1 frame-shift deletion associated with psychosis in a single family may not be a pathogenic mutation. Mol Psychiatry. 2006;11:798–799. doi: 10.1038/sj.mp.4001853. [DOI] [PubMed] [Google Scholar]
- 199.Chiang CH, et al. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry. 2011;16:358–360. doi: 10.1038/mp.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Brandon NJ, et al. Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci. 2004;25:42–55. doi: 10.1016/j.mcn.2003.09.009. [DOI] [PubMed] [Google Scholar]