

Abstract
A dynamic kinetic resolution (DKR) of allylic sulfoxides has been demonstrated by combining the Mislow [2,3]-sigmatropic rearrangement with catalytic asymmetric hydrogenation. The efficiency of our DKR was optimized by using low pressures of hydrogen gas to decrease the rate of hydrogenation relative to the rate of sigmatropic rearrangement. Kinetic studies reveal that the rhodium complex acts as a dual-role catalyst and accelerates the substrate racemization while catalyzing olefin hydrogenation. Scrambling experiments and theoretical modeling support a novel mode of sulfoxide racemization which occurs via a rhodium π-allyl intermediate in polar solvents. In non-polar solvents, however, the substrate racemization is primarily uncatalyzed. Computational studies suggest that the sulfoxide binds to rhodium via O–coordination throughout the catalytic cycle for hydrogenation.
INTRODUCTION
Sulfoxides are valuable targets in medicinal chemistry (e.g., Esomeprazole, Modafinil) and useful intermediates in organic synthesis.1 In addition, they act as both chiral auxiliaries2 and as chiral ligands for asymmetric synthesis.3–5 As ligands, they can bind to a metal-center through O-coordination and/or S-coordination. In contrast to other heteroatom functional groups (e.g., hydroxyl, amino), however, the use of sulfoxides to direct stereoselective transformations remains relatively unexplored. We have shown that a sulfoxide directed hydroacylation proceeds with high diastereoselectivity.6 In the following article, we study the ability of sulfoxides to direct Rh-catalyzed hydrogenations and demonstrate a novel concept for dynamic kinetic resolution (DKR) of sulfoxides by the classic Mislow-Evans rearrangement.
Back in 1966, Mislow reported that enantioenriched allylic sulfoxides undergo spontaneous racemization via an achiral sulfenate ester.7–9 Although the equilibrium generally favors the sulfoxide, Evans subsequently showed that the sulfenate ester could be reduced in situ to generate alcohols.10 This Mislow- Evans transformation has been used for the stereoselective construction of allylic alcohols in many natural product syntheses. Considering the ability of sulfoxides to act as ligands, however, we envisioned a new use for the Mislow-Evans rearrangement (Scheme 1). Rather than trapping the sulfenate ester with stoichiometric reductants, we propose a sulfoxide-directed catalytic transformation of the olefin. This proposed DKR features the rare use of a sigmatropic rearrangement as the key mechanism for racemization of the starting reagent.11– 13 Transforming the olefin prevents epimerization of the sulfoxide product, thus leading to enantioenriched sulfoxides.
Scheme 1.

Proposed DKR of allylic sulfoxides
As proof of this concept, we herein demonstrate the synthesis of enantioenriched chiral sulfoxides by enantioselective reduction, rather than conventional oxidation methods.1 Kinetic experiments point to a novel rhodium-catalyzed epimerization of allylic sulfoxides. Of note, catalytic antibodies have been observed to accelerate this rearrangement,14 while metal catalysis has only been predicted theoretically.15 Complementary theoretical studies provide mechanistic insights that will more broadly impact the development of other sulfoxide directed metal catalyzed transformations, catalytic sigmatropic methods, and DKR strategies.
RESULTS AND DISCUSSION
Initial result
While various olefin functionalizations, including hydroacylation, can be pursued, we chose asymmetric olefin hydrogenation for initial investigation due to the wide-range of catalysts known for this reduction. To achieve an efficient DKR, the chiral catalyst must preferentially transform one enantiomer of the allylic sulfoxide (Scheme 1). In addition, the racemization of the allylic sulfoxide must be fast relative to the desired hydrogenation.16 Consequently, controlling the relative rate of hydrogenation and racemization is critical. Mislow and coworkers determined that allyl p-tolyl sulfoxide racemizes with a half-life of 4.4 hours at 40 °C in benzene. Considering that certain metals are known to catalyze sigma-tropic rearrangements, we anticipated that catalysis of the substrate racemization might be possible.
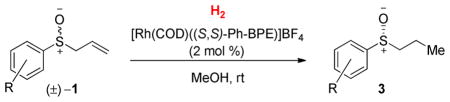
After evaluating a series of chiral bisphosphine ligands with a cationic Rh(I) source, we identified Ph-BPE17 as a promising ligand for the DKR of allylic sulfoxides (Scheme 2). The chiral sulfoxide was produced in 90% ee after complete consumption of the starting material in the presence of this ligand. Propyl sulfenate 4a was generated as a byproduct (2.4 : 1 sulfoxide : sulfenate) from hydrogenation of the transient allyl sulfenate ester 2a that is in equilibrium with 1a.
Scheme 2.

DKR under non-polar and polar conditions.
When using methanol as the solvent, we found that the undesired formation of propyl sulfenate ester significantly diminished to a 92:8 sulfoxide 3a: sulfenate ester 4a ratio, presumably because the equilibrium concentration of the allylic sulfenate ester 2 is lower in polar solvents.8 High enantioselectivity (88% ee) could be achieved at full substrate conversion and in 73 % isolated yield of the sulfoxide product by lowering the H2 pressure to 0.1 atm.
Kinetics of racemization
To better understand the effect of reaction conditions on the rate of racemization, we performed a series of kinetic experiments. Enantioenriched phenyl allyl sulfoxide (S)-1b was synthesized using the method of Pelotier et al.18–20 The rate of racemization was measured by monitoring the decay of optical activity in a polarimeter or by monitoring the enantiomeric excess by SFC analysis (see supporting information for details).
Based on the ability of Pd(II) salts to catalyze related sigmatropic rearrangements,21–24 we wondered whether catalysis of the Mislow-Evans rearrangement would be possible. Pd(0) catalysis has been predicted theoretically.15 Indeed, we found that PdCl2(PhCN)2 accelerated the rate of racemization in 1,2-dichloroethane (DCE) by a factor of 17 (Table 1, entry 2). A dual catalyst system using both rhodium and palladium, however, did not prove optimal for the DKR hydrogenation. Using PdCl2(PhCN)2 (5 mol%) in addition to the rhodium catalyst in the hydrogenation reaction resulted in recovery of starting material, presumably due to coordination of the benzonitrile ligands to rhodium. Use of PdCl2(COD) resulted in formation of palladium black and substrate decomposition.
Table 1.
Racemization Rate Constants for Phenyl Allyl Sulfoxide
| ||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst (2 mol%) | k (hr−1)a | τ1/2 (h) |
| 1b | 1,2-DCE | None | 0.0105 (3) | 66 |
| 2b | 1,2-DCE | Pd(PhCN)2Cl2 | 0.181 (4) | 3.8 |
| 3c | MeOH | None | 0.0022 (1) | 315 |
| 4c | MeOH | [Rh(L)]BF4 | 0.072 (5) | 9.6 |
| 5c | 9:1 tol:DCM | None | 0.0870 (2) | 8.0 |
| 6c | 9:1 tol:DCM | [Rh(L)]BF4 | 0.0949(5) | 7.3 |
Rate constant for racemization, see Reist et al.26 for definitions. All rate constants are mean values of 3 kinetic runs. Values in parenthesis are sample standard deviations in the final decimal place.
Measured by the polarimetry method.
Measured by the SFC method. L = COD + (S,S)-Ph-BPE
In accordance with studies by Mislow, we found racemization to be slow in polar solvents, with a racemization half-life of 315 h in methanol (Table 1, entry 3). In a less polar solvent, (9:1 toluene:DCM),25 the rate of racemization was faster than in methanol by a factor of 40 (compare entries 5 and 3). Our best hydrogenation catalyst [Rh((S,S)-Ph-BPE)(COD)]BF4 (2 mol%) accelerated the racemization in methanol by a factor of 33 (compare entries 3 and 4). On the other hand, addition of [Rh((S,S)-Ph-BPE)]BF4 (2 mol%) in 9:1 toluene:DCM resulted in only a modest racemization rate enhancement of 10% (entries 5 and 6).
The kinetic experiments suggest that sulfoxide racemization in the DKR hydrogenation predominantly proceeds through an uncatalyzed [2,3]-sigmatropic rearrangement under non-polar solvent conditions (toluene:DCM mixtures). However, the allylic sulfoxide undergoes a rhodium-catalyzed racemization pathway under polar solvent (MeOH) conditions. This suggests that the rhodium complex can catalyze both the hydrogenation and epimerization processes over the course of the DKR. Therefore, the catalyst concentration will impact the relative rates of hydrogenation versus racemization differently, depending upon the solvent choice.
Mechanism of rhodium-catalyzed racemization
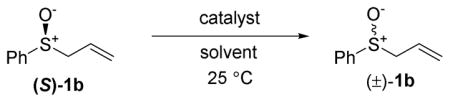
We propose two possible mechanisms for the Rh(I)-catalyzed racemization of allylic sulfoxides. First, the metal could act as a Lewis acid and bind the olefin, thus facilitating the [2,3]-sigmatropic rearrangement by stabilizing negative charge buildup at the 2-position of the allyl group (Scheme 3a). In this case, the metal is catalyzing the reversible formation of the achiral sulfenate ester. This mechanism is analogous to soft Lewis acid (such as Pd(II) and Hg(II)) catalysis of similar sigmatropic rearrangements.21–24
Scheme 3.
Two proposed mechanisms for rhodium-catalyzed racemization in MeOH
On the other hand, the metal could undergo oxidative addition to form a Rh-π-allyl intermediates II (Scheme 3b). Rotation of the sulfenate ligand followed by reductive elimination leads to overall sulfoxide epimerization. This mechanism is analogous to Pd(0) catalysis of related sigmatropic rearrangements.22,27
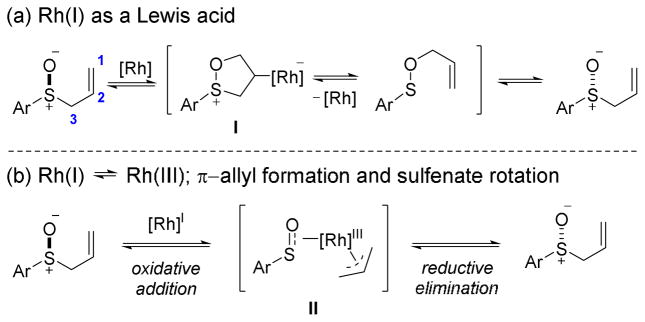
Deuterium scrambling experiments with deuterated allylic sulfoxide 1a–D were used to distinguish between the Lewis acid and π-allyl type mechanisms (Scheme 4). If a Lewis acid mechanism is operative, no scrambling of the deuterium should be observed since transposition of the deuterium labels in the formation of allyl sulfenate ester 2a–D is reversed when the allylic sulfoxide is reformed. However, if a rhodium π-allyl species II is an intermediate in the racemization and rotation of the π-allyl ligand is facile, then the deuterium label should scramble to the α- and γ-positions of allylic sulfoxide 1a–D.
Scheme 4.
Possible deuterium scrambling scenarios during allylic sulfoxide racemization
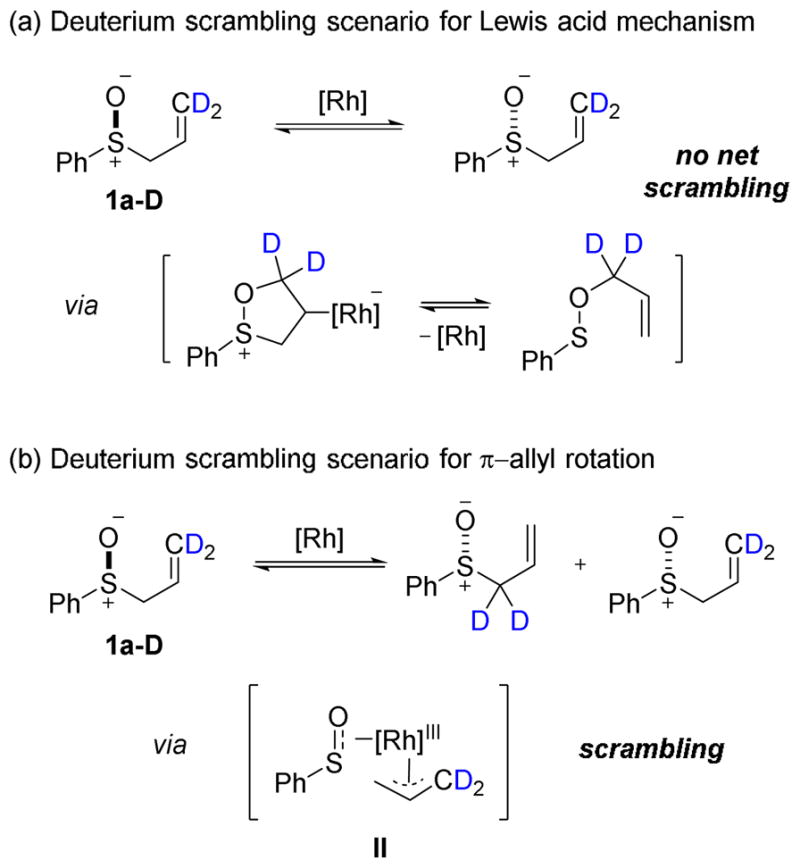
Exposure of 1a–D to hydrogenation under polar solvent conditions in methanol led to scrambling of the deuterium label in both the recovered starting material and the product (Scheme 5). This result suggests the intermediacy of a rhodium π-allyl species in the mechanism of racemization. Note that complete (1:1 α:γ) scrambling in the product would not be expected because the starting material is labeled exclusively at the outset of the reaction and only scrambles as the reaction proceeds. The extent of deuterium scrambling in the product (40:60 α:γ) is consistent with a racemization occurring exclusively through a rhodium π-allyl intermediate. On the other hand, when the reaction is performed in 9:1 toluene:DCM, only 12% deuterium scrambling was observed in the propyl sulfoxide product 3a–D. The extent of scrambling is intimately related to the product ee because these two quantities result from the same pathway. Based on kinetic modeling, 20% scrambling is expected given 50% product ee. Since the observed scrambling is significantly less than this prediction, we conclude that the Rhcatalyzed racemization is only partially responsible for the substrate racemization and that uncatalyzed substrate racemization is also occurring. This proposal is consistent with our racemization kinetics results (vide supra).
Scheme 5.

Hydrogenation of deuterium labeled allylic sulfoxide
To better understand the racemization process in MeOH, we conducted theoretical studies on the π-allyl mechanism. In a study of metal catalyzed aza-Claisen rearrangements, Bosnich proposed that cationic Rh(I) bisphosphine catalysts could undergo oxidative addition to generate a rhodium π-allyl intermediate with the amide anion still bound to rhodium.22 Analogously, we considered oxidative addition of the allylic sulfoxide to generate a rhodium π-allyl sulfenate complex. A relevant model system was designed to minimize computational cost while maintaining relevance to the experimental system. Dimethylphosphinoethane was modeled as the ligand to match the electronic and geometrical parameters of Ph-BPE. All calculations were performed with Gaussian 09.28 Structures were optimized in the gasphase with M0629 and a mixed basis set of SDD for rhodium and 6-311+G** for all other atoms. The need for a large basis set to accurately calculate the thermodynamics of the 2,3-rearrangement of allylic sulfoxides has previously been noted in several studies,15,30–32 and we confirmed this in our own benchmarking studies. Frequency calculations were performed to check the nature of the stationary points and obtain free energies. A solvation energy correction was added using the SMD33 method with methanol as solvent. In cases where multiple conformations of the substrate or ligand were stable minima, the lowest energy conformer is shown.
The energy profile for this racemization pathway is illustrated in Figure 1. Oxidative addition of the allylic sulfoxide occurs through a six-membered transition state where the sulfoxide is bound to rhodium through oxygen (6TS), with an energy of 16.4 kcal/mol. The resulting rhodium π-allyl intermediate contains an η-2 bound sulfenate ligand (7). Overall racemization requires rotation of the sulfenate ligand, which occurs through an η-1 O-bound isomerization transition state (8TS). A transition state for inversion of the allyl group (10TS) was located which involves rotation of the allyl group by 180°, with a transition state energy of 16.6 kcal/mol. The transition structure for this isomerization was found to be a simple rotation with a slight lengthening of one of the Rh–C bonds (2.42 versus 2.14 Å), indicating a small degree of π-allyl to σ-allyl interconversion in the transition state.34 Reductive elimination from intermediate 11 results in the enantiomer of 5.35 This model system with an achiral ligand provides a useful description of the overall process. The true system contains a chiral ligand, however, and thus the binding affinity of either enantiomer of substrate to the catalyst will not be equal in energy.
Figure 1.

The free energy (kcal/mol) profile for the Rh-catalyzed racemization of allylic sulfoxides.
In this pathway, due to rotation of the allyl group, a net transposition of the allylic termini has occurred. This theoretical prediction is consistent with the deuterium scrambling observed experimentally. Scrambling occurs as a direct result of the formation of a rhodium π-allyl sulfenate intermediate and would not be expected to occur in a simple uncatalyzed [2,3]-sigmatropic rearrangement.
The highest barrier in this Rh-catalyzed racemization pathway has an energy of 16.6 kcal/mol, which is significantly lower than the uncatalyzed [2,3]-sigmatropic rearrangement transition state energy of 23.5 kcal/mol (See SI for details). Therefore this rhodium π-allyl type mechanism22,36 is consistent with the observed racemization rate enhancement as well as the deuterium labelling study. However, the calculated barrier for hydrogenation (18.4 kcal/mol, vide infra) is higher than that for racemization (16.6 kcal/mol). Taking into account the concentration of dissolved hydrogen (~0.004 M for 1 atm),37 this predicts that racemization should be faster than hydrogenation. The truncation of the ligand (chiral Ph-BPE to achiral bis-dimethylphosphinoethane) and substrate (aryl allyl sulfoxide to methyl allyl sulfoxide) may play a role in altering these rates. Additionally, the difficulty in obtaining accurate energies for reactions involving change in oxidation states at sulfur has been noted previously.15,30–32,38
Hydrogen pressure effects on enantioselectivity
Given this mechanistic insight, we examined the effect of H2 pressure and catalyst concentration on enantioselectivity (Table 2). Using 1 atm H2 and methanol as solvent, the product was obtained in only 27% ee at full conversion. This poor enantioselectivity reflects the slow rate of racemization in methanol relative to the rate of olefin hydrogenation. Reducing the H2 pressure resulted in an increase in the enantiomeric excess of the product at full conversion. By simply decreasing the pressure of H2 to 0.1 atm, 88% ee was achieved at full conversion (73% isolated yield). A more efficient DKR is possible because lowering the pressure of H2 decreases the rate of hydrogenation, while not affecting the rate of racemization. Furthermore, the catalyst loading did not significantly impact the ee (compare entries 2,3 and 5,6). These results support the idea that rhodium catalyzes both hydrogenation and racemization. The sulfenate ester byproduct 4 was formed in only 1–10% under these conditions. 39
Table 2.
Effect of Catalyst Loading and Hydrogen Pressure
| |||||
|---|---|---|---|---|---|
| Entry | Loading (mol %) | p(H2) (atm) | conv 3a (%)a | ee 3a (%) | Yield 4b (%)a |
| 1 | 2 | 1.0 | 100 | 27 | 1 |
| 2 | 2 | 0.44 | 100 | 53 | 1 |
| 3 | 4 | 0.44 | 100 | 48 | 1 |
| 4 | 2 | 0.29 | 100 | 74 | 6 |
| 5 | 2 | 0.10 | 100 (73) | 88 | 8 |
| 6 | 8 | 0.10 | 100 | 88 | 3 |
| 7 | 3 | 0.06 | 100 | 89 | 10 |
| 8b | 1 | 1.0 | 6 | 93 | - |
| 9c | 1 | 0.1 | 7 | 94 | - |
NMR yield (isolated yield in parentheses).
Reaction time = 5 min.
Reaction time = 1 h
In our DKR, the H2 pressure effect is related to the relative rates of interconversion of enantiomeric substrates versus productive hydrogenation. We observe lower enantioselectivities at higher pressures of H2 because hydrogenation is relatively fast compared to racemization of the substrate. As shown in Table 2 (entries 8 and 9), the H2 pressure does not affect the ee at low conversion. In a classic study, Halpern observed lower enantioselectivities at higher pressures in his study of the hydrogenation of α-acylaminoacrylic acid derivatives. However this inverse relationship occurred for a different reason: the interconversion of diastereomeric catalyst-substrate complexes was slow relative to hydrogenation.40
Substrate scope
With this protocol for the DKR of 1a, we investigated the effect of substituents on the aromatic ring (Table 3). Electron-neutral 1b resulted in only 50 % ee at full conversion of starting material (entry 2). Having electron withdrawing groups at the ortho-position of the aromatic ring was necessary to achieve high enantioselectivity, presumably since they increase the rate of allylic sulfoxide racemization. Nitro (1e, entry 5) and trifluoromethyl (1f, entry 6) groups were tolerated; however the best enantioselectivities were obtained with ester substituents (entries 1, 3 and 4). The steric effect of the ortho- substituent may also contribute to enantioinduction. 41 Reduced sulfenate ester 4 and the corresponding disulfide were observed as by-products in these reactions, accounting for most of the mass balance.
Table 3.
Substrate scope for the asymmetric hydrogenation of allylic sulfoxides
| |||||
|---|---|---|---|---|---|
| Entrya | R | PH2 (atm) | t (h) | yield 3 (%) | ee 3 (%) |
| 1 | 2-CO2Me (1a) | 0.1 | 48 | 73 | 88 |
| 2 | H (1b) | 0.1 | 48 | 65 | 50 |
| 3 | 2-CO2tBu (1c) | 0.1 | 48 | 69 | 84 |
| 4 | 2-CO2nC6H13 (1d) | 0.1 | 48 | 84 | 86 |
| 5 | 2-NO2 (1e) | 1 | 24 | 55 | 72 |
| 6b | 2-CF3 (1f) | 0.1 | 48 | 53 | 88 |
Full substrate conversion was achieved in all cases.
4 mol % Rh-catalyst used
Mechanism of hydrogenation
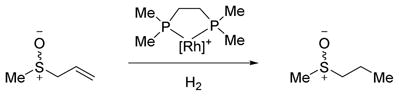
The mechanism of the rhodium catalyzed hydrogenation was investigated with DFT calculations to study sulfoxide coordination. Sulfoxides are known to bind to transition metals through either oxygen or sulfur and low-valent Rh(I) complexes with both S-bound and O–bound sulfoxides have been isolated.1,42,43 Coordination through oxygen is generally favored for hard metals, while coordination through sulfur is favored for soft metals. However, these preferences can be overridden if one binding mode is strained or otherwise geometrically unfavorable. In our system, S-coordination would likely produce more strain in the relevant hydrogenation intermediates than O–coordination. In this model study (eq 2), the ligand is achiral, 44 while the substrate contains a chiral center. Considering the facial approach of H2 to the square planar Rh-substrate complex, and two possible orientations of H2 oxidative addition, there are eight diastereomeric pathways each for O–coordination and S–coordination. Landis previously modeled a similarly complex set of pathways for the [Rh(DuPHOS)]+ catalyzed hydrogenation of enamides.45,46
 |
(2) |
The lowest energy O–bound and S–bound pathways are shown in Figure 1.47 Molecular H2 first binds to the catalyst-substrate complex (5) with a weak ion-induced dipole interaction (13). The dihydrogen molecule than isomerizes (14TS) to a η-2 complex 15, followed by oxidative addition (16TS) to generate the octahedral dihydride complex 17. After olefin insertion (18TS), a square pyramidal alkyl hydride complex 19 is formed. Reductive elimination (20TS) furnishes the hydrogenated product. Pathways where the phosphine is trans to the olefin were generally more favorable than pathways where a hydride is trans to the olefin, presumably due to the high trans effect of the olefin and hydride ligands. The product is predicted to bind to the Rh-complex preferentially through sulfur, as has been observed experimentally for most Rhsulfoxide complexes.1,42 All catalytic intermediates and transition states, however, are higher in energy for the S-bound pathways versus the O–bound pathways, presumably due to strain in the S–bound chelated intermediates. The most favorable S–bound pathway had a rate-determining transition state energy of 23.3 kcal/mol, while the most favorable O–bound pathway was 18.4 kcal/mol. Therefore, we conclude that the hydrogenation is directed by O–coordination.
CONCLUSION
We have demonstrated a DKR of allylic sulfoxides by asymmetric hydrogenation using a dual role catalyst. The chiral rhodium complex catalyzes both the sulfoxide directed olefin hydrogenation and sulfoxide racemization in methanol. A deuterium scrambling experiment supports a rhodium π-allyl species as an intermediate in the substrate racemization. Modeling of the rhodium catalyzed racemization suggests allylic sulfoxide oxidative addition generates a rhodium π-allyl sulfenate intermediate, which can undergo sulfenate rotation followed by reductive elimination to obtain the epimerized sulfoxide. This catalysis of racemization was further supported by deuterium labeling experiments. In non-polar solvents, however, racemization is primarily uncatalyzed. Computational studies of the hydrogenation pathway implicate the sulfoxide as a directing group through O–coordination. The mechanistic insights gained from this study will enable the development of other sulfoxide-directed metal processes49 that are amenable to DKR and dual role catalysts. These new transformations will add molecular complexity to the alkene while setting the sulfoxide stereocenter. In addition, future work fill focus on developing new catalysts and strategies for promoting allylic sulfoxide racemization.
Supplementary Material
Figure 2.

Free energy profile for the most favorable O– and S– bound pathways in the rhodium catalyzed hydrogenation of methyl allyl sulfoxide. 5 = starting material; 13 = ion-induced dipole complex; 14TS = ion-induced dipole transition state;48 15 = molecular hydrogen complex; 16TS = oxidative addition transition state; 17 = dihydride complex; 18TS = insertion transition state; 19 = alkyl hydride complex, 20TS = reductive elimination transition state; 21 = product.
Acknowledgments
K.N.H. thanks the National Science Foundation (CHE-0548209) and V.M.D thanks the National Institute of Health (GM105938) and Eli Lilly for financial support. P.K.D. is grateful for an NSERC Vanier award and an NSERC Michael Smith Foreign Study Supplement. K.G.M.K. is grateful for an NSERC CGS-D award. We thank Dr. Peng Liu for helpful discussions. Computations were performed on the IDRE Hoffman2 cluster at UCLA and the HPCVL cluster at Queen’s University, Kingston.
Footnotes
Full synthetic and characterization details, energies and optimized geometries of all calculated species. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Fernández I, Khiar N. Chem Rev. 2003;103:3651–3706. doi: 10.1021/cr990372u. [DOI] [PubMed] [Google Scholar]
- 2.For select examples of sulfoxides used as chiral auxiliaries, see: Adrio J, Carretero JC. J Am Chem Soc. 1999;121:7411–7412.Maezaki N, Sakamoto A, Nagahashi N, Soejima M, Li YX, Imamura T, Kojima N, Ohishi H, Sakaguchi K, Iwata C, Tanaka T. J Org Chem. 2000;65:3284–3291. doi: 10.1021/jo9919409.Carreño MC, Des Mazery R, Urbano A, Colobert F, Solladié G. Org Lett. 2004;6:297–299. doi: 10.1021/ol036299i.
- 3.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]
- 4.Mariz R, Luan X, Gatti M, Linden A, Dorta R. J Am Chem Soc. 2008;130:2172–2173. doi: 10.1021/ja710665q. [DOI] [PubMed] [Google Scholar]
- 5.Bügi JJ, Mariz R, Gatti M, Drinkel E, Luan X, Blumentritt S, Linden A, Dorta R. Angew Chem Int Ed. 2009;48:2768–2771. doi: 10.1002/anie.200900429. [DOI] [PubMed] [Google Scholar]
- 6.Coulter MM, Dornan PK, Dong VM. J Am Chem Soc. 2009;131:6932–6933. doi: 10.1021/ja901915u. [DOI] [PubMed] [Google Scholar]
- 7.Rayner DR, Miller EG, Bickart P, Gordon AJ, Mislow K. J Am Chem Soc. 1966;88:3138–3139. [Google Scholar]
- 8.Bickart P, Carson FW, Jacobus J, Miller EG, Mislow K. J Am Chem Soc. 1968;90:4869–4876. [Google Scholar]
- 9.Tang R, Mislow K. J Am Chem Soc. 1970;92:2100– 2104. [Google Scholar]
- 10.Evans DA, Andrews GC. Acc Chem Res. 1974;7:147– 155. [Google Scholar]
- 11.Akai S, Tanimoto K, Kanao Y, Egi M, Yamamoto T, Kita Y. Angew Chem Int Ed. 2006;45:2592–2595. doi: 10.1002/anie.200503765. [DOI] [PubMed] [Google Scholar]
- 12.Akai S, Hanada R, Fujiwara N, Kita Y, Egi M. Org Lett. 2010;12:4900–4903. doi: 10.1021/ol102053a. [DOI] [PubMed] [Google Scholar]
- 13.Levin S, Nani RR, Reisman SE. J Am Chem Soc. 2011;133:774–776. doi: 10.1021/ja110192b. [DOI] [PubMed] [Google Scholar]
- 14.Zhou ZS, Flohr A, Hilvert D. J Org Chem. 1999;64:8334–8341. doi: 10.1021/jo991299a. [DOI] [PubMed] [Google Scholar]
- 15.Kleimark J, Prestat G, Poli G, Norrby PO. Chem -Eur J. 2011;17:13963–13965. doi: 10.1002/chem.201102937. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura M, Tokunaga M, Noyori R. J Am Chem Soc. 1993;115:144–152. [Google Scholar]
- 17.Pilkington CJ, Zanotti-Gerosa A. Org Lett. 2003;5:1273–1275. doi: 10.1021/ol0341952. [DOI] [PubMed] [Google Scholar]
- 18.Pelotier B, Anson MS, Campbell IB, Macdonald SJF, Priem G, Jackson RFW. Synlett. 2002:1055–1060. [Google Scholar]
- 19.Substrate (S)-1b could be stored neat for several months in the freezer without significant racemization.
- 20.For other examples of asymmetric sulfide oxidation, see: Pitchen P, Dunach E, Deshmukh MN, Kagan HB. J Am Chem Soc. 1984;106:8188–8193.Dai W, Li J, Chen B, Li G, Lv Y, Wang L, Gao S. Org Lett. 2013;15:5658–5661. doi: 10.1021/ol402612x. and references therein.
- 21.Overman LE. Angew Chem Int Ed Eng. 1984;23:579– 586. [Google Scholar]
- 22.Schenck TG, Bosnich B. J Am Chem Soc. 1985;107:2058–2066. [Google Scholar]
- 23.Lee EE, Batey RA. J Am Chem Soc. 2005;127:14887–14893. doi: 10.1021/ja054161k. [DOI] [PubMed] [Google Scholar]
- 24.Bao H, Qi X, Tambar UK. J Am Chem Soc. 2011;133:1206–1208. doi: 10.1021/ja110500m. [DOI] [PubMed] [Google Scholar]
- 25.A small amount of dichloromethane was used in the solvent mixture in order to ensure solubility of the catalyst throughout the experiment.
- 26.Reist M, Testa B, Carrupt PA, Jung M, Schurig V. Chirality. 1995;7:396–400. [Google Scholar]
- 27.Ikariya T, Ishikawa Y, Hirai K, Yoshikawa S. Chemistry Letters. 1982:1815–1818. [Google Scholar]
- 28.Frisch MJ, et al. Gaussian 09. Gaussian Inc; Wallingford, CT: 2010. [Google Scholar]
- 29.Zhao Y, Truhlar DG. Theor Chem Acc. 2007;120:215– 241. [Google Scholar]
- 30.Jones-Hertzog DK, Jorgensen WL. J Am Chem Soc. 1995;117:9077–9078. [Google Scholar]
- 31.Amaudrut J, Pasto DJ, Wiest O. J Org Chem. 1998;63:6061–6064. doi: 10.1021/jo980547k. [DOI] [PubMed] [Google Scholar]
- 32.Freeman F, Bathala RM, Cavillo JE, Huang AC, Jackson TK, Lopez-Mercado AZ, Phung S, Suh J, Valencia DO. Int J Quantum Chem. 2006;106:2390– 2397. [Google Scholar]
- 33.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 34.Faller JW, Thomsen ME, Mattina MJ. J Am Chem Soc. 1971;93:2642–2653. [Google Scholar]
- 35.A rhodium catalyzed racemization pathway without allylic transposition could occur if reductive elimination occurred directly from intermediate 9. This reductive elimination was found to have a barrier of 19.5 kcal/mol.
- 36.Consiglio G, Waymouth RM. Chem Rev. 1989;89:257– 276. [Google Scholar]
- 37.Katayama T, Nitta T. J Chem Eng Data. 1976;21:194– 196. [Google Scholar]
- 38.Turec]ek F. J Phys Chem A. 1998;102:4703–4713. [Google Scholar]
- 39.The amount of sulfenate ester generated was found to be higher using the low pressure conditions. To explain this observation, we note that allyl sulfenate ester 2 is a transient intermediate in the racemization process, and not observable by 1H NMR. Under conditions where the racemization is fast relative to hydrogenation, the ratio of allyl sulfenate ester 2 to sulfoxide 1 can be maintained at thermodynamic equilibrium. This scenario is relevant for entry 7, where high ee and 10% sulfenate was observed. However if the racemization is slow relative to hydrogenation, then formation of allyl sulfenate ester 2 from (R)-1 or (S)-1 will also be slow. As the hydrogenation proceeds and the initial equilibrium concentration of 2 is consumed, the regeneration of 2 is slow and therefore the concentration of 2 will be lower than expected based on equilibrium constants. Consequently propyl sulfenate ester 4 will be formed to a lesser extent over the course of the reaction. This scenario is relevant for entry 1, where low ee and only 1% sulfenate ester was observed.
- 40.Halpern J. Science. 1982;217:401–407. doi: 10.1126/science.217.4558.401. [DOI] [PubMed] [Google Scholar]
- 41.While metal coordination of the ortho substituent (in the case of the esters) is also possible, we don’t believe this contributes to enantioinduction since comparable selectivities can be obtained with the trifluoromethyl substituent.
- 42.Calligaris M, Carugo O. Coord Chem Rev. 1996;153:83–154. [Google Scholar]
- 43.Dorta R, Rozenberg H, Milstein D. Chem Commun. 2002:710–711. doi: 10.1039/b200321j. [DOI] [PubMed] [Google Scholar]
- 44.We have calculated the energies of the two diastereomeric catalyst-substrate complexes with Rh[(S,S)-PhBPE]+ and (R) or (S)-phenyl allyl sulfoxide. The complex leading to the observed major product enantiomer was calculated to be more stable by 3.1 kcal/mol. See SI for details. Further studies are needed to explore the entire reaction coordinate in order to fully understand the origins of enantioselectivity.
- 45.Feldgus S, Landis CR. J Am Chem Soc. 2000;122:12714–12727. [Google Scholar]
- 46.Landis CR, Feldgus S. Angew Chem Int Ed. 2000;39:2863–2866. doi: 10.1002/1521-3773(20000818)39:16<2863::aid-anie2863>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 47.See supporting information for discussion of the higher energy pathways.
- 48.The solvent corrected energy of the ion induced dipole transition state for the O-bound pathway is lower than the energy of the molecular hydrogen complex. This occurs because the structure was optimized on the gas phase potential energy surface, followed by addition of a single point solvent correction. Indeed in the gas phase, the transition state is higher in energy than the molecular hydrogen complex by 0.4 kcal/mol.
- 49.25% yield, 25% ee was achieved in the linear selective hydroacylation of phenyl allyl sulfoxide with salicylaldehyde.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.