

Abstract
Resistance to antiangiogenic therapies is a critical problem that has limited the utility of antiangiogenic agents in clinical settings. However, the molecular mechanisms underlying this resistance have yet to be fully elucidated. In this study, we established a novel xenograft model of acquired resistance to bevacizumab. To identify molecular changes initiated by the tumor cells, we performed human-specific microarray analysis on bevacizumab-sensitive and -resistant tumors. Efficiency analysis identified 150 genes upregulated and 31 genes downregulated in the resistant tumors. Among angiogenesis-related genes, we found upregulation of fibroblast growth factor-2 (FGF2) and fibroblast growth factor receptor-3 (FGFR3) in the resistant tumors. Inhibition of the FGFR in the resistant tumors led to the restoration of sensitivity to bevacizumab. Furthermore, increased FGF2 production in the resistant cells was found to be mediated by overexpression of upstream genes phospholipase C (PLCg2), frizzled receptor-4 (FZD4), chemokine [C-X3-C motif] (CX3CL1), and chemokine [C-C motif] ligand 5 (CCL5) via extracellular signal-regulated kinase (ERK). In summary, our work has identified an upregulation of a proangiogenic signature in bevacizumab-refractory HNSCC tumors that converges on ERK signaling to upregulate FGF, which then mediates evasion of anti-VEGF therapy. These findings provide a new strategy on how to enhance the therapeutic efficacy of antiangiogenic therapy.
Implication Statement
Novel xenograft model leads to the discovery of FGF as a promising therapeutic target in overcoming the resistance of antiangiogenic therapy in HNSCC.
Keywords: Acquired Resistance, Bevacizumab, Biomarker, FGF, HNSCC
Introduction
Resistance to antiangiogenic therapies limits the clinical benefit of these agents in cancer patients. The single-agent response rate to antiangiogenic drugs such as bevacizumab (a monoclonal antibody to VEGF-A) is less than 10%, and even in patients who do respond, the duration of response is typically less than 3 months (1–3). Similar responses are seen in HNSCC (4–6), where bevacizumab is being evaluated in phase III clinical trials (NCT00588770). Available evidence suggests that tumors can adapt to the effects of VEGF blockade by acquiring alternative signaling pathways that sustain growth and survival. Studies using relevant preclinical models that identify these escape mechanisms will help develop reliable biomarkers of resistance and allow the development of co-targeting approaches to overcome resistance.
In recent years, there have been an increasing number of reports that described potential mechanisms of resistance to antiangiogenic therapy (7–11). In a xenograft model of Wilms tumor, vascular remodeling was seen in tumors that relapsed due to prolonged anti-VEGF therapy. In addition, remodeled vessels were associated with increased expression of ephrinB2 and PDGF-β (12). Angiogenic revascularization was observed in pancreatic-islet tumors, after a transient decrease in microvessel density (MVD) with anti-VEGFR2 antibody. This revascularization was accompanied by increased expression of members of the FGF family (13). In murine tumor models, infiltrating myeloid cells produced proangiogenic factors including Bv8 and tumor cells secreted HGF, which were shown to mediate intrinsic resistance to anti-VEGF antibody and sunitinib respectively (14, 15). Increased plasma IL-8 expression was reported in renal cell carcinoma xenografts that acquired resistance to sunitinib (16). Our previous work has also demonstrated IL-8 as a contributor of innate resistance to bevacizumab in HNSCC xenograft models(17). Upregulated stromal EGFR and FGFR have been implicated in bevacizumab-resistant NSCLC xenografts (18). High expression of proinflammatory factors has been associated with increased aggressiveness of bevacizumab-resistant pancreatic tumors (19).
Above studies indicate that tumors can rely on multiple mechanisms of resistance using a variety of angiogenic proteins secreted by both tumor cells and stromal cells. Hence, selection of one critical mediator of resistance for cotargeting with VEGF remains a daunting task. To address this issue, preclinical models that test combinatorial therapies along with VEGF inhibitors are needed to assist selection of suitable cotargets. Also, studies that elucidate the mechanism of upregulation of these resistance-contributing proteins will enable identification of functional networks that integrate additional upstream genes as potential contributors of resistance.
In the present study, we established a novel HNSCC xenograft model of acquired resistance to bevacizumab and identified upregulation of FGF signaling in resistant tumors. Angiogenesis-related genes PLCg2, FZD4, CX3CL1, and CCL5 regulated increased expression of FGF2 via increased ERK signaling. Co-targeting VEGF and FGFR sensitized resistant tumors to bevacizumab by disrupting angiogenesis. Overall, our results indicate that bevacizumab-refractory HNSCC tumors utilize FGF signaling as a path of least resistance using a battery of proangiogenic genes that converge on ERK signaling to upregulate its expression and mediate resistance to anti-VEGF therapy.
Materials and Methods
Cell lines and reagents
Tu138 cells were kindly provided by Dr. Jeffrey N. Myers (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA) (20). Tu138 cells and the bevacizumab-resistant isogenic clones were maintained in DMEM/F12 medium supplemented with 10% FBS. Both cell lines were validated by genotyping using short tandem repeat analysis within 12 months of their use. Bevacizumab (Avastin, Genentech Inc., South San Francisco, CA, USA) was purchased from the University of Pittsburgh Pharmacy. PD173074 was purchased from Selleck Chemicals (Houston, TX, USA).
Animal Studies
Model of acquired resistance
Five- to six-week-old female athymic nude-Foxn1nu mice were purchased from Harlan Laboratories (Indianapolis, IN) and were maintained under guidelines provided by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC). To generate the preclinical model, Tu138 cells (3×106 cells/ mouse) were inoculated in mice (n=10). After formation of subcutaneous tumors (~2 weeks), these mice were randomized to receive vehicle or bevacizumab and treated biweekly by intraperitoneal injection. Tumors were also measured biweekly and mean tumor volumes were computed as 3.14/6 × length × width2. Bevacizumab was administered at a moderate dose of 4mg/kg followed by incrementing the dose by 4mg/kg with every subsequent increase in tumor volume. Drug concentration was increased upto the maximum-tolerated dose in patients (20mg/kg). Mice were sacrificed if the tumors exceeded 20mm in diameter. Resistant tumors were excised and small tumor fragments (~ 1mm in diameter) were reimplanted into new mice (n=2) to propagate the model. Mice were treated with saline or bevacizumab (increasing concentrations, 8mg/kg–20mg/kg) for a period of two months.
Validation experiments
Small fragments from the resistant tumor (TuR3) were implanted to generate xenografts (n=8) for validation in a separate in vivo study with a short-term treatment regime (4 weeks). Parental Tu138 cells were also inoculated in mice (n=8) as a positive control for sensitivity to bevacizumab. Two weeks after tumor cell inoculation, the mice were randomized to receive vehicle or bevacizumab (4mg/kg).
Combination experiments
For the combination treatment study, small fragments from the resistant tumor were implanted in mice (n=12). Mice were randomized into four treatment groups receiving saline, bevacizumab, PD173074 or a combination of bevacizumab and PD173074. Bevacizumab and PD173074 were administered intraperitoneally at 8mg/kg (biweekly) and 25mg/kg (daily) respectively. Tumors were measured daily and tumor growth was assessed for two weeks.
Immunohistochemistry and immunofluorescence
Immunohistochemical staining for CD31 and immunofluorescence staining for CD31/TUNEL was performed on frozen tumor sections as previously described (21). Vessels completely stained with anti-CD31 antibodies were counted in 10 random 0.04-mm2 fields with a 20× objective and mean MVD was expressed as number of vessels per square millimeter. Quantification of CD31+/ TUNEL+ staining was done as the average percentage of apoptotic endothelial cells in 10 random 0.01-mm2 fields using a 40× objective.
Microarray
Total RNA was extracted from frozen tumors using TRIzol reagent (Invitrogen/Life Technologies Grand Island, NY, USA) and purified using the RNeasy Kit (Qiagen, Germantown, MD, USA). RNA amplification and biotin labeling was done using Illumina Total Prep RNA Amplification Kit (Ambion/ Life Technologies Grand Island, NY, USA). BiotinylatedcRNA was hybridized to human HT-12 v4 BeadChips (Illumina Inc., San Diego, CA, USA) and scanned using an IlluminaBeadChip Array Reader.
Efficiency analysis was used to determine the optimal methods for data normalization, transformation, and feature selection that produced the most internally consistent gene set(22). Raw data were normalized using a log2 and z-transformation and differentially expressed genes were identified using J5 test. This test computes a J5-score by comparing the mean difference in expression intensity between two groups for any gene to the average mean group difference over the whole array. Gene expression changes were considered to be statistically significant for genes bearing a J5-score higher than the threshold value 8.0. Gene expression pattern grids were generated for differentially expressed genes with the GEDA web application(23).
A pathway level impact analysis (24), was performed to provide both statistical and biological significance in suggesting the potential pathways affected by the observed changes in gene expression. Differentially expressed genes between bevacizumab-sensitive and -resistant tumors were also subjected to the functional interaction network analysis using ingenuity pathway analysis (IPA) software.
Real-time RT-PCR
Real-time RT-PCR was performed using taqMan one-step RT-PCR master mix kit and taqman gene expression assay kits (Applied Biosystems/ Life Technologies Grand Island, NY, USA) on a 7900HT Real-Time PCR system (Applied Biosystems/ Life Technologies Grand Island, NY, USA). Samples were prepared in triplicates in a 20ul reaction volume containing 200ng input RNA. RT-negative controls were run on each plate to ensure no amplification in the absence of input RNA. Standard cycling conditions were programmed as: 95°C for 12 minutes, 40 cycles of: 95°C for 15 seconds, 60°C for 1 minute. b-Actin was used as endogenous control. Following gene-specific taqman gene expression assay kits were used; FGF2: Hs00266645_m1, FGFR3: Hs00179829_m1, and PLCg2: Hs00182192_m1.
Western
Parental Tu138 and bevacizumab-resistant cells were plated in 10cm dishes. The following day complete medium was replaced with serum free medium. After 24 hrs, cells were treated with MEK inhibitor U0126 for 6 hrs and whole cell lysates were prepared and resolved on 10% SDS-page gels. Following transfer onto nitrocellulose membranes, antibody staining was done using: pERK (Thr202/Tyr204), ERK, FGF2 and β-Actin (Cell Signaling Technology Inc., Danvers, MA, USA). Reactive bands were detected by chemiluminiscence using ECL plus western blotting detection kit (Amersham Biosciences, Piscataway, NJ, USA). Similarly, immunoblots were performed using untreated cells and siRNA transfected cells using the following antibodies; FGF1, FGFR1, FGFR3, pPLCg1 (Tyr783), PLCg1, pPLCg2 (Tyr759), PLCg2, pSrc (Tyr416), Src, pAKT (S473), AKT, CCL5 (Cell Signaling Technology Inc., Danvers, MA, USA) FGFR2, FGFR4, FZD4, and CX3CL1 (Abcam, Cambridge, MA, USA).
ELISA
CCL5 was measured in cell culture supernatants from siRNA-transfected cells using ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA) as per the manufacturer’s instructions. Cytokine concentration is represented as pg/ml normalized to total protein measured in supernatant using Bradford assay. Human FGF2 was also measured in plasma from mice bearing bevacizumab-sensitive and -resistant tumors using ELISA.
siRNA transfection
Parental Tu138 and bevacizumab-resistant cells were transfected with siRNA targeting PLCg2, FZD4, CX3CL1, CCL5 and negative control siRNA (non-targeting) using Opti-MEM media, lipofectamine-2000 (Invitrogen/ Life Technologies, Grand Island, NY, USA). After 4 hrs, the media was replaced with complete medium and cells were incubated at 37°C for 1 hr. Following incubation, transfected cells were replated in 6-cm plates for western blot analysis after 48 hrs of transfection. Following gene-specific siRNAs were used; CX3CL1: s12629, FZD4: s15840, PLCg2: s10634, and CCL5: s12575 (Invitrogen/ Life Technologies, Grand Island, NY, USA).
Results
Generation of preclinical model of acquired resistance to bevacizumab
In order to establish xenograft model of acquired resistance, we inoculated mice with HNSCC cell line Tu138, which has been previously shown to be highly sensitive to bevacizumab in vivo(17). After generation of subcutaneous tumors, these mice were randomized to receive vehicle or bevacizumab at an initial dose of 4mg/kg followed by incrementing the dose by 4mg/kg with every subsequent increase in tumor volume. Drug concentration was increased up to the maximum-tolerated dose in patients (20mg/kg). Such an escalating dosing regimen eliminated the sensitive tumor cells and sequentially selected for the resistant clones. Initially, 3/5 xenograft tumors showed resistance quite early in the treatment cycle with growth rates comparable to the saline control (Figure 1A). Mean tumor volumes for the resistant xenografts TuR1, TuR2 and TuR3 were 2351.2mm3, 1329.4mm3 and 1194.0mm3 respectively.
Figure 1. Generation of HNSCC xenograft model of acquired resistance to bevacizumab.
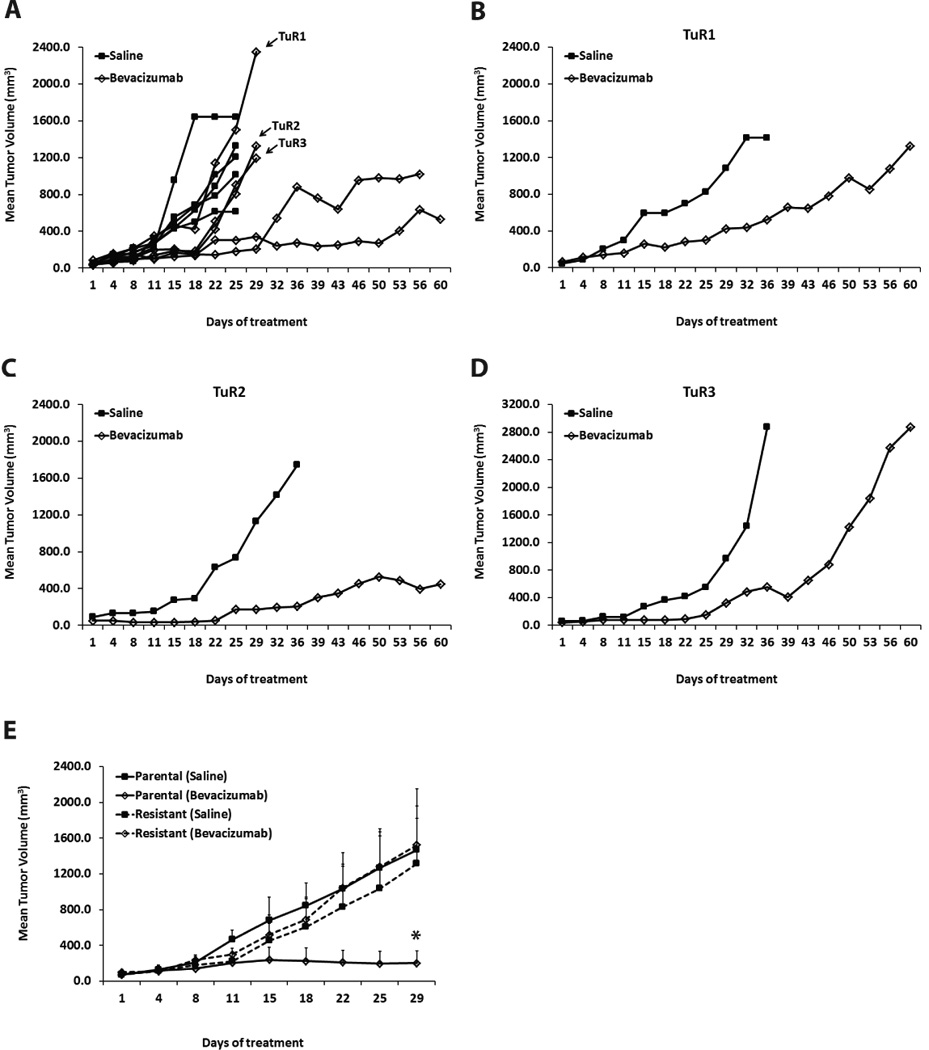
(A) Growth curve of Tu138 xenografts (n=5 per group) treated with saline or bevacizumab at an initial dose of 4mg/kg followed by incrementing the dose by 4mg/kg with every subsequent increase in tumor volume. 3/5 xenograft tumors (TuR1, TuR2 and TuR3) showed resistance with growth rates comparable to the saline control. (B–D) Resistant xenografts were excised and small tumor fragments were reimplanted into new mice (n=2) to propagate the model. Reimplanted tumors were subjected to a second phase of treatment with saline or bevacizumab (increasing concentrations, 8mg/kg–20mg/kg). Emergence of resistance was observed in bevacizumab-treated tumor TuR1 and TuR3 while TuR2 remained fairly sensitive. (E) Validation of acquired resistance in reimplanted TuR3 resistant tumors by treating xenografts (n=4 per group) with saline or bevacizumab. Parental Tu138 tumors were sensitive to bevacizumab resulting in 88% growth inhibition (* P=0.0171). In contrast, the resistant tumors showed no significant reduction in tumor growth.
The resistant xenografts were excised and small tumor fragments were reimplanted into new mice to propagate the model (Figure 1B–1D). These reimplanted tumors were subjected to a second phase of treatment where the resistant tumor-bearing mice were exposed to saline or bevacizumab (increasing concentrations, 8mg/kg–20mg/kg) for a period of two months. We observed a slow emergence of resistance in bevacizumab-treated tumor TuR1 with mean tumor volumes equivalent to the respective saline control (Figure 1B). Bevacizumab-treated tumor TuR2 remained moderately sensitive throughout the treatment suggesting that it failed to retain the initial resistance under incremental drug- selective pressure (Figure 1C). However, we observed a steep increase in tumor growth in the bevacizumab-treated tumor TuR3 beyond day 36 (Figure 1D) indicating emergence of resistance. Mean tumor volumes at the end of the treatment were 2571.1mm3 as compared to 1326.7mm3 in tumor TuR1. Hence, we selected tumor TuR3 to validate the acquisition of resistance.
In the validation study, we inoculated parental Tu138 cells as a positive control for sensitivity to bevacizumab and implanted small fragments from the bevacizumab-treated tumor TuR3 to generate xenografts (Figure 1E). The parental tumors were sensitive to bevacizumab, as expected, resulting in 88% growth inhibition. In contrast, TuR3 tumors showed complete resistance to bevacizumab as the growth rate of bevacizumab-treated tumors was almost identical to the saline-treated tumors.
Bevacizumab-refractory tumors exhibit sustained angiogenesis and resistance to endothelial cell apoptosis
To characterize the preclinical model of acquired resistance, we assessed MVD and endothelial cell apoptosis in the bevacizumab-sensitive and -resistant xenografts (from the validation study) (Figure 2A). The parental tumors showed significant decrease in MVD and increase in endothelial apoptosis in response to bevacizumab treatment. However, treatment of resistant tumors with bevacizumab did not result in statistically significant changes in MVD or endothelial apoptosis (Figure 2B, C). These results suggest that the bevacizumab-resistant tumors were able to maintain tumor angiogenesis and prevent endothelial apoptosis despite the sequestration of VEGF by bevacizumab.
Figure 2. Bevacizumab-refractory tumors exhibit sustained angiogenesis and resistance to endothelial cell apoptosis.
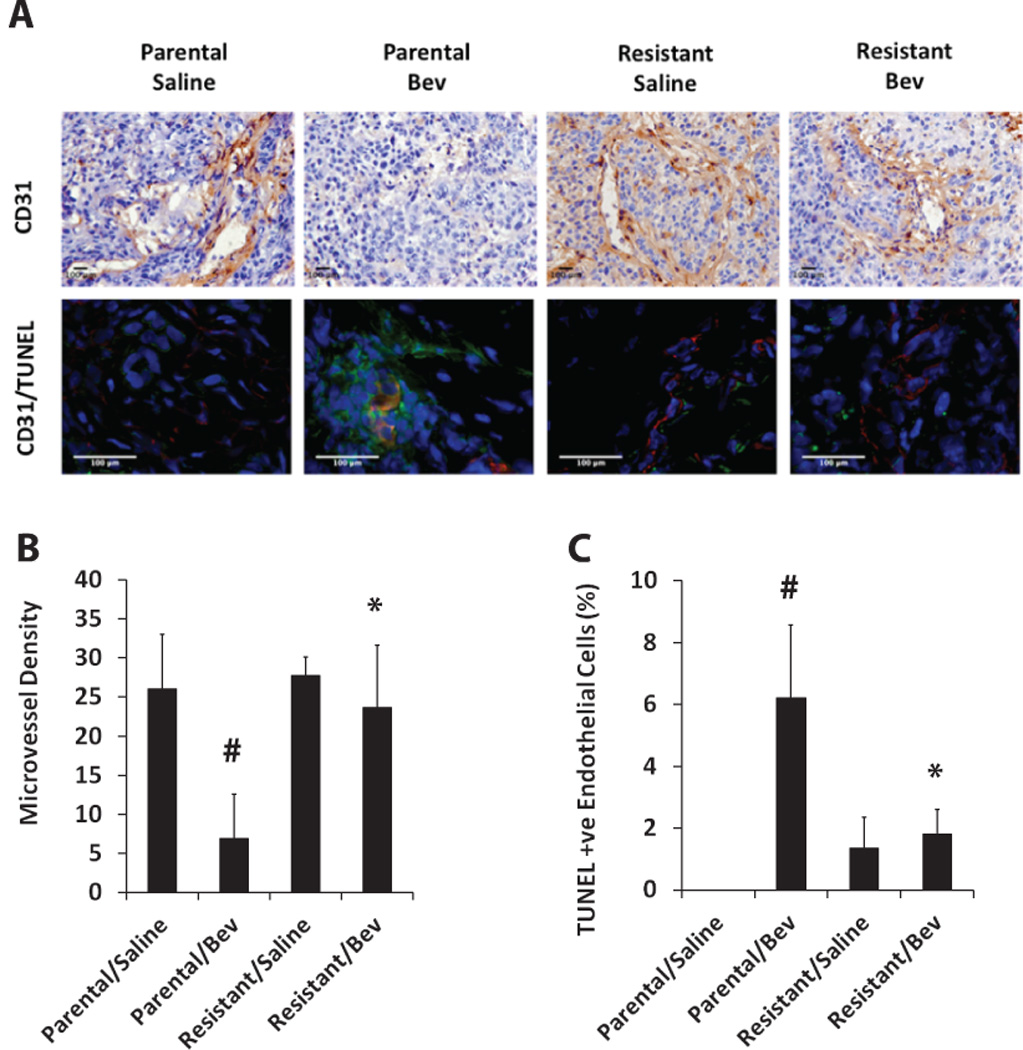
(A) CD31 staining (brown) in parental and resistant tumor sections using immunohistochemistry (upper panel). Immunofluorescence double staining of CD31 (red) and TUNEL (green) was performed to assess endothelial cell-specific apoptosis (lower panel). (B) Bar graph represents quantification of microvessel density in parental and resistant tumors treated with saline or bevacizumab. (C) Quantitative analysis of CD31+/TUNEL+ cells represented as percentage of apoptotic endothelial cells. Bevacizumab-resistant tumors showed statistically significantly higher microvessel density along with reduced endothelial cell apoptosis compared to parental tumors treated with bevacizumab (*bevacizumab-treated resistant tumors vsbevacizumab-treated parental tumors, p<0.05; #bevacizumab-treated parental tumors vs saline-treated parental tumors, p<0.05).
Bevacizumab-resistant tumors upregulate angiogenesis genes in response to chronic anti-VEGF therapy
To identify molecular changes initiated by the tumor cells to mediate bevacizumab resistance, we performed whole genome microarray analysis. Bevacizumab-sensitive and -resistant isogenic tumor models were compared for differences in gene expression using HumanHT-12 v4 BeadChips. Efficiency analysis was used to determine the best statistical method and the differentially expressed genes were identified using J5 test.
We found 150 genes upregulated and 31 genes downregulated in the resistant tumors (Figure 3A, Table S1). Using these differentially expressed genes, we carried out a pathway level impact analysis and found several cancer-related pathways to be affected by the observed changes in gene expression (Table 1). Among the angiogenesis-related genes, we found upregulation of FGF2, FGFR3, PLCg2, FZD4, CX3CL1, and CCL5 in the resistant tumors (Table 2). We further analyzed the functional interaction network involving differentially expressed genes using ingenuity pathway analysis tool, and observed FGF2 as a highly connected nodal gene with upregulated proangiogenic genes FGFR3, PLCg2, TNFSF10, CASP1 and BGN (Figure 3B). Interestingly, three of these genes FGF2, FGFR3 and PLCg2 belonged to the FGF signaling pathway, which suggests that this axis might be involved in bevacizumab-associated acquired resistance in our HNSCC xenograft model.
Figure 3. Bevacizumab-resistant tumors upregulate angiogenesis genes in response to chronic anti-VEGF therapy.

(A) Gene expression grid displaying 181 differentially expressed genes between bevacizumab-resistant tumors (n=4) (left column/ A) and bevacizumab-sensitive tumors (n=4) (right column/ B) indicates that higher number genes are upregulated than downregulated in the resistant tumors. Also, there is relatively less heterogeneity in gene expression (black color) among the replicates within each group. (B) Functional interaction network analysis involving differentially expressed genes between bevacizumab-resistant tumors and bevacizumab-sensitive tumors using IPA tool.
Table 1.
Pathway level impact analysis using genes differentially expressed in resistant/bevacizumab tumors compared to parental/bevacizumab tumors.
| S.No. | Pathway Name | Genes Upregulated |
Impact Factor |
P-value |
|---|---|---|---|---|
| 1 | Phosphatidylinositol signaling system | PLCg2, ARHGEF16 | 25.009 | 3.58E-10 |
| 2 | Pathways in cancer | FGF2, FGFR3, PLCG2, FZD4, IGFBP6, IRS1, GRB10, CCNB2, CCND3, CDC42EP4, TP63, CAPN1 | 8.601 | 1.77E-4 |
| 3 | Cytokine-cytokine receptor interaction | CXCR7, CX3CL1, CCL5, IL20RB | 5.551 | 2.5E-2 |
| 4 | Cell cycle | CCNB2, CCND3, CDC42EP4 | 5.163 | 3.53E-2 |
| 5 | Apoptosis | IRF1, TNFSF10, PARP9, CRABP2, CASP1 | 4.866 | 4.52E-2 |
Table 2.
Angiogenesis-related genes upregulated in resistant/bevacizumab tumors compared to parental/bevacizumab tumors.
| S.No. | Entrez Gene Name | J5-score | Accession # |
|---|---|---|---|
| 1 | Insulin-like growth factor binding protein 6 (IGFBP6) | 27.040 | NM_002178.2 |
| 2 | Frizzled homolog 4 (Droshophila) (FZD4) | 18.239 | NM_012193.2 |
| 3 | Chemokine (C-X3-C motif) ligand 1 (CX3CL1) | 13.329 | NM_002996.3 |
| 4 | Chemokine (C-X-C motif) receptor 7 (CXCR7) | 13.142 | NM_020311.1 |
| 5 | Phospholipase C, gamma 2 (phosphatidylinositol-specific) (PLCG2) | 12.871 | NM_002661.1 |
| 6 | Biglycan (BGN) | 12.165 | NM_001711.3 |
| 7 | Growth factor receptor-bound protein 10(GRB10) | 11.518 | NM_005311.3 |
| 8 | Chemokine (C-C motif) ligand 5(CCL5) | 10.718 | NM_002985.2 |
| 9 | Fibroblast growth factor 2 (basic) (FGF2) | 9.741 | NM_002006.3 |
| 10 | Caspase 1, apoptosis-related cysteine peptidase (CASP1) | 9.477 | NM_033292.2 |
| 11 | Insulin receptor substrate 1 (IRS1) | 8.817 | NM_005544.1 |
| 12 | Fibroblast growth factor receptor 3(FGFR3) | 8.424 | NM_000142.2 |
Upregulation of FGF signaling in resistant xenografts
To test our hypothesis that FGF signaling contributes to bevacizumab resistance, we first validated the upregulation of FGF pathway genes in the resistant tumors by real-time RT-PCR and western blotting. We confirmed a 6-fold increase in the expression of FGF2 mRNA in bevacizumab-treated resistant tumors compared to bevacizumab-treated parental tumors (Figure 4A). Similarly, we observed a 4.5-fold increase in FGFR3 mRNA and a 8-fold increase in PLCg2 mRNA (Figure 4B–C). Human FGF2 protein levels were significantly higher in plasma from mice bearing resistant/ bevacizumab tumors compared to mice bearing parental/ bevacizumab tumors as shown by ELISA (Figure 4D). We also confirmed increased protein levels of these genes in tumor cells expanded from the bevacizumab-treated resistant xenografts compared to the parental Tu138 cell line (Figure 4E). Since, we observed an increased expression of members of the FGF pathway including FGF2, FGFR3 and downstream protein PLCg2 in the resistant cells, we also assessed the expression of other members of the FGF pathway for increased signaling (Figure 4F). We observed increased expression of FGF2 and not FGF1 in the resistant cells. Among the four FGFR receptors, FGFR1, 2 and 3 were upregulated and FGFR4 was downregulated in the resistant cells. Among the downstream proteins, higher levels of phospho PLCg1, total PLCg1, phospho PLCg2, total PLCg2, phospho AKT and phospho ERK were seen in the resistant cells.
Figure 4. Upregulation of FGF signaling in resistant xenografts.
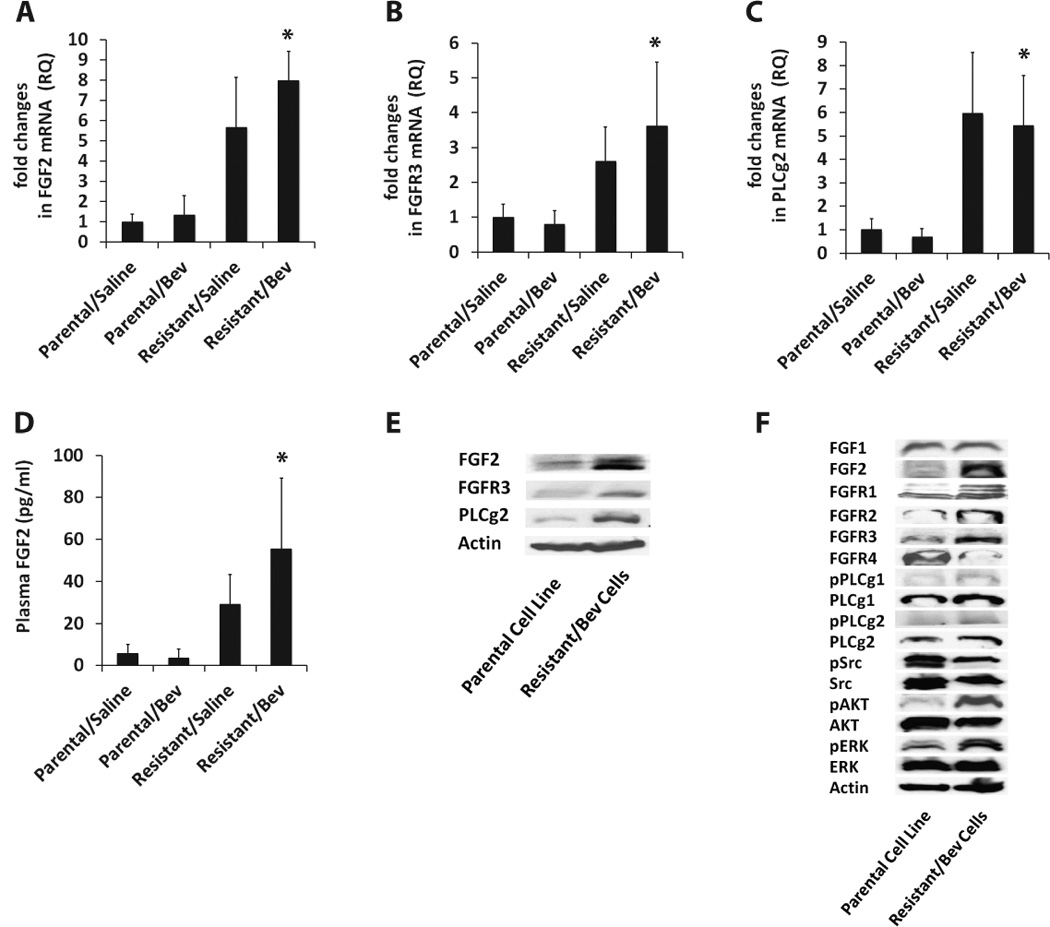
(A–C) qRT-PCR analysis of FGF2, FGFR3 and PLCg2 mRNA levels reveal 6-fold, 4.5-fold and 8-fold increase in expression in resistant/bevacizumab tumors compared to parental/bevacizumab tumors respectively (* FGF2; P=0.0002, FGFR3; P=0.0439, PLCg2; P=0.0160). (D) Plasma FGF2 protein levels were significantly higher in resistant/bevacizumab tumors compared to parental/bevacizumab tumors (* P=0.0441) as shown by ELISA. (E) Western blot analysis confirms increased protein levels of FGF2, FGFR3 and PLCg2 in tumor cells expanded from bevacizumab-treated resistant xenografts compared to the parental Tu138 cell line. (F) Increased expression of FGF2 ligand, FGFR1–3 receptors and downstream proteins, phospho PLCg1, total PLCg1, phospho PLCg2, total PLCg2, phospho AKT and phospho ERK in resistant cells.
Increased ERK activation upregulates FGF2 expression in resistant cells
Studies have shown that activated ERK can positively regulate FGF levels (25). We next examined if increased expression of FGF2 was due to increased ERK activation in the bevacizumab-resistant cells. Parental and resistant cells were treated with the MEK inhibitor U0126 to block ERK activation and FGF2 expression was then examined by western blotting and ELISA (Figure 5A–B). We observed a complete abrogation of ERK activation in the parental and resistant cells within 6hrs of U0126 treatment. Furthermore, the inhibition of ERK resulted in a significant decrease in FGF2 expression as measured by ELISA. These results suggest that increased activation of ERK in the resistant cells regulates increased expression of FGF2.
Figure 5. Increased ERK activation upregulates FGF2 expression in resistant cells.

(A–B) Parental and resistant cells were treated with the MEK inhibitor U0126 to block ERK activation and FGF2 expression was examined by western blotting (A) and ELISA (B). Complete abrogation of ERK activation was observed within 6hrs of inhibitor treatment and a significant decrease in FGF2 expression (* P=0.0003).
Upregulated angiogenesis genes induce increased expression of FGF2 in resistant cells by activating ERK
Based on the above finding, we investigated other upregulated genes in the microarray that were known to activate ERK. Using the IPA tool, we generated a functional gene-interaction network, and found high J5-score-bearing differentially expressed genes PLCg2, FZD4, CX3CL1, CCL5, and FGFR3 as known activators of ERK (Figure 6A). We hypothesized that in the resistant cells these upregulated genes lead to increased activation of ERK and subsequent increase in FGF2 expression. To test this, we first validated overexpression of these upstream genes in the resistant cells (Figure 6B–C). We then used siRNA approach to downregulate PLCg2, FZD4, CX3CL1, and CCL5 in resistant cells and examined the effect on pERK and FGF2 expression (Figure 6D–G). We observed that downregulation of these genes resulted in a significant decrease in ERK activation and a corresponding decrease in FGF2 expression.
Figure 6. Upregulated angiogenesis genes induce increased expression of FGF2 in resistant cells by activating ERK.
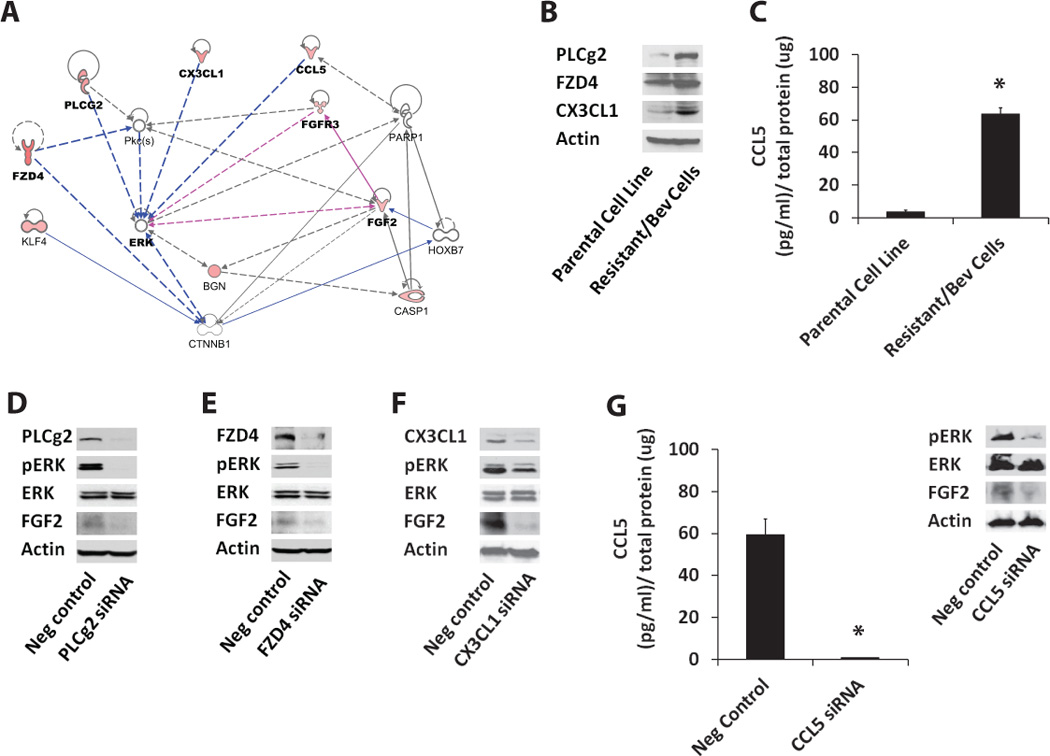
(A) Using IPA tool, we identified potential activators of ERK based on at least one literature reference. These included angiogenesis-related genes with high J5-scores such as FDZ4, PLC-g2, CX3CL1, CCL5, and FGFR3, which were found to be upregulated in the resistant tumors using microarray analysis. (B) Western blot analysis shows increased protein levels of PLCg2, FZD4 and CX3CL1 in tumor cells expanded from bevacizumab-treated resistant xenografts compared to the parental Tu138 cell line. (C) Increased expression of CCL5 observed in resistant cells using ELISA (* P<0.0001). (D–G) siRNA targeting of PLCg2 (D), FZD4 (E), CX3CL1 (F), and CCL5 (G) (* P=0.0002) in resistant cells and its effect on pERK and FGF2 expression.
Co-targeting VEGF and FGFR sensitize HNSCC tumors to bevacizumab
To test the contribution of FGF signaling in bevacizumab-associated acquired resistance, we inhibited FGFRs in the resistant xenografts using PD173074 small molecule inhibitor in combination with bevacizumab (Figure 7A). Treatment with FGFR inhibitor alone resulted in a modest but statistically significant decrease in tumor growth. However, cotargeting VEGF and FGFR completely abrogated tumor growth in these resistant xenografts. Further, CD31 staining in the tumor sections revealed a significant decrease in MVD in the combination group compared to bevacizumab or PD173074 alone (Figure 7B–C). These data suggest that co-targeting VEGF and FGFR sensitize resistant tumors to bevacizumab by disrupting angiogenesis.
Figure 7. Co-targeting VEGF and FGFR sensitize HNSCC tumors to bevacizumab.

(A) Resistant xenografts (n=12) were randomized into four treatment groups receiving saline, bevacizumab, PD173074 or a combination of both. Bevacizumab and PD173074 were administered intraperitoneally at 8mg/kg and 25mg/kg respectively. Treatment with PD173074 alone resulted in a modest but statistically significant decrease in tumor growth (# P=0.0369). Combined knockdown of VEGF and FGFR completely abrogated tumor growth (* P=0.0427, combination vsbevacizumab alone; P=0.0451, combination vs PD173074 alone). (B–C) CD31 staining in resistant xenografts showed significantly reduced microvessel density in the combination group compared to bevacizumab or PD173074 alone (* P=0.012, combination vsbevacizumab alone; P=0.0364, combination vs PD173074 alone).
Discussion
HNSCC is the eighth leading cancer by incidence worldwide. Although there have been significant advances in surgery and chemoradiotherapy for HNSCC in the past 50 years, the overall survival has remained unchanged. Hence, there is a pressing need to develop new therapeutic strategies in HNSCC. Preclinical studies have shown antiangiogenic therapy to be a promising therapeutic strategy. However, clinical use of antiangiogenic therapy has been hampered by the phenomenon of resistance. Furthermore, there are no reliable predictive biomarkers to identify those patients who are likely to benefit or show resistance to this therapeutic approach. Studies using relevant preclinical models that identify mechanisms of resistance to antiangiogenic agents will help meet these challenges.
There is growing evidence that suggests that the FGF/FGFR axis influences the sensitivity of tumors to antiangiogenic therapy (13, 18, 19, 26). Recent studies have shown that dual inhibition of VEGFR and FGFR in preclinical models can overcome anti-VEGF therapy resistance (27, 28). However, there are no reports that describe the underlying mechanisms that drive the overexpression of the FGF pathway in response to treatment with antiangiogenic agents. Knowledge of such regulatory mechanisms is necessary to discover functional networks that can integrate additional upstream genes as potential contributors of resistance.
In the present study, we established a novel HNSCC xenograft model of acquired resistance to bevacizumab and identified upregulation of FGF signaling in resistant tumors. Increased expression of FGF2 was regulated by upstream genes including PLCg2, FZD4, CX3CL1, and CCL5 via increased ERK signaling. We also showed that modulation of FGF signaling in the resistant xenografts regulated the sensitivity to bevacizumab.
FGFs are known to play an important role in a variety of cellular processes including differentiation, cell proliferation, apoptosis, angiogenesis and inflammation (29–31). These FGF signaling-mediated functions can greatly contribute to the process of tumorigenesis. Accumulating evidence highlights the deregulation of FGF/FGFRs in cancer through different mechanisms, including aberrant expression, mutations, and gene amplifications (32). There are 4 known FGFRs, FGFR1 through FGFR4. These receptors differ in the distribution patterns of specific isoforms on tumor cells and stromal cells, collectively mediating autocrine and paracrine signaling in tumors. Studies have shown that FGF2/FGFRs autocrine signaling contributes to EGFR-TKI resistance in NSCLC lines (33, 34).
In our HNSCC xenograft model of acquired resistance to bevacizumab, we have shown upregulation of FGF signaling specifically in the tumors cells. Several members of the FGF pathway were found to be overexpressed including FGF2, FGFR1–3, downstream proteins phospho PLCg1, PLCg1, phospho PCLg2, PLCg2, phospho AKT and phospho ERK. Lastly, we showed that the co-targeting of VEGF and FGF pathways resulted in restoration of sensitivity to anti-VEGF therapy in the resistant tumors. Taken together, our data suggests that the upregulation of FGF/FGFR autocrine signaling may be one of the ways by which bevacizumab-resistant tumors cells circumvents VEGF inhibition.
We next addressed the molecular basis for overexpression of FGF2 in the resistant tumors. We observed that the resistant cells had higher levels of pERK and that the expression of FGF2 was dependent at least in part to ERK activity. Using the IPA tool we then found several high J5-score bearing differentially expressed genes including PLCg2, FZD4, CX3CL1 and CCL5, which are well-known activators of ERK. Downregulation of these upstream genes in resistant cells resulted in a significant decrease in activation of ERK and a corresponding decrease in FGF2 levels. Although FGF has been implicated, as a mediator of resistance to antiangiogenic therapy in a handful of other reports, our study is the first to demonstrate a mechanism by which the overexpression of FGF occurs in resistant tumors (13, 27, 28). This mechanism involved ERK as a central regulator of FGF expression, but there may be other potential contributors such as HOXB7, CTNNB1 and PKC, which merit further study (Figure 6A)(35, 36).
The mechanism by which FGF allows evasive resistance to VEGF inhibition remains to be elucidated further. However, our observation that the resistant tumors were able to maintain tumor vasculature and resist endothelial cell apoptosis despite bevacizumab treatment suggests that the primary effects of FGF may be on tumor endothelium. Although we have not analyzed the levels of FGFRs on the endothelial cells, a recent report by Cascone et al., described upregulation of stromal FGFR in a NSCLC xenograft model of acquired resistance to bevacizumab (18). It is well known that FGF is a potent angiogenic cytokine (37–41). Studies have shown that FGF pathway may interact with the VEGF pathway in regulating tumor angiogenesis. Initial studies using in vitro models showed that addition of both FGF and VEGF resulted in greater tubule formation by endothelial cells compared to either cytokines alone (41). Furthermore, in vivo studies have shown that tumor overexpressing both FGF and VEGF have higher growth rate and MVD compared to tumors that were engineered to overexpress either cytokine alone (42). Lastly, it has been shown that VEGFR2 antagonists can inhibit both VEGF and FGF induced angiogenesis suggesting interplay between the two pathways (43).
Our study puts forward an important concept in the phenomenon of acquired resistance. In response to anti-VEGF therapy, our HNSCC tumors showed an increased expression of a number of angiogenesis-related genes. Despite changes in the expression of many different signaling proteins, the cumulative effect of these changes appears to feed through a common protein or pathway to exert its effect. In our acquired model, the overexpression of a proangiogenic signature comprising of PLGg2, FZD4, CX3CL1, and CCL5 has the common effect of ERK activation, which eventually leads to the overexpression of FGF2.
Based on this observation, we can put forth a hypothesis that although treatment with antiangiogenic agents lead to pleiotrophic changes to the tumor, therapeutic attempts to reverse the resistance may not require the inhibition of all these differentially expressed proteins but rather one or two integral pathways that these changes mainly feeds through. In our model, that common pathway appeared to be ERK and eventually FGF2. These data also provide an added rationale for the use of ERK inhibitors in conjunction with bevacizumab as a means to overcome resistance to anti-VEGF therapy. A multicenter phase I trial of the ERK Inhibitor BAY 86–9766 in patients with advanced cancer indicated some evidence of clinical benefit across a range of tumor types (44), an encouraging trend for future clinical trials.
In our model of acquired resistance, we observed increased expression of FGF2 and not FGF1 in the resistant cells. In our previous study, which focused on the mechanisms of bevacizumab resistance in the intrinsic models we observed overexpression of FGF1 in the resistant cells (17). Also, IL-8 was the primary mechanism of bevacizumab resistance in these intrinsic models. In contrast, IL-8 was downregulated in the acquired resistant tumors, suggesting that the tumors excluded this mechanism to maintain angiogenesis and sustain tumor growth in presence of VEGF blockade (Figure 3B, Table S1). These findings indicate that there might be an inherent difference in the underlying mechanisms of intrinsic and acquired resistance to antiangiogenic therapies. These distinct mechanisms could correspond to different patient subpopulations. Identification of these subsets can help improve pretreatment patient selection for personalized medicine and enhance the therapeutic efficacy of antiangiogenic therapies.
In summary, our work has identified a proangiogenic signature including PLCg2, FZD4, CX3CL1, and CCL5 genes that converge on ERK signaling to upregulate FGF2 expression, which mediates resistance to anti-VEGF therapy. Knowledge of these regulatory networks provides a stronger mechanistic rationale for co-targeting VEGF and FGF in future clinical trials.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Jeffrey N. Myers for kindly providing the Tu138 cell line and Dr. Robert L. Ferris for review of the manuscript. We thank the University of Pittsburgh Genomics and Proteomics Core Laboratories (GPCL) and Debby Hollingshead for generating microarray data. We also thank GPCL-Bioinformatics Analysis Core and Drs. James Lyons-Weiler and Tamanna Sultana for providing assistance with microarray data analysis.
Financial support: This work was supported by the National Institutes of Health [K08 DE019201], the American College of Surgeons/ American Head and Neck Society Physician Scientist Career Development Award, and the UPMC Endowed Assistant Professorship in Otolaryngology to Seungwon Kim.
Footnotes
Conflict of Interest:
The authors declare no conflict of interest.
References
- 1.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A Randomized Trial of Bevacizumab, an Anti-Vascular Endothelial Growth Factor Antibody, for Metastatic Renal Cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-Carboplatin Alone or with Bevacizumab for Non-Small-Cell Lung Cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 3.Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539–1544. doi: 10.1200/JCO.2006.09.6305. [DOI] [PubMed] [Google Scholar]
- 4.Vokes EE, Cohen EEW, Mauer AM, Karrison TG, Wong SJ, Skoog-Sluman LJ, et al. A phase I study of erlotinib and bevacizumab for recurrent or metastatic squamous cell carcinoma of the head and neck (HNC) J Clin Oncol (Meeting Abstracts) 2005;23:5504-. [Google Scholar]
- 5.Karamouzis MV, Friedland D, Johnson R, Rajasenan K, Branstetter B, Argiris A. Phase II trial of pemetrexed (P) and bevacizumab (B) in patients (pts) with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC): An interim analysis. J Clin Oncol (Meeting Abstracts) 2007;25:6049-. [Google Scholar]
- 6.Cohen EEW, Davis DW, Karrison TG, Seiwert TY, Wong SJ, Nattam S, et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: a phase I/II study. The Lancet Oncology. 2009;10:247–257. doi: 10.1016/S1470-2045(09)70002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis LM, Hicklin DJ. Pathways Mediating Resistance to Vascular Endothelial Growth Factor,ÄìTargeted Therapy. Clinical Cancer Research. 2008;14:6371–6375. doi: 10.1158/1078-0432.CCR-07-5287. [DOI] [PubMed] [Google Scholar]
- 9.Crawford Y, Ferrara N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends in Pharmacological Sciences. 2009;30:624–630. doi: 10.1016/j.tips.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Eikesdal HP, Kalluri R. Drug resistance associated with antiangiogenesis therapy. Seminars in Cancer Biology. 2009;19:310–317. doi: 10.1016/j.semcancer.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Soffer SZ, Kim ES, McCrudden KW, Huang J, New T, et al. Vascular Remodeling Marks Tumors That Recur During Chronic Suppression of Angiogenesis. Molecular Cancer Research. 2004;2:36–42. [PubMed] [Google Scholar]
- 13.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotech. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 15.Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70:10090–10100. doi: 10.1158/0008-5472.CAN-10-0489. Epub 2010 Oct 15. [DOI] [PubMed] [Google Scholar]
- 16.Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–1071. doi: 10.1158/0008-5472.CAN-09-3965. Epub 2010 Jan 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gyanchandani R, Sano D, Ortega Alves MV, Klein JD, Knapick BA, Oh S, et al. Interleukin-8 as a modulator of response to bevacizumab in preclinical models of head and neck squamous cell carcinoma. Oral Oncol. 2013;49:761–770. doi: 10.1016/j.oraloncology.2013.03.452. [DOI] [PubMed] [Google Scholar]
- 18.Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest. 2011;121:1313–1328. doi: 10.1172/JCI42405. Epub 2011 Mar 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carbone C, Moccia T, Zhu C, Paradiso G, Budillon A, Chiao PJ, et al. Anti-VEGF Treatment,ÄìResistant Pancreatic Cancers Secrete Proinflammatory Factors That Contribute to Malignant Progression by Inducing an EMT Cell Phenotype. Clinical Cancer Research. 2011;17:5822–5832. doi: 10.1158/1078-0432.CCR-11-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and Initial Characterization of a Panel of 85 Genomically Validated Cell Lines from Diverse Head and Neck Tumor Sites. Clinical Cancer Research. 2011;17:7248–7264. doi: 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim S, Yazici YD, Calzada G, Wang ZY, Younes MN, Jasser SA, et al. Sorafenib inhibits the angiogenesis and growth of orthotopic anaplastic thyroid carcinoma xenografts in nude mice. Mol Cancer Ther. 2007;6:1785–1792. doi: 10.1158/1535-7163.MCT-06-0595. [DOI] [PubMed] [Google Scholar]
- 22.Jordan R, Patel S, Hu H, Lyons-Weiler J. Efficiency analysis of competing tests for finding differentially expressed genes in lung adenocarcinoma. Cancer Inform. 2008;6:389–421. doi: 10.4137/cin.s791. Epub 2008 Jul 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel S, Lyons-Weiler J. caGEDA: a web application for the integrated analysis of global gene expression patterns in cancer. Appl Bioinformatics. 2004;3:49–62. doi: 10.2165/00822942-200403010-00007. [DOI] [PubMed] [Google Scholar]
- 24.Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. Epub 2007 Sep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sales KJ, Boddy SC, Williams AR, Anderson RA, Jabbour HN. F-prostanoid receptor regulation of fibroblast growth factor 2 signaling in endometrial adenocarcinoma cells. Endocrinology. 2007;148:3635–3644. doi: 10.1210/en.2006-1517. Epub 2007 May 3. [DOI] [PubMed] [Google Scholar]
- 26.Fernando NT, Koch M, Rothrock C, Gollogly LK, D'Amore PA, Ryeom S, et al. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin Cancer Res. 2008;14:1529–1539. doi: 10.1158/1078-0432.CCR-07-4126. [DOI] [PubMed] [Google Scholar]
- 27.Bello E, Colella G, Scarlato V, Oliva P, Berndt A, Valbusa G, et al. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 2011;71:1396–1405. doi: 10.1158/0008-5472.CAN-10-2700. Epub 011 Jan 6. [DOI] [PubMed] [Google Scholar]
- 28.Allen E, Walters IB, Hanahan D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin Cancer Res. 2011;17:5299–5310. doi: 10.1158/1078-0432.CCR-10-2847. Epub 011 May 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 30.Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8:1439–1452. doi: 10.1158/1541-7786.MCR-10-0168. Epub 2010 Oct 13. [DOI] [PubMed] [Google Scholar]
- 31.Andres G, Leali D, Mitola S, Coltrini D, Camozzi M, Corsini M, et al. A pro-inflammatory signature mediates FGF2-induced angiogenesis. J Cell Mol Med. 2009;13:2083–2108. doi: 10.1111/j.1582-4934.2008.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.d'Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 1998;9:71–78. [PubMed] [Google Scholar]
- 33.Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, et al. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS One. 2010;5:e14117. doi: 10.1371/journal.pone.0014117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. Epub 2008 Oct 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Care A, Silvani A, Meccia E, Mattia G, Stoppacciaro A, Parmiani G, et al. HOXB7 constitutively activates basic fibroblast growth factor in melanomas. Mol Cell Biol. 1996;16:4842–4851. doi: 10.1128/mcb.16.9.4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moffett J, Kratz E, Myers J, Stachowiak EK, Florkiewicz RZ, Stachowiak MK. Transcriptional regulation of fibroblast growth factor-2 expression in human astrocytes: implications for cell plasticity. Mol Biol Cell. 1998;9:2269–2285. doi: 10.1091/mbc.9.8.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broadley KN, Aquino AM, Woodward SC, Buckley-Sturrock A, Sato Y, Rifkin DB, et al. Monospecific antibodies implicate basic fibroblast growth factor in normal wound repair. Lab Invest. 1989;61:571–575. [PubMed] [Google Scholar]
- 38.Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000;60:7163–7169. [PubMed] [Google Scholar]
- 39.Gualandris A, Urbinati C, Rusnati M, Ziche M, Presta M. Interaction of high-molecular-weight basic fibroblast growth factor with endothelium: biological activity and intracellular fate of human recombinant M(r) 24,000 bFGF. J Cell Physiol. 1994;161:149–159. doi: 10.1002/jcp.1041610118. [DOI] [PubMed] [Google Scholar]
- 40.Javerzat S, Auguste P, Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med. 2002;8:483–489. doi: 10.1016/s1471-4914(02)02394-8. [DOI] [PubMed] [Google Scholar]
- 41.Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189:824–831. doi: 10.1016/0006-291x(92)92277-5. [DOI] [PubMed] [Google Scholar]
- 42.Giavazzi R, Sennino B, Coltrini D, Garofalo A, Dossi R, Ronca R, et al. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162:1913–1926. doi: 10.1016/S0002-9440(10)64325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tille JC, Wood J, Mandriota SJ, Schnell C, Ferrari S, Mestan J, et al. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit VEGF- and basic fibroblast growth factor-induced angiogenesis in vivo and in vitro. J Pharmacol Exp Ther. 2001;299:1073–1085. [PubMed] [Google Scholar]
- 44.Weekes CD, Von Hoff DD, Adjei AA, Leffingwell DP, Eckhardt SG, Gore L, et al. Multicenter Phase I Trial of the Mitogen-Activated Protein Kinase 1/2 Inhibitor BAY 86–9766 in Patients with Advanced Cancer. Clin Cancer Res. 2013;19:1232–1243. doi: 10.1158/1078-0432.CCR-12-3529. Epub 2013 Feb 22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.