

Abstract
The homeostasis of insulin-like growth factor-1 (IGF-1) is essential for metabolism, development and survival. Insufficient IGF-1 is associated with poor recovery from wounds whereas excessive IGF-1 contributes to growth of tumours. We have shown that cyclic glycine-proline (cGP), a metabolite of IGF-1, can normalise IGF-1 function by showing its efficacy in improving the recovery from ischemic brain injury in rats and inhibiting the growth of lymphomic tumours in mice. Further investigation in cell culture suggested that cGP promoted the activity of IGF-1 when it was insufficient, but inhibited the activity of IGF-1 when it was excessive. Mathematical modelling revealed that the efficacy of cGP was a modulated IGF-1 effect via changing the binding of IGF-1 to its binding proteins, which dynamically regulates the balance between bioavailable and non-bioavailable IGF-1. Our data reveal a novel mechanism of auto-regulation of IGF-1, which has physiological and pathophysiological consequences and potential pharmacological utility.
Insulin-like growth factor 1 (IGF-1) plays an essential role in development1, metabolism and survival2. Its function is mediated through activating IGF-1 receptors for initiating downstream signalling pathways3,4. The homeostasis of IGF-1 function is dynamically regulated through reversible binding to circulating and tissue associated IGF-1 binding proteins (IGFBPs)5. Both insufficient and excessive amounts of IGF-1 are related to medical conditions, such as poor recovery from injuries2 and tumorigenesis6, respectively.
The unbound IGF-1 can be enzymatically metabolized to des-(1-3) IGF-1 (des-IGF-1) and glycine-proline-glutamate (GPE), which is cleaved from its N-terminal7,8. GPE is enzymatically unstable9,10 and is rapidly metabolized into single amino acids and dipeptides including cyclic glycine-proline (cGP)9. Similar to IGF-111, both GPE and cGP are neuroprotective in the treatment of hypoxic-ischemic (HI) brain injury in rats10,12,13. While the neuroprotective effect of IGF-1 is clearly mediated by IGF-1 receptors11, GPE does not interact with IGF-1 receptors12,13,14,15. The mode of action of GPE and cGP still remains unknown. The neuroprotective effects of GPE and cGP after intracerebroventricular (icv) administration are dose-dependent, suggesting receptor-mediated pharmacodynamics12. The neuroprotective effects of IGF-116and GPE17,18 also involve reduced vascular damage and improve vascular remodelling. This prompted us to investigate whether IGF-1-mediated vascular remodelling contributes to cGP associated neuroprotection in a rat model of unilateral ischemic brain injury. The investigation led to the identification of cGP as a novel mechanism regulating the bioavailability of IGF-1.
Results and Discussion
Vascular protection of cGP was associated with IGF-1
Hypoxic ischemic (HI) injury to the brain was induced unilaterally in the hemisphere ipsilateral to the ligated carotid artery17,19. Compared to the contralateral side (Control side), HI injury induced a significant loss of capillaries in the ligated side (Injured side) of the hippocampus (Fig. 1a, p < 0.05). The loss of capillaries in the striatum was mild and not significant (Fig. 1b). A single dose of cGP (0.2 μg/rat) that is known to only partially prevent neuronal damage13, completely restored the density of capillaries in the hippocampus (p < 0.01) and striatum compared to the controls (vehicle, Fig. 1a, b). These vascular effects of cGP may contribute to its neuroprotective properties13 as a preserved vascular network is central to neuroprotection17,20.
Figure 1. cGP prevents vascular loss by promoting IGF-1 associated vascular remodelling.

(a), Vascular density in the hippocampus after either vehicle (open bars, n = 12) or cGP (black bars, n = 13) treatments. (b), Vascular density in the striatum after either vehicle or cGP treatments. (c), Photograph shows the distribution of capillary (red) and IGF-1 receptor (green) (bar = 100 μm) (d), Photograph shows the morphology of capillary (red) and IGF-1 receptor (green) (bar = 20 μm). (e), The number of capillaries with IGF-1 receptor expression in the hippocampus after either vehicle or cGP treatment. (f), The number of capillaries with IGF-1 receptor expression in the striatum after either vehicle or cGP treatment. (g), The number of capillaries with phosphorylated IGF-1 receptor expression in the hippocampus after either vehicle or cGP treatment. (h), The number of capillaries with phosphorylated IGF-1 receptor expression in the striatum after either vehicle or cGP treatment. Error bars show SEM, #p < 0.05, ##p < 0.01 by two-way ANOVA, indicate the difference between the injured and control hemispheres; *p < 0.05, **p < 0.01 by two-way ANOVA, indicate the difference between the vehicle and cGP treatments.
In addition to morphological injuries to the ipsilateral hemisphere, HI injury also induces biological changes in both hemispheres, which can be dominant changes in brain regions where the morphological injury is either mild or even absent10. To determine the potential role for IGF-1 in cGP- associated vascular protection, we evaluated the expression of both inactivated and phosphorylated IGF-1 receptors in the capillaries. Using double labelling, we found morphologically that while the neuronal expression of the phosphorylated receptors was more evenly distributed in most neurons (Fig. 1c), the vascular expression of the activated IGF-1 receptors (green) was strongly expressed in a small population of capillaries (red, Fig. 1d). The expression of IGF-1 receptors was not on the endothelial cells, but was on morphologically identified pericytes (Fig. 1d), which play a key role in vascular remodelling through expressing angiogenic factors and their receptors21. This suggests that those capillaries expressing activated IGF-1 receptors may have a role in vascular remodelling. Thus, the increase in these ‘angiogenic capillaries' may contribute to the recovery of the brain from ischemic injury22.
IGF-1 accumulates in the blood vessels of damaged brain regions within a few hours after HI injury23 and glial cells start to produce IGF-1 locally thereafter, for several days23. However, the mRNA transcripts encoding IGF-1 receptors either remain the same or decrease after brain injury23,24, suggesting that an insufficiency of endogenous IGF-1 facilitates brain recovery25. Enumeration of capillaries that were positive for IGF-1 receptors revealed that HI injury induced a profound loss of angiogenic capillaries in the injured hemispheres (Fig. 1e–h, p < 0.01, open bars). Therefore, IGF-1-mediated vascular remodelling may be compromised. In contrast, the treatment with cGP significantly restored the number of angiogenic capillaries in the injured side of the hippocampus (p < 0.001) and striatum (Fig. 1e–h, p < 0.01). The effects of cGP treatment on phosphorylated IGF-1 receptors were similar to that of IGF-1 receptors in all brain regions examined. The data indicate that cGP promotes IGF-1-induced vascular remodelling when the endogenous IGF-1 is insufficient to prevent neuronal and vascular damage. Furthermore, this observation suggested that the protective effects of cGP may be mediated through IGF-1.
The expression of IGF-1 receptors and the phosphorylated IGF-1 receptors were more complex in the contralateral. While cGP moderately increased angiogenic capillaries in the non-damaged (Control) hippocampus (Fig. 1e, g, p < 0.05), we found a reduction of angiogenic capillaries in the striatum (Fig. 1f, h, p < 0.01). The data suggested a potential biphasic effect of cGP on normalising IGF-1 activity. Nevertheless, these changes in IGF-1 associated angiogenesis did not alter the vascular density in the brain regions with no vessel damage, suggesting that cGP associated vascular remodelling was injury-specific.
cGP can either inhibit or stimulate IGF-1 induced angiogenesis
We speculated that cGP had effects on the homeostatic mechanisms of IGF-1-induced vascular remodelling. Using a Human Microvascular Endothelial Cell line (HMEC-1)26, we first evaluated the temporal effects of cGP on IGF-1-induced endothelial cell growth in response to either serum (0.2%) or serum withdrawal. With the presence of serum, IGF-1 (100 nM) continuously stimulated endothelial cell growth during 6 days of culture compared to the phosphate buffered saline (PBS) control (Fig. 2a, p < 0.01). In contrast, the treatment with cGP alone (100 nM) did not promote endothelial cell growth (Fig. 2a). However, the combined treatment of IGF-1 and cGP (both at 100 nM) produced a similar growth rate to that of IGF-1 alone during the first 4 days of culture, then showed a significant inhibition at day 6 when compared to the treatment with IGF-1 alone (Fig. 2a, p < 0.01). We interpret these data to indicate that cGP only inhibits IGF-1 function when its bioactivity is sustained above a threshold. To confirm this finding, we evaluated the formation of tubes in endothelial cells using growth factor reduced Matrigel26. Similarly, the combined treatment of cGP and IGF-1 significantly inhibited the total number of tubes formed after the treatment with IGF-1 alone (Fig. 2b, p < 0.01). The inhibitory effects also required 6 days of the same treatment to elevate IGF-1 induced angiogenesis above ‘the threshold' (Fig. 2b, p < 0.01). We also performed the experiments after withdrawal of serum. Compared with PBS controls, IGF-1 did not stimulate endothelial cell growth in the absence of serum (Fig. 1c). In contrast, the combined treatment of IGF-1 and cGP promoted the proliferation of endothelial cells when the treatment with IGF-1 alone was ineffective (Fig. 2c, p < 0.001). Together, these data support our finding that cGP have a biphasic interaction with IGF-1 depending on whether IGF-1 activities are elevated or insufficient.
Figure 2. cGP regulates IGF-1 associated angiogenesis in stimulatory, inhibitory and static manners.

(a), Effects of IGF-1 (n = 3), cGP (n = 3), combined treatment of cGP + IGF-1 (n = 3) and PBS control, (n = 3) on endothelial cell growth in serum. Combination treatment of cGP + IGF-1 significantly inhibited IGF-1-induced cell growth only when it was elevated at day 6. Treatment with cGP did not alter the growth compared to control group. ##/**p < 0.01 by two-way ANOVA. The experiments were repeated 8 times. (b), The effects of IGF-1 (n = 15), combined treatment of cGP + IGF-1 (n = 15) and PBS (n = 10) on tube formation. Similarly, the combination treatment (cGP + IGF-1) reduced IGF-1-induced tube formation. ##/**p < 0.01 by one way ANOVA. The experiments were repeated 3 times. (c), The effects of IGF-1 (n = 3), combined treatment of cGP + IGF-1 (n = 3) and PBS control (n = 3) on endothelial cell growth after serum withdrawal. The combination treatment increased total cell counts when IGF-1 failed to be effective. **p < 0.01, ***p < 0.001 by two-way ANOVA. The experiments were repeated 4 times. (d, e, f), The effects of IGF-1 alone (black bar, n = 6), combined treatments of cGP + IGF-1 (grey bars, n = 6) and PBS (while bars, n = 6) on cell viability. Compared to IGF-1 alone treatment the combined treatment of cGP + IGF-1 either inhibited (d), or remained (e) or stimulated (f) IGF-1 function. One-way ANOVA with repeated measures were used for analysing the data. Data are presented as mean ± SEM, where #p < 0.05, ##p < 0.01 indicate the difference between the Controls and IGF-1; *p < 0.05, **p < 0.01, indicate the differences between IGF-1 alone and the combination treatments. These experiments were repeated 3 times.
We then further evaluated these biphasic effects of cGP using a different experimental paradigm, in which we tested the effects of different concentration ratios of cGP:IGF on IGF-1-induceed cell survival at a single time point of 24 hours after the treatments. IGF-1, at a higher concentration (6.5 nM) promoted cell viability compared to the PBS control (Fig. 2d, p < 0.001). The combined treatment with cGP (1–20 nM) and IGF-1 (6.5 nM) significantly reduced cell viability compared to IGF-1 alone (Fig. 2d, p < 0.01). In contrast, the same dose range of cGP failed to alter IGF-1 function when the concentration of IGF-1 was reduced fivefold (1.3 nM, Fig. 2e). Thus, the inhibitory effects of cGP were negated when IGF-1 was low, possibly due to the ratio of cGP:IGF-1 being close to equimolar. However, when the concentration of cGP was increased to 50–100 nM in the presence of 1.3 nM IGF-1 we found a moderate stimulation of cell viability (Fig. 2e, p < 0.05), which was clearly due to the increase in the concentration ratio of cGP:IGF-1. To further explore this stimulatory effect of cGP, we then treated cells with cGP alone, such that cGP dominated endogenous IGF-1 in the culture system. We observed a dose-dependent stimulatory effect of cGP (5–20 nM) compared to PBS treated controls (Fig. 2f, p < 0.01). These data from multiple experimental paradigms and various assays demonstrated that the efficacy of cGP on endothelial cells was the result of interacting with IGF-1, which normalises homeostasis by inhibiting or stimulating or maintaining IGF-1 activity, depending on whether the IGF-1 induced angiogenesis was excessive, insufficient or within a moderate, perhaps physiological range, respectively.
The role of IGF-1 receptor and IGFBP in cGP efficacy
To investigate the role of IGF-1 receptors and IGFBPs on the interaction between cGP and IGF-1, we evaluated the effects of cGP on IGF-1 induced angiogenesis using endothelial cells transfected with an IGF receptor-α expression plasmid or siRNA knock-down plasmids27,28,29, and an antibody to inhibit IGFBP-3. To minimise the interactive effects of cGP on endogenous IGF-1, we used serum free conditions.
IGF-1 (1 nM) failed to promote the survival of endothelial cells after mock transfection of the control siRNA (psiU6-CK) compared to the PBS controls (Fig. 3a). The loss of the effectiveness of IGF-1 might be related to down-regulation of IGF-1 receptors after the control mock transfection (data not show). As expected, the combined treatment of IGF-1 (1 nM) and cGP (10 nM) promoted cell survival while IGF-1 alone was ineffective (Fig. 3a, p < 0.001). However, knockdown of the IGF-1 receptors by transfection with the psiU6-IGF1R siRNA plasmid completely negated the stimulatory effect of combined IGF-1 and cGP on cell viability (Fig. 3a). Overexpression of IGF-1 receptor-α by gene transfection promoted IGF-1-induced cell viability compared to the PBS treatment (Fig. 3b, p < 0.001). While the combined treatment of cGP and IGF-1 did not affect cell viability in the si-Vec controls (Fig. 3b, pIRESneo3-/Vec), the overexpression of IGF-1 receptors to elevate IGF-1 function, the combined treatment of cGP and IGF-1 inhibited the viability of cells (Fig. 3b, p < 0.01). Using western blotting we also found that the treatment with IGF-1 alone increased the expression of activated IGF-1 receptors in the endothelial cells, which was inhibited after a combined treatment of cGP and IGF-1 (Supplementary Fig. 3b). We do not have direct evidence whether or not cGP interacts directly with the IGF-1 receptor. However, we did observe that the effective dose of cGP when given as combined treatment failed to further stimulate cell growth when given alone compared to the control treatment (Fig. 2a & f and supplementary Fig. 2 & 3). These data suggest that the presence of IGF-1 is essential for cGP to be effective and that cGP is unlikely to activate the IGF-1 receptor directly. Furthermore, use of a competitive binding assay shows that GPE does not displace IGF-1 or des-IGF-1 binding to rat brain IGF-1 receptors7. A separate study also demonstrated the inhibitory effect of cGP on IGF-1-induced viability of C2C12 myoblasts (Supplementary Fig. 2b). These data provide further evidence that cGP modulates the actions of IGF-1.
Figure 3. The role for IGF-1 receptor and IGFBP-3 in cGP-associated efficacy.

(a, b), The effects of IGF-1 alone (black bar, n = 3) and combination treatment (grey bars, n = 3) on cell viability were compared after knock down of IGF-1 receptors via siRNA (a, n = 3) or overexpression (b, n = 3). (c), Compared the treatments without IGFBP-3 antibody (without IGFBP3AB, n = 3–6), treatment with IGFBP-3 antibody (with IGFBP3AB, n = 3–6) also shifted the effects of cGP + IGF-1 (grey bars) on cell viability from no change and stimulatory to inhibitory (c) when compared to IGF-1 alone treatment (black bars). These experimenters were repeated four times. (d), The percentage of unbound IGF-1 with (black bars, n = 6) and without the presence of cGP (white bars, n = 6). Data were analysed using two-way ANOVA and presented as Mean ± SEM, where #p < 0.05, ##p < 0.01 indicate the difference between the controls and IGF-1 alone; *p < 0.05, **p < 0.01, indicate the differences between the treatments with and without cGP.
IGFBPs have higher affinity for IGF-1 than do IGF-1 receptors, and the conversion of IGF-1 to des-IGF-1 is associated with a low binding protein affinity5. The major carrier protein for IGF-1 in the plasma is IGFBP-330,31. The binding of IGF-1 to IGFBPs prolongs the half-life of circulating IGF-132 and modulates the availability and activity of free IGF-1 to maintain homeostasis6,30,31. We compared the effects of cGP on IGF-1 induced cell viability with and without the presence of an antibody to IGFBP-3 (218 nM, IGFBPAB). Blockade of IGFBP-3, to increase free IGF-1, changed the effects of cGP on IGF-1 (1 nM) mediated cell viability from both homeostatic (no change, 1 nM) and stimulatory (10 nM) to inhibitory (Fig. 3c). Our data suggest that cGP mediates the bioavailability of IGF-1 via an interaction with IGFBP-3.
Given that the binding of IGF-1 to IGFBPs is reversible and that the ratio of cGP:IGF-1 appeared to determine the efficacy of cGP, we tested the possibility for cGP to alter the binding of IGF-1 to IGFBP-3. Using an in vitro peptide-peptide interaction assay and HPLC analysis13, we compared the percentage of unbound IGF-1 between the co-incubation of IGF-1 and IGFBP-3 either with or without the presence of cGP. We found a dose-dependent change in unbound IGF-1 after incubating IGFBP-3 (0.1 molar-ratio) with IGF-1 (0.01 to 10 molar-ratio, Fig. 3d). The co-incubation in the presence of cGP (1 molar-ratio) increased the amount of free IGF-1 at a high cGP:IGF-1 molar ratio (1:0.01–0.1), but reduced the amount of free IGF-1 when the cGP:IGF-1 molar ratio was low (1:5–10), and did not alter the amount of free IGF-1 when the ratio was equivalent (Fig. 3d). These data may not explain entirely the complex biological interaction of IGF-1 and cGP to IGFBPs. Nevertheless, the ratio of cGP:IGF-1 influences IGF-1 function possibly through interfering with the binding of IGF-1 to IGFBP-3.
As an N-terminal tripeptide of IGF-1, GPE is a major binding site for IGFBPs15 as des-IGF-1 has low affinity for IGFBPs5. IGFBPs can inhibit the formation of GPE by preventing free IGF-1 from being metabolised32. Given that GPE is enzymatically unstable33, does not exist in tissues15 and shares the biological characteristics with cGP13 (Supplementary Fig. 1), as well as being transformed to cGP, there may be a possibility for cGP, a biologically stable isoform, to interact with IGFBPs and, thereby create a complex auto-regulatory loop.
It is possible that the deficiency of IGF-1 may be associated with a higher cGP/IGF-1 ratio and administration of cGP may normalise IGF-1 function through increasing the availability of free IGF-1. In contrast, excessive IGF-1 function may be associated with a lower cGP:IGF-1 ratio and the administration of cGP may restore the equilibrium of IGF-1 and cGP. Our interpretation of our data is that the release of the enzymatic product of free IGF-1, cGP maintains and/or restores equilibrium between free and bound IGF-1 by interfering with the binding of IGF-1 to IGFBPs. Therefore, cGP may play a role in the auto-regulation of IGF-1 action. The mechanism of cGP on IGF-1 binding to IGFBPs is entirely unknown, which needs to be determined in future.
Mathematical modelling analysis
We extended a previous model of IGF-1 dynamics34 to include the competitive interaction between IGF-1 and cGP for binding proteins as observed experimentally. A schematic diagram of the interactions described in the mathematical model is shown in Fig. 4. The model fitted well with the data (R2 = 0.83), and suggested that the effects of cGP on IGF-1 function, via the competitive binding characterized by the model equations, can describe a significant portion of the complex interaction between cGP and IGF-1 on cell viability.
Figure 4. Schematic diagram of the eight concentration compartments in the mathematical model.

Each box represents a compartment, and the arrows represent transport between compartments (Supplementary information). The red compartments/arrows represent cGP (q6) being introduced as a competitor to free IGF-1 (q2) for the free binding proteins (q1).
We also validated our mathematical model using an independent set of data that measured the effects of IGFBP-3 antibody on the interaction between cGP and IGF-1 on cell growth (R2 = 0.77) (Supplementary Fig. 5a–d).
cGP inhibits tumour growth
We evaluated whether cGP inhibited IGF-1-dependent tumour growth, to confirm the ability of cGP to normalize the pathological consequences of elevated IGF-1 function. The mouse EL-4 lymphoma naturally secretes IGF-1 and has high affinity for IGF-1 receptors35. We treated mice with cGP (25 mg/kg/day) either at the time of tumour cell inoculation to determine its preventive effect on tumour formation, or after the tumour was established to determine whether it would cause the regression of the tumour. In the prevention experiment, all saline treated mice developed tumours at day 10, whereas only half of the mice treated with cGP developed palpable tumours 16 days after inoculation (Fig. 5c). The tumours of the control mice were significantly larger than those of cGP treated mice (1.05 vs. 0.2 cm in diameter (p < 0.01), Fig. 5a, b).
Figure 5. cGP treatment delayed the onset of and inhibited tumour growth.

(a), A photograph shows a large tumour from a mouse in saline-treated group. (b), A photograph shows a mouse from the cGP treated group, which had a smaller tumour. (c), Compared treatment effects between the saline (open squares, n = 6) and cGP (closed squares, n = 6) when given at the time of tumour inoculation. (d), Compared the treatment effects between the saline (open squares, n = 6) and cGP (closed squares, n = 6) when given after the onset of tumour inoculation. Data were analysed using two-way ANOVA with repeated measures and presented as mean ± SEM; **p < 0.01, indicate the differences between cGP and saline treatments.
For treating established tumours, we initiated cGP (25 mg/kg/day) treatment when tumours were approximately 0.3 cm in diameter. cGP treatment of established tumours was less effective than the prevention of tumour formation. Nevertheless, the size of tumours was 20% smaller at day 22, compared to the saline-treated control mice (1.33 vs 1.67 cm, p < 0.01) (Fig. 5d).
Excessive IGF-1 activity promotes neoplasia6. Given that IGF-1 protects EL-4 cells from apoptosis36 our results suggest that cGP antagonizes IGF-1-mediated protection of EL-4 cells from apoptosis. The inhibition of cGP on IGF-1 associated tumour growth in vivo suggests that exogenous cGP normalises the autocrine function of IGF-1 under pathological conditions.
Taken together, these diverse observations suggests that cGP can normalise IGF-1 function under pathophysiological states of both functional deficiency, as illustrated by improving brain recovery from injury and inhibiting excessive bioactivity as implied by inhibiting tumour growth. Enhanced IGF-1 function has indeed been suggested for treating neurological conditions36,37,38 and inhibiting IGF-1 activity is a strategy for cancer treatment39. Both conditions might have unavoidable adverse effects by non-specific stimulation or inhibition of IGF-1 function38. Our data suggest that cGP is an additional component in the axis regulating IGF-1 homeostasis, and that cGP has potential therapeutic applications for a long list of medical conditions where IGF-1 homeostasis is impaired.
Methods
All experimenters were blind to the experimental groups.
Human microvascular endothelial cell line
All experimental procedures for the human microvascular endothelial cell line (HMEC-1) assays have been previously reported26.
Total cell count assay for temporal effects
A total cell count assay was used to determine time-depended effects of cGP and/or IGF-1 under conditions of either serum enrichment (0.2% FBS, Fig. 2a & b) or serum withdraw (Fig. 2c). The assay enables an evaluation of temporal changes over a 6 day period at multiple time points. The total number of cells was determined by seeding cells in 6-well plates at a density of 15,000 cells/well and the assays were set up in triplicate. After overnight incubation in complete medium, cells were washed twice with PBS and compounds in 0.2% serum containing media were added to the wells. The compounds used were IGF-1 alone (100 nM, Genentech, San Francisco, CA, USA), cGP alone (100 nM, Bachem AG, Switzerland), IGF-1 + cGP (100 nM), GPE alone (100 nM, Sigma), GPE + IGF-1 (100 nM) and IGF-1 + cGP + GPE (100 nM). Media and compounds were replaced every 48 h. Cells were trypsinised and counted every 24 h from day 4 to day 6. Cell counting was done using a hematocytomter. Ten μl of cell suspension was pipetted onto each grid and all 9 squares were counted. Eight counts were performed for each well and cells/ml was calculated using the formula: cells/ml = (Average of counts/9) × 10,000.
In-vitro tube formation assay
To determine the formation of tubes in vitro, 70,000 cells were seeded per 25 cm2 flask and pre-treated with PBS, IGF-1 (100 nM) and cGP + IGF-1 (100 nM) in 0.2% serum media for 6 days before the cells were transferred onto growth factor-reduced Matrigel (GFR-Matrigel, BD, Franklin Lakes, NJ, USA). The treatment protocol used for the tube formation assay is same as that for the total cell count assay. Each treatment was carried out in triplicate and media was changed every 48 h. Cells were trypsinised and transferred onto Matrigel on day 6 in serum-free media containing the compounds.
GFR-Matrigel was thawed at 4°C overnight and 100 μl/well was added to 24-well plates precooled to −20°C. The plate was incubated at 37°C in a humidified incubator for 30 minutes to allow the Matrigel to solidify. Cells from 25 cm2 flasks were trypsinised and 40,000 cells in 1 ml serum free medium containing the compounds were added to each well. Cells were incubated at 37°C in a humidified incubator for 18 h and 5 photos were taken per well at 100× magnification using the OLYMPUS IX71 Inverted Laboratory Systems Microscope (Olympus Optical Co., Tokyo, Japan). Images were processed using ImageJ version 1.43u (National Institute of Health) and tube formation parameters analysed using ImagePro Plus version 6.0.0.260 (Media Cybernetics, Inc.). The number of tubes was analysed and compared between the groups.
Cell viability assay
A cell viability assay was used to evaluate the efficacy of cGP on IGF-1 induced cell survival at 24 hours after treatments. Cells were seeded in pre-coated 96-well plates (0.1% gelatine) at a density of 10,000 cells/well for HMEC-1. The assays were set up with six replicates per treatment group. Cells were left to attach in complete medium (10% FBS, 1% penicillin/streptomycin, 1% glutamine) overnight. Once fully attached, cells were incubated with serum-free medium (MCDB 131 medium) for 24 h. On the following day, cells were washed twice with PBS and serum free medium containing cGP (1–100 nM, Bachem AG, Switzerland) and/or IGF-1(1–6.5 nM, Genentech, San Francisco, CA, USA) were added to each well to determine the efficacy of cGP on IGF-1 induced cell viability. To determine the role for IGFBP-3 in cGP efficacy, IGFBP-3 antibody (278 nM, mouse, monoclonal, sc-135947 – Santa Cruz Biotechnology) were also added to the wells accordingly. The plates were incubated for 72 h with media and compounds being replaced at 48 h. After 72 h, 10 μl of 1 × Alarmar Blue (Invitrogen Life Technologies, Carlsband, CA, USA) was added, incubated for 4 h and absorbance was measured (530 nm ex/590 nm em) using a Synergy 2 Multi-mode Microplate Reader and Gen5 data analysis software (Biotek, Winooski, VT USA).
IGF-1 receptors knock-down and overexpression
To determine the role of IGF-1 receptors, we evaluated the efficacy of cGP in the HMEC-1 after either know-down or overexpression of IGF-1 receptors. For knock-down of IGF-1 receptors, plasmid-based small interfering RNA (siRNAs)29 that target the α-subunit of IGF-1 receptor mRNA sequences AACGACUAUCAGCAGCUGAAG and AACAAUGAGUACAACUACCGC were used as previously described27. A negative control siRNA targeted mRNA sequence AACUACCGUUGUUAUAGGUGU (Ambion)27,28. For IGF-1 receptor overexpression, the expression plasmid pIRESneo3-IGF-1 receptor containing cDNA encoding IGF-1 receptor from the plasmid pCVN-IGF1R was used to overexpress IGF1 receptors40. Western blotting was used to determine the expression of IGF-1 receptors in the HMEC-1 cells. We observed a 50% decrease in the abundance of IGF-1 receptors after IGF-1 receptor siRNA knock-down and 35% increase after IGF-1 receptor overexpression.
Animal experiments
The Animal Ethics Committee, the University of Auckland has approved all experimental procedures (R913 for conducting mice lymphoma experiment and R157 for conducting ischemic brain injury in rats) in accordance with Animal welfare Act 1999 and the Code of ethics conduct, New Zealand. Every effort was made to minimize animal suffering and to reduce the number of animals used for the study.
cGP administration after HI injury in rats
The procedures of HI injury and central administration of peptides have been reported previously13. Briefly, Wistar rats (male, 280–310 g, 60–70 days old) from the Animal Resources Unit, the University of Auckland were used. Rats were anaesthetised using 3% Halothane (3%)/O2. For intracerebralventricular administration a guide cannular was fixed stereotaxically at the coordinates of 1.5 mm from the midline and +7.5 mm anterior-posterior (A–P) from the inter-aural zero plane on top of the dura.
The rats were re-anaesthetized 3 days late. The right common carotid artery was surgically exposed and ligated two times using sutures. Two hours after the ligations a brief hypoxia (15 min, 6 ± 0.2%) was induced in an incubator where the relative humidity (90 ± 5%) and temperature (31 ± 0.5°C) were controlled.
The rats were randomly divided into two groups for receiving either cGP or saline treatment 2 h after the hypoxia. Rats were re-anaesthetized for peptide administration. A needle, connected to a Hamilton syringe (100 μl) by a catheter was inserted through the pre-fixed cannula. Either cGP (0.2 μg/rat, n = 12, Sigma), dissolved in saline (20 μl) or saline only (n = 12) was delivered to the right lateral ventricle (1 μl/min infusion rate) using a microdialysis pump (CMA/100, Carnegie Medicin, Stockholm, Sweden). The rats were killed for tissue collections 5 days later. All experimenters were blind to the treatments.
Immunohistochemical staining and assessments
Brain tissue collections, preparations, staining and assessments have been described previously20,41,42. Briefly, rats were deeply anaesthetized using an overdose of pentobarbital (125 mg/kg, intraperitoneally). The brains were perfused with saline, followed by neutral buffered formalin (10%). The brains were collected and further fixed in same fixatives in situ for at least 48 hours, then processed and embedded in paraffin.
Six sequential sections (every 20th sections) were collected from approximately 6 mm, for striatum and from 4.5 mm and 4.2 mm AP inter-aural zero plane for the hippocampus and mounted on pre-coated histology slides.
The sections were de-paraffinised in xylene, rehydrated in an ethanol series and washed with Phosphate buffer saline (0.1 M) with triton X-100 (0.2%, PBST). The sections were pre-treated with H202 (1%) in methanol (50%) for 20 min before antigen retrieval. For the antigen retrieval, the slides were immersed in sodium citrate buffer (10 mM, pH 6.0, 95°C) for 20 min. The sections were then washed and incubated with either monoclonal mouse anti-IGF-1 receptors (Sigma, 1:1000) or rabbit anti-phosphorylated IGF-1 receptors (pY1161, Abcam, Sapphire Bioscience (NZ) Ltd., Hamilton, NZ, 1:100) at 4°C for 48 hours. The sections were washed before being incubated with secondary antibodies at 4°C overnight accordingly, and then with ExtrAvidin® (Sigma-Aldrich Pty, Ltd., Sydney, AU) at room temperature for 3 hours after being washed, developed in 3, 3, diaminobenzidine tetrahydrochoride (DAB, Sigma-Aldrich Pty, Ltd.) and cover slipped with DPX. Isolectin B4 biotin-conjugated (1:4000, Sigma, St. Louis, MO, U.S.A.) was used as a marker for capillaries18. The sections were pre-treated with 1% H2O2 in 50% methanol after being deparaffinized, then incubated overnight at 4°C with the isolectin B-4 in Tris buffered saline before being developed in DAB.
For double labelling, Alexa Fluor® 568 streptavidin conjugate (Invitrogen, 1:250) was used for visualising vascular staining (IB4), donkey anti-mouse (Alexa Fluor® 488 conjugate, Invitrogen, 1:250) and donkey anti-rabbit Alexa Fluor 488 conjugate (Invitrogen) diluted in 1% normal horse serum in PBS was used for visualising immunostatining of IGF-1 receptors. Photomicrographs were taken using a Zeiss LSM 510 confocal microscope at 10 and 100 magnifications.
The density of capillaries was measured using image analysis17,18,43. Briefly the number of capillaries in the striatum and hippocampus was measured using Image J43, the number of capillaries with positive IGF-1 receptors was counted in the entire striatum and hippocampus in both hemispheres directly under microscopy. The areas used for counting were measured using Sigma scan v5.0. The density of IGF-1 receptors containing capillaries was calculated as total number/mm2. The persons involved in data assessment and analysis were blind to the treatment groups.
cGP administration in a mice model of lymphoma
Experiment 1
Experimental procedures have been previously described44. To determine the preventive effects of cGP in tumour growth, 12 mice (8 week old, male C57BL/c) were obtained from the Vernon Jansen Unit, University of Auckland. The mice were randomly assigned to the control and cGP groups (each group had 6 mice). The mice were anaesthetized by intraperitoneal injection of ketamine/xylazine. An incision (5–10 mm) was made at the mid-scapular region on the back of each mouse and a subcutaneous pocket was created. An ALZET mini-osmotic pump filled with 100 μl of cGP peptide solution or vehicle was inserted into the pocket, and the incision was closed with wound clips. The drug was delivered by the pumps at a dose of 25 mg/kg/day. While the mice were under anaesthetic condition, EL-4 thymic lymphoma cells (2 × 105 suspended in 50 μl of PBS) were inoculated subcutaneously into the right flank of the mice. The sites of inoculation were checked daily for the presence of palpable tumours, the diameter of tumours were measured.
Experiment 2
To determine the effect of cGP on tumour growth, 12 mice (8 week old, male C57BL/c) were randomly assigned to the control- and cGP-treated groups (each group had 6 mice). The mice were anaesthetized by intraperitoneal injection of ketamine/xylazine. EL-4 thymic lymphoma cells (2 × 105 suspended in 50 μl of PBS) were inoculated subcutaneously into the right flank of the mice. An ALZET mini-osmotic pump filled with 100 μl of cGP peptide solution or vehicle was inserted into the pocket at 10 days after inoculation when the average tumour size was approximately 0.3 cm in diameter. The incision was closed with wound clips. The drug was delivered by the pumps at a dose of 25 mg/kg/day. The site of tumour inoculation was checked daily for the presence of palpable tumours, the diameter of which was measured. Experimenters were blind to the treatment groups.
IGFBP-3 binding assays
The assay has been previously reported13. To establish the baseline of IGF-I binding to IGFBP-3, we incubated IGFBP-3 in a range of concentrations of IGF-I (0.5, 2, 10, 50 and 100 μg/ml) in phosphate buffer saline (PBS, pH 7.4), giving a final IGF-I:IGFBP-3 molar ratio of 0.01, 0.1, 1, 5 and 10:0.1. To evaluate the effects of cGP on IGF-I/IGFBP-3 binding we added 1 molar ratio of cGP to each IGF-I/IGFBP-3 sample to give cGP:IGF-I molar ratio from 1:0.01 to 1:10 with 0.1 molar ratio of IGFBP-3. To establish the baseline of total IGF-1 concentration (Ct), the same dose range of IGF-I solutions were incubated with PBS in the absence of IGFBP-3. After 4 hour incubation at 37°C, the samples were centrifuged using ultrafiltration spin columns (Vivaspin with 10,000 Da cutoff; GE Healthcare, UK) at 2500 × g for 30 min. The filtrate (unbound IGF-I) of each individual sample in the ultrafiltrate (Cu) was analysed by RP-HPLC. The samples from different concentration ratios were process and analysed at the same time.
The percentage of unbounded IGF-I was calculated using the following equation:
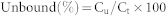 |
Ct = IGF-I from the samples without the presence of IGFBP-3; Cu = the unbound IGF-I with the presence of IGFBP-3.
The analytical chromatography, used for quantitating the amount of IGF-1 was performed by a high performance liquid chromatography (HPLC) assay. The assay ran on an Agilent 1260, using a Jupiter C18 column (250 × 4.6 mm; 300 Ǻ pore size; Phenomenex, Torrance, CA, USA), a flow rate of 1 ml/min, an injection of 50 μl. The column temperature was maintained at 45°C and the detection was monitored at 214 nm. The mobile phase consisted of buffer (A), methanol (B) and Milli Q water (C). Mobile phase A was prepared by dissolving 6.71 g of di-sodium phosphate and 2.21 g of sodium-1-heptane sulfonate in 1 litre Milli Q water. All the mobile phase was filtered and degassed prior to use. All the assays were performed under gradient conditions as described in Supplementary Table 2.
Mathematical analysis
For mathematical modelling, the Boroujerdi et al. (1997)34 model was extended to include cGP dynamics and was solved in MATLAB. Nonlinear optimisation was used to compare model simulations to data (see Supplementary information).
Author Contributions
J.G., P.I., designed the research project and the experiments and wrote the manuscript. P.G. provided scientific guidance throughout the projects and edited the manuscript. P.Y. a postgraduate student, conducted the experiment for testing cGP after ischemic brain injury in rats. G.K. was involved in the experimental design to test cGP in mouse models of cancer, and critiqued the manuscript. Dr X.S. a postdoctoral fellow, conducted the experiment which tested cGP in mouse models of cancer. Y.Z. and J.W. designed and conducted the peptide binding experiments. C.M. and M.T. designed and conducted the experiment for testing cGP on muscle cells and edited the manuscript. G.P., a postgraduate student conducted the data analysis using the mathematical model. P.S. and G.W. designed and supervised the data analysis using the mathematical model. W.C. a postgraduate student, conducted the endothelial cell assays. A.R. and S.M. conducted western blotting and endothelial cell assays. D.L. was involved in the experimental design of the in vitro experiments and edited the manuscript. All authors discussed the results and reviewed the manuscript.
Supplementary Material
Cyclic glycine-proline regulates IGF-1 homeostasis by altering the binding of IGFBP-3 to IGF-1
Acknowledgments
The project was funded by the Ministry of Business, Innovation and Employment, New Zealand, Gravida National Centre for Growth and Development, New Zealand, and the Norman F Barry Foundation, New Zealand. The authors thank Dr. Renato Baserga (Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, USA) for providing human IGF1R cDNA.
References
- Sara V. R. & Carlsson-Skwirut C. The role of the insulin-like growth factors in the regulation of brain development. [Review] [64 refs]. Prog Brain Res 73, 87–99 (1988). [DOI] [PubMed] [Google Scholar]
- Gluckman P. D. et al. A role for IGF-1 in the rescue of CNS neurons following hypoxic- ischemic injury. Biochem Biophys Res Commun 182, 593–599 (1992). [DOI] [PubMed] [Google Scholar]
- Adem A. et al. Insulin-like growth factor 1 (IGF-1) receptors in the human brain: quantitative autoradiographic localization. Brain Res 503, 299–303 (1989). [DOI] [PubMed] [Google Scholar]
- Drakenberg K. et al. Identification of IGF-1 receptors in primitive vertebrates. Regul Pept 43, 73–81 (1993). [DOI] [PubMed] [Google Scholar]
- Sara V. R. & Hall K. Insulin-like growth factors and their binding proteins. [Review] [265 refs]. Physiol Rev 70, 591–614 (1990). [DOI] [PubMed] [Google Scholar]
- Mohanraj L. & Oh Y. Targeting IGF-I, IGFBPs and IGF-I receptor system in cancer: the current and future in breast cancer therapy. Recent pat anti-canc 6, 166–177 (2011). [DOI] [PubMed] [Google Scholar]
- Sara V. R. et al. Truncated IGF-1 in the CNS. Ann NY Acad Sci 175th, 24 (1992). [Google Scholar]
- Yamamoto H. & Murphy L. J. Enzymatic conversion of IGF-I to des(1-3)IGF-I in rat serum and tissues: A further potential site of growth hormone regulation of IGF-I action. J Endocrinol 146, 141–148 (1995). [DOI] [PubMed] [Google Scholar]
- O'Han M. K., Baxter R. C. & Schedlich L. J. Effects of endogenous insulin-like growth factor binding protein-3 on cell cycle regulation in breast cancer cells. Growth Factors 27, 394–408, 10.3109/08977190903185032 (2009). [DOI] [PubMed] [Google Scholar]
- Guan J. et al. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic-ischemic adult rats. Neuropharmacology 47, 892–903 (2004). [DOI] [PubMed] [Google Scholar]
- Guan J., Williams C., Gunning M., Mallard C. & Gluckman P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J Cereb Blood Flow Metab 13, 609–616 (1993). [DOI] [PubMed] [Google Scholar]
- Guan J. & Gluckman P. D. IGF-1 derived small neuropeptides and analogues: a novel strategy for the development of pharmaceuticals for neurological conditions. Br J Pharmacol 8, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J. et al. Peripheral administration of a novel diketopiperazine, NNZ 2591, prevents brain injury and improves somatosensory-motor funcion following hypoxia-ischemia in adult rats. Neuropharmacology 53, 749–762 (2007). [DOI] [PubMed] [Google Scholar]
- Saura J. et al. Neuroprotective effects of Gly-Pro-Glu, the N-terminal tripeptide of IGF-1, in the hippocampus in vitro. Neuroreport 10, 161–164 (1999). [DOI] [PubMed] [Google Scholar]
- Sara V. R. et al. Indentification of Gly-Pre-Glu(GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncted in brain, as a novel neuroaction peptide. Biochemi Biophysi Res Commun 165, 766–771 (1989). [DOI] [PubMed] [Google Scholar]
- Imrie H. et al. Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61, 2359–2368, 10.2337/db11-1494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedin P. et al. Delayed peripheral administration of a GPE analogue induces astrogliosis and angiogenesis and reduces inflammation and brain injury following hypoxia-ischemia in the neonatal rat. Dev Neurosci 29, 393–402 (2007). [DOI] [PubMed] [Google Scholar]
- Shapira S., Mathai S., Zhang R. & Guan J. Delayed peripheral administration of the N-terminal tripeptide of IGF-1 (GPE) reduces brain damage following microsphere induced embolic damage in young adult and aged rats. Neurosci Lett 454, 53–57 (2009). [DOI] [PubMed] [Google Scholar]
- Guan J., Waldvogel H. J., Faull R. L., Gluckman P. D. & Williams C. E. The effects of the N-terminal tripeptide of insulin-like growth factor- 1, glycine-proline-glutamate in different regions following hypoxic- ischemic brain injury in adult rats. Neuroscience 89, 649–659 (1999). [DOI] [PubMed] [Google Scholar]
- Uchiyama Y., Maxson M. M., Sawada T., Nakano A. & Ewing A. G. Phospholipid mediated plasticity in exocytosis observed in PC12 cells. Brain Res 1151, 46–54, 10.1016/j.brainres.2007.03.012 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner O. et al. Time- and cell type-specific induction of platelet-derived growth factor receptor-beta during cerebral ischemia. Brain Res Mol Brain Res 113, 44–51 (2003). [DOI] [PubMed] [Google Scholar]
- Zhu W. et al. Insulin growth factor-1 gene transfer enhances neurovascular remodeling and improves long-term stroke outcome in mice. Stroke 39, 1254–1261, 10.1161/strokeaha.107.500801 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz E. J. et al. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Brain Res Mol Brain Res 59, 119–134 (1998). [DOI] [PubMed] [Google Scholar]
- Gaddipati J. P. et al. Picroliv modulates the expression of insulin-like growth factor (IGF)-I, IGF-II and IGF-I receptor during hypoxia in rats. Cell Mol Life Sci 56, 348–355 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J., Miller O. T., Waugh K. M., McCarthy D. & Gluckman P. D. Insulin-like growth factor-1 improves somatosensory function and reduces the extent of cortical infarction and ongoing neuronal loss after hypoxia-ischemia in rats. Neuroscience 105, 299–306 (2001). [DOI] [PubMed] [Google Scholar]
- Brunet-Dunand S. E. et al. Autocrine human growth hormone promotes tumor angiogenesis in mammary carcinoma. Endocrinology 150, 1341–1352 (2009). [DOI] [PubMed] [Google Scholar]
- Rochester M. A., Riedemann J., Hellawell G. O., Brewster S. F. & Macaulay V. M. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer gene ther 12, 90–100, 10.1038/sj.cgt.7700775 (2005). [DOI] [PubMed] [Google Scholar]
- Liu D. X. & Lobie P. E. Transcriptional activation of p53 by Pitx1. Cell Death Differ 14, 1893–1907, 10.1038/sj.cdd.4402209 (2007). [DOI] [PubMed] [Google Scholar]
- Kang J. et al. Artemin is oncogenic for human mammary carcinoma cells. Oncogene 28, 2034–2045, 10.1038/onc.2009.66 (2009). [DOI] [PubMed] [Google Scholar]
- Butler A. A., Gallaher B. W., Ambler G. R., Gluckman P. D. & Breier B. H. IGF-I and IGF-binding protein-3 in plasma of GH-deficient rats. J Endocrinol 150, 67–76 (1996). [DOI] [PubMed] [Google Scholar]
- Baxter R. C. Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab 278, E967–976 (2000). [DOI] [PubMed] [Google Scholar]
- Bourguignon J. P. & Gerard A. Role of insulin-like growth factor binding proteins in limitation of IGF-1 degradation into the N-methyl-D-aspartate receptor antagonist GPE: evidence from gonadotrophin-releasing hormone secretion in vitro at two developmental stages. Brain Res 847, 247–252 (1999). [DOI] [PubMed] [Google Scholar]
- Baker A. M. et al. Central penetration and stability of N-terminal tripeptide of insulin-like growth factor-I, glycine-proline-glutamate in adult rat. Neuropeptides 39, 81–87 (2005). [DOI] [PubMed] [Google Scholar]
- Boroujerdi M. A., Jones R. H., Sonksen P. H. & Russell-Jones D. L. Simulation of IGF-I pharmacokinetics after infusion of recombinant IGF-I in human subjects. Am J Physiol 273, E438–447 (1997). [DOI] [PubMed] [Google Scholar]
- Weigent D. A. & Arnold R. E. Expression of insulin-like growth factor-1 and insulin-like growth factor-1 receptors in EL4 lymphoma cells overexpressing growth hormone. Cell immunol 234, 54–66, 10.1016/j.cellimm.2005.04.016 (2005). [DOI] [PubMed] [Google Scholar]
- Guan J., Bennet L., Gluckman P. D. & Gunn A. J. Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobiol 70, 443–462 (2003). [DOI] [PubMed] [Google Scholar]
- Riikonen R. Insulin-like growth factor delivery across the blood-brain barrier. Potential use of IGF-1 as a drug in child neurology. Chemotherapy 52, 279–281, 10.1159/000095957 (2006). [DOI] [PubMed] [Google Scholar]
- Torres-Aleman I. Toward a comprehensive neurobiology of IGF-I. Dev Neurobiol 70, 384–396 (2010). [DOI] [PubMed] [Google Scholar]
- Sachdev D. Targeting the type I insulin-like growth factor system for breast cancer therapy. Curr Drug Targets 11, 1121–1132 (2010). [DOI] [PubMed] [Google Scholar]
- Reiss K., D'Ambrosio C., Tu X., Tu C. & Baserga R. Inhibition of tumor growth by a dominant negative mutant of the insulin-like growth factor I receptor with a bystander effect. Clin Cancer Res 4, 2647–2655 (1998). [PubMed] [Google Scholar]
- Gluckman P. D. et al. The role of the insulin-like growth factor system in neuronal rescue. Ann N Y Acad Sci 692, 138–148 (1993). [DOI] [PubMed] [Google Scholar]
- Guan J. et al. The window of opportunity for neuronal rescue with insulin-like growth factor-1 after hypoxia-ischemia in rats is critically modulated by cerebral temperature during recovery. J Cereb Blood Flow Metab 20, 513–519 (2000). [DOI] [PubMed] [Google Scholar]
- Guan J. et al. Vascular degeneration in Parkinson's disease. Brain Pathol 23, 154–164, 10.1111/j.1750-3639.2012.00628.x (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Vale M., Jiang X., Gupta R. & Krissansen G. W. Antisense HIF-1alpha prevents acquired tumor resistance to angiostatin gene therapy. Cancer gene ther 17, 532–540, 10.1038/cgt.2010.7 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cyclic glycine-proline regulates IGF-1 homeostasis by altering the binding of IGFBP-3 to IGF-1