

Significance
Recent genomic studies have revealed more than 1,000 coding sites in the human genome that are subject to A-to-I editing at the RNA level, but it is unclear whether these RNA editing events, many of which are nonsynonymous, are generally advantageous. Analyzing the frequencies and levels of RNA editing at groups of coding sites with different functional importance, we provide unequivocal evidence that human coding RNA editing is generally nonadaptive. We propose that the vast majority of the observed coding RNA editing results from tolerable promiscuous targeting by RNA editing enzymes. Our finding has important implications for understanding the physiological consequences of normal and abnormal coding RNA editing.
Keywords: deleterious, neutral, synonymous
Abstract
Impairment of RNA editing at a handful of coding sites causes severe disorders, prompting the view that coding RNA editing is highly advantageous. Recent genomic studies have expanded the list of human coding RNA editing sites by more than 100 times, raising the question of how common advantageous RNA editing is. Analyzing 1,783 human coding A-to-G editing sites, we show that both the frequency and level of RNA editing decrease as the importance of a site or gene increases; that during evolution, edited As are more likely than unedited As to be replaced with Gs but not with Ts or Cs; and that among nonsynonymously edited As, those that are evolutionarily least conserved exhibit the highest editing levels. These and other observations reveal the overall nonadaptive nature of coding RNA editing, despite the presence of a few sites in which editing is clearly beneficial. We propose that most observed coding RNA editing results from tolerable promiscuous targeting by RNA editing enzymes, the original physiological functions of which remain elusive.
First discovered 28 y ago (1), RNA editing refers to posttranscriptional alterations of RNA molecules through insertion, deletion, or modification of nucleotides, not including RNA processing events such as splicing, capping, or polyadenylation (2, 3). RNA editing results in differences between genomic sequences and the corresponding RNA sequences. The predominant type of RNA editing in animals is the conversion of adenosine (A) to inosine (I), catalyzed by a family of adenosine deaminases that act on RNA (2). This editing is also known as A-to-G editing because inosine in RNA is interpreted as guanosine (G) by the translational machinery (2). Another well-documented type of RNA editing in animals is cytidine-to-uridine (C-to-U) editing, catalyzed by the activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like family of deaminase, but it is much rarer than A-to-G editing (2).
Before the genomic era, only about a dozen human coding sites had been reported to be subject to A-to-G RNA editing (4), and a few of them were extensively characterized functionally (2, 5). For example, A-to-G editing in GRIA2 (glutamate receptor, ionotropic, AMPA 2) changes the genomically encoded glutamine to arginine at position 607, altering the calcium permeability of the channel (6). Mice deficient in editing at this site exhibit seizures and early death (7, 8). Such examples, albeit small in number (9), led to the widely held view that coding RNA editing offers an “extreme advantage” (10). It is commonly stated that coding RNA editing expands transcriptome diversity such that the same gene codes for proteins of different functions, which could be deployed in different tissues or at different times (10, 11). Some researchers suggest that RNA editing also serves as a safeguard because it can reverse harmful genomic mutations in corresponding RNA transcripts (12, 13).
In the last few years, genomic studies in humans have uncovered an astonishingly large number of RNA editing sites (4, 14–20). Although editing apparently occurs primarily in noncoding regions, especially in Alu repeat elements (15, 19, 20), the number of edited coding sites also exceeds 100 times the previously known number (14). However, in contrast to the previously identified editing sites, which typically have high editing levels, most of the newly identified editing sites are edited in only a small percentage of RNA molecules (14–18). These new genomic findings raise an important question (3, 18): What fraction of coding RNA editing is advantageous? Here we address this question by analyzing all reported high-quality A-to-G editing sites in human coding regions; C-to-U editing is omitted because of the small sample size, which prevents meaningful statistical analysis. Because the functional consequences of the vast majority of coding RNA editing have not been experimentally investigated (5), we take a comparative genomic approach by examining the relative frequencies and levels of RNA editing at large groups of sites with known differences in functional importance and by comparing the phylogenetic variations of edited and unedited sites.
Results
Nonsynonymous Editing Is Rarer Than Synonymous Editing.
We compiled A-to-G RNA editing sites in human coding regions from six genomic datasets (4, 14–18). After removing redundant and potentially false-positive sites, we retrieved a total of 1,783 A-to-G editing sites in protein coding regions (Dataset S1). Among these 1,783 sites, editing causes a nonsynonymous change (i.e., amino acid change) at 1,183 sites (n) and causes a synonymous change at the remaining 600 sites (s). Of all human coding regions, 7,112,448 A sites (N) would have nonsynonymous changes if edited to G, whereas 2,258,040 A sites (S) would have synonymous changes if edited to G. Thus, the frequency of nonsynonymous editing is fn = n/N = 1.66 × 10−4, whereas the frequency of synonymous editing is fs = s/S = 2.66 × 10−4; the difference is statistically significant (P = 5.2 × 10−21, χ2 test; Fig. 1A). Because of the lack of effect on protein sequences (except when splicing is altered), synonymous editing is likely to be more or less selectively neutral. The finding that fn is ∼38% lower than fs suggests that a substantial fraction of potential editing at nonsynonymous sites is harmful and is purged by purifying selection. Note that although a recent study (12) also suggested that fn/fs < 1, the finding was unreliable because the author considered all possible nucleotide changes rather than only A-to-G changes when calculating N and S. Because A-to-G changes are transitions, which tend to be synonymous (21), the author overestimated N/S and, subsequently, underestimated fn/fs.
Fig. 1.

Frequencies and editing levels of synonymous (syn) and nonsynonymous (nonsyn) A-to-G editing in humans. (A) Fraction of nonsynonymous A sites edited is significantly lower than that of synonymous A sites edited. Error bars show one SD. P value is from a χ2 test. (B) The editing level is significantly lower at the nonsynonymous editing sites than the synonymous editing sites. The values of upper quartile, median, and lower quartile are indicated in each box, whereas the bars outside the box show the fifth and 95th percentiles. P value is obtained from a two-tail Mann–Whitney U test.
The observed nonsynonymous editing events must be either beneficial or slightly deleterious, if we ignore the rare type of strict neutrality. If they are slightly deleterious, their editing levels are expected to be lower than those of synonymous editing events because higher nonsynonymous editing levels would impose greater harm. In contrast, if they are beneficial, the opposite is expected because higher nonsynonymous editing levels would confer greater benefits. We discovered that the editing levels are significantly lower for nonsynonymous editing than for synonymous editing (P = 0.04, two-tail Mann–Whitney U test; Fig. 1B), suggesting that even among the observed nonsynonymous editing sites, editing is so deleterious that only those with relatively low editing levels are selectively permitted.
Because the few coding RNA editing sites that have been functionally characterized are all related to neural functions, it is often stated that RNA editing is of special importance to brain function (9, 11, 22). To examine whether our results, obtained from multiple tissues, also apply specifically to the brain, we analyzed the editing sites identified from the brain and other tissues in the largest of the six datasets used (14). The ratio between the number of nonsynonymous editing sites and the number of synonymous editing sites is slightly lower for brain-specific editing (n/s = 370/211 = 1.75) than for editing found in other tissues (260/119 = 2.18), although their difference is not significant (P = 0.13, χ2 test). Further, the median editing level of nonsynonymous editing divided by that of synonymous editing is 30.4% lower in the brain than in other tissues (P = 0.037, one-tail bootstrap test). These results suggest that, if anything, RNA editing is more deleterious in the brain than in other tissues, which is consistent with the fact that brain-specific genes tend to be more conserved in protein sequence than other tissue-specific genes during human evolution (23–25).
RNA Editing Is Rarer in Essential Genes Than in Nonessential Genes.
If nonsynonymous RNA editing is generally disadvantageous, we should see less nonsynonymous editing and lower nonsynonymous editing levels in functionally more important genes than in less important ones. To verify these predictions, we classified human genes into essential and nonessential on the basis of the essentiality information of their one-to-one orthologs in mouse. Essential genes are those that cause infertility or death before puberty when deleted (26); all other genes are considered nonessential. Consistent with our prediction, the fraction of coding sites edited is significantly lower in essential genes than in nonessential genes (P = 0.02, χ2 test; Fig. 2A). This deficit of editing in essential genes is entirely contributed by nonsynonymous editing (P = 4.8 × 10−3, χ2 test; Fig. 2B), whereas synonymous editing is virtually identical between essential and nonessential genes (P = 0.96, χ2 test; Fig. 2C). Further, the editing level of nonsynonymous editing is significantly lower than that of synonymous editing in essential genes (P = 0.03, two-tail Mann–Whitney U test; right bar in Fig. 2D), but not so in nonessential genes (P = 0.33; left bar in Fig. 2D). As predicted, the ratio between the median nonsynonymous editing level and the median synonymous editing level is significantly lower for essential genes than for nonessential genes (P = 0.02; one-tail bootstrap test; Fig. 2D). A previous study showed that gene essentiality is not completely conserved between human and mouse (27), rendering the inference of human gene essentiality from mouse less reliable. However, the fact that significant differences in editing are still observed between the inferred essential and nonessential genes suggests that the true differences should be greater than observed. Thus, our results are conservative.
Fig. 2.
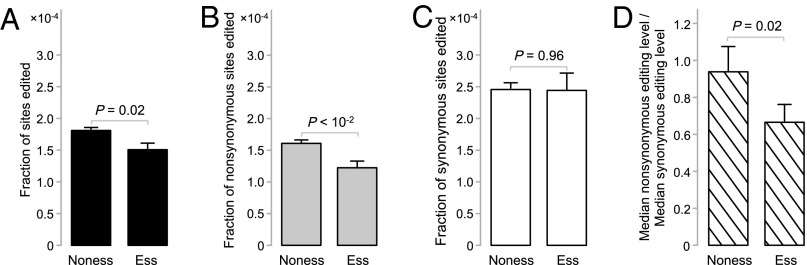
A-to-G coding site editing in essential (ess) and nonessential (noness) genes. (A) The fraction of A sites edited is significantly lower in essential than in nonessential genes. (B) The fraction of nonsynonymous A sites edited is significantly lower in essential than in nonessential genes. (C) The fraction of synonymous A sites edited is similar between essential and nonessential genes. (D) The ratio of the median nonsynonymous editing level and the median synonymous editing level is lower in essential than in nonessential genes. In all panels, error bars indicate one SD. P values are from χ2 tests in A–C and from a one-tail bootstrap test in D.
RNA Editing Is Rarer in Genes Under Stronger Functional Constraints.
The functional constraint of a gene can be measured by the ratio of the nonsynonymous nucleotide substitution rate (dN) to the synonymous rate (dS) during its evolution (28). The lower the dN/dS ratio, the greater the functional constraint (28). Our hypothesis that nonsynonymous RNA editing is generally deleterious predicts that both the frequency and editing level of nonsynonymous editing decrease as the dN/dS ratio of a gene decreases. These predictions are strongly supported by our data. Specifically, we ranked all human genes by their dN/dS ratios, calculated using one-to-one orthologs from the human and mouse, and assigned the bottom half of the genes to a low dN/dS bin and the top half to a high dN/dS bin. We found a significantly smaller fn for the lower dN/dS bin than for the higher dN/dS bin (P = 0.03, χ2 test; Fig. 3A), whereas fs is comparable between the two bins (P = 0.20, χ2 test; Fig. 3A). To further analyze the relationship between RNA editing and dN/dS, we divided all genes into 20 equal-size bins according to their dN/dS. There is a significant positive correlation between fn/fs for the genes in a bin and the median dN/dS of the bin (Spearman’s rank correlation ρ = 0.71; P = 7.1 × 10−4; Fig. 3B). Similarly, we observed a positive correlation between the ratio of the median nonsynonymous editing level and the median synonymous editing level for a bin and the median dN/dS of the bin (ρ = 0.46; P = 0.04; Fig. 3C).
Fig. 3.

Reduced RNA editing in genes with low nonsynonymous to synonymous rate ratios (dN/dS). (A) Fractions of synonymous and nonsynonymous sites edited in the 50% of genes with low dN/dS (median dN/dS = 0.055) and the 50% of genes with high dN/dS (median dN/dS = 0.250). Error bars show one SD. P value is from χ2 test. (B) The fraction of nonsynonymous sites edited relative to the fraction of synonymous sites edited (fn/fs) increases with dN/dS. Each dot represents 5% of all human genes with dN/dS estimates. (C) The median nonsynonymous editing level relative to the median synonymous editing level increases with dN/dS. Each dot represents 5% of all human genes with edited sites and editing level information. In B and C, ρ and P value are from Spearman’s rank correlation based on the binned data. Error bars in B and C show SDs estimated from 1,000 bootstrap samples.
For multiple reasons that have begun to be elucidated (29–32), high gene expression imposes strong evolutionary constraint in organisms ranging from bacteria to mammals (29, 33, 34). If nonsynonymous editing is generally deleterious and synonymous editing is more or less neutral, fn/fs should decrease with the rise of gene expression level. Indeed, after grouping genes into 20 equal-size bins according to their expression levels, we found a significant negative correlation between the median expression of a gene group and fn/fs for the group (ρ = −0.57; P = 0.01; Fig. 4A). Further, nonsynonymous editing level (ρ = −0.45; P = 0.04; Fig. 4B), but not synonymous editing level (ρ = −0.31; P = 0.18; Fig. 4C), decreases significantly as gene expression level rises.
Fig. 4.

Reduced nonsynonymous RNA editing in genes with high expressions. (A) The fraction of nonsynonymous sites edited relative to the fraction of synonymous sites edited (fn/fs) decreases with gene expression level. Each dot represents 5% of human genes with expressions in lymphoblastoid cells. (B) The median nonsynonymous editing level decreases with gene expression level. (C) The median synonymous editing level shows no significant correlation with gene expression level. In B and C, each dot represents 5% of all human genes with edited sites and editing level information. In all panels, gene expression levels were estimated by mRNA sequencing in units of reads per kilobase per million mapped reads. ρ and P value are from Spearman’s rank correlation based on the binned data. Error bars show SDs estimated from 1,000 bootstrap samples.
Edited As Are More Likely Than Unedited As to Be Replaced with Gs in Evolution.
Enhancing transcriptome diversity is often cited as a major advantage of nonsynonymous A-to-G editing compared with the direct use of genomic Gs at the same positions (4, 10, 11). This adaptive hypothesis predicts that compared with unedited As, edited As are less likely to be replaced with Gs during evolution because such replacements would lower transcriptome diversity and fitness. In contrast, if RNA editing is nonadaptive, such that it is fixed and observed only at positions where transcriptomic Gs are tolerated but not beneficial, edited As should be more likely than unedited As to be replaced with Gs during evolution because, on average, Gs are more acceptable at the former sites than at the latter sites. To distinguish between the two hypotheses, we compared edited and unedited A sites from all human genes with observed coding A-to-G editing. We focused on the As with orthologous sites that exist in both mouse and dog and that are also As in dog. Given the phylogenetic relationships among human, mouse, and dog (Fig. 5), the common ancestor of the three species likely had As at these sites. The key question is, What fraction of these ancestral As are replaced with Gs in mouse? We first examined first and second codon positions, where A-to-G editing is all nonsynonymous. We found this fraction to be higher for edited As (6.92%) than for unedited As (2.98%) (P = 7.2 × 10−3, χ2 test; Fig. 5A), which refutes the adaptive hypothesis and supports the nonadaptive hypothesis. As a control, we examined the fraction of ancestral As that become T or C in mouse. We found this fraction to be independent of whether the As are edited or not (P = 0.31; Fig. 5A), confirming that the edited and unedited As under comparison have similar rates of substitution to other nucleotides and, thus, are comparable. As another control, we did the same analysis for third codon positions, where almost all A-to-G editing is synonymous (except for ATA codons). As expected, no significant difference is observed between edited and unedited As in the frequency of changes to G or T/C (Fig. 5B).
Fig. 5.

Frequencies with which an ancestral A is replaced with G or with T/C in mouse, in relation to whether the A is edited in human. Results for As at (A) first and second codon positions and (B) third codon positions. Error bars show one SD. P values are from χ2 tests.
Phylogenetic Variation of Nonsynonymously Edited Sites.
The pattern of phylogenetic variation of a trait often offers significant insights into its function or functional importance. To this end, we retrieved from the genome sequences of 44 nonhuman vertebrates the orthologous nucleotides and corresponding codons of each human nonsynonymous editing site. A total of 143 sites have sufficient phylogenetic coverage, and they can be divided into four types on the basis of their phylogenetic variations: 43 “conserved” sites, 16 “hardwired” sites, 36 “unfound” sites, and 48 “diversified” sites (Fig. 6A; Dataset S2). At each conserved site, only the human genomically encoded amino acid is present in all species examined. At each hardwired site, either the human genomically encoded amino acid or the human edited amino acid is observed in all species. At each unfound site, the human edited amino acid is not found in the genome of any species, but the human preedited amino acid and at least another amino acid are found. At each diversified site, the human preedited, edited, and at least one other amino acid are found in nonhuman species.
Fig. 6.

Four different types of phylogenetic variation of A-to-G nonsynonymous editing sites and a comparison of their editing levels. (A) Editing sites are divided into conserved, hardwired, unfound, and diversified groups, according to the evolutionary variations among human and 44 other vertebrate species. The observed nucleotides and corresponding codons and amino acids at each site are shown for one example of each group, with the number of identified cases in each group provided in parentheses. The four listed examples are GRIK1 (edited position chr21: 30953750), CYFIP2 (chr5: 156726808), STXBP5 (chr6: 147636753), and RALBP1 (chr18: 9522377). The tree topology follows http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phyloP46way/. The branches are not drawn to scale; our analysis does not depend on the accuracy of the tree. (B) Editing levels of the four groups of edited sites. The values of upper quartile, median, and lower quartile are indicated in each box, whereas the bars outside the box show the fifth and 95th percentiles. P value is from two-tail Mann–Whitney U test.
If the observed nonsynonymous editing is mostly advantageous, we predict that the editing level at diversified sites will be the lowest among all four types because editing would have the smallest advantage at diversified sites, given that these sites allow many different amino acids. In contrast, if the observed nonsynonymous editing is mostly slightly deleterious, we predict that the editing level will be the highest at diversified sites because editing would cause the smallest harm at diversified sites, given their tolerance for many different amino acids. We found that the median editing level at diversified sites (0.317) is significantly greater than that (0.200) at the other three types of sites (P = 0.001, Mann–Whitney U test; Fig. 6B), supporting the slightly deleterious hypothesis.
As mentioned, the safeguard hypothesis asserts that nonsynonymous A-to-G editing is beneficial because it can reverse, at the RNA level, the deleterious genomic mutation from G to A (12, 13). The 16 hardwired sites we identified appear to be consistent with this hypothesis (Fig. 6A). Nevertheless, this hypothesis predicts a higher editing level for hardwired sites than for the other types of sites because a low editing level cannot fully reverse the deleterious genomic mutation at the RNA level. Contrary to this prediction, we found the median editing level at hardwired sites (0.191) to be lower than the median at the other three types of sites (0.250), although the difference is not statistically significant because of the limited number of hardwired sites. Thus, the safeguard hypothesis is not supported by our data.
Discussion
Using comparative genomic analysis, we showed that a substantial fraction of nonsynonymous A-to-G editing is too deleterious to survive purifying selection and be detected. Of those nonsynonymous editing sites that are detected, most are slightly deleterious, rather than beneficial. If coding A-to-G editing is generally harmful, why does it exist? RNA editing evolved multiple times in the history of life via the recruitment of various enzyme genes (35). Covello and Gray proposed a three-stage neutral model that explains the origins of various types of RNA editing (36). First, duplication of a preexisting metabolic enzyme gene, followed by random mutations, creates a gene with a product capable of RNA editing. Second, with the emergence of A-to-G RNA editing, sites that need to be Gs in RNAs can tolerate As and be replaced with As in the genome. Third, RNA editing becomes selectively maintained because a failure to convert these genomic As to Gs in RNAs would be harmful. Given our finding that ∼38% of potential nonsynonymous A-to-G editing is deleterious, the origin of A-to-G editing would have been harmful rather than neutral. To be established evolutionarily, A-to-G editing must have had one or more beneficial functions to at least offset its harm in nonsynonymous editing. It is likely that coding site editing results from promiscuous targeting by the editing enzyme and is a deleterious byproduct of the unknown beneficial functions of RNA editing. In addition to coding sequence editing, A-to-G editing also occurs in repetitive noncoding sequences (e.g., Alu elements), microRNAs, and viral RNAs (10). Related to these activities, a number of physiological functions of A-to-G editing have been proposed (10). For instance, it may be used to suppress the proliferation of transposons (20) or to inhibit viral replication (37). These host defense functions fit the relatively unspecific targeting of A-to-G editing (10). RNA editing has also been suggested to play roles in marking RNAs for degradation (10), regulating alternative splicing (10), and modulating nuclear retention of RNAs (10). The difficulty, however, is in identifying the initial beneficial functions prompting the establishment of A-to-G editing because the initial functions may or may not exist at present.
Regardless of the initial benefits of A-to-G editing, the evolutionary maintenance of the editing machinery in present-day organisms may well be a few As in coding regions that must be edited to Gs to be functional (35, 36). This notion is not inconsistent with our finding that most observed editing events are slightly deleterious because a handful of highly beneficial editing events are sufficient to maintain the editing machinery when strongly deleterious editing has already been avoided after millions of years of natural selection.
That important genes/sites have less frequent and lower levels of RNA editing raises the question of how these genes/sites escape RNA editing. Although the exact determinants of the presence/absence of RNA editing at an A site are still unclear, it has been suggested that the sequence flanking the edited site and the mRNA structure are important (10, 38). It is possible that at important genes/sites, natural selection has led to the avoidance of sequence motifs and mRNA structures conducive to RNA editing. It is also possible that RNA editing is not reduced at important genes/sites but that the edited mRNAs are prone to degradation. At any rate, molecular mechanisms allowing the escape of deleterious editing at specific sites will be a subject of great interest.
Our finding that observed coding RNA editing is largely deleterious, rather than beneficial, helps understand some enigmatic phenomena. For instance, Chen recently reported that of all the As in the human genome that were not As in the common ancestor of human and chimpanzee, edited As are more likely than unedited As to be ancestrally Gs (12). She found this phenomenon “most surprising” and named it “editing-mediated RNA memory of evolution,” as if there is a memory in RNA of the ancestral nucleotide in the genome. She suggested that this “memory” is advantageous as a mechanism for correcting deleterious genomic G-to-A mutations. Given our finding that observed RNA editing reflects tolerance for slightly deleterious editing, rather than adaptation, her observation is fully expected. That is, compared with an average unedited A position, an edited A position is more tolerable for G. Thus, genomic Gs are more likely to be accepted at the orthologous positions of edited As than at the orthologous positions of unedited As (Fig. 5).
Our results suggest that of the four phylogenetic types of nonsynonymous A-to-G editing, the unfound group is especially harmful because G is most likely not tolerated at these positions. With this in mind, it is not surprising that an increase in the editing level of a serine position in antizyme inhibitor 1 causes hepatocellular carcinoma (39). This A-to-G editing leads to a Ser-to-Gly change. In all 45 vertebrate genomes examined, many amino acids, including Ser, Arg, Gln, His, Asn, and Lys, are found at the site, but no Gly is observed. These findings call for special attention to the unfound group of A-to-G editing sites in the study of editing-related human diseases.
Materials and Methods
Genomic Data.
A-to-G RNA editing sites in human coding regions were retrieved from six publicly available datasets (4, 14–18). Editing sites in one dataset (4) were identified from seven different tissues of an individual by tailored target capture, followed by massively parallel DNA sequencing, whereas those in the other five datasets were identified by analysis of transcriptome and genomic DNA data from a single individual (15–18) or by analysis of transcriptome data from multiple individuals (14). The original authors of these datasets took several steps to minimize false-positives. For instance, potential RNA editing sites located in mapping-error prone regions (e.g., paralogous regions), heterozygous nucleotide positions, or known SNP positions were excluded (14–18). Further, in the first dataset (4), >5% editing level in at least two tissues was required to call an editing site. In three other datasets (16–18), at least three different RNA reads with the focal position as “G” (mismatched reads) were required. In the fifth dataset (15), at least two mismatched reads for sites in Alu regions and at least three mismatched reads for sites in non-Alu regions were required. In the sixth dataset (14), at least one read with a base quality score ≥25 and a mapping quality score ≥20 were required.
All six datasets provided the chromosomal location, strand, and editing level of each edited site. Most of these datasets also provided annotations of synonymous or nonsynonymous editing. For datasets without such annotations, we annotated the editing effects by searching for the corresponding codons using Ensembl (version 70) (40). Chromosomal coordinates in two datasets (4, 18) were updated from version NCBI36 to GRCh37 using the liftOver process (http://genome.ucsc.edu/cgi-bin/hgLiftOver). If the same site appeared in multiple datasets or various tissues, the highest reported editing level was used in subsequent analysis. Some sites in one dataset (16) were suggested by another (18) to be false-positives, and they were excluded from further analyses. Edited sites identified from cancer cell lines (17) were also removed. One study reported a large number of editing sites, including many previously unknown types (41). Because the validity of this study was controversial (15, 18, 42–46), we did not use its data. The dS and dN values between human and mouse orthologs were retrieved from Ensembl, using BioMart (www.ensembl.org/biomart/martview). Fig. 4 is based on human gene expression levels measured by mRNA sequencing in the lymphoblastoid cell line GM12878 (47), which was used to identify RNA editing sites in ref. 15.
Human Essential and Nonessential Genes.
Ensembl IDs of 2,618 mouse essential genes were obtained from the Online Gene Essentiality Database (http://ogeedb.embl.de), and 2,471 of them have one-to-one orthologs in humans based on the Ensembl ortholog database. These human orthologs were regarded as essential genes, and all other human protein-coding genes were regarded as nonessential. This treatment likely misassigned many human essential genes as nonessential, and some human nonessential genes as essential. That we still detected significant differences in editing between essential and nonessential genes suggests that their true differences are even greater than observed.
Potential Synonymous and Nonsynonymous Editing Sites.
Coding sequences of all canonical transcripts in humans were downloaded from Ensembl, and those with premature stop codons were excluded from further analysis. For each coding sequence, the potential number of synonymous editing sites (S) is the total number of As that would cause a synonymous change if edited to Gs, and the potential number of nonsynonymous editing sites (N) is the total number of As that would cause a nonsynonymous change if edited to Gs.
Phylogenetic Variation of Nonsynonymous Editing Sites.
For each human nonsynonymous editing site, we retrieved the codon in which the editing site resides and the homologous codons in 44 other vertebrate species from the Ensembl Compara (version 70) database, using Ensembl Perl API. We found that for a large number of editing sites, there are no homologous codons in most of the species. According to our definition, as long as the homologous codons encode for at least one more type of amino acid in addition to the human preedited and edited amino acids, we classify this site as diversified. For the other three groups, we require representatives from at least two different orders in addition to primates.
Evolution of Edited and Unedited Sites.
Coding sequences of human genes that contain at least one edited site (version GRCh37), as well as their one-to-one orthologous sequences from mouse (version GRCm38) and dog (version CanFam3.1), were retrieved from Ensembl using custom Perl scripts. MUSCLE (48) with default options was used to align the three sequences on the basis of their translated protein sequences, and the corresponding DNA sequence alignment was then created accordingly using PAL2NAL (49). In the alignment of human, mouse, and dog sequences of each gene, there are both edited and unedited As. We isolated human As that are also As in dog. We calculated the fraction of such sites that have been replaced with Gs (or Ts/Cs) in mouse for edited and unedited As, respectively.
Supplementary Material
Acknowledgments
We thank Chuan Li for stimulating discussion; Jian-Rong Yang for assistance in statistical analysis; and Xiaoshu Chen, Chuan Li, Xinzhu Wei, Jian-Rong Yang, and two anonymous reviewers for valuable comments. This work was supported in part by research grants from the National Institutes of Health (R01GM103232, to J.Z.) and the National Natural Science Foundation of China (31100170, to G.X.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321745111/-/DCSupplemental.
References
- 1.Benne R, et al. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46(6):819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 2.Nishikura K. Editor meets silencer: Crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7(12):919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farajollahi S, Maas S. Molecular diversity through RNA editing: A balancing act. Trends Genet. 2010;26(5):221–230. doi: 10.1016/j.tig.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li JB, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324(5931):1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 5.Maas S. Posttranscriptional recoding by RNA editing. Adv Protein Chem Struct Biol. 2012;86:193–224. doi: 10.1016/B978-0-12-386497-0.00006-2. [DOI] [PubMed] [Google Scholar]
- 6.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67(1):11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 7.Feldmeyer D, et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2(1):57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 8.Brusa R, et al. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270(5242):1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 9.Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006;3(1):1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gommans WM, Mullen SP, Maas S. RNA editing: A driving force for adaptive evolution? Bioessays. 2009;31(10):1137–1145. doi: 10.1002/bies.200900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L. Characterization and comparison of human nuclear and cytosolic editomes. Proc Natl Acad Sci USA. 2013;110(29):E2741–E2747. doi: 10.1073/pnas.1218884110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian N, Wu X, Zhang Y, Jin Y. A-to-I editing sites are a genomically encoded G: Implications for the evolutionary significance and identification of novel editing sites. RNA. 2008;14(2):211–216. doi: 10.1261/rna.797108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramaswami G, et al. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods. 2013;10(2):128–132. doi: 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramaswami G, et al. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods. 2012;9(6):579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Z, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30(3):253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 17.Park E, Williams B, Wold BJ, Mortazavi A. RNA editing in the human ENCODE RNA-seq data. Genome Res. 2012;22(9):1626–1633. doi: 10.1101/gr.134957.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinman CL, Adoue V, Majewski J. RNA editing of protein sequences: A rare event in human transcriptomes. RNA. 2012;18(9):1586–1596. doi: 10.1261/rna.033233.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DD, et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14(9):1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2(12):e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA. 1998;95(7):3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenthal JJ, Seeburg PH. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron. 2012;74(3):432–439. doi: 10.1016/j.neuron.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi P, Bakewell MA, Zhang J. Did brain-specific genes evolve faster in humans than in chimpanzees? Trends Genet. 2006;22(11):608–613. doi: 10.1016/j.tig.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Kuma K, Iwabe N, Miyata T. Functional constraints against variations on molecules from the tissue level: Slowly evolving brain-specific genes demonstrated by protein kinase and immunoglobulin supergene families. Mol Biol Evol. 1995;12(1):123–130. doi: 10.1093/oxfordjournals.molbev.a040181. [DOI] [PubMed] [Google Scholar]
- 25.Duret L, Mouchiroud D. Determinants of substitution rates in mammalian genes: Expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 2000;17(1):68–74. doi: 10.1093/oxfordjournals.molbev.a026239. [DOI] [PubMed] [Google Scholar]
- 26.Liao B-Y, Zhang J. Mouse duplicate genes are as essential as singletons. Trends Genet. 2007;23(8):378–381. doi: 10.1016/j.tig.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Liao BY, Zhang J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci USA. 2008;105(19):6987–6992. doi: 10.1073/pnas.0800387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- 29.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JR, Zhuang SM, Zhang J. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol Syst Biol. 2010;6:421. doi: 10.1038/msb.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JR, Liao BY, Zhuang SM, Zhang J. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci USA. 2012;109(14):E831–E840. doi: 10.1073/pnas.1117408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park C, Chen X, Yang JR, Zhang J. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 2013;110(8):E678–E686. doi: 10.1073/pnas.1218066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pál C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158(2):927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23(11):2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- 35.Gray MW. Evolutionary origin of RNA editing. Biochemistry. 2012;51(26):5235–5242. doi: 10.1021/bi300419r. [DOI] [PubMed] [Google Scholar]
- 36.Covello PS, Gray MW. On the evolution of RNA editing. Trends Genet. 1993;9(8):265–268. doi: 10.1016/0168-9525(93)90011-6. [DOI] [PubMed] [Google Scholar]
- 37.Taylor DR, Puig M, Darnell ME, Mihalik K, Feinstone SM. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol. 2005;79(10):6291–6298. doi: 10.1128/JVI.79.10.6291-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rieder LE, Staber CJ, Hoopengardner B, Reenan RA. Tertiary structural elements determine the extent and specificity of messenger RNA editing. Nat Commun. 2013;4:2232. doi: 10.1038/ncomms3232. [DOI] [PubMed] [Google Scholar]
- 39.Chen L, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19(2):209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flicek P, et al. Ensembl 2013. Nucleic Acids Res. 2013;41(Database issue):D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li M, et al. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333(6038):53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piskol R, Peng Z, Wang J, Li JB. Lack of evidence for existence of noncanonical RNA editing. Nat Biotechnol. 2013;31(1):19–20. doi: 10.1038/nbt.2472. [DOI] [PubMed] [Google Scholar]
- 43. Pickrell JK, Gilad Y, Pritchard JK (2012) Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335(6074):1302, author reply 1302. [DOI] [PMC free article] [PubMed]
- 44. Lin W, Piskol R, Tan MH, Li JB (2012) Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335(6074):1302, author reply 1302. [DOI] [PubMed]
- 45. Kleinman CL, Majewski J (2012) Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335(6074):1302, author reply 1302. [DOI] [PubMed]
- 46.Schrider DR, Gout J-F, Hahn MW. Very few RNA and DNA sequence differences in the human transcriptome. PLoS ONE. 2011;6(10):e25842. doi: 10.1371/journal.pone.0025842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37(18):e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suyama M, Torrents D, Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(Web Server issue):W609-12. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.