

Abstract
Isogenic stem cell populations display cell-to-cell variations in a multitude of attributes including gene or protein expression, epigenetic state, morphology, proliferation and proclivity for differentiation. The origins of the observed heterogeneity and its roles in the maintenance of pluripotency and the lineage specification of stem cells remain unclear. Addressing pertinent questions will require the employment of single-cell analysis methods as traditional cell biochemical and biomolecular assays yield mostly population-average data. In addition to time-lapse microscopy and flow cytometry, recent advances in single-cell genomic, transcriptomic and proteomic profiling are reviewed. The application of multiple displacement amplification, next generation sequencing, mass cytometry and spectrometry to stem cell systems is expected to provide a wealth of information affording unprecedented levels of multiparametric characterization of cell ensembles under defined conditions promoting pluripotency or commitment. Establishing connections between single-cell analysis information and the observed phenotypes will also require suitable mathematical models. Stem cell self-renewal and differentiation are orchestrated by the coordinated regulation of subcellular, intercellular and niche-wide processes spanning multiple time scales. Here, we discuss different modeling approaches and challenges arising from their application to stem cell populations. Integrating single-cell analysis with computational methods will fill gaps in our knowledge about the functions of heterogeneity in stem cell physiology. This combination will also aid the rational design of efficient differentiation and reprogramming strategies as well as bioprocesses for the production of clinically valuable stem cell derivatives.
Keywords: Human embryonic stem cells, Induced pluripotent stem cells, Heterogeneity, Single-cell analysis, Time-lapse microscopy, Flow cytometry, Multiple displacement amplification, Mass cytometry, Stochastic multiscale model, Gene expression noise
1. Introduction
Phenotypic diversity is an intrinsic feature observed in isogenic populations of prokaryotic and eukaryotic cells. This diversity is also observed in stem cells including embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and mesenchymal stem cells (MSCs) (Enver et al., 2009; Graf and Stadtfeld, 2008; Narsinh et al., 2011; Phinney, 2012; Young et al., 2012). It is becoming increasingly clear that population variation contributes substantially to the variability observed in stem cell responses to their microenvironment and factors inducing their self-renewal or lineage commitment (Losick and Desplan, 2008). Within individual stem cell lines, heterogeneity can arise from several sources including stochastic fluctuations in gene regulatory networks (GRNs) (Arias and Hayward, 2006; Kaern et al., 2005), the kinetics of protein synthesis and degradation, partitioning of cellular material during division (Huh and Paulsson, 2011; Wu and Tzanakakis, 2012), asynchronous or asymmetric cell proliferation, allelic regulation of gene expression (Miyanari and Torres-Padilla, 2012), and spatial gradients of soluble cues and matrix factors in the extracellular milieu (Park et al., 2009; Parmar et al., 2007; Suslov et al., 2002).
Although heterogeneity is commonly observed in stem/progenitor cell ensembles, its sources and roles in stem cell biology and engineering have only recently attracted greater attention. Such heterogeneity can be manifested at the population level with the existence of phenotypically distinct cells (e.g. pluripotent and committed cells) and is termed ‘macro-heterogeneity’ (Huang, 2009). In contrast, ‘micro-heterogeneity’ refers to variations, for example, in gene or protein expression displayed by a particular subpopulation. Additional concepts related to the variations within stem cell populations can be found in Table 1. The discrimination of these two types of inhomogeneity is important because each has different biological implications. Macro-heterogeneity is typically easier to observe and may suggest a bistability/multistability feature of the system while micro-heterogeneity simply refers to the dispersion in the distribution of a trait (e.g. mRNA or protein). Furthermore, the mechanisms that cells employ to maintain macro-heterogeneity or transition between multiple stable states remain to be elucidated.
Table 1.
Terms used in the analysis of stem cell population heterogeneity.
| Gene expression heterogeneity | Cell-to-cell variation in the expression level of a gene or genes in an isogenic cell population |
| Micro-heterogeneity | The dispersion in the distribution of a trait (e.g. specific protein content) within a state (attractor) |
| Macro-heterogeneity | The multimodal distribution of a trait |
| Cell-fate heterogeneity | Varying lineage specification potential among cells exposed to the same environmental condition(s) |
| Gene expression noise | Fluctuations in gene expression due to stochasticity in pertinent biochemical reactions, e.g. random promoter activation, transcription bursts, and mRNA/protein degradation |
| Extrinsic noise | Noise contributed from sources affecting cell properties globally, e.g. asynchronous proliferation and asymmetric division |
It is becoming increasingly clear that studies on the diversity of stem/progenitor cell populations will require methodologies with single-cell and even single-molecule resolutions. Routine biochemical assays such as reverse transcription-polymerase chain reaction (RT-PCR), and western blotting provide population-level information with property averages (mainly gene or protein expression levels) masking micro-/macro-heterogeneity. This makes imperative the development of novel methods for the high-throughput analysis of single cells. In this review, we discuss the adaptation of existing methods as well as emerging technologies for the genomic, transcriptomic and proteomic profiling of single stem cells within populations.
The high content and complexity of information generated by single-cell analytical methods necessitates synergistic efforts in parallel with mathematical and computational modeling. Stem cell specification is an intricate process entailing multilevel interactions among extensive GRNs, extracellular signals and intercellular cross-talk. Deeper understanding of these interactions is often confounded by the multiple sources of ‘noise’ present in stem cell processes. Equally important is the fact that those processes span multiple temporal and physical scales. For example, DNA transcription and translation transpires in seconds to minutes or faster whereas cell division occurs every 10-30 h. Diversity in cell populations is also affected by subcellular actions (e.g. by transcription factor networks or signal transduction), cell-cell and cell-substrate processes (e.g. paracrine effects, extracellular matrix components), and population-wide regulation (e.g. interactions among phenotypically dissimilar subpopulations). Here, we review quantitative frameworks for the analysis of stem cells. Models commonly fall within two categories: those describing (temporally evolving) traits within an individual cell (or groups of identical cells), and those simulating whole ensembles of cells (even with single cell resolution). The latter provide a vista of heterogeneity at the level of the population. Approaches based on the landscape model of cell states introduced by Waddington (1957)) are also presented.
2. Experimental methods for the analysis of stem cell population heterogeneity
Traditional experimental methods such as western blotting and quantitative PCR (qPCR) commonly employed for cell analysis yield end-point, population-average information. As such their utility is limited for addressing questions about the origins and role of heterogeneity on the evolving properties and fate specification of stem/progenitor cell ensembles. Instead, real-time analysis at single-cell resolution is necessary to investigate cell-to-cell variability (Table 2). Techniques routinely used for analyzing single cells include mainly flow cytometry and fluorescence-activated cell sorting (FACS) (Chang et al., 2006, 2008; Hayashi et al., 2008; Kalmar et al., 2009), and fluorescence microscopy (Davidson et al., 2012; Kalmar et al., 2009) including time-lapse microscopy (Eden et al., 2011; Smith et al., 2010). Yet, recent advances reviewed below have brought forward powerful new methods for high-throughput analysis of the genome, transcriptome and proteome of single cells (Fig. 1). The obtained data allow for the visualization of cell properties not just as mean values but as probability density distributions providing researchers with new insights about stem cell heterogeneity and associated mechanisms.
Table 2.
Comparison between population-level and single-cell analyses.
| Single-cell analysis | Population-level analysis | |
|---|---|---|
| Resolution | Single cell, subcellular | Cell populations |
| Data pattern | Distribution/scatter | Average value, standard deviation (based on number of experimental replicates) |
| Dimensionality | Multiple (O(10–103)a) markers can be assessed at genomic and mRNA levels, more limited (O(10)) at protein level | Multiple marker (up to O(104) or higher) assessment is possible |
| Downstream data processing | Significantly higher | Lower |
| Required detection sensitivity | Higher | Lower |
| Instrumentation requirements | High | Relatively low |
O(): order of magnitude.
Fig. 1.
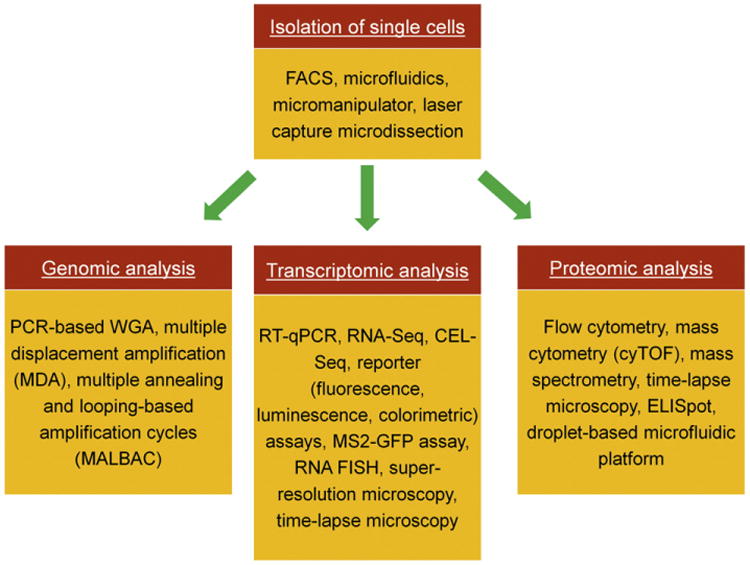
Methods for single-cell analysis. After isolation of single cells, various techniques are available for profiling the genome, proteome (and metabolome) or transcriptome of single cells in a population.
Various methods in this review are available for single cell analysis depending on the desired type of profiling (e.g. genomic) as shown in Fig. 1. The typical workflow entails the culture of cells in monolayers (e.g. single cells or colonies) or three-dimensional (3D) structures (e.g. aggregates), subsequent dispersion into single cells and selection followed by acquisition of pertinent data. Cell isolation is accomplished by FACS, micromanipulation or the use of microfluidic devices in addition to laser capture microdissection, which can be used for the isolation of cells from tissues as well. Some methods such as live-cell time-lapse microscopy, which provides real-time information for individual cells, require the genetic modification of cells with reporter transgenes (Giudice and Trounson, 2008) under particular promoters. Furthermore, cells are maintained in chambers or microfluidic devices with controlled conditions (e.g. humidity, gas, pH, temperature) allowing continuous microscopic observation. Besides the relatively high capital costs associated with these setups, cells can only be maintained for short periods (typically a few hours) compared to the duration of differentiation experiments (days). Computational image processing (e.g. segmentation algorithms Chirieleison et al., 2011; Fero and Pogliano, 2010) increases the throughput by reducing the time for analysis and eliminating user bias. However, other methods may be more straightforward technically but at the expense of providing only static rather than dynamic information. Flow cytometry and fluorescence microscopy of fixed cells are implemented to obtain ‘snapshots’ of populations with single-cell resolution at different time points. Flow cytometry analysis can be performed on live cells expressing a transgene or incubated with a non-cytotoxic dye (e.g. carboxylfluorescein diacetate succinimidyl ester (CFSE)), and on fixed cells after immunostaining. Live-cell flow cytometry can be combined with FACS for narrowing down to subpopulations of interest for the acquisition of data over time. Here, cell manipulations including repetitive sorting may have adverse effects on stem cell viability and differentiation propensity. Moreover, both flow cytometry and fluorescence microscopy require labeling cellular components with high specificity. Therefore, label-free methods based on differential characteristics (e.g. morphology, adhesion Singh et al., 2013) for the analysis of single cells are highly desirable. To that end, microfluidic platforms present an appealing technology for developing tools for isolating and analyzing single cells under defined microenvironments (‘niches’) (Chung et al., 2005; Zare and Kim, 2010; Zhong et al., 2008).
2.1. Single-cell analysis by time-lapse microscopy
Time-lapse microscopy has been utilized to monitor protein expression dynamics in live single cells within populations (Fig. 2). Typically, cells are transfected or incubated with a fluorescent reporter (e.g. GFP, CFSE) prior to their observation. Previously, time-resolved microscopy was applied to bacteria and yeast carrying two reporters (e.g. YFP, CFP) flanked by identical promoters to demonstrate stochasticity in gene expression (Fig. 3). Elowitz et al. (2002) utilized this dual reporter assay to discern noise (Table 1) due to the low copy number of transcripts and random promoter activation (intrinsic noise) from other sources including asynchronicity among cells (extrinsic noise) in Escherichia coli bacterial populations. Detailed mathematical analysis of the intrinsic and extrinsic noise concepts can be found elsewhere (Swain et al., 2002).
Fig. 2.
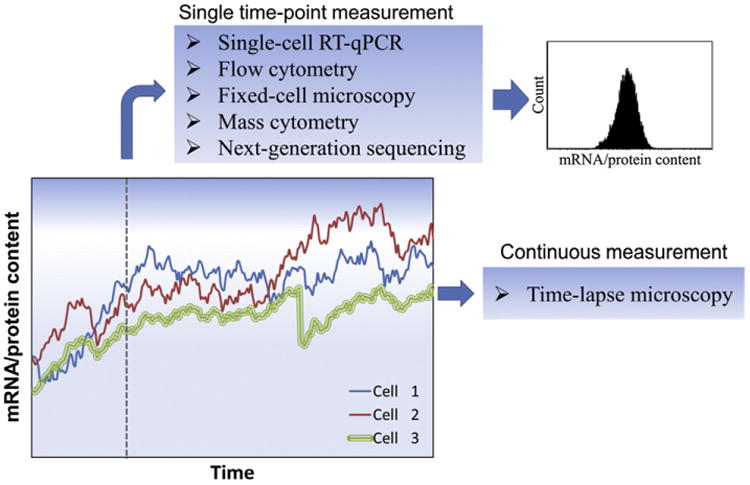
Static and dynamic methods for investigating stem cell population heterogeneity. The levels of mRNA molecules and proteins fluctuate over time with significant variation among individual cells. Data from single-cell analysis assays yield distributions of the measured properties. However, only a subset of these assays (e.g. time-lapse microscopy) is suitable for dynamic monitoring of cellular properties of interest.
Fig. 3.
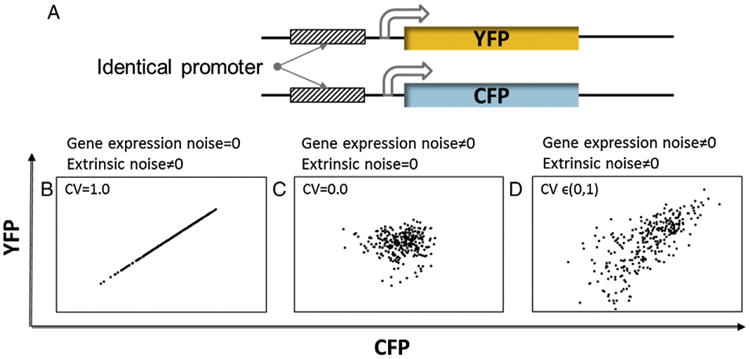
Measurement of gene expression noise with a dual-reporter system. (A) Two fluorescent reporters such as CFP and YFP are expressed from identical promoters of the gene of interest. Differences in the CFP and YFP signals from a single cell are attributed to gene expression (intrinsic) noise whereas extrinsic noise is manifested as differences in the fluorescence among cells in the population. Scatter plots of protein level obtained from a cell population assuming the presence of (B) only extrinsic noise, or (C) gene expression noise, or (D) both types of noise. Because the extrinsic noise affects protein levels globally, the signatures from the two reporters are strongly correlated as quantified by the coefficient of variation (CV). However, the effect of gene expression noise is random indicating no correlation between the levels of the two reporters. Dispersion that is perpendicular to the diagonal in the scatter plot is attributed to gene expression noise.
The division kinetics and cell cycle distribution of rhesus monkey ESCs has also been studied by time-lapse microscopy (Fluckiger et al., 2006). Cells were transduced with a lentiviral vector carrying the gene for the enhanced GFP (eGFP) downstream of the human elongation factor 1α (EF1α) promoter. The results showed a variable cycle duration among 32 individual cells ranging from 12 to 21 h with a median of ∼15 h. Combined with population-level data, these findings led to the conclusion that primate ESCs possess a rapid G1 phase progression with a limited or absent checkpoint control mechanism.
Bistability in the expression of Nanog in mESC populations was also observed by microscopy (Kalmar et al., 2009). Mouse ESCs engineered to express GFP from the nanog locus (Chambers et al., 2007) exhibited a bimodal Nanog (GFP) distribution. The low Nanog (LN) expression state was characterized by a shorter residence time since LN mESCs transitioned to a high Nanog (HN) expression state within several cell cycles while the reverse switching was less frequent. Additionally, FACS-isolated cultured LN mESCs exhibited a higher propensity for differentiation than HN cells.
High-resolution time-lapse visualization was also deployed in the tracing of single mouse embryonic fibroblasts (mEFs) after being reprogrammed to iPSCs over two weeks (Smith et al., 2010). Mouse fibroblasts with doxycycline-inducible expression of pou5f1 (Oct4), sox2, klf4 and myc (c-myc) transgenes (Wernig et al., 2008) were transduced with lentiviruses carrying fluorescent reporters (GFP, YFP, RFP, or CFP fused to β-actin) before initiation of reprogramming. Reporter expression allowed tracking of single mEFs and their corresponding progeny by image acquisition and processing using the Cell Profiler software (Carpenter et al., 2006). Successfully reprogrammed cells underwent a rapid shift in their proliferation rate. Besides the identification of proliferation and morphological attributes preceding the emergence of pluripotency markers, the use of live-cell microscopy made it possible to discern among proposed models of reprogramming (Hanna et al., 2009; Yamanaka, 2009). For example, the normal distribution of colony compaction times from microscopy data of cells during iPSC generation is consistent with a model in which a series of (not stochastically timed) steps in a lineage leads progressively to a reprogrammed state.
Miyanari and Torres-Padilla (2012) recently reported an adaptation of the dual-reporter system (Fig. 3) to mESCs for monitoring the allelic expression of nanog. The expression pattern for Nanog was strikingly distinct from that of other ‘stemness’ genes (e.g. Pou5f1 (Oct4)) which are transcribed from both alleles. In early embryos and cultured mESCs, nanog levels are heterogeneous with only 30% of mESCs demonstrating biallelic expression. Imaging of blastocysts and cultured mESCs from transgenic mice carrying fast-degrading variants of two reporters (eGFP, mCherry) each embedded in a single nanog allele, revealed unexpected allelic switching of nanog expression depending on the stage of development or on the culture conditions. Besides RNA fluorescence in situ hybridization (RNA FISH) and RT-qPCR, live cells were tracked microscopically over time. The data indicated that the majority of cells (over 98%) switching between patterns of allelic Nanog expression did so over multiples of the cell cycle time. Interestingly, enhanced biallelic nanog expression and less heterogeneous distribution of the Nanog protein were observed when mESCs were cultivated with inhibitors of MEK (also known as mitogen activated protein kinase kinase (MAPKK)) and GSK3β (‘2i’ condition). Taken together, stochastic Nanog expression at the chromosomal level is a source of NANOG expression heterogeneity in stem cell populations. This was also shown recently based on mathematical modeling (Wu and Tzanakakis, 2013).
Davidson et al. (2012) reported that activation of Wnt/β-catenin signaling varied among hESCs of the same population exposed to particular stimuli (Wnt3a ligand, GSK3 inhibitor CHIR99021, etc.). For this purpose, a β-catenin activated reporter (BAR) featuring a TCF/LEF binding element repeat (12×) upstream of the Venus or luciferase gene was used to create stable hESC lines. The construct also contained a selectable marker (DsRED) constitutively expressed from a ubiquitin promoter. DsRED+ hESCs exhibited a distribution of Venus signal indicative of the intrinsic varied activity of canonical Wnt signaling. The authors confirmed that reporter heterogeneity was due to variations in levels of β-catenin signaling and not because of transgene silencing. For this purpose, DsRED+ cells were sorted by FACS and transcriptionally analyzed by RT-qPCR It is deduced that this cell-to-cell variation in Wnt signaling contributes to the differential response to factors (including Wnt-related molecules) promoting stem cell commitment and the resulting heterogeneous progeny.
Fluorescence microscopy has also been utilized to study the heterogeneity of ESC and progenitor cell populations with respect to their expression of other markers. Stewart et al. tracked SSEA-3+ hESCs after sorting and reported a faster proliferation than that of their SSEA-3− counterparts (Stewart et al., 2006). Moreover, the two subpopulations displayed differences in the expression levels of stem cell transcription factors, clonogenic capacity, morphology and cell cycle attributes.
A recently developed tool (Sakaue-Sawano et al., 2008), the fluorescent ubiquitination-based cell cycle indicator (FUCCI), allows monitoring the transition of live cells through different cell cycle phases. The assay principle is based on the variable expression of the Cdc10-dependent transcript 1 (Cdt1) and geminin proteins during cell cycle. Cdt1 participates in the formation of a ‘prereplicative complex’ during the late M-to-G1 phases recruiting DNA helicase for initiation of DNA replication. With replication commenced during G1/S, Cdt1 is targeted by E3 ligases for ubiquitin/proteasome degradation. Geminin, which is expressed during the S/G2/early M phases, binds Cdt1 preventing aberrant reactivation of DNA replication. Degradation of geminin takes place during mitosis and early G1 removing the inhibition on Cdt1, which licenses a new round of DNA replication. Shortened versions of the Cdt1 (30-120 amino acid region mediating ubiquitination) and geminin (amino acids 1-110) are fused to fast-folding variants of fluorescent reporters for use in the FUCCI assay. Cells transfected or transduced with expression vectors of the DNA encoding for these fused proteins exhibit changes in fluorescence indicating their phase in the cell cycle. Stable expression of the FUCCI constructs is preferable due to the division-induced dilution in copy number of non-integrated transgenes.
The FUCCI construct is used to tag cells at different cell cycle stages and track them during culture with live cell imaging and/or sort them via flow cytometry. Information about the cell cycle stage is particularly important for stem cells as changes in the length of different phases has been linked to the propensity of cells to self-renew or adopt particular fates. For instance, the duration of the G1 phase lengthens from 8 h at the start of neurogenesis to 18 h by day E16 in mouse embryos (Takahashi et al., 1995). Using the FUCCI technology, mESCs were shown to have a very short G1 phase (Roccio et al., 2013). Based on this observation, neural stem cells (NSCs) were induced to a pluripotent state at a higher proportion by selecting FUCCI-NSCs which were about to enter the G1 phase at the early stage of reprogramming. A software suite (FUCCIJ) was also developed, based on the ImageJ software, for the automated analysis of cell cycle phases in single cells from time-lapse fluorescence microscopy images.
Software-based analysis is becoming an integral part of dynamic fluorescent microscopy. Desai et al. (2013) studied the osteogenic differentiation in adipose-derived stem cells through fluorescence microscopy utilizing MATLAB and CellProfiler modules for image segmentation. The cells were tagged non-destructively with molecular beacons targeting mRNA sequences for alkaline phosphatase, type I collagen and osteocalcin. A temporal gene expression profile was constructed from repeated measurements in differentiating cells over 21 days. The differentiation response of adipose-derived stem cells cultured in osteogenic medium became uniform after 2 weeks indicating that cell commitment becomes temporally synchronized.
Observation of fixed cells or tissue sections by fluorescence microscopy is a common method for analysis of single cells and populations presenting fewer challenges than time-resolved microscopy of live specimens but does not permit the continuous monitoring of cellular processes. Still, high-content information such as cell morphology and marker expression levels can be obtained from fixed cells. Suh et al. (2007) generated Sox2-GFP transgenic mice and observed two Sox2-expressing but morphologically distinct populations in the subgranular zone in vivo. Further analysis showed that a subpopulation of Sox2+ cells gives rise to neural stem cells to maintain the constant size of the neural stem cell pool in vivo.
2.2. Single-cell analysis by flow cytometry/FACS
Flow cytometry provides DNA, RNA or protein (e.g. transcription factor, cell surface receptor) content or enzymatic activity information at single-cell resolution commonly through the use of appropriate fluorescent probes. Typical applications include the quantitation of DNA content for cell cycle phase distribution and aneuploidy, tagging of proliferating cells after incorporation of bromodeoxyuridine (BrdU) or 5-ethynyl-2′-deoxyuridine (EdU) (Salic and Mitchison, 2008), determining differences in proliferation kinetics with CFSE staining (Koche et al., 2011), measuring changes in intracellular calcium flux, and identifying (e.g. side population) cells through efflux of fluorescent dyes. The multiparametric profiling of cells achieved by flow cytometry may be more representative of the developmental and functional states of stem cells and their progeny than transcriptome-level information since changes in mRNA may not be directly translated to the protein level (Lu et al., 2009). Moreover, at least 104–105 cells (104 cells/s) can be processed with most current flow cytometry systems yielding results which are representative of the entire cell population. Besides their analysis, desired subpopulations can be further maintained for studies after being sorted by FACS. In most instances, this requires either live-cell staining limited to cell-surface markers or the engineering of cell lines expressing fluorescent reporters under appropriate regulation.
However, there are also challenges in the analysis of stem cell heterogeneity by flow cytometry. First, instrument calibration, most often repetitive, is necessary for consistency between experiments (Mittag and Tarnok, 2009; Zenger et al., 1998). Second, unlike the analysis of the expression in single cells by qPCR of 10–20 or more genes, routine flow cytometry measurements are typically limited to 2–4 species based on the availability of excitation sources and emission detectors. And although flow cytometry systems with up to 16 channels are available for resolving fluorescence for sample analysis and sorting, compensation problems should be carefully addressed (Herzenberg et al., 2006). Lastly, sample analysis by flow cytometry is commonly performed as an end-point assay at distinct time points rather than in a continuous fashion limiting the use of the method for investigating the dynamics of fast-rate processes.
Nonetheless, flow cytometric analysis has been pivotal in studying the heterogeneity of stem cell populations based on the expression of several markers. For example, the profile of expressed Sca-1 (also known as Ly6a) in mouse hematopoietic stem cells was shown to span over three orders of magnitude via flow cytometry (Chang et al., 2008). Further isolation by FACS and culture of Sca-1low and Sca-1high cells revealed that both fractions restore the distribution of Sca-1 to its initial state pointing to the existence of mechanisms for robust maintenance of population heterogeneity (Fig. 4). Upon differentiation, Sca-1low cells gave rise to erythroid cells whereas Sca-1high cells yielded myeloid cells suggesting that Sca-1 heterogeneity is linked to variable proclivities for cell specification. Similarly, robust heterogeneity has been demonstrated in ESCs expressing Nanog, SSEA-3 and Stella (Chang et al., 2008; Hayashi et al., 2008; Kalmar et al., 2009). In fact, a bimodal distribution was shown for Nanog in mESCs and hESCs by flow cytometry (Chambers et al., 2007; Fischer et al., 2010).
Fig. 4.
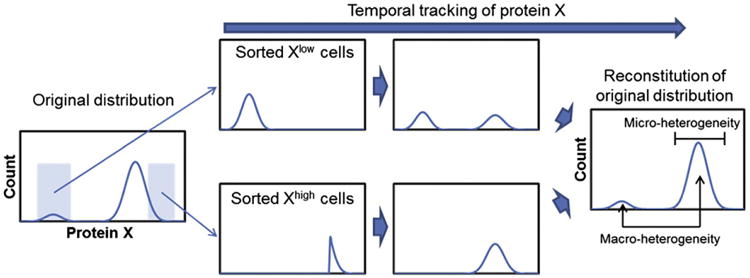
Robustness of stem cell population heterogeneity exhibited in protein content measured via flow cytometry/FACS. Protein X conforms (for the purpose of illustration) to a bimodal distribution in the original population. However, cells with extreme (high or low) expression levels of the protein can be sorted and the respective subpopulations tracked over time. Their signature of protein X converges to the original bimodal distribution and heterogeneity. Macro-heterogeneity refers to the existence of multiple distinct peaks whereas the heterogeneity within a subpopulation or state is termed micro-heterogeneity (Huang, 2009).
2.3. Single-cell analysis beyond time-lapse fluorescence microscopy and flow cytometry/FACS
In this section, recent advances are presented in methods for genomic, transcriptomic and proteomic characterizations of single cells. These methods are based on the same principles as the corresponding assays for multicellular specimens but have been modified to various extents for adaptation to one-cell samples. The assay workflow entails the isolation of single cells from tissues or tissue cultures, their lysis and component extraction, and performance of the appropriate reaction(s) such as reverse transcription (RT), polymerase chain reaction (PCR), etc. The relatively small amount of material obtained from a single cell becomes a significant workflow bottleneck. This can be further compounded by sample dilution, mixing and biased pre-amplification (e.g. of RNA). These challenges have led to advances in molecular biology protocols with the development and use of enzymes with high-processivity beyond that of traditional polymerases. Similarly, microfluidic technologies have been adopted for on-chip culture, isolation and analysis of single cells.
2.3.1. Genomic analysis of single cells
A subset of various whole-genome analysis (WGA) methods developed so far has been adapted to the genomic investigation of single cells. In PCR-based WGA random primers are utilized for exponential amplification of the DNA. Variations to the typical PCR scheme have been reported, including degenerate oligonucleotide-primed PCR (Cheung and Nelson, 1996) and ligation-mediated PCR (Klein et al., 1999), which increase the amplification efficiency and genome coverage. Nonetheless, the coverage of PCR-based WGA can generally be limited due to the sequence-dependent bias from exponential amplification with random primers. This issue has been largely tackled with the deployment of multiple displacement amplification (MDA) in which random primers are used in conjunction with isothermal amplification by the bacteriophage φ29-derived strand-displacing DNA polymerase (Dean et al., 2001; Spits et al., 2006). MDA has also been combined with microfluidic platforms (digital MDA) for the absolute quantification of DNA. Using digital MDA, Quake and colleagues demonstrated that background amplification arises from high molecular weight DNA contaminants (Blainey and Quake, 2011). More recently, a new WGA method termed multiple annealing and looping-based amplification cycles (MALBAC) was described involving a quasi-linear pre-amplification step to minimize the bias due to nonlinear amplification (Zong et al., 2012). The method reproducibly yielded 85–93% genome coverage of single SW480 human cancer cells while allowing for the identification of single-nucleotide variations (SNVs) over successive clonal passaging of the cells. Employing MALBAC to reveal SNVs and other genetic alterations in cultured hPSCs will be of great interest in gaining a deeper understanding of the effects of expansion conditions on stem cells.
2.3.2. Single-cell transcriptome analysis
Stem cell population heterogeneity is largely reflected in transcriptome differences among individual isogenic cells. Analysis of the transcriptional profile of single cells aims to make connections between their genotype and phenotype. Gene expression in single cells displays temporal fluctuations with significant ramifications for cell fate decisions. In addition to the stochastic nature of the transcription of a single gene from one or two allelic templates of genomic DNA, heterogeneityingene expression among cells with identical genotype is imparted by the differences in cell cycle, microenvironment, and epigenetic states. Thus, single-cell transcriptional profiling is critical for gaining a deeper insight of heterogeneity and its role in stem cell self-renewal and differentiation.
Transcriptome analysis of single cells has yielded new information regarding the heterogeneity of self-renewing and differentiating stem cell populations. The heterogeneity of hESCs expressing transcription factors such as Nanog, Oct4 and Sox2 was demonstrated by carrying out single-cell RT-qPCR for 100 cells collected by glass capillaries (Stahlberg et al., 2009). Up to 100-fold differences were observed in the expression of these genes among different cells. Such variations are significant for ESC lineage specification given that even smaller perturbations cause dramatic changes in the transcriptional regulatory net-works of mESCs and hESCs (Boyeretal., 2005; Luetal., 2009; MacArthur et al., 2012). In fact, single-cell transcript analysis revealed the co-expression of pluripotency and differentiation markers at the onset of differentiation (Gibson et al., 2009). During their incubation with activin A, BMP4 and serum, ∼20 differentiating hESCs were selected during a 7-day experiment and analyzed for the expression of various genes with low density array cards. Pluripotency (e.g. POU5F1, NANOG, SOX2) and differentiation (e.g. EOMES, T, FOXA2, AFP) genes were not only expressed simultaneously but their profiles could be classified as uniform, absent and sporadically detected. These expression patterns were linked to the chromatin states of respective promoters. Lastly, single-cell qPCR has revealed increased heterogeneity in several human iPSCs lines compared to hESC lines (Narsinh et al., 2011). These studies exemplify the applicability of single-cell transcriptomics in expanding our knowledge of processes for the maintenance of pluripotency, lineage commitment and reprogramming and are expected to facilitate the development of more efficient differentiation and reprogramming protocols.
Analysis of transcripts in single cells is mainly based on the conversion of RNA into complementary DNA (cDNA) by RT and either exponential amplification by PCR (Brady et al., 1990) or linear amplification by in vitro transcription (IVT) with a T7 RNA polymerase (Eberwine et al., 1992). Generating cDNA from whole cell lysates by RT entails the use of oligo(dT) primers whereas random primers tend to amplify the more abundant rRNA and tRNA (mRNA accounts for only ∼1% of the 10 pg of total RNA per cell). Even with the use of oligo(dT) primers the majority of amplified cDNA corresponds to (3′ end) RNA fragments of up to 3 kb but it should be noted that approximately 35% of genes correspond to longer mRNAs.
Amplification by PCR is relatively fast and the protocols for single cell analysis have undergone significant optimizations in recent years. Nonetheless, the increase of dimers and other non-specific products particularly during the late cycles and the amplification bias toward more plentiful moieties present challenges for PCR-based analysis of single cells. These issues are largely addressed with IVT of cDNA which is characterized by linear amplification and reduction of non-specific species. This method however is more laborious than PCR, results in less than 1000-fold amplification and the amplified transcripts are shorter compared to the template mRNAs (Livesey, 2003).
The combination of single-cell cDNA amplification methods (Kurimoto et al., 2007) with next generation sequencing has largely overcome the issue of short amplicons (Tang et al., 2009). RNA sequencing (RNA-Seq) protocols have proven effective in analyzing the transcriptome of single ESCs (Tang et al., 2010). The expression of 10,815 genes was detected by RNA-Seq compared to ∼6800 genes reported by single-ESC cDNA microarrays (Kurimoto et al., 2006). Deep sequencing analysis of single-cell cDNA has a dynamic range of over 5 logs but should be accompanied by sufficient computational power for data processing with available bioinformatics software (Pepke et al., 2009). RNA-Seq has also been combined with IVT for the transcriptional analysis of single cells (Hashimshony et al., 2012). The protocol, dubbed CEL-Seq for Cell Expression by Linear amplification and Sequencing, starts with the RT of mRNA from single cells. The RT reaction for each cell is supplemented with a primer containing a unique barcode upstream of a polyT, a 5′ adaptor and the T7 promoter. After synthesis of the second strand, the samples are pooled to yield sufficient RNA amounts (above ∼400 pg) for subsequent IVT. The resulting RNA is fragmented at a second adaptor ligated at the 3′ end and reverse-transcribed to DNA. The fragments containing both adaptors and the barcodes (each corresponding to an individual cell) are selected by PCR and the resulting library is analyzed by pair-end sequencing for the identification of mRNA transcripts from individual cells. CEL-Seq is not only suitable for multiplexing but also highly sensitive and reproducible as runs with single mESCs and differentiating cells from Caenorhabditis elegans embryos have shown. Nonetheless, miRNAs (or non-polyadenylated transcripts) cannot be detected and alternative splice forms cannot be discerned due to the strong 3′ bias.
In addition to direct measurement of transcript levels, other methods allow for indirect determination of mRNA in single cells. Such assays include the use of reporter molecules such as luciferase (Castano et al., 1996; de Wet et al., 1987), β-galactosidase and destabilized fluorescent proteins (Li et al., 1998) with computational microscopy. Reporter gene expression may be driven by the same promoter as the gene of interest, for instance, by knocking in the reporter gene in the same gene locus or through delivery to cells of an expression vector featuring the relevant promoter with the reporter gene. The result is the generation of transgene transcripts quantitatively representative of the target mRNA. Reporter proteins have also been fused with RNA-interacting proteins for mRNA analysis in single cells. One such protein has been derived from the bacterio-phage MS2 and binds a specific RNA motif. The MS2 protein–RNA complexes can be visualized after fusion of the MS2 with GFP (Chubb et al., 2006; Tyagi, 2009). However, this approach requires the insertion of the cognate motif to the RNA of interest and the transfection (or homologous recombination) of cells with the engineered cDNA. It is unclear if the engineered transcript retains the characteristics of the native mRNA target. Moreover, GFP fluorescence is present irrespective of whether the MS2-GFP is bound to the target RNA. This results in high background necessitating the meticulous exclusion of the unbound MS2-GFP prior to sample analysis.
RNA FISH has also been employed for transcriptome analysis in single cells (Levsky et al., 2002) illustrating the stochasticity in gene expression (Raj et al., 2006; Wijgerde et al., 1995) which is an important determinant of the cell phenotype. Current RNA FISH protocols require the use of multiple probes complementing each target mRNA molecule (single molecule FISH) facilitating its detection by fluorescence microscopy. The method has been adapted to multiplexing with the simultaneous use of multiple oligonucleotide probes tagged with fluorophores (barcodes) for the detection of transcripts from several genes in single cells. In fact, the measurement of 32 genes concurrently in single Saccharomyces cerevisiae cells was reported recently (Lubeck and Cai, 2012). The visualization of single mRNA molecules is enabled with the use of super-resolution microscopy which allows the imaging of structures with 10–20 nm lateral resolution compared to 200 nm for confocal laser scanning microscopy (Schermelleh et al., 2010). Spatial barcoding relies on the tiling in a particular order of small oligonucleotides tagged with various fluorophores on transcripts. This approach requires high resolution due to the localized hybridization patterns and linearization of the coded molecule. Transcript recognition and quantitation however may also be based solely on distinct color patterns (spectral barcoding). In this way, probes with lower fluorescence intensity can be used as the required resolution is lower than for spatial barcoding. Advantages of the spatial/spectral barcoding over other transcriptional profiling methods include the preservation of spatial information within and among cells, the exclusion of bias due to RNA extraction and cDNA synthesis, and the rapid imaging of multiple samples and cells scaling up the throughput of the method considerably. An important barrier in the implementation of the method is the high cost of super-resolution microscopy systems and probe sets but this is expected to drop with wide adoption of this tool by the research community. It should be noted that with further development in 3D microscopy and the synthesis of far-infrared fluorophores the technique can be scaled to identify over 18,000 genes simultaneously.
Absolute copy numbers can be obtained with single-molecule FISH in single cells (Raj et al., 2008) without the need for spiking the sample with known amounts of distinct mRNA. From a functionality viewpoint however, the concentration of mRNA rather than its absolute copy number may be more important indicating a requirement for size determination of individual cells (Baserga, 2007). Moreover, current state-of-the-art methods for transcriptome analysis involve the lysis or fixation of cells limiting our ability to study gene network dynamics (i.e. actively transcribed/translated mRNAs), for instance, over the course of stem cell differentiation or cell reprogramming. Live-cell imaging with single-cell next generation sequencing will dramatically aid efforts in this direction. Achieving a major objective of single-cell analysis, i.e. linking cell genotype with phenotype and the environment, will also entail the development of improved protocols for the simultaneous analysis of the genome and transcriptome of single cells (Klein et al., 2002).
2.3.3. Analysis of single-cell proteome and metabolome
In addition to flow cytometry/FACS, a recent development for single-cell proteomics is mass cytometry or cytometry by flight-of-time (cyTOF) (Bendall et al., 2011). Cells prepared for cyTOF are stained with metal-conjugated probes and individually atomized and ionized rendering this an ‘end-point’ assay. The probes contain isotopes that are not commonly found in biological specimens. These isotopes from each atomized cell are separated by mass yielding characteristic time-of-flight signatures. Signal intensity is proportional to the number of antibodies originally bound per cell. CyTOF was recently implemented for the concurrent detection of 30 surface markers and 9 cytokines in human peripheral blood mononuclear cells (PBMCs) (Bodenmiller et al., 2012). For this purpose, seven tags were used to analyze 14 phosphorylation sites in 14 PBMC types under 96 conditions. This study showed that cyTOF displays higher multiplexing capacity than flow cytometry. Yet, there is still room for improvements especially in the throughput (currently around 103 cells/s) of cyTOF and the sample volumes required for proper analysis (Bendall et al., 2012). As of this writing, there were not many published reports of cyTOF analysis of stem cells but several such investigations are underway and the findings are forthcoming.
Mainstream proteomic analysis of biological samples is carried out by mass spectrometry (MS), which undergone several refinements resulting in increased resolution, specificity and sensitivity. Unlike with antibody-based assays, prior knowledge of protein targets is not necessary for MS analysis. To that end, large-scale studies have been reported to better understand complex mechanisms of pluripotency by analyzing the ESC proteome (Nagano et al., 2005; Schulz et al., 2007), including post-translation modifications and comparisons with the iPSC proteome (Brill et al., 2009; Phanstiel et al., 2011). Application of MS technology has also been reported in comparative studies between cell states and particularly during differentiation (Phanstiel et al., 2008). Analysis of histone and other chromatin modifications showed that self-renewing stem cells retain a more open and accessible chromatin than differentiated cells. MS has also been used for identification of interactions (e.g. after immunoprecipitation) of proteins such as Nanog (Wang et al., 2006), Oct4 (Pardo et al., 2010) and Sox2 (Mallanna et al., 2010). Studies like these have brought forward key proteomic components of networks involved in pluripotency and lineage commitment expediting directed research.
From recent studies on the metabolic profiling of stem cells and their derivatives it becomes increasingly clear that metabolism is a defining characteristic of the cell state. Particularly for cultured cells, metabolomics may provide information about the consumption and secretion rates of compounds in supernatants or media, and metabolite changes measured in cell extracts and intact cells (Khoo and Al-Rubeai, 2007). Like in cellular proteome analysis, metabolomic studies have relied mostly on the use of MS, which is more sensitive that other available methods and requires lower initial capital investment and equipment maintenance costs. Using MS, Yanes et al. reported a reduction in the abundance of unsaturated metabolites (phospholipids) as ESCs undergo commitment to cardiac muscle cells and neurons (Yanes et al., 2010) conjecturing that stem cells differentiate in response to oxidative processes such as inflammation. Differences in polyunsaturated phosphati-dylcholines and other metabolic compounds between mouse iPSCs and mESCs and between iPSCs and the parent fibroblasts undergoing reprogramming (Meissen et al., 2012). In addition, analysis of mESC extracts showed dependence of cell proliferation in vitro on threonine catabolism (Wang et al., 2009a). Others have also proposed the preference for glycolysis and anaplerosis of glucose at the cost of effective ATP production by complete oxidative phosphorylation in hematopoietic stem cells (Simsek et al., 2010), hESCs and during reprogramming of somatic cells to hiPSCs (Panopoulos et al., 2012) although findings contradicting this proposal have also been described (Birket et al., 2011).
Nuclear magnetic resonance spectroscopy (NMRS), which is based on the identification of chemical shifts of resonance frequencies of nuclear spins in an externally applied magnetic field, is the most commonly used method after MS for metabolomics analysis. For example, stem cells implanted in the circle of Willis of Parkinson patients, were monitored via NMRS in a clinical magnetic resonance imaging (MRI) scanner (Brazzini et al., 2010). Moreover, mobile lipids have been detected via proton magnetic resonance in ESCs and ESC- and iPSC-derived neural stem cells (Loewenbruck et al., 2011). Choline-containing compounds linked to cell proliferation have also been monitored via NMRS in self-renewing and differentiating stem cells. A decrease in the ratio of phosphocholine to glycerophosphocholine was noted during fate specification of mESCs and mouse embryonal carcinoma cells (Romanska et al., 2009). Compared to MS, NMRS has lower sensitivity, and relatively higher instrumentation cost but it does not require the destruction (e.g. cell lysis) of samples and may be used in a quantitative high-throughput fashion.
Optical spectroscopy combining traditional spectroscopy methods such as Raman (based on inelastic scattering) or Fourier-transform infrared (relying on absorption) with microscopy have also been used for stem cell metabolome analysis. This category of technologies has the potential to tie spatial information (e.g. the position of cells within a tissue-like structure) to metabolomics data without requiring labeling of cells yet its use so far has been mostly for qualitative rather than quantitative investigations. To that end, mESCs and hESCs lower their RNA-to-protein ratio during neural and cardiac differentiation in culture, respectively (Chan et al., 2009; Ghita et al., 2012). Raman microspectroscopy (RMS) has also been employed to determine the cell cycle phase, differentiation status and intracellular glycogen content in populations of hESCs providing tangible evidence about the potential of RMS for stem cell analysis (Konorov et al., 2011, 2013; Schulze et al., 2010).
These reports also indicate that we are closer to obtaining single-cell read-outs by RMS and MS-based methods. Indeed, despite their broad use with multicellular samples, efforts to adapt MS and related methods to the analysis of single cells are now bearing fruits. In a recent study, peptides and metabolites at femtomole quantities from single cells were quantified by matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) MS (Rubakhin and Sweedler, 2008). More recently, Ibanez et al. (2013) spotted single yeast cells on transparent glass microarrays and analyzed them individually via MS to observe changes in glycolysis particularly in the presence of chemical (glycolysis inhibitor 2-deoxy-d-glucose) and genetic perturbations (deletion of the PFK2 gene coding for a phosphofructo-kinase isoenzyme). Analysis of multiple single cells suggested that the wild-type yeast population is metabolically heterogeneous comprising two main subpopulations with high or low fructose-1,6-bisphosphate, respectively. The results were characterized by low coverage with only 26 moieties observed (21 correctly identified) pointing to one of the challenges for future metabolomic investigation of single cells, including stem cells. As these report showed, single-cell analysis will benefit from the integration of MS with microfluidic platforms affording analyte extraction with low dilution, rapid cell lysis, sharp separation and sensitive detection.
Other methods capitalizing on recent advances in microfluidic technologies for single-cell proteome profiling include the enzyme linked immunosorbent spot (ELISPOT) assay (Moodie et al., 2010), the single-cell barcode chip (Ma et al., 2011), and the droplet-based detection of molecules secreted by single cells (Konry et al., 2011).
3. Mathematical and computational modeling for stem cell population heterogeneity
Stem cell fate decisions rely on regulatory circuits of transcription factors, signaling pathways and extracellular molecules. One can argue that these networks control the specification of stem cells deterministically. This can readily be concluded by the dose-dependent response of stem cells to differentiation stimuli. The molecular processes however underlying regulatory networks are characterized by random fluctuations imparting stochasticity to the specification of stem cells. This was demonstrated early on by Till et al. (1964) for the proliferation of spleen colony-forming cells (‘colony-forming units’ or CFUs). The portion of CFUs varied greatly from colony to colony akin to a ‘birth’ (self-renewal) and ‘death’ (differentiation) process demonstrating the stochasticity of cell fate decisions. The combination of stochastic and deterministic controls confounds studies of the behavior of stem cell populations based solely on experimental methods. Parallel implementation of mathematical modeling can enhance our understanding of the molecular partners participating in cell fate changes and relevant control mechanisms.
Isogenic stem cell populations are heterogeneous with differential propensities to exit self-renewal or commit along a particular lineage, diverse epigenetic states and varying levels of markers such as NANOG, Stella, SSEA-3 and Oct4 (Chambers et al., 2007; Hayashi et al., 2008; Stewart et al., 2006; Toyooka et al., 2008). Stochasticity in gene expression is a major contributor of the observed population diversity affecting greatly the adoption of phenotypes by individual cells. Reducing this heterogeneity is often without merit except maybe at the initial stages of a study where experimental convenience and rapid evaluation using traditional whole-sample methods such as qRT-PCR or western blot analysis are desirable. The irreducible inhomogeneity makes modeling of stem cell systems challenging.
The stochastic nature of processes mediating self-renewal and differentiation imparts randomness in stem cell ensembles also posing difficulties when quantitative descriptions are sought. Although reducing randomness, for instance by employing a set of (typically time-dependent) ordinary differential equations (ODEs), leads to more tractable deterministic models and possible inference of some properties of the system, stochastic fluctuations are still present. That is, even in large ensembles in which stochastic effects may be less prominent, gene expression and the communication among cells and with the rest of their environment remain noisy. Therefore, randomness also is an irreducible attribute of stem cell populations and pertinent models.
Depending on the desirable granularity of the model, interactions among different components are also a complicating factor. Such interactions can be considered at the molecular level, for example, between transcription factors or other proteins and DNA (e.g. via RNA-chromatin immunoprecipitation or RNA-ChIP), or among cells via signaling. The latter is evident, for example, in co-cultures of stem cells with endothelial cells, pancreatic anlage or Sertoli cells coaxing them to adopt a cardiomyocyte, endocrine islet cell or neuronal cell fate, respectively (Leon-Quinto et al., 2004; Mummery et al., 2007; Yue et al., 2006).
The mathematical and computational approaches employed for stem cell dynamics can be broadly classified into three categories. The first encompasses models describing the dynamics in an individual cell of one or more properties such as genes, transcripts, proteins, and signaling intermediates. Typically, these models are based on a series of relations among genes of interest (gene networks) or signal transducers (signaling cascades) represented by biochemical reactions or sometimes as circuits. Models of population dynamics fall within the second category focusing on stem/progenitor cell ensembles transitioning among phenotypically, transcriptionally or epigenetically distinct states. Cells within population models may be considered individually or in groups. The last type covers ‘thermodynamic’ models based mainly on the concept of an epigenetic landscape introduced by Waddington (1957). Each state or attractor is represented by a valley in the landscape and cells moving between states must overcome an ‘energy’ or quasi-potential (U) barrier. The second and third model types share the concept of cell states but the former encompasses kinetic models whereas time dependence is largely absent in the thermodynamic theorization of stem cell states and transitions.
The distinct characteristics of each modality may be illustrated in a simple example of gene expression bistability (Fig. 5). A single-cell gene expression model usually entails reactions (interactions) relevant to the target gene or protein. A positive auto-regulation is assumed to be the source of bistability in this system. Then, protein X fluctuations in a single cell are dictated by chemical reaction kinetics and gene expression noise may cause the switch between states. If predictions about heterogeneity of the population are desirable, repetitive simulations corresponding to different cells will yield the distribution of protein X, an approach that falls within the scope of cell ensemble models. Population-based models may also treat different subgroups in the population by assigning specific properties to each subtype and describing interactions among them (transitions between attractors). Cellular activities affecting population heterogeneity such as proliferation, phenotype switching and apoptosis characterized by specific kinetics can be embedded in population-based models. This not only allows for the prediction of the temporal evolution of particular properties but also the estimation of the relative contributions of each process to the observed inhomogeneity. Lastly, a landscape model can be implemented for protein (or other trait) X under equilibrium among various states. The driving force behind the transition between different attractors depends on the gradient of the potential U(x), which is a function of the probability distribution of the steady states Pss. This framework informs whether a passage between states is permissible and if so under what conditions but not about the time needed for the transition to transpire.
Fig. 5.
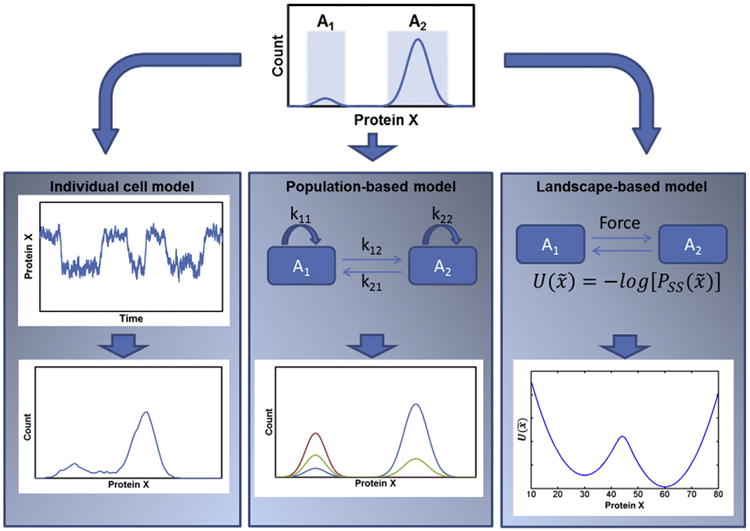
Individual-cell, population and landscape models. A gene with bistable expression profile is considered. A biochemical reaction network is constructed to describe all the production of protein X in a cell. In this model, a cell switches between the A1 and A2 states by virtue of gene expression stochasticity. In the population model, each subtype of cells is assigned specific kinetics for processes such as proliferation (e.g. kinetic parameters k11 and k22), attractor switching (e.g. k12 and k21) and apoptosis in addition to the net rate of generating protein X. A map of the potential energy U(x) is calculated in the landscape model given the steady-state probability distribution (Pss(x)) of the protein. Stem cells require certain force to drive the transition between different attractors.
3.1. Individual-cell models
Models for individual cells typically describe the evolution of gene or protein levels in conjunction with the activity of gene networks or signal transduction pathways. Modeling of GRNs has gained significant interest recently as pertinent genomic analysis methods are becoming commonplace and the amounts of available gene expression data are increasing. Dynamic fluctuations in the expression of genes/proteins are the net effect of biochemical reactions such as transcription, translation and mRNA/protein degradation. Different modalities include Bayesian (Friedman, 2004) and Boolean network models (Shmulevich et al., 2002). Still, the temporal evolution of the amount (e.g. concentration) C(t) of protein or mRNA in a cell can be simply described, for example, with Eq. (1) where r(C) represents the rate of reactions linked to protein/mRNA. The equation can be written in a vector form to include multiple species within a single cell depending on the size of the network under consideration. Various expressions can be employed as rate functions taking into account the production and degradation of moieties. These rates are usually modeled with first-order (linear dependence on substrate), Michaelis-Menten (Wang et al., 2009b) and Hill type kinetics (Kalmar et al., 2009). Moreover, the positive and/or negative feedback loops can be introduced for GRNs as deemed appropriate (Mettetal et al., 2006; Thattai and van Oudenaarden, 2001; Weinberger et al., 2005).
| (1) |
Chickarmane et al. (2006) developed a model describing a network of Oct4, Sox2, and Nanog in ESCs. The model identified a bistable switch in the network, caused by several positive feedback loops under the influence of signals (e.g. environmental cues). The study examined various motifs of interaction among the three partners showing how ESCs can achieve self-renewal intrinsically without extracellular signal support. This model was further extended (Chickarmane and Peterson, 2008) to include modules for ESC lineage commitment orchestrated by Cdx2, Gata6 and the germ cell nuclear factor (GCNF). An excess of Cdx2 over Oct4 results in specification to the trophoectoderm lineage whereas a surplus of Gata6 compared to Nanog leads to ESC differentiation to endodermal progeny.
Although ODE systems are suitable to describe how stem cell fate decisions are affected by certain factors deterministically (e.g. in an all-or-none pattern), they fail to capture the stochastic nature of these phenomena. This has prompted the use of stochastic differential equations (SDEs) for the study of the heterogeneity in cell populations. SDEs combine deterministic terms with a noise term as shown in Eq. (2). Here, ε(t) can be a white noise function with zero mean and standard deviation σ as the noise magnitude. In this form, SDEs have been used for modeling individual stem cells (Kalmar et al., 2009; Wang et al., 2009b), despite the fact that noise quantification is still experimentally challenging. Glauche et al. (2010) constructed a theoretical SDE system to describe the bimodal distribution in Nanog expression. The model, which considered the interaction between Nanog and Oct4, was used to test whether the observed bimodal distribution of Nanog arises from expression noise (‘fluctuation scenario’) or oscillations in the expression of Nanog (‘oscillation scenario’).
Besides the simple SDE described above, other SDE forms have been used for modeling biological systems including the Langevin equation (Gillespie, 2000) (Eq. (3)). In this case, the function g(C) determines the scaling of the noise term dependent on the concentration, unlike the constant-level noise in Eq. (2). A relevant type is the Fokker-Planck partial differential equation (Hasty et al., 2000) (Eq. (4)) in which P(C,t) represents a density function describing the probability that the random variable C will assume a particular value at time t. This formalism has also been employed in the investigation of noise-induced transitions during stem cell differentiation (Wang et al., 2010). On the right-hand side, the first term represents deterministic drift and the second term is the diffusion term. Numerical methods to solve SDEs are based on stochastic variable calculus and more information can be found elsewhere including treatment of Fokker-Planck problems (Gillespie, 2000; Kloeden and Platen, 1999; Risken and Frank, 1996).
| (2) |
| (3) |
| (4) |
Regardless of whether SDEs or ODEs are chosen for the evolution of properties in individual cells, model construction requires several kinetic parameters. This presents a substantial challenge because current experimental methodologies are not suitable for direct determination of all parameter values. As a result, parametric quantities are typically estimated or guessed necessitating relevant sensitivity analysis to ensure robustness of the model. The same is true for the measurement of noise-related parameters (e.g. noise magnitude) which has not been demonstrated explicitly to date for single stem cells. Finally, single-cell models consider only intrinsic noise whereas the effects on heterogeneity arising from population interactions (extrinsic noise) are absent.
3.2. Cell population-based models
Individual-cell models in some cases can be considered building blocks of cell ensemble models. In these quantitative frameworks, a cell population is deconstructed into multiple subpopulations (sometimes down to unicellular groups), each of which retains specific kinetics of cellular activities such as proliferation and differentiation. The overall model focuses on the macro-heterogeneity of the population and its effects on development, differentiation and growth in response to relevant cues. Unlike single-cell models however, population-based models apply over considerably longer physical (i.e. intracellular vs. intercellular) scales. The same is true regarding the temporal span of these model modalities. That is, changes at the population level (e.g. differentiation) become evident over multiple cell cycles while gene expression in individual cells is dependent on biochemical reaction kinetics with much shorter time frames. For example, a stem cell population growth model was reported incorporating proliferative heterogeneity (Deasy et al., 2003) by considering two subpopulations, i.e. mitotic cells and quiescent cells, exhibiting distinct differentiation and loss/apoptosis kinetics.
A simple way to scale single-cell models to populations is via cell ensemble models (originally applied to bacterial systems Domach and Shuler, 1984) and agent-based models (e.g. for differentiation of stem cells in 3D aggregates White et al., 2013). Such models are based on assemblies of single-cell equations describing attributes of interest (e.g. level of a particular marker). Viswanathan et al. (2005) modeled the clonal evolution of 1000 mESCs and their differentiated progeny by coupling model results to cell numbers and Oct4+ cell fractions. Differentiation of individual cell was assessed in each cell cycle by comparing the number of LIF signaling complexes to a self-renewal threshold, and the level of Oct4 expression to 50% of that of undifferentiated cells. Initial conditions for each cell were randomly set so as to introduce heterogeneity. Moreover, LIF receptor numbers to newborn cells were stochastically assigned after division. Stem cell fate decision depended on fast molecular activities such as Oct4 expression and over several cell-cycle intervals. However, LIF signaling was assumed to immediately achieve steady-state and remain constant until the next decision-making point.
Cell ensemble models are straightforward to construct and computationally efficient but the embedding of intracellular activities and detailed GRNs is still lacking. The inclusion of processes such as differentiation, division and death occurring in hours or days, will enhance not only the complexity of cell ensemble models but their prediction potential as well.
A universal dynamics model was described (Kussell and Leibler, 2005) for a clonal population in an environment that fluctuates in time stochastically (Eq. (5)). The model assumes a population composed of m different phenotypes and N(t) is the population vector correspondingly containing m elements each representing the specific phenotype size. The matrix k(E) depends on the set of environment conditions (E). The diagonal elements kii (i = 1,2,…,m) correspond to growth rates whereas the off-diagonal elements kij (i ≠ j) represent the rates for switching between states i and j.
| (5) |
where
This scheme has been employed to investigate the heterogeneous expression of Sca-1 observed in mouse hematopoietic stem cells (HSCs) (Chang et al., 2008). The distribution of HSCs into Sca-1high and Sca-1low fractions was demonstrated to be only a one-dimensional projection of transcriptome-wide heterogeneity. Interactions of Sca-1low with Sca-1high cells were modeled taking into account the proliferation and transition kinetics between the two states. A bimodal distribution of HSCs based on Sca-1 expression was shown although the molecular programs participating in the generation and maintenance of the observed heterogeneity in Sca-1 expression remain to be elucidated.
Similarly, Roeder and Loeffler (2002) developed a HSC differentiation model by considering two HSC niches: one supporting HSC proliferation and another that is permissive to HSC differentiation. Each cell in the model was described by theoretical variables to specify its commitment potential while symmetric duplication was assumed. Stochasticity originated from the transition between the two environments with transition intensities dependent on the affinity and cell numbers in the system. Differentiation was considered but not along a specific type. A lineage propensity variable was later included (Glauche et al., 2007) with cells adopting a phenotype by randomly increasing one lineage propensity out of all proclivities modeled. The comparative differentiation of paired daughter cells, lineage contribution of single differentiating cells and lineage specification in differentiating HSC cultures were simulated as case studies and compared to published experimental data. Stem cell commitment along different lineages was purported to result from the competition between priming and the dominance of one lineage under the governance of environmental stimuli.
Population balance equation (PBE) models, which have been applied to microbial and animal cell systems (Fredrickson et al., 1967; Mantzaris and Daoutidis, 2004; Ramkrishna et al., 1968; Sherer et al., 2008; Nielsen et al., 1998), are an appealing modeling framework for stem cell ensembles (Jing et al., 2011; Kehoe et al., 2010). A general cell PBE, which is essentially a cell mass or number balance, can be written as:
| (6) |
where N(x̃, t)dx̃ represents the number of cells (per unit of culture volume) having a physiological state representation between x̃ and x̃ + dx̃ at time t (Fig. 6). The physiological state vector x̃ reflects the diversity of the stem cell population in terms of variables such as cell mass, age, DNA, RNA protein, differentiation status, and expression level of particular markers, and x̃max contains the maximum values of these variables.
Fig. 6.
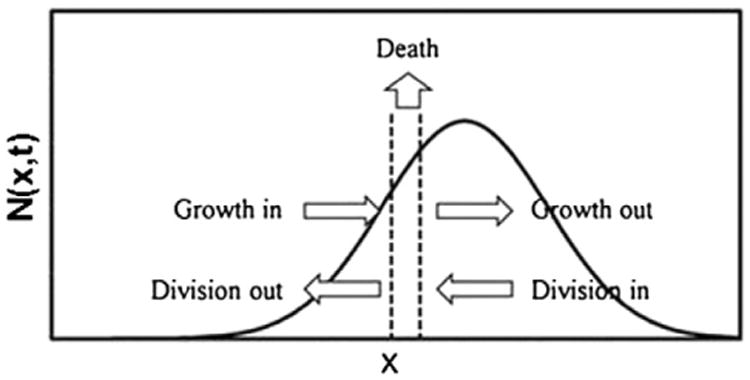
Illustration of single-variable cell PBE model. N(x,t) is the probability density function corresponding to the fraction of cells with property x at time t. The contribution of processes such as cellular growth, proliferation and death are also shown.
The functions Γ(x̃, C̃), Θ(x̃, C̃) and D(x̃, C̃) correspond to the cell division, death and differentiation intensities. The birth state distribution p(z̃, x̃, C̃) describes how the cellular material is partitioned among daughter cells. The quantity r(x̃, C̃) is the single-cell rates of growth or production of the elements inx. For example, r (x̃, C̃) includes the growth rate of single cells (e.g. see Eakman et al., 1966) if the physiological state vector contains the size (mass or volume) of individual cells. Stem cell PBEs can be coupled to equations describing the variations of the elements of ˜ in the culture or tissue (e.g. substrate addition/consumption or growth factor supplementation).
The differentiation of stem/progenitor cell populations has been modeled with multiple coupled PBEs, each corresponding to a particular cell subpopulation (Jing et al., 2011). A mass-structured PBE-based model was constructed for the osteogenic differentiation of mesenchymal stem cells (Pisu et al., 2007). The model comprised balance equations for three density functions mapping the fractions of undifferentiated mesenchymal stem cells (MSCs), chondrocytes and osteoblasts. These were coupled to expressions for the temporal profiles of extracellular matrix components (glycosaminoglycans, collagen) and growth factors involved in the differentiation (BMPs and TGF-β ligands). The model provided an integrated view of MSC growth, division, differentiation and extracellular matrix production and could offer insights in prescribing experimental conditions for studying the role of MSCs in pathologic conditions (e.g. infection, arthritis).
The PBE framework was also employed in the analysis of noise as a driving force for differentiation (Hoffmann et al., 2008). In this study, the differentiation potential was modeled after a state variable a and stochastic differentiation was included in the model. Simulation results were compared to data from cultures of differentiating promyelotic precursor cells concluding that state-specific noise modulation by external stimuli regulates the population dynamics.
Recently, the heterogeneity in NANOG expression in hESCs was investigated through a combination of experiments and stochastic PBE modeling (Wu and Tzanakakis, 2012). Both stochastic partitioning of cellular material at hESC mitosis and gene expression noise were taken into account as factors contributing to the distribution of NANOG among hESCs. The model provided information for the changes in NANOG and size for each cell in the population in line with flow cytometry data from cultured hESCs. Synchronization of the population by nocodazole or colcemid treatment led to a 39% increase in the average NANOG content. More importantly, stochastic partitioning accounted for 17% of the total noise in the profile of NANOG in undifferentiated cells. It should be noted that a simple single-gene model was implemented as part of the PBE framework in this report although more complex GRNs can be incorporated. Yet, integrating GRNs present challenges mainly because of the lack of methods for accurate quantitative, real-time measurements of GRN activity.
Although PBE models are multiscale and afford multivariable description of self-renewing or differentiating stem cell populations their application in this field has been limited in large part due to the difficulties in the collection of pertinent experimental data and the development/execution of suitable numerical methods. For instance, the selection of cell mass as a physiological state vector variable (Pisu et al., 2007) is advantageous because the first moment of the population density function is the biomass, which is straightforward to measure. Moreover, the mass distribution in a cell population can be determined by flow cytometry but to date, there is little published information on stem cell size (or mass) distribution which can be used in conjunction with PBE modeling. Acquiring reliable mass-based distributions of stem/progenitor cell populations can also be challenging due to the propensity of cells for aggregation. Nonetheless, there are efforts to address this issue with advances in flow cytometry and microfluidic technologies (Grover et al., 2011; Son et al., 2012; Tzur et al., 2011). Similarly, state variables corresponding to the expression of pluripotency and/or lineage markers requires highly specific antibodies and the engineering of stem cell lines carrying reporter genes under the control of phenotype-specific promoters. In this regard, the role of high-throughput methods for single-cell analysis reviewed above will be pivotal in the generation of experimental measurements for PBE model development and implementation.
As already mentioned, application of PBE frameworks hinges on the associated computational requirements, which grow as the dimensionality of the state vector increases. Pertinent numerical schemes are based on finite discretization methods (Mantzaris et al., 2001a, 2001b), multi-level discretization (Pinto et al., 2007) and MC approaches (e.g. Gillespie, 1976; Hatzis et al., 1995; Shah et al., 1976). Several notable improvements in stochastic simulation algorithms and tools (Gillespie, 2001; Li et al., 2008; Salis and Kaznessis, 2005) and the increasingly available computational power are expected to aid the wider adoption of PBE-based models for the investigation and control of stem cell systems including the development and optimization of bioprocesses for the production of stem cell progeny for clinical use (Kehoe et al., 2010).
3.3. Landscape-based models
The landscape model proposed by Waddington (1957) provides a simple illustration of the differentiation process. Stem/progenitor cells are considered as rolling balls in a landscape comprising hills and basins. Each basin represents an attractor state in which cells reside. Borrowing a term from chemical reaction thermodynamics, a cell must overcome a ‘potential energy barrier’ for switching from one attractor to another (Fig. 7). In this view, traveling between attractors is assumed to be irreversible. Stem cells however exhibit distinct phenotypes and are able to maintain macro-heterogeneity. More importantly, the recent advances in reprogramming challenge the irreversibility in the transition between attractors suggesting that improvements are necessary to this model to account for the plasticity of stem/progenitor cells. Self-renewing cells (‘A’) can reciprocate between attractors A1 and A2 (blue line) representing distinct phenotypes (‘1’ and ‘2’). A stem cell ‘A’ may also differentiate into cell type ‘B’ (green line). Cells in A1 and A2 have significantly different gene expression profiles and thus their potential for transitioning to attractor B will also vary. In this example, cells in A2 would reach B more easily compared to cells in A1 based on the energy level map (right panel). It should be noted however, that this simple model does not account for the molecular underpinnings of stem cell commitment. For example, the heterogeneous expression of particular genes leads to variable responses by members of the stem cell population thereby influencing fate decisions.
Fig. 7.
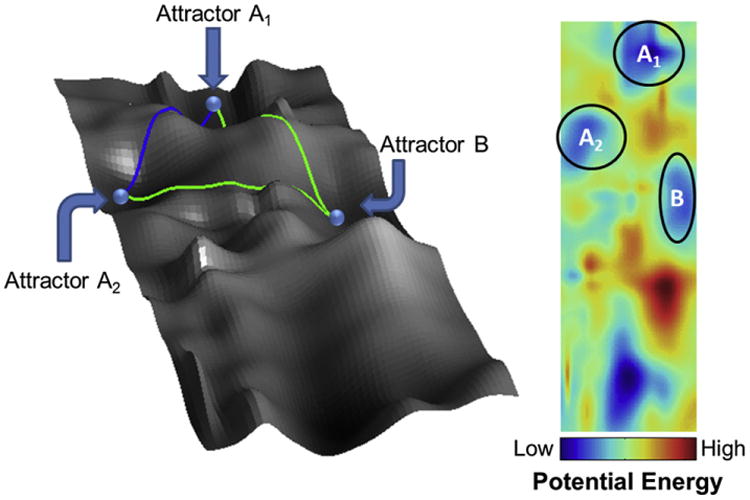
Landscape view of stem cell differentiation. The phenotype of a stem cell (or its progeny) is indicated by its position in the landscape while the height (z-axis) represents the “energy level” of this phenotype. This may be better visualized by projecting the hills and valleys onto a two-dimensional heat map. The color bar indicates the energy level. The attractors A and B represent two different cell types and the subscripts of A1 and A2 represent phenotypic diversity among the same cell type. Stem cells may switch between states without differentiating (blue line) or undergo lineage commitment (green line).
The Waddington landscape has been accompanied by computational models focusing mainly on the calculation of potential energy changes for stem cells transitioning from one attractor to another. For example, in a landscape model developed by Wang et al. (2011) the potential U(x̃) (Eq. (7)) was correlated with the steady-state probability of the system Pss(x̃) where x̃ is the vector of model variables. The driving force vector F(x̃) (Eq. (8)) dictates the transition of single cells from one attractor to another and consists of two terms: the gradient of potential energy U(x̃) scaled by a diffusion coefficient D, and the curl flux force in which Jss is the steady-state probability flux.
| (7) |
| (8) |
Therefore, both the gradient of U(x̃) and the curl force on the landscape are regulators of developmental processes. Differentiation in the reverse direction (e.g. B → A1) is not necessarily bound to the same biological path as the original commitment process (e.g. A1 → A2 → B). The landscape itself may also be subjected to reshaping due to possible changes in the components of the driving force (Wang et al., 2010). An increase of the diffusion coefficient D, for instance, lowers the U(x̃) barrier between attractors facilitating stem cell commitment. This outcome is suggested in differentiation experiments of myeloid progenitor cells where PU.1/GATA1 and PU.1/GATA2 interactions matched this attribute of the proposed landscape model (Huang et al., 2007).
A landscape framework was casted to describe the distribution of Nanog in mESCs (Luo et al., 2013). The model focused on equilibrium state distributions under different culture conditions and simulation results were coupled to flow cytometric data. The study concluded that a landscape with three attractors (low Nanog-LN, middle Nanog-MN, high Nanog-HN) fit the experimental observations with the least error, inferring three stable states for Nanog levels. Moreover, the noise associated with each state and the width of each attractor were computed along with shifts in these features brought about by the different culture conditions tested. Lastly, thermodynamic or attractor-based models have also been reported for transcription factor activity (Chen et al., 2008) and as Boolean networks (Serra et al., 2010).
4. Concluding remarks
Heterogeneity has been a commonly observed but overlooked attribute of stem/progenitor cell populations. The emerging roles of cell-to-cell variability on the differentiation trajectories, molecular and functional characteristics of stem cells and their progeny point to the need for the development of methodologies permitting the multivariable profiling of ensembles at single-cell resolution. To that end, rapid advances in methods for obtaining genomic, transcriptomic, proteomic and metabolomic information from single cells bring us closer to the identification of causal associations between observed genotypes and phenotypes. This knowledge will be pivotal in the design of highly efficient protocols for guiding the fate of stem cells in vitro or in vivo along specific lineages in therapeutically relevant quantities.
Single-cell analyses deployed for studying the phenotypic diversity in stem/progenitor cell populations is expected to generate large amounts of complex data necessitating the development of appropriate mathematical and computational models. The construction of such models should take into account the particular attributes of stem cells, including interweaving stochastic and deterministic processes and the multiscale nature of self-renewal and differentiation.
An important challenge has been the lack of clearly defined nomenclature associated with stem cell population analysis and modeling. Heterogeneity, for example, has been loosely used to describe variability in a wide range of (most often intimately related) characteristics such as proliferation, gene and protein expression, genetic background (e.g. among iPSC lines), epigenetic state and function. This loose use of the term ‘heterogeneity’ can be confusing when compiling and comparing outcomes from studies of different sources of stem cell population diversity. The terminology for gene expression noise may also be confounding partly because the tools for measuring and controlling noise experimentally are still under development. And while gene expression noise is typically termed intrinsic noise, the definition of extrinsic noise is not clear. Extrinsic noise has usually been defined simply as noise other than intrinsic noise. This statement leaves open the contribution from many often unspecified elements such as the extracellular environment in 3D culture (Suslov et al., 2002) and the cell-cycle dependent expression of genes (Orford and Scadden, 2008). Yet, the importance of extrinsic noise cannot be overstated necessitating clearer definitions and distinctions of different noise types. For instance, reconstitution of a stem/progenitor cell population from a subpopulation with extreme expression level of a certain gene (Sca-1, Nanog) takes ten to twenty cell cycles, a time scale long enough for significant influences from extrinsic noise sources (Chang et al., 2008; Kalmar et al., 2009).
The abundance of data from the analysis of single cells at different levels (DNA, RNA, proteins, metabolites) provides an unprecedented level of multivariable portraying of stem cell populations and their progeny. When combined with appropriate modeling approaches, such knowledge is expected to shine light on how heterogeneity modulates fate specification under different conditions. The relative contribution of stochasticity to stem cell fate decisions also remains quantitatively ambiguous given the precise response of stem cells to pluripotency/differentiation signals in spite of gene expression fluctuations. And as already mentioned, sources of noise and their effects, for example, on stem cell differentiation are still largely unaccounted for. The integration of single-cell analysis methods with modeling approaches will boost progress in our understanding of stem cell physiology. This will enable the rational design ofstrategies for efficient stem cell differentiation and somatic cell reprogramming aiming at the engineering of therapeutics for regenerative medicine.
Acknowledgments
Funding support has been provided by the National Institutes of Health (NHLBI, R01HL103709) and the New York Stem Cell Science Trust (NYSTEM, contract C024355) to EST. JW is the recipient of a Mark Diamond Research Fund award. This work was performed in part at the University at Buffalo's Center for Computational Research.
Contributor Information
Jincheng Wu, Email: jincheng@buffalo.edu.
Emmanuel S. Tzanakakis, Email: emtzan@buffalo.edu.
References
- Arias AM, Hayward P. Filtering transcriptional noise during development: concepts and mechanisms. Nat Rev Genet. 2006;7:34–44. doi: 10.1038/nrg1750. [DOI] [PubMed] [Google Scholar]
- Baserga R. Is cell size important? Cell Cycle. 2007;6:814–6. doi: 10.4161/cc.6.7.4049. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler's guide to cytometry. Trends Immunol. 2012;33:323–32. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birket MJ, Orr AL, Gerencser AA, Madden DT, Vitelli C, Swistowski A, et al. A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J Cell Sci. 2011;124:348–58. doi: 10.1242/jcs.072272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, Quake SR. Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res. 2011;39:e19. doi: 10.1093/nar/gkq1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–67. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady G, Barbara M, Iscove NN. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol Cell Biol. 1990;2:17–25. [Google Scholar]
- Brazzini A, Cantella R, De la Cruz A, Yupanqui J, Leon C, Jorquiera T, et al. Intraarterial autologous implantation of adult stem cells for patients with Parkinson disease. J Vasc Interv Radiol. 2010;21:443–51. doi: 10.1016/j.jvir.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Brill LM, Xiong W, Lee KB, Ficarro SB, Crain A, Xu Y, et al. Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell. 2009;5:204–13. doi: 10.1016/j.stem.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano JP, Kineman RD, Frawley LS. Dynamic monitoring and quantification of gene expression in single, living cells: a molecular basis for secretory cell heterogeneity. Mol Endocrinol. 1996;10:599–605. doi: 10.1210/mend.10.5.8732690. [DOI] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–4. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Chan JW, Lieu DK, Huser T, Li RA. Label-free separation of human embryonic stem cells and their cardiac derivatives using Raman spectroscopy. Anal Chem. 2009;81:1324–31. doi: 10.1021/ac801665m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Oh PY, Ingber DE, Huang S. Multistable and multistep dynamics in neutrophil differentiation. BMC Cell Biol. 2006;7 doi: 10.1186/1471-2121-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–7. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Zhu XG, Zhong S. Selection of thermodynamic models for combinatorial control of multiple transcription factors in early differentiation of embryonic stem cells. Genomics. 2008;9(Suppl. 1):S18. doi: 10.1186/1471-2164-9-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci U S A. 1996;93:14676–9. doi: 10.1073/pnas.93.25.14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickarmane V, Peterson C. A computational model for understanding stem cell, trophectoderm and endoderm lineage determination. PLoS One. 2008;3:e3478. doi: 10.1371/journal.pone.0003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickarmane V, Troein C, Nuber UA, Sauro HM, Peterson C. Transcriptional dynamics of the embryonic stem cell switch. PLoS Comput Biol. 2006;2:e123. doi: 10.1371/journal.pcbi.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirieleison SM, Bissell TA, Scelfo CC, Anderson JE, Li Y, Koebler DJ, et al. Automated live cell imaging systems reveal dynamic cell behavior. Biotechnol Progr. 2011;27:913–24. doi: 10.1002/btpr.629. [DOI] [PubMed] [Google Scholar]
- Chubb JR, Trcek T, Shenoy SM, Singer RH. Transcriptional pulsing of a developmental gene. Curr Biol. 2006;16:1018–25. doi: 10.1016/j.cub.2006.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BG, Flanagan LA, Rhee SW, Schwartz PH, Lee AP, Monuki ES, et al. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip. 2005;5:401–6. doi: 10.1039/b417651k. [DOI] [PubMed] [Google Scholar]
- Davidson KC, Adams AM, Goodson JM, McDonald CE, Potter JC, Berndt JD, et al. Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc Natl Acad Sci U S A. 2012;109:4485–90. doi: 10.1073/pnas.1118777109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–37. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11:1095–9. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deasy BM, Jankowski RJ, Payne TR, Cao B, Goff JP, Greenberger JS, et al. Modeling stem cell population growth: Incorporating terms for proliferative heterogeneity. Stem Cells. 2003;21:536–45. doi: 10.1634/stemcells.21-5-536. [DOI] [PubMed] [Google Scholar]
- Desai HV, Voruganti IS, Jayasuriya C, Chen Q, Darling EM. Live-cell, temporal gene expression analysis of osteogenic differentiation in adipose-derived stem cells. Tissue Eng Part A. 2013;19:40–8. doi: 10.1089/ten.tea.2012.0127. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Domach MM, Shuler ML. A finite representation model for an asynchronous culture of E. coli. Biotechnol Bioeng. 1984;26:877–84. doi: 10.1002/bit.260260810. [DOI] [PubMed] [Google Scholar]
- Eakman JM, Fredrickson AG, Tsuchiya HH. Statistics and dynamics of microbial cell populations. Chem Eng Prog Symp Ser. 1966;62:37–49. [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, et al. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A. 1992;89:3010–4. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, et al. Proteome half-life dynamics in living human cells. Science. 2011;331:764–8. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–6. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. 2009;4:387–97. doi: 10.1016/j.stem.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Fero M, Pogliano K. Automated quantitative live cell fluorescence microscopy. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer Y, Ganic E, Ameri J, Xian X, Johannesson M, Semb H. NANOG reporter cell lines generated by gene targeting in human embryonic stem cells. PLoS One. 2010;5:e12533. doi: 10.1371/journal.pone.0012533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluckiger AC, Marcy G, Marchand M, Negre D, Cosset FL, Mitalipov S, et al. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006;24:547–56. doi: 10.1634/stemcells.2005-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson AG, Ramkrishna D, Tsuchiya HM. Statistics and dynamics of procaryotic cell populations. Math Biosci. 1967;1:327–74. [Google Scholar]
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- Ghita A, Pascut FC, Mather M, Sottile V, Notingher I. Cytoplasmic RNA in undifferentiated neural stem cells: a potential label-free Raman spectral marker for assessing the undifferentiated status. Anal Chem. 2012;84:3155–62. doi: 10.1021/ac202994e. [DOI] [PubMed] [Google Scholar]
- Gibson JD, Jakuba CM, Boucher N, Holbrook KA, Carter MG, Nelson CE. Single-cell transcript analysis of human embryonic stem cells. Integr Biol. 2009;1:540–51. doi: 10.1039/b908276j. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. General method for numerically simulating stochastic time evolution of coupled chemical-reactions. J Comput Phys. 1976;22:403–34. [Google Scholar]
- Gillespie DT. The chemical Langevin equation. J Chem Phys. 2000;113:297–306. [Google Scholar]
- Gillespie DT. Approximate accelerated stochastic simulation of chemically reacting systems. J Chem Phys. 2001;115:1716–33. [Google Scholar]
- Giudice A, Trounson A. Genetic modification of human embryonic stem cells for derivation of target cells. Cell Stem Cell. 2008;2:422–33. doi: 10.1016/j.stem.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Glauche I, Cross M, Loeffler M, Roeder I. Lineage specification of hematopoietic stem cells: mathematical modeling and biological implications. Stem Cells. 2007;25:1791–9. doi: 10.1634/stemcells.2007-0025. [DOI] [PubMed] [Google Scholar]
- Glauche I, Herberg M, Roeder I. Nanog variability and pluripotency regulation of embryonic stem cells—insights from a mathematical model analysis. PLoS One. 2010;5:e11238. doi: 10.1371/journal.pone.0011238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T, Stadtfeld M. Heterogeneity of embryonic and adult stem cells. Cell Stem Cell. 2008;3:480–3. doi: 10.1016/j.stem.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Grover WH, Bryan AK, Diez-Silva M, Suresh S, Higgins JM, Manalis SR. Measuring single-cell density. Proc Natl Acad Sci U S A. 2011;108:10992–6. doi: 10.1073/pnas.1104651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–73. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Hasty J, Pradines J, Dolnik M, Collins JJ. Noise-based switches and amplifiers for gene expression. Proc Natl Acad Sci U S A. 2000;97:2075–80. doi: 10.1073/pnas.040411297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis C, Srienc F, Fredrickson AG. Multistaged corpuscular models of microbial growth: Monte Carlo simulations. Biosystems. 1995;36:19–35. doi: 10.1016/0303-2647(95)01524-o. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7:681–5. doi: 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Chang HH, Huang S, Ingber DE, Loeffler M, Galle J. Noise-driven stem cell and progenitor population dynamics. PLoS One. 2008;3:e2922. doi: 10.1371/journal.pone.0002922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–62. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Guo YP, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev Biol. 2007;305:695–713. doi: 10.1016/j.ydbio.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Huh D, Paulsson J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nat Genet. 2011;43:95–100. doi: 10.1038/ng.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez AJ, Fagerer SR, Schmidt AM, Urban PL, Jefimovs K, Geiger P, et al. Mass spectrometry-based metabolomics of single yeast cells. Proc Natl Acad Sci U S A. 2013;110:8790–4. doi: 10.1073/pnas.1209302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing D, Parikh A, Tzanakakis ES. Stem cell bioprocessing for regenerative medicine. In: Stachowiak MK, Tzanakakis ES, editors. Stem cells: from mechanisms to technologies. New Jersey: World Scientific; 2011. pp. 197–229. [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–64. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kalmar T, Lim C, Hayward P, Munoz-Descalzo S, Nichols J, Garcia-Ojalvo J, et al. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe DE, Jing D, Lock LT, Tzanakakis EM. Scalable stirred-suspension bioreactor culture of human pluripotent stem cells. Tissue Eng Part A. 2010;16:405–21. doi: 10.1089/ten.tea.2009.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo SH, Al-Rubeai M. Metabolomics as a complementary tool in cell culture. Biotechnol Appl Biochem. 2007;47:71–84. doi: 10.1042/BA20060221. [DOI] [PubMed] [Google Scholar]
- Klein CA, Schmidt-Kittler O, Schardt JA, Pantel K, Speicher MR, Riethmuller G. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci U S A. 1999;96:4494–9. doi: 10.1073/pnas.96.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, et al. Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol. 2002;20:387–92. doi: 10.1038/nbt0402-387. [DOI] [PubMed] [Google Scholar]
- Kloeden PE, Platen E. Numerical solution of stochastic differential equations. Third. New York: Springer; 1999. [Google Scholar]
- Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konorov SO, Schulze HG, Atkins CG, Piret JM, Aparicio SA, Turner RF, et al. Absolute quantification of intracellular glycogen content in human embryonic stem cells with Raman microspectroscopy. Anal Chem. 2011;83:6254–8. doi: 10.1021/ac201581e. [DOI] [PubMed] [Google Scholar]
- Konorov SO, Schulze HG, Piret JM, Blades MW, Turner RF. Label-free determination of the cell cycle phase in human embryonic stem cells by Raman microspectroscopy. Anal Chem. 2013 doi: 10.1021/ac400310b. [DOI] [PubMed] [Google Scholar]
- Konry T, Dominguez-Villar M, Baecher-Allan C, Hafler DA, Yarmush ML. Droplet-based microfluidic platforms for single T cell secretion analysis of IL-10 cytokine. Biosens Bioelectron. 2011;26:2707–10. doi: 10.1016/j.bios.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, et al. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006;34:e42. doi: 10.1093/nar/gkl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Ohinata Y, Saitou M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat Protoc. 2007;2:739–52. doi: 10.1038/nprot.2007.79. [DOI] [PubMed] [Google Scholar]
- Kussell E, Leibler S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 2005;309:2075–8. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- Leon-Quinto T, Jones J, Skoudy A, Burcin M, Soria B. In vitro directed differentiation of mouse embryonic stem cells into insulin-producing cells. Diabetologia. 2004;47:1442–51. doi: 10.1007/s00125-004-1458-8. [DOI] [PubMed] [Google Scholar]
- Levsky JM, Shenoy SM, Pezo RC, Singer RH. Single-cell gene expression profiling. Science. 2002;297:836–40. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–5. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- Li H, Cao Y, Petzold LR, Gillespie DT. Algorithms and software for stochastic simulation of biochemical reacting systems. Biotechnol Progr. 2008;24:56–61. doi: 10.1021/bp070255h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ. Strategies for micro array analysis of limiting amounts of RNA. Brief Funct Genomic Proteomic. 2003;2:31–6. doi: 10.1093/bfgp/2.1.31. [DOI] [PubMed] [Google Scholar]
- Loewenbruck KF, Fuchs B, Hermann A, Brandt M, Werner A, Kirsch M, et al. Proton MR spectroscopy of neural stem cells: does the proton-NMR peak at 1.28 ppm function as a biomarker for cell type or state? Rejuvenation Res. 2011;14:371–81. doi: 10.1089/rej.2010.1102. [DOI] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–8. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Markowetz F, Unwin RD, Leek JT, Airoldi EM, MacArthur BD, et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature. 2009;462:358–62. doi: 10.1038/nature08575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods. 2012;9:743–8. doi: 10.1038/nmeth.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Lim CL, Nichols J, Martinez-Arias A, Wernisch L. Cell signalling regulates dynamics of Nanog distribution in embryonic stem cell populations. J R Soc Interface. 2013;10:20120525. doi: 10.1098/rsif.2012.0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–43. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur BD, Sevilla A, Lenz M, Muller FJ, Schuldt BM, Schuppert AA, et al. Nanog-dependent feedback loops regulate murine embryonic stem cell heterogeneity. Nat Cell Biol. 2012;14:1139–47. doi: 10.1038/ncb2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallanna SK, Ormsbee BD, Iacovino M, Gilmore JM, Cox JL, Kyba M, et al. Proteomic analysis of Sox2-associated proteins during early stages of mouse embryonic stem cell differentiation identifies Sox21 as a novel regulator of stem cell fate. Stem Cells. 2010;28:1715–27. doi: 10.1002/stem.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantzaris NV, Daoutidis P. Cell population balance modeling and control in continuous bioreactors. J Process Control. 2004;14:775–84. [Google Scholar]
- Mantzaris NV, Daoutidis P, Srienc F. Numerical solution of multi-variable cell population balance models. III. Finite element methods. Comput Chem Eng. 2001a;25:1463–81. [Google Scholar]
- Mantzaris NV, Daoutidis P, Srienc F. Numerical solution of multi-variable cell population balance models: I. Finite difference methods. Comput Chem Eng. 2001b;25:1411–40. [Google Scholar]
- Meissen JK, Yuen BT, Kind T, Riggs JW, Barupal DK, Knoepfler PS, et al. Induced pluripotent stem cells show metabolomic differences to embryonic stem cells in polyunsaturated phosphatidylcholines and primary metabolism. PLoS One. 2012;7:e46770. doi: 10.1371/journal.pone.0046770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettetal JT, Muzzey D, Pedraza JM, Ozbudak EM, van Oudenaarden A. Predicting stochastic gene expression dynamics in single cells. Proc Natl Acad Sci U S A. 2006;103:7304–9. doi: 10.1073/pnas.0509874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag A, Tarnok A. Basics of standardization and calibration in cytometry — a review. J Biophotonics. 2009;2:470–81. doi: 10.1002/jbio.200910033. [DOI] [PubMed] [Google Scholar]
- Miyanari Y, Torres-Padilla ME. Control of ground-state pluripotency by allelic regulation of Nanog. Nature. 2012;483:470–3. doi: 10.1038/nature10807. [DOI] [PubMed] [Google Scholar]
- Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Lower M, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–501. doi: 10.1007/s00262-010-0875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery CL, Ward D, Passier R. Differentiation of human embryonic stem cells to cardiomyocytes by coculture with endoderm in serum-free medium. Current protocols in stem cell biology. 2007;Chapter 1:F.2.1–F.2.14. doi: 10.1002/9780470151808.sc01f02s2. Unit 1. [DOI] [PubMed] [Google Scholar]
- Nagano K, Taoka M, Yamauchi Y, Itagaki C, Shinkawa T, Nunomura K, et al. Large-scale identification of proteins expressed in mouse embryonic stem cells. Proteomics. 2005;5:1346–61. doi: 10.1002/pmic.200400990. [DOI] [PubMed] [Google Scholar]
- Narsinh KH, Sun N, Sanchez-Freire V, Lee AS, Almeida P, Hu S, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011;121:1217–21. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen LK, Bender JG, Miller WM, Papoutsakis ET. Population balance model of in vivo neutrophil formation following bone marrow rescue therapy. Cytotechnology. 1998;28:157–62. doi: 10.1023/A:1008098118491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–28. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–77. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, et al. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell. 2010;6:382–95. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Kim SK, Woo DH, Lee EJ, Kim JH, Lee SH. Differentiation of neural progenitor cells in a microfluidic chip-generated cytokine gradient. Stem Cells. 2009;27:2646–54. doi: 10.1002/stem.202. [DOI] [PubMed] [Google Scholar]
- Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104:5431–6. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6:S22–32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, et al. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:4093–8. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8:821–7. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney DG. Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem. 2012;113:2806–12. doi: 10.1002/jcb.24166. [DOI] [PubMed] [Google Scholar]
- Pinto MA, Immanuel CD, Doyle FJ. A feasible solution technique for higher-dimensional population balance models. Comput Chem Eng. 2007;31:1242–56. [Google Scholar]
- Pisu M, Concas A, Cao G. A novel simulation model for stem cells differentiation. J Biotechnol. 2007;130:171–82. doi: 10.1016/j.jbiotec.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–9. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkrishna D, Fredrickson AG, Tsuchiya HM. On relationships between various distribution functions in balanced unicellular growth. Bull Math Biophys. 1968;30:319–23. [Google Scholar]
- Risken H, Frank T. The Fokker–Planck equation: methods of solution and applications. 2nd. New York, NY: Springer-Verlag; 1996. [Google Scholar]
- Roccio M, Schmitter D, Knobloch M, Okawa Y, Sage D, Lutolf MP. Predicting stem cell fate changes by differential cell cycle progression patterns. Development. 2013;140:459–70. doi: 10.1242/dev.086215. [DOI] [PubMed] [Google Scholar]
- Roeder I, Loeffler M. A novel dynamic model of hematopoietic stem cell organization based on the concept of within-tissue plasticity. Exp Hematol. 2002;30:853–61. doi: 10.1016/s0301-472x(02)00832-9. [DOI] [PubMed] [Google Scholar]
- Romanska HM, Tiziani S, Howe RC, Gunther UL, Gulzar Z, Lalani el N. Nuclear magnetic resonance detects phosphoinositide 3-kinase/Akt-independent traits common to pluripotent murine embryonic stem cells and their malignant counterparts. Neoplasia. 2009;11:1301–8. doi: 10.1593/neo.09850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubakhin SS, Sweedler JV. Quantitative measurements of cell-cell signaling peptides with single-cell MALDI MS. Anal Chem. 2008;80:7128–36. doi: 10.1021/ac8010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–98. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105:2415–20. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H, Kaznessis Y. Accurate hybrid stochastic simulation of a system of coupled chemical or biochemical reactions. J Chem Phys. 2005;122 doi: 10.1063/1.1835951. [DOI] [PubMed] [Google Scholar]
- Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–75. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TC, Swistowska AM, Liu Y, Swistowski A, Palmarini G, Brimble SN, et al. A large-scale proteomic analysis of human embryonic stem cells. BMC Genomics. 2007;8:478. doi: 10.1186/1471-2164-8-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze HG, Konorov SO, Caron NJ, Piret JM, Blades MW, Turner RF. Assessing differentiation status of human embryonic stem cells noninvasively using Raman microspectroscopy. Anal Chem. 2010;82:5020–7. doi: 10.1021/ac902697q. [DOI] [PubMed] [Google Scholar]
- Serra R, Villani M, Barbieri A, Kauffman SA, Colacci A. On the dynamics of random Boolean networks subject to noise: attractors, ergodic sets and cell types. J Theor Biol. 2010;265:185–93. doi: 10.1016/j.jtbi.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Shah BH, Borwanker JD, Ramkrishna D. Monte-Carlo simulation of microbial-population growth. Math Biosci. 1976;31:1–23. [Google Scholar]
- Sherer E, Tocce E, Hannemann RE, Rundell AE, Ramkrishna D. Identification of age-structured models: cell cycle phase transitions. Biotechnol Bioeng. 2008;99:960–74. doi: 10.1002/bit.21633. [DOI] [PubMed] [Google Scholar]
- Shmulevich I, Dougherty ER, Kim S, Zhang W. Probabilistic Boolean networks: a rule-based uncertainty model for gene regulatory networks. Bioinformatics. 2002;18:261–74. doi: 10.1093/bioinformatics/18.2.261. [DOI] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Suri S, Lee T, Chilton JM, Cooke MT, Chen W, et al. Adhesion strength-based, label-free isolation of human pluripotent stem cells. Nat Methods. 2013;10:438–44. doi: 10.1038/nmeth.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Nachman I, Regev A, Meissner A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat Biotechnol. 2010;28:521–6. doi: 10.1038/nbt.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son S, Tzur A, Weng Y, Jorgensen P, Kim J, Kirschner MW, et al. Direct observation of mammalian cell growth and size regulation. Nat Methods. 2012;9:910–2. doi: 10.1038/nmeth.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I, et al. Whole-genome multiple displacement amplification from single cells. Nat Protoc. 2006;1:1965–70. doi: 10.1038/nprot.2006.326. [DOI] [PubMed] [Google Scholar]
- Stahlberg A, Bengtsson M, Hemberg M, Semb H. Quantitative transcription factor analysis of undifferentiated single human embryonic stem cells. Clin Chem. 2009;55:2162–70. doi: 10.1373/clinchem.2009.131433. [DOI] [PubMed] [Google Scholar]
- Stewart MH, Bosse M, Chadwick K, Menendez P, Bendall SC, Bhatia M. Clonal isolation of hESCs reveals heterogeneity within the pluripotent stem cell compartment. Nat Methods. 2006;3:807–15. doi: 10.1038/nmeth939. [DOI] [PubMed] [Google Scholar]
- Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 2007;1:515–28. doi: 10.1016/j.stem.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci U S A. 2002;99:14506–11. doi: 10.1073/pnas.212525299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain PS, Elowitz MB, Siggia ED. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc Natl Acad Sci U S A. 2002;99:12795–800. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15:6046–57. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Bao S, Lee C, Nordman E, Wang X, et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6:468–78. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thattai M, van Oudenaarden A. Intrinsic noise in gene regulatory networks. Proc Natl Acad Sci U S A. 2001;98:8614–9. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till JE, McCulloch EA, Siminovitch L. A stochastic model of stem cell proliferation, based on the growth of spleen colony-forming cells. Proc Natl Acad Sci U S A. 1964;51:29–36. doi: 10.1073/pnas.51.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka Y, Shimosato D, Murakami K, TakahashiK, Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–18. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods. 2009;6:331–8. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- Tzur A, Moore JK, Jorgensen P, Shapiro HM, Kirschner MW. Optimizing optical flow cytometry for cell volume-based sorting and analysis. PLoS One. 2011;6:e16053. doi: 10.1371/journal.pone.0016053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S, Davey RE, Cheng D, Raghu RC, Lauffenburger DA, Zandstra PW. Clonal evolution of stem and differentiated cells can be predicted by integrating cell-intrinsic and -extrinsic parameters. Biotechnol Appl Biochem. 2005;42:119–31. doi: 10.1042/BA20040207. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The strategy of the genes; a discussion of some aspects of theoretical biology. London: Allen & Unwin; 1957. [Google Scholar]
- Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–8. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009a;325:435–9. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Walker BL, Iannaccone S, Bhatt D, Kennedy PJ, Tse WT. Bistable switches control memory and plasticity in cellular differentiation. Proc Natl Acad Sci U S A. 2009b;106:6638–43. doi: 10.1073/pnas.0806137106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu L, Wang E, Huang S. The potential landscape of genetic circuits imposes the arrow of time in stem cell differentiation. Biophys J. 2010;99:29–39. doi: 10.1016/j.bpj.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang K, Xu L, Wang E. Quantifying the Waddington landscape and biological paths for development and differentiation. Proc Natl Acad Sci U S A. 2011;108:8257–62. doi: 10.1073/pnas.1017017108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–82. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Wernig M, Lengner CJ, Hanna J, Lodato MA, Steine E, Foreman R, et al. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol. 2008;26:916–24. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DE, Kinney MA, McDevitt TC, Kemp ML. Spatial pattern dynamics of 3D stem cell loss of pluripotency via rules-based computational modeling. PLoS Comput Biol. 2013;9:e1002952. doi: 10.1371/journal.pcbi.1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijgerde M, Grosveld F, Fraser P. Transcription complex stability and chromatin dynamics in vivo. Nature. 1995;377:209–13. doi: 10.1038/377209a0. [DOI] [PubMed] [Google Scholar]
- Wu J, Tzanakakis ES. Contribution of stochastic partitioning at human embryonic stem cell division to NANOG heterogeneity. PLoS One. 2012;7:e50715. doi: 10.1371/journal.pone.0050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tzanakakis ES. Distinct allelic patterns of nanog expression impart embryonic stem cell population heterogeneity. PLoS Comput Biol. 2013;9:e1003140. doi: 10.1371/journal.pcbi.1003140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S. Elite and stochastic models for induced pluripotent stem cell generation. Nature. 2009;460:49–52. doi: 10.1038/nature08180. [DOI] [PubMed] [Google Scholar]
- Yanes O, Clark J, Wong DM, Patti GJ, Sanchez-Ruiz A, Benton HP, et al. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol. 2010;6:411–7. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MA, Larson DE, Sun CW, George DR, Ding L, Miller CA, et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10:570–82. doi: 10.1016/j.stem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cui L, Johkura K, Ogiwara N, Sasaki K. Induction of midbrain dopaminergic neurons from primate embryonic stem cells by coculture with sertoli cells. Stem Cells. 2006;24:1695–706. doi: 10.1634/stemcells.2005-0409. [DOI] [PubMed] [Google Scholar]
- Zare RN, Kim S. Microfluidic platforms for single-cell analysis. Annu Rev Biomed Eng. 2010;12:187–201. doi: 10.1146/annurev-bioeng-070909-105238. [DOI] [PubMed] [Google Scholar]
- Zenger VE, Vogt R, Mandy F, Schwartz A, Marti GE. Quantitative flow cytometry: Inter-laboratory variation. Cytometry. 1998;33:138–45. doi: 10.1002/(sici)1097-0320(19981001)33:2<138::aid-cyto8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Zhong JF, Chen Y, Marcus JS, Scherer A, Quake SR, Taylor CR, et al. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab Chip. 2008;8:68–74. doi: 10.1039/b712116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman R, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–6. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]