

Abstract
The modern era of drug development for Alzheimer’s disease began with the proposal of the cholinergic hypothesis of memory impairment and the 1984 research criteria for Alzheimer’s disease. Since then, despite the evaluation of numerous potential treatments in clinical trials, only four cholinesterase inhibitors and memantine have shown sufficient safety and efficacy to allow marketing approval at an international level. Although this is probably because the other drugs tested were ineffective, inadequate clinical development methods have also been blamed for the failures. Here we review the development of treatments for Alzheimer’s disease during the past 30 years, considering the drugs, potential targets, late-stage clinical trials, development methods, emerging use of biomarkers and evolution of regulatory considerations in order to summarize advances and anticipate future developments. We have considered late-stage Alzheimer’s disease drug development from 1984 to 2013, including individual clinical trials, systematic and qualitative reviews, meta-analyses, methods, commentaries, position papers and guidelines. We then review the evolution of drugs in late clinical development, methods, biomarkers and regulatory issues. Although a range of small molecules and biological products against many targets have been investigated in clinical trials, the predominant drug targets have been the cholinergic system and the amyloid cascade. Trial methods have evolved incrementally: inclusion criteria have largely remained focused on mild to moderate Alzheimer’s disease criteria, recently extending to early or prodromal Alzheimer disease or ‘mild cognitive impairment due to Alzheimer’s disease’, for drugs considered to be disease modifying. The duration of trials has remained at 6 to 12 months for drugs intended to improve symptoms; 18- to 24-month trials have been established for drugs expected to attenuate clinical course. Cognitive performance, activities of daily living, global change and severity ratings have persisted as the primary clinically relevant outcomes. Regulatory guidance and oversight have evolved to allow for enrichment of early-stage Alzheimer’s disease trial samples by using biomarkers and phase-specific outcomes. In conclusion, validated drug targets for Alzheimer’s disease remain to be developed. Only drugs that affect an aspect of cholinergic function have shown consistent, but modest, clinical effects in late-phase trials. There is opportunity for substantial improvements in drug discovery and clinical development methods.
Keywords: Alzheimer, dementia, drug development, clinical trial, treatment
Introduction
The 9th Key Symposium marks an opportunity to examine the 30-year history of drug development and clinical trials for Alzheimer’s disease. In this review we will examine and assess recent therapeutic endeavours in order to understand the evolution of the drug development paradigms and anticipate future directions. Discovery and clinical development of Alzheimer’s disease treatments have been propelled by an enormous clinical need and a potentially huge world market. Consequently, drug development in this area has become a major political, academic and industrial effort. Despite considerable advances in knowledge of the pathogenesis of Alzheimer’s disease and in medicinal chemistry, no practical treatments have been introduced over the past quarter of a century.
Only four cholinesterase inhibitors and memantine have been marketed for the treatment of Alzheimer’s disease and none since 2002 in Europe and 2003 in the USA. Two reviews in 2008 identified over 100 [1] and 172 [2] drug development failures in the field of Alzheimer’s disease. The Pharmaceutical Research and Manufacturers of America, PhRMA, an industry trade group, identified 101 failures and three successes since 1998 [3]. Drug discovery in neuroscience in general is complicated, lengthy and uncertain, with an overall failure rate greater than 95%. Any given development programme may continue for 10 to 15 years from discovery to marketing approval [3].
The modern era of clinical trials for Alzheimer’s disease began with the advent of the cholinesterase inhibitors as a cognitive therapeutic during the late 1970s and early 1980s [4, 5]. This early work led to a proposed multicentre trial of orally administered physostigmine to be funded by the US National Institute on Aging (NIA). This proposal led to a trial of the cholinesterase inhibitor tacrine after the latter caused controversy and came to the attention of US politicians [6]. A two-stage design trial involving an initial crossover dose-finding phase, followed by a 6-week parallel group was funded by both the NIA and Warner Lambert Pharmaceuticals [7]. Other cholinesterase inhibitors under development at the time, including velnacrine and a sustained-release formulation of physostigmine, employed a similar paradigm [8, 9].
These trials used a simple, seven-point clinician’s global impression of change (CGIC) [10] and the Alzheimer’s Disease Assessment Scale – cognitive subscale (ADAS-cog) [11] as endpoints. Trial methods were facilitated in 1984 by the advent of the National Institute of Neurological and Communicative Disorders and Stroke- Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) consensus criteria for Alzheimer’s disease, known as the McKhann et al. criteria [12] that were soon incorporated into the 3rd revised edition of the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R) [13].
US Food and Drug Administration (FDA) advisory committees in 1989, 1991 and 1993 that discussed trial methods, as well as a new drug application for tacrine and unofficial unpublished FDA guidelines in 1990 [14], helped to further shape the process under which a drug could be approved for treatment of Alzheimer’s disease. In 1993 the cholinesterase inhibitor tacrine, branded Cognex®, was the first drug approved ‘for the treatment of mild to moderate dementia of the Alzheimer’s type’.
Methods
We selectively reviewed late-stage drug development and trials for Alzheimer’s disease from 1984 to 2013, including individual clinical trials, systematic and qualitative reviews, meta-analyses, methods, commentaries, position papers and guidelines. We focused on the methods, trends and results of phase 2 and 3 trials with the goal of summarizing advances and anticipating future developments. Finally we considered the evolution of drugs in late-stage clinical development, along with methodology, use of biomarkers, the contributions of regulators and future directions.
Historical review of drug development and trials
In 1974 Drachman and Leavitt suggested that memory was related to the cholinergic system and was age dependent [15], a notion that is still considered valid today. Around the same time two British groups independently demonstrated that the pathology of Alzheimer’s disease was associated with a severe loss of central cholinergic neurons; more precisely, the severity of dementia was correlated with the extent of cholinergic loss in the nucleus basalis of Meynert [16, 17]. Alzheimer’s disease was conceptualized as a cholinergic disease, similar to the way that Parkinson’s disease is considered a dopaminergic disease [18].
The cholinergic hypothesis drove drug development and trials throughout the 1980s and 1990s. It continues to provide a basis for current development efforts with neuronal nicotinic receptor modulators and other small molecules that have effects on cholinergic function, including muscarinic and nicotinic agonists, partial agonists and allosteric modulators, and 5-hydroxytryptamine (5-HT) receptor subtype-specific molecules [4].
Although other themes for therapeutic agents (e.g. neuroprotective, anti-inflammatory and nutritional/metabolic interventions) and targets for Alzheimer’s disease emerged in the early 1990s, drug development has been most influenced by the cholinergic hypothesis and the amyloid cascade hypothesis (see below).
The amyloid cascade hypothesis
The amyloid cascade hypothesis has become the most-researched conceptual framework for Alzheimer’s disease since its proposal in 1991 [19]. It has been the dominant influence on the development of targets and therapeutic agents for Alzheimer’s disease [20, 21]. The essence of the hypothesis is that amyloid-β peptide (Aβ) deposition is an early pathological process that drives tau phosphorylation, neurofibrillary tangle formation and neuron death; and that both the pathology and clinical expression of Alzheimer’s disease result from the increased production or impaired clearance of particular toxic Aβ species, particularly oligomers, produced by sequential β- and γ-secretase cleavage of the transmembrane protein amyloid precursor protein (APP).
This has led to the development of drugs to disrupt the cascade and to clinical trials from the late 1990s onwards to test them. Although simple in concept, the validation and development of amyloid drug targets has been complex in practice. For example, oligomers, protofibrils and amyloid plaques may have distinct toxicities. Oligomers may be toxic at synapses, and plaque-associated Aβ fibrils may be pro-inflammatory and neurotoxic in their local (interstitial) environment, thus constituting separate drug targets. Despite extraordinary, extensive preclinical research and substantial clinical research, a validated drug target has not been developed for Alzheimer’s disease based on the amyloid cascade. Specifically, it is not at all clear that inhibition or modulation of APP secretases, inhibition of Aβ fibril aggregation or the use of antibodies targeting various Aβ forms are valid strategies. The first late-stage trials specifically with ‘anti-amyloid’ drugs were not conducted until 2001; notably these trials investigated the use of an aggregated human Aβ vaccine with a conjugate (AN1792) [22].
Early symptomatic trials (approximately 1986–1996)
Despite initial dose titration, dose-ranging and crossover studies designed to find the ideal individualized doses for cholinesterase inhibitors, the basis for regulatory approval of the cholinesterase inhibitors (and subsequently for memantine), were the requirement of a minimum of one 3-month and one 6-month parallel-group, randomized placebo-controlled trials showing safety and effectiveness. The regulatory authorities initially permitted two 3-month trials but this was extended to a 6-month trial after an FDA advisory committee in 1989 suggested that 6 months was the minimum time needed to demonstrate a clinically meaningful effect [23].
Full and partial agonists at the M1 muscarinic receptor were also under development using similarly designed 3-month and 6-month trials of drugs including cevimeline (AF102B), milameline, sabcomeline (SB 202026), talsaclidine, xanomeline and alvameline (LU 25–109) [24]. Muscarinic agonists in phase 2 and 3 studies generally showed measurable efficacy in terms of cognition but with considerable, acute parasympathetically mediated adverse effects, including gastrointestinal symptoms, salivation, sweating and frequent urination. These adverse effects, which overwhelmed potential clinical utility, may have been due to insufficient selectivity of the drugs explored for the M1 receptor subtype [24].
The most important drugs investigated in 3-month and 6-month clinical development programmes during this period were the cholinesterase inhibitors including tacrine [7, 25, 26], velnacrine [8], sustained-release physostigmine [9, 27], eptastigmine [28, 29], metrifonate [30], donepezil [31], rivastigmine [32] and galantamine [33]. These drugs were sometimes combined with the acetylcholine precursor lecithin in an effort to enhance clinical effects [34, 35]. In addition, during this period several non-cholinergic drugs were tested that did not show efficacy in 6-month clinical trials, including ergoloid mesylates (Hydergine®) [36], D-cycloserine [37] and selegiline [38].
Six-month trials (approximately 1990–2001)
Development programmes were based almost exclusively on the original tacrine programme, and most subsequent cholinesterase inhibitors that were advanced to phase 2b or 3 trials showed cognitive efficacy in both 3- and 6-month trials in patients with mild to moderate Alzheimer’s disease. Three cholinesterase inhibitors in addition to tacrine were marketed in the USA: donepezil in 1996 (1997 in the UK), rivastigmine in 2000 (1998 in Europe) and galantamine in 2001 (2000). Other cholinesterase inhibitors such as velnacrine, physostigmine, eptastigmine and metrifonate showed efficacy as well, but were not marketed because of adverse events, safety and patent issues.
The general efficacy of the marketed cholinesterase inhibitors was considered modest with effects of about 2 to 3 points on the standard cognitive outcome, the ADAS-cog, and similarly small effects on global assessments and activities of daily living (ADL) over the course of 6 months [39]. The cognitive effects of the three currently marketed cholinesterase inhibitors are depicted in Fig. 1.
Fig. 1.

Forest plots for 6-month trials of the three currently marketed cholinesterase inhibitors showing the general consistency of effect, mean drug–placebo differences on the ADAS-cog and 95% confidence intervals. The overall mean effect is 2.37 ADAS-cog points. Reproduced with permission from Birks [39].
Although the effects of cholinesterase inhibitors in 6-month trials were statistically robust, the small size of the effects can be appreciated by considering that patients’ baseline scores on the ADAS-cog are about 22 to 24 and so the effect represents about a 10 –12% improvement. Health economists and systematic reviewers nevertheless recognized that ‘donepezil, rivastigmine, and galantamine can delay cognitive impairment’ based on the results of the 6-month trials [40, 41]. However, as branded medications in the early 2000s they were not considered cost-effective for mild to moderate Alzheimer’s disease by some health economists [40, 41]. The fact that donepezil and other cholinesterase inhibitors are now available as generics has substantially mitigated the economic issues of their use.
Development programes for dozens of other drugs have been initiated based on the cholinesterase inhibitor protocol of 6-month pivotal trials [2, 5]. Nearly all failed on the basis of insufficient cognitive efficacy. Some may have demonstrated detectable cognitive effects, such as the muscarinic agonists discussed above [24], but the effects were slight, not supported by other clinical ratings or not validated by further confirmatory trials.
The N-methyl-D-aspartate (NMDA) receptor antagonist memantine followed this development pattern for both mild to moderate and for moderate to severe Alzheimer’s disease. Efficacy in two of three 6-month trials was observed for moderate to severe Alzheimer’s disease and the drug subsequently received marketing approval in 2002 in Europe and in 2003 in the USA. Memantine was not shown to be effective in three, mild to moderate Alzheimer’s disease trials and is not approved for mild Alzheimer’s disease in Europe or the USA [42].
Twelve-month trials (approximately 1994–2010)
One possibility that may explain why the 6-month trials were successful for drugs such as the cholinesterase inhibitors is that they had clear positive effects showing improvement in cognition over the short term compared to baseline; in addition, the combination of this slight positive effect for the drug and a slight worsening for the placebo group was sufficient to detect a statistically significant change. However, a large number of drugs entered clinical development for which absolute cognitive improvement was not expected but attenuation or halting of cognitive decline was the expected therapeutic effect.
These drugs included those that might act as anti-inflammatory and neuroprotective agents and metabolic enhancers. Trials in mild to moderate Alzheimer’s disease were extended to 12 months of follow-up in order to be able to detect a stabilization effect compared to the continuing decline for the placebo group. These phase 2 and 3 trials assessed prednisone [43], conjugated oestrogens [44], transdermal selegiline [45] rofecoxib [46], naproxen [47], celecoxib [48], acetyl-L-carnitine [49, 50], ginkgo biloba extract [51], idebenone [52], nicergoline [53], and propentofylline [54]. The first phase 2 trials of an Aβ vaccine, AN1792 [22], and the first of a γ-secretase modulator or Aβ42-lowering agent, tarenflurbil [55], were 12-month trials. More recently phase 2 and pivotal 12-month trials were conducted with drugs in development for the treatment of Alzheimer’s disease, including the growth hormone secretagogue MK-677 [56], latrepirdine (data unpublished) and the neuroprotective agent T-817 [57].
Compared to the 6-month trials in mild to moderate Alzheimer’s disease, those with a 12-month duration reliably showed progression of cognitive impairment in the placebo group. The fact that cholinesterase inhibitors were marketed based on 6-month trials provided little incentive for their evaluation in longer studies. The one exception was a 12-month trial of donepezil that, although showing drug–placebo differences on the Mini-Mental State Examination (MMSE) [58] favouring donepezil, illustrated that patients no longer showed cognitive improvement after 12 months, continued to decline on other rating scales and had high discontinuation or non-compliance rates of about one-third over the 12 months [59].
One adaptation to the 12-month trial design was the random allocation of patients with more moderate impairment and their follow-up to a predetermined level of clinical or functional decline as the study endpoint, exemplified by a trial of donepezil [60]. An earlier example, was a 2-year trial of selegiline and vitamin E intended to delay loss of functional activities, nursing home placement or death [61].
In summary, the 12-month trials were successful in that their methods clearly demonstrated that the various test drugs with the exception of donepezil did not alter the decline in cognition that is reliably observed with placebo treatment. These methods continue to be used in late-phase drug development.
Mild cognitive impairment trials (approximately 2000–2005)
The interest in treating early Alzheimer’s disease evolved from consideration that a risk state can be identified before the onset of the dementia syndrome. The concept of incipient cognitive impairment as detected by the mild cognitive decline stage of the Global Deterioration Scale [62], the ‘questionable dementia’ or the 0.5 stage of the Clinical Dementia Rating scale (CDR) [63], was established in the 1980s. This stage may represents very early Alzheimer’s disease before the onset of dementia, i.e. not fulfilling the McKhann et al. criteria.
Subsequently, mild cognitive impairment (MCI) and amnestic MCI were characterized in the late 1990s as progressive memory impairment similar to that seen in patients with early Alzheimer’s disease but with less impairment in other cognitive and functional domains. Such individuals were shown to decline at a greater rate than non-impaired control subjects, such that about half developed Alzheimer’s disease (dementia) within 3 years [64]. The idea that amnestic MCI could be a clinical target for early treatment interventions was facilitated by an FDA advisory committee in 2001 [65] that coincided with some longer-term clinical trials of MCI. These trials used continuous treatment over 2 to 4 years with the main outcome of progression to Alzheimer’s disease dementia and secondary outcomes of change in cognitive rating scales. The drugs investigated included the marketed agents rivastigmine, galantamine, donepezil, vitamin E, vitamin B complex and rofecoxib [66–70]. The nootropic piracetam was used in a small 1-year MCI pilot trial [71] and donepezil in larger 6-month [72] and 12-month trials [73].
The results of all the trials in MCI were negative, with no significant benefit on progression or onset of Alzheimer’s disease dementia. The rates of progression from MCI to Alzheimer’s disease tended to be lower than expected based on prior cohort studies of MCI [64]. The different rates of progression could have been due to variations in sample ascertainment, heterogeneity of patients and different definitions of MCI and of outcomes. Of particular importance to drug development, the trial assessing the anti-inflammatory rofecoxib showed significant cognitive-impairing effects compared to placebo, but these effects were explainable, post hoc, by the fact that non-steroidal anti-inflammatory drugs (NSAIDs) are associated with cognitive impairment and delirium and that smaller phase 2 trials had not been performed to demonstrate proof of concept or assess safety. Rofecoxib was later withdrawn from the market because of cardiotoxic events, but at the time the MCI trial was conducted, the sponsors presumably did not think that phase 2 proof of concept or safety studies were necessary. Yet the planning and execution of the trial foreshadowed a current trend in drug development for Alzheimer’s disease to bypass phase 2 studies on the basis that phase 3 would better demonstrate efficacy and safety.
Individual MCI trials are reviewed, contrasted and compared elsewhere [71, 74]. Fig. 2 summarizes the cognitive and functional outcomes from these trials, showing also the broad heterogeneity of outcomes.
Fig. 2.

Outcomes of clinical trials of cholinesterase inhibitors for mild cognitive impairment showing a general lack of cognitive and global effects in trials from 2 to 4 years in duration (except for one 24-week trial). The data show the point estimates and 95% confidence intervals of the drug–placebo differences. The Z-cognitive is a composite of the neuropsychological test battery used in the rivastigmine trial. Reproduced with permission from Raschetti et al. [74].
Regulatory considerations for trials of MCI
In 2001 an FDA advisory committee clarified drug development for Alzheimer’s disease by suggesting several requirements for the marketing approval of drugs for MCI [65]: (i) MCI must be clearly defined in the clinical setting; (ii) there are valid criteria for its diagnosis; (iii) it is either distinguished from or defined in terms of future onset of Alzheimer’s disease; (iv) appropriate outcomes are used in trials; and (v) trials should be designed to assess rating scale changes, onset of dementia and clinically meaningful effects.
Post hoc analyses of these trials variously suggested that one subgroup or another had potentially benefited from cholinesterase inhibitor treatment. However, as post hoc analyses these findings did not gain acceptance and confirmatory trials were not conducted to test the hypotheses. The failures of MCI trials led to the consideration at the time that MCI was a heterogeneous at-risk state that did not necessarily lead to Alzheimer’s disease; indeed it might be too early in the disease course for the then current drugs to be effective. The European Medicines Agency (EMA) clearly stated that MCI could not be recognized as a clinical entity for which target drugs could be developed, and more work on characterization of diagnostic criteria including the role of aetiological subtypes and the development of appropriate assessment tools had to be done [75]. Moreover, the fact that a marketing claim could not be achieved for delaying onset of Alzheimer’s disease or disease modification, but rather only for the existing claim, ‘for the treatment of Alzheimer’s disease’, probably contributed to the lack of interest. Thus, there was little commercial incentive to pursue MCI as a clinical target.
Eighteen-month and disease-modifying trials (approximately 2001–2013)
Limited experience with 12-month trials led to the notion that longer trials could be helpful in showing sufficient progression in the placebo group so that the effect of a drug that only attenuated decline (or lost its positive effect early) could be better detected. It was also reasoned that the temporal resolution of the ADAS-cog should be better at 18 months than at 12 months, although this may not be the case [76]. These longer trials represented a departure from testing drugs that improved function to a model in which attenuation of decline was the desired outcome.
As with the 12-month trials, anti-inflammatory and neuroprotective agents were initially tested in 18-month trials of hydroychloroquine [77], the cholesterol-reducing 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors atorvastatin [78] and simvastatin [79], food supplements such as vitamin B combinations to lower homocysteine [80] and docosahexaenoic acid [81]. Except for two very large phase 3 trials with the neuroprotective 5-HT1A agonist xaliproden [82], the early 18-month trials investigated marketed drugs or available food supplements. More recent phase 2 and 3 trials involved almost entirely investigational drugs to test the amyloid hypothesis. These included an Aβ aggregation inhibitor tramiprosate [83], two phase 3 trials of a gamma-secretase modulator to lower Aβ levels, tarenflurbil [84], the Aβ aggregation inhibitor scyllo-inositol (ELND005) [85], two trials of the gamma-secretase inhibitors semagacestat [86]and one of avagacestat [87], a RAGE inhibitor (PF 04494700 or TTP 488), infusions of Aβ antibodies including phase 2 and 3 trials of bapineuzumab [88] [unpublished], two phase 3 trials of solanezumab [unpublished], and immunoglobulin G (IVIG) [unpublished]. All these trials yielded null results with respect to their main outcomes. Other 18-month trials are ongoing.
The 18-month trials have been similar in design to earlier trials, using the same inclusion criteria and outcomes, except that usually cholinesterase inhibitors and memantine have been allowed as concomitant medications. In fact, 80–90% of participants in later trials have been maintained on these drugs. Although most included mild to moderate Alzheimer’s disease (allowing a maximum MMSE score of 26), the lower limit defining ‘moderate’ severity has varied from a score of 12 to 20 such that some trials focused exclusively on ‘mild’ Alzheimer’s disease (i.e. restricting the MMSE score to 20 to 26). Patient groups in these trials are comparable to groups in the 6-, 12- and 18-month trials with respect to age, gender, apolipoprotein E (APOE) genotype distribution, education and clinical rating scale scores [76].
Recent enhancements to 18-month trial designs have been the addition of cerebrospinal fluid (CSF) and brain imaging measures of amyloid deposition, CSF assays of tau concentration and volumetric magnetic resonance imaging (MRI) to detect neurodegeneration. These biomarkers were first used in subsets of patients in the hope that any clinical effects could be better explained, or that a change in a marker during a trial would be considered as supportive evidence of a disease-modifying effect. More recently, and perhaps influenced by EMA guidelines on sample enrichment with biomarkers [89, 90], these biomarkers have been used to help ‘enrich’ or restrict clinical trial samples to patients with evidence of amyloid pathology. The rationale is that biomarker enrichment will more accurately identify trial participants with Alzheimer’s disease pathology and perhaps select those more likely to respond to the investigational drug, although this is without empirical support [89].
The design of these longer trials has been influenced by expert group recommendations that the most well-established scales be used, i.e. the ADAS-cog, Alzheimer’s Disease Cooperative Study - Activities of Daily Living (ADCS-ADL) scale and CDR, because of a lack of suitable alternatives; in addition, a 2-point drug–placebo group difference on the ADAS-cog would be the minimal mean change required to denote a clinically meaningful effect [91, 92]. Notably, this change is less than that observed with cholinesterase inhibitors and less than the 4-point difference recommended by an FDA advisory committee in 1989 [23].
For an 18-month trial to better detect efficacy using the clinical outcomes above, the placebo group must decline to a substantial extent, while the drug group still needs to improve slightly over baseline to offset the large variances in the change in rating scales. Initial sample sizes need to be large enough to gain this precision and counter the effects of large numbers of dropouts. Indeed, the sample sizes of 18-month trials have increased from about 400 to 1200 patients for industry-sponsored phase 3 trials in mild to moderate Alzheimer’s disease, and to 2100 for a trial limited to participants with mild Alzheimer’s disease treated with the Aβ antibody solanezumab (http://www.clinicaltrials.gov/ct2/show/NCT01900665?term=LZAX&rank=1). This trial should be able to detect less than a 1.5- and 2-point drug–placebo difference on the 11-item and 14-item ADAS-cog, respectively.
Although most ADL scales may not be accurate or able to assess the ‘functional domain’ in a clinically meaningful way, they are considered relevant for helping to determine disease modification and are consistent with patients’, caregivers’ and health economists’ perspectives with regard to effective treatment. In view of this, even an effect as small as a 1.5-point change on the ADCS-iADL over 18 months might be considered sufficient in early Alzheimer’s disease.
Many characteristics of these trials, such as the modest decline of placebo group rating scores, heterogeneity and variability of clinical course, imprecision of the outcomes, and the expectation that new drugs will only attenuate decline, when taken together, diminish the likelihood of discovering modestly effective drugs using these trial designs. Therefore, as above, biomarker enhancement, large sample sizes and more diligent training of clinical raters are expected to provide greater efficiency.
In summary, 18-month, placebo-controlled trials have become a de facto standard for trials of mild and moderate Alzheimer’s disease even though no 18-month trial has shown statistically significant primary outcomes favouring the test drug in a priori analyses. The use of biomarkers such as CSF Aβ and tau, volumetric MRI and amyloid positron emission tomography (PET) may allow demonstration of disease modification, depending on the mechanism of action of the drug.
Prodromal Alzheimer’s disease trials (approximately 2010–2014)
The earlier MCI trials used slightly differing definitions for MCI, including amnestic MCI in one trial [66] which later became ‘MCI due to Alzheimer’s disease’ for the Alzheimer Disease Neuroimaging Initiative and for the new research diagnostic guidelines for Alzheimer’s disease in 2011 [93]. The advancement of trials for prodromal Alzheimer’s disease followed proposed research criteria from an international workgroup [94], i.e. with the intent to diagnose Alzheimer’s disease before dementia onset, and to enrich clinical trial samples in terms of increasing confidence that patients actually had Alzheimer’s disease pathology and presumably would advance to dementia after a relatively brief interval [95]. The inclusion of patients with prodromal disease also allows individuals to be treated earlier in their illness and, hypothetically, at a time when some drugs may be more effective than they would be at a later stage.
The current ‘MCI due to Alzheimer’s disease’ and prodromal Alzheimer’s disease trials have characteristics similar to the 18-month trials described above and include a requirement that participants have positive Aβ biomarkers, such as low CSF Aβ42 concentrations or increased retention of an Aβ-binding ligand on amyloid PET scan, in order to enhance the likelihood that participants have Alzheimer’s disease pathology. These trials are different from previous MCI trials in having somewhat shorter treatment periods of 18 to 24 months and in the use of biomarkers both for entry criteria and as indices of change that might be interpreted as supportive of disease modification. A number of these trials are listed in Table 1.
Table 1.
Phase 2–3 prodromal or early Alzheimer’s disease (AD) trials
| Avagacestat | ACC-001 | NewGam, IVIG | Gantenerumab | BAN2401 | BIIB 037 | |
|---|---|---|---|---|---|---|
| Sponsor | Bristol-Myers Squibb | Janssen Alzheimer Immunotherapy | Octapharma, Sutter Health | Roche | Eisai | Biogen Idec |
| Clinicaltrials.gov EudraCT number | NCT00890890 | NCT01227564 | NCT01300728 | NCT01224106 ECT2010-19895-66 | NCT01767311 | NCT01677572 |
| Diagnosis | Prodromal AD | Early AD | MCI | Prodromal AD | MCI due to AD or early AD | Prodromal or mild AD |
| Sample size; number of sites | 270; 72 | 63; 35 | 50; 1 | 770; 159 | 800; 67 | 160; 12 |
| Clinical criteria | Memory complaint, MMSE 24–30, CDR 0.5, impairment on WMS Logical Memory II or FCSRT | Concern about cognition, MMSE ≥25, CDR 0.5, not dementia | Amnestic MCI, MMSE 24–30 CDR 0.5 | Age 50–85 years, worsening memory, MMSE ≥ 24, impairment on Logical Memory II or FCSRT | Age 50–90 years, MMSE 23–30, CDR 0.5, impairment on Logical Memory II | MMSE 20–30, including cut off on FCSRT <27 CDR 0.5 or 1 |
| Biomarker criteria | CSF Aβ42 <200 pg/mL or t- tau/Aβ42 ≥ 0.39 | Positive Aβ PET scan | Moderate or severe cortical or hippocampal atrophy | Positive Aβ PET scan | Positive Aβ PET scan | Positive Aβ PET scan |
| Duration | 2 years | 2 years | 2 years | 2 years | 1.5 years | 2.5 years |
| Outcomes | Primary: safety, CSF markers | Primary: brain Aβ, safety, tolerability; secondary: biomarkers, immunogenicity, clinical outcomes | MRI ventricular volume, AD, clinical ratings | Primary: CDR-SB and brain Aβ; secondary: ADAS- cog, FAQ, safety, pharmacokinetics | Primary: composite clinical score at 1 year; secondary: hippocampal volume, brain amyloid | Primary: safety, tolerability; secondary: Aβ PET, PK, immunogenicity |
Other drugs in prodromal Alzheimer’s disease trials include the BACE-1 inhibitor MK-8931
Data sources: www.clinicaltrials.gov; www.clinicaltrialsregister.eu; and http://www.medicine.ox.ac.uk/alois.
MCI: Mild Cognitive Impairment; MMSE: Mini Mental State Examination; CDR: Clinical Dementia rating scale; CRD-SB: Clinical Dementia rating scale, sum of boxes; CSF: cerebrospinal fluid; Aβ42: amyloid-beta 1–42; t-tau: total tau; PET: positron emission tomography; MRI: magnetic resonance imaging; ADAS-cog: Alzheimer Disease Assessment Scale-Cognitive; IVIG: intravenous immunoglobulin; WMS: Wechsler Memory Scale; FCSRT: Free and Cued Selective Reminding Test; FAQ: Functional Activities Questionnaire.
As an example, a phase 2/3 trial in patients with prodromal Alzheimer’s disease uses a cognitive test threshold requirement followed by an amyloid biomarker requirement to define prodromal Alzheimer’s disease. In this trial the Aβ antibody gantenerumab or placebo are given intravenously over a 2-year period. Anticipating the FDA draft guidelines for early-stage Alzheimer’s disease [96], the CDR-sum of boxes (CDR-SB) is used as the sole primary outcome, supported by cognitive, functional and biomarker secondary outcomes. As with earlier trials, if the primary outcome is not statistically robust and supported by most secondary outcomes, interpretation of the drug’s clinical meaning will be challenging.
Prevention trials (approximately 1996–2014)
Prevention trials are logical extensions of MCI and prodromal trials and address the fact that the pathology of Alzheimer’s disease is progressing perhaps two decades before the onset of dementia [97]. The shift towards recognizing presymptomatic and preclinical phases of Alzheimer’s disease, before the prodromal or MCI stage [95], has furthered the need for prevention treatment trials.
There have been only a handful of completed prevention trials, in large part because they require large samples, prolonged follow-up, are complicated and expensive to conduct, and until recently were hindered by the lack of compelling and safe drugs or interventions to test [98]. Known and presumed risk and protective factors for Alzheimer disease strongly influence the design and prosecution of these trials despite the fact that Alzheimer dementia and Alzheimer neuropathology do not necessarily involve the same risk factors [99]. Prevention trials have tended to select participants with risk factors such as family history, cardiovascular disease, relatively greater age or existing MCI in an effort to increase the numbers of individuals who progress to dementia over a typical 5-year follow-up.
Dementia prevention trials have generally used available, marketed or safe drugs and food supplements, and often as an add-on or nested within a larger trial for another condition, such as the preventive effects of anti-hypertension medications, conjugated oestrogens, an HMG-CoA reductase inhibitor or vitamin E and selenium [100]. There have been two prevention trials using Ginkgo biloba standardized extract EGb 761 [101, 102], and one using two NSAIDs [103]. These as well as prevention trials of antihypertensive agents [104] did not show protective effects on cognitive function or dementia onset and, indeed, in some cases may have shown the opposite [98].
Recent advances in prevention trials include enrolling elderly people with an increased risk of Alzheimer’s disease based on a biomarker or genetic marker, and the use of multi-domain interventions composed of concurrent management of risk factors based on lifestyle changes and marketed pharmacological products. The trials include the Finnish Geriatric Intervention Study to Prevent Cognitive impairment and Disability (FINGER), Multi-domain Alzheimer Prevention Study (MAPT) and Prevention of Dementia by Intensive Vascular Care (PreDIVA) [105]. Additionally, there are a number of planned trials involving structured physical activity interventions that are discussed elsewhere in this issue [105].
The current, single-drug, pharmaceutical company-sponsored prevention trials are components of proprietary drug development programmes in which participants must have an Aβ biomarker as an enrichment or risk factor, or a genetic risk marker or specific PSEN 1 or APP mutation that determines their eligibility. These studies include the Alzheimer’s Prevention Initiative (API), the Dominantly Inherited Alzheimer Network (DIAN), the Anti-amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) and the ApoE/TOMM40 trials; the first three tested Aβ antibodies and the latter a very low-dose of the thiazolidinedione pioglitazone [105].
The two trials in patients with autosomal dominantly inherited Alzheimer’s disease are unique in that there is no question that all participants will develop cognitive and neurological impairment and will have Alzheimer’s disease pathology. As participants are symptom free, they can be considered to have preclinical Alzheimer’s disease. Both trials are investigating the efficacy of Aβ antibodies – crenezumab in the API and gantenerumab and solanezumab in the DIAN study – and so are also tests of the amyloid hypothesis. One consideration is that Aβ42-reducing approaches may have a particular effect in familial Alzheimer’s disease where Aβ42 formation is increased, compared to sporadic Alzheimer’s disease where Aβ42 formation is not affected. The A4 trial is testing the concept of treating participants who are amyloid PET positive, without notable cognitive symptoms (i.e. who are at statistical risk of sporadic, late-onset Alzheimer’s disease), with the Aβ antibody solanezumab.
The pioglitazone trial is also unique in that it includes approximately 5000 non-cognitively impaired participants whose increased risk of Alzheimer’s disease is determined by a biomarker comprising age, particular variants of the TOMM40 gene and APOE genotypes; the efficacy of low dose pioglitazone to delay the onset of MCI due to Alzheimer’s disease will be evaluated over the course of 6 years in the high-risk biomarker group.
In summary, prevention trials select participants based on enhanced risk of Alzheimer’s disease or preclinical Alzheimer’s disease status, use marketed medications, food supplements, environmental interventions, Aβ antibodies or pioglitazone in randomized, placebo-controlled trials with 3- to 6-year follow-up periods, and both cognitive change and time to onset of cognitive impairment or dementia as endpoints.
The evolution of pharmacological targets and the new symptomatic phase (approximately 2007–2014)
The amyloid cascade hypothesis [19] has dominated drug development for the past two decades. Targets were developed for individual steps in the cascade: secretase inhibitors and modulators, passive and active immunization with antibodies against various epitopes of Aβ monomers, oligomers and fibrils, fibrilization inhibitors, anti-aggregants and other approaches. As a result of recent development failures the currently active phase 2 and 3 anti-amyloid approaches involve three antibodies with an emphasis on mild, prodromal and preclinical Alzheimer’s disease; Aβ vaccines, and beta-site amyloid precursor protein cleaving enzyme 1 (BACE-1) or β-secretase inhibitors. There are several examples of vaccines in development, including CAD-106, ACC-001, V950, AC-24, AD01 and AD02, all composed of peptides or fragments that mimic Aβ42.
The BACE-1 competitive inhibitor MK-8931 is being assessed in prodromal and mild to moderate Alzheimer’s disease populations in two phase 2 trials for 2 years and 18 months, respectively, with the latter enrolling over 1900 patients (NCT01739348). Other BACE-1 inhibitors are in development, although there have been failures due to toxicity.
Interest in tau-based approaches has led to putative inhibitors of enzymes involved in tau phosphorylation (e.g. GSK-3β), aggregation inhibitors, microtubule stabilizers and inhibitors of tau N-glcNAcylation. Agents in development include small molecules, monoclonal antibodies and vaccines. Most advanced is a formulation of methylene blue, methylthioninium chloride, (TRx0237) (TauRx), that may act as a tau aggregation inhibitor. It showed uncertain outcomes in a 6-month phase 2 trial but adequate safety [106] and is currently being tested in two phase 3 trials, a 12-month trial in mild to moderate Alzheimer’s disease with 833 patients, and an 18-month trial in mild Alzheimer’s disease with 500 patients. Anti-tau approaches in clinical development are listed in Table 3.
Table 3.
Alzheimer’s disease (AD)treatment: recent therapeutic approaches in phase 2 or 3. Putative disease-modifying drugs
| Main mechanism of action | RCTs completed (examples) | RCTs ongoing (examples) |
|---|---|---|
| Anti-amyloid: ↓ Aβ production | ||
| β-secretase inhibitors | Development limited by difficulties in identifying molecules able to cross the BBB and safety issues. Thiazolidinediones: rosiglitazone, tested in three phase 3 RCTs in mild–moderate AD (up to 1 year, ≈ 3800 participants); and pioglitazone, phase 2 RCT in mild–moderate AD, no benefits [135] |
Pioglitazone: phase 2 RCT in MCI (6 months, ≈ 300 participants) New compounds with increased ability to pass the BBB tested: MK-8931: Phase 2/3 RCT in mild–moderate AD (18 months, ≈ 1960 participants) |
|
| ||
| γ-secretase modulators (GSMs) | Tarenflurbil: negative phase 3 RCT (≈ 1684 participants, 18 months; another RCT in 900 subjects interrupted) in mild AD [84]. New compounds (e.g. CHF 5074, EVP-0962) are under development | EVP-0962: phase 2 RCT in healthy subjects, MCI and early AD cases (14 days, ≈ 50 participants) |
|
| ||
| γ-secretase inhibitors (GSIs) | GSI: development hampered by safety issues (i.e. interference with Notch signalling). Notch-sparing GSI under development Semagacestat: failures of phase 3 RCTs in 2600 participants with mild–moderate AD, halted because of lack of efficacy and adverse events, including ↑ skin cancer risk and infections [86] |
|
|
| ||
| Avagacestat: a 6-month, phase 2 RCT in 209 patients with mild–moderate AD: ↑ adverse events for high dosages, pharmacodynamics poorly characterized, no significant effects on CSF/MRI markers [87] | Avagacestat: phase 2 in prodromal AD, 2 years, 270 participants; arms with higher doses already discontinued due to safety issues | |
|
| ||
| α-secretase activators | Etazolate: ↑ α-secretase activity and modulates GABA-A receptors, 3 months, phase 2 RCT in 159 cases of mild–moderate AD, reported acceptable safety and tolerability [136] | Epigallocatechin gallate: ↑ α-secretase activity and ↓ Aβ aggregation, Phase 2/3 RCT in early AD, 18 months, ≈ 50 participants |
|
| ||
|
Anti-amyloid: ↓ Aβ aggregation or oligomerization |
Tramiprosate: negative phase RCT in 1052 cases of mild–moderate AD,18 months [137]. Another phase 3 RCT, 930 subjects, interrupted based on the latter results PBT2: negative 12-week phase 2 RCT, 78 participants with mild AD with positive secondary outcome [138] Scyllo-inositol: 18 months, phase 2 RCT in 353 cases of mild–moderate AD, showed no evidence of clinical benefits, and with high doses stopped because of unexpected death and infections [85] |
PBT2: phase 2 trial in 40 patients with prodromal or mild AD and PiB PET positive treated for 12 months, PiB PET as primary outcome (ACTRN12611001008910) |
|
| ||
|
Anti-amyloid: ↑ Aβ clearance, active immunotherapy |
Vaccines: contain different Aβ-derived epitopes with various adjuvants and mechanisms of delivery (e.g. adenoviruses, DNA vaccine, single- chain antibody fragments) AN1792: first anti-Aβ vaccine tested in a 12- month, phase 2 trial of 372 patients with mild– moderate AD, stopped because of aseptic meningoencephalitis due to cytotoxic T cells and/or autoimmune reactions [22] CAD-106 (Novartis and Cytos): a phase 2, 12- month RCT (plus 16-month open-label extension), 120 subjects with mild AD, showed good safety, tolerability and antibody response [139] |
ACI-24: a phase 1/2 12-month RCT to evaluate safety and efficacy in ≈ 186 patients with mild– moderate AD |
|
| ||
|
Affitopes: short peptides mimicking native Aβ. AD02 and AD03 in phase 1 in mild–moderate AD, ≈ 76 individuals, 12 months [140] ACC-001: two phase 2 RCTs in ≈ 520 subjects with early or mild–moderate and prodromal AD over 24 months (results unpublished) |
AD02 (Affiris/GSK): Phase 2 RCT in 300 individuals with early AD (18 months); additionally, extended follow-up of subjects who participated in the phase I RCT is ongoing (total follow-up of 2 years) AD03 (Affiris): an RCT is ongoing to follow-up subjects who participated in the phase I RCT |
|
|
| ||
|
↑ Aβ clearance, passive immunotherapy |
Monoclonal antibodies: target different epitopes of Aβ, and differ in the ability to bind different Aβ conformations. Some side effects (i.e. ARIA-E, ARIA-H) are more frequent compared to active immunotherapy Bapineuzumab: mainly targets fibrillar forms of Aβ. Two phase 3 RCTs in mild–moderate AD, ≈ 2400 subjects, did not report clinical benefits after 18 months. Two other phase 3 RCTs interrupted based on the latter results [141] Solanezumab: mainly targets soluble, monomeric Aβ. Two phase 3 RCTs in mild–moderate AD, ≈ 2050 subjects, did not show benefits in the primary outcomes after 18 months. Questionable benefit reported in mild AD [142] |
Gantenerumab, solanezumab: phase 2/3 RCT in carriers of genetic mutations for autosomal dominant AD with normal cognition, or MCI, or mild AD, 2+3 years, ≈ 240 participants Solanezumab is in a phase 2/3 RCT in older adults with positive Aβ PET scans and without cognitive impairment (2+3 years, ≈ 1500 participants); and a phase 3 trial of mild AD, with ≈ 2100 participants |
|
| ||
|
Gantenerumab: mainly targets Aβ plaques. 4- week phase 1 RCT in 18 patients with mild– moderate AD, reduction in brain Aβ; high doses showed adverse effects (ARIA-E) [143] Crenezumab: mainly targets monomeric or oligomeric forms of Aβ BAN2401: monoclonal antibody against Aβ oligomers, phase 2a, 60 AD participants (completed) |
Gantenerumab: in a phase 2/3 RCT in prodromal AD, 2 years, ≈ 770 participants Crenezumab: phase 2 in mild–moderate AD, 2 years, ≈ 375 participants; phase 2/3 RCT in PS 1 mutation carriers with normal cognition, 5 years, ≈ 300 participants BAN2401: an 18-month phase 2 RCT in 800 participants with MCI due to AD or mild AD, NCT01767311 |
|
|
| ||
| Ponezumab PF-04360365: phase 2 RCT, 175 participants, completed in 2011 (not published; development discontinued) | B11B 037: phase 2 RCT in prodromal and early AD, 160 participants (NCT01677572) | |
|
| ||
| IVIG polyclonal antibodies includeing anti- Aβ antibodies: 6 months, phase 2 RCT in 24 participants with mild–moderate AD reported cognitive improvement [144]; 6-month, phase 2 RCT in 58 with mild–moderate AD showed no effect on cognition or AD biomarkers [145] 18-month, phase 3 RCT in mild–moderate AD (≈ 390 subjects) showed no significant effect [146] | IVIG: phase 2 RCT in aMCI, 2 years, ≈ 50 participants | |
|
| ||
|
Anti-tau: ↓ p-tau production GSK-3 inhibitors: ↓ tau phosphorylation; microtubule stabilizers |
Lithium: too toxic in a 10-week RCT with 71 patients with mild AD [147]; 12- month RCT in 45 subjects with aMCI showed decrease in CSF p-tau and some cognitive improvement [148] Valproate: 2-year, phase 3 RCT in 313 patients with moderate AD negative for cognitive effects and prevention of behavioural problems; treatment associated with decreased brain volumes [149] |
|
|
| ||
| Tideglusib (NP031112, NP-12): a 20-week, phase 1/2 RCT in 30 patients with mild–moderate AD showed an acceptable safety profile; frequent asymptomatic liver function test abnormalities [150]. Another phase 2 RCT, 6 months, evaluated the drug in 306 subjects with mild–moderate AD (not published) | ||
|
| ||
|
Anti-tau: ↓ tau fibrillization or ↓ deposition |
Methylene blue: tau anti-aggregant, 6-month phase 2 RCT in mild–moderate AD with 321 subjects showed uncertain results [106] Davunetide (AL-108, NAP): ↓ p-tau production and ↓ tau deposition. In a 12-week phase 2 RCT in 144 aMCI patients davunetide had some benefits on memory functions [134] |
LMTX: methylene blue formulation with improved bioavailability, in two phase 3 mild– moderate AD RCTs, ≈ 1333 subjects (12 and 18 month) [151, 152] |
CSF, cerebrospinal fluid; GSK-3, glycogen-synthase-kinase-3; P-tau, phosphorylated-tau; ARIA-E, amyloid-related imaging abnormalities with parenchymal oedema; ARIA-H, amyloid-related imaging abnormalities with intracerebral microhaemorrhage IVIG, intravenous immunoglobulin; aMCI: amnestic mild cognitive impairment; MCI: mild cognitive impairment; RCT: randomized controlled trial; BBB: blood brain barrier; MRI: magnetic resonance imaging; ↑: increase ; ↓: decrease; GABA: γ-Aminobutyric acid
Despite the interest in amyloid-targeted drugs for longer-term, disease-modification trials, there has been continued interest in small molecules that would have relatively quick and symptomatic effects targeting a range of receptor complexes that result in cholinergic enhancement, or modulate monoamine and other neurotransmitter systems. Success was reported with these investigational drugs in phase 2, 6-month trials using the ADAS-cog as the primary clinical outcome, and in particular with the 5-HT6 antagonist Lu AE58054 (Lundbeck) and the α7 nicotinic modulators EVP6124 (En Vivo), ABT-126 (Abbvie) and MK-7622 (Merck). Thus the pursuit of the cholinergic hypothesis has continued and cholinergic pathways remain viable drug targets.
Monoamine oxidase (MAO)-B inhibitors are being revisited since the early trials of selegiline [38] and derivatives in the 1990s. One MAO-B inhibitor, RO4602522 (EVT 302) (Roche), is being tested in a phase 2 dose-ranging trial including 495 patients with moderate Alzheimer’s disease, i.e. MMSE from 13 to 20, treated for 12 months (NCT01677754).
Some so-called ‘symptomatic drugs’ are in development to improve cognition in patients with cognitive impairment associated with schizophrenia and in those with attention deficit disorder. Examples include phosphodiesterase-9 inhibitors and agents to increase activation of the NO/cGMP pathway. For the latter class, preclinical evidence suggests an effect similar to that of acetylcholinesterase inhibitors, and clinical data are now being collected in phase 2a trials. These drugs are being developed in 6-month trials in mild to moderate Alzheimer’s disease using the same framework as for the cholinesterase inhibitors, but cholinesterase inhibitor treatment is allowed as background therapy in some trials.
In summary, anti-amyloid and tau-based approaches are being advanced in 18-month trials in patients with mild and prodromal Alzheimer’s disease, and small molecule, cholinergic approaches are entering phase 3 in 6-month trials in patients with mild to moderate dementia. The cholinergic approaches are better supported by ‘proof of concept’ trials and appear to improve clinical outcomes.
Diagnoses for clinical trials in Alzheimer’s disease
The McKhann et al. criteria for possible, probable and definite Alzheimer’s disease published in 1984 [12] provided a clear framework and an important step forward for advancing clinical trials. Subsequently, severity levels of Alzheimer’s disease for the purpose of clinical trials were conventionally defined by reference to MMSE scores: mild to moderate, 10–26; moderate to severe, ≤14; severe, less than 10 or 12; and mild disease, ≥20.
The evolution of inclusion criteria in clinical trials reflects the change in clinical diagnosis towards aetiologically directed diagnostic approaches as compared with more clinically and phenotypically based inclusion criteria. The extent to and speed with which the proposed 2011 research diagnostic criteria for Alzheimer’s disease [107] might replace the 1984 criteria [12] for trials is not yet clear. The essential difference between the two sets of criteria is that biomarkers such as CSF Aβ and tau concentrations, cerebral glucose metabolism and amyloid burden assessed by PET, and brain volume biomarkers are integrated into the new diagnostic criteria for probable and possible Alzheimer’s disease dementia [107]. This represents a paradigm shift from conceptualizing Alzheimer’s disease as being definitively diagnosable only post-mortem, with the diagnosis being predicted with variable certainty in vivo (i.e. possible and probable Alzheimer’s disease), to the construct of Alzheimer’s disease as being reliably diagnosable in life, even early in the course of illness, using prognostic biomarkers.
Early on, trial inclusion criteria for MCI or amnestic MCI followed either the Mayo clinic criteria [64] or other criteria that generally required, in part, a CDR global score of 0.5, and a New York University NYU paragraph recall score or Auditory Verbal Learning Test AVLT score below a certain critical level. The former MCI criteria were somewhat more precisely defined, requiring memory impairment using the Wechsler Memory Scale Logical Memory II test at the same threshold limit as required for Alzheimer’s disease dementia. All MCI criteria as used in clinical trials further required relative preservation of other aspects of cognitive function and ADL. In retrospect, the Mayo clinic criteria for amnestic MCI have been recognized as being relatively stringent in their requirements for memory impairment and led to identification of subjects with relatively rapid rates of progression to dementia. The criteria for ‘MCI due to Alzheimer’s disease’ as used in one recent trial [66] are the same as the new 2011 MCI due to Alzheimer’s disease criteria except that the latter can be further enriched with a range of Alzheimer-related biomarkers [93]. By including one of the above-mentioned biomarkers, the latter criteria become essentially equivalent to the criteria for ‘prodromal Alzheimer’s disease’ of an international workgroup [95].
It remains unclear how the new definitions of minor and major neurocognitive disorders in the recently introduced 5th edition of the DSM will be employed in Alzheimer’s disease drug development. For example, the term minor neurocognitive disorder defines a population that overlaps with MCI due to Alzheimer’s disease but also allows for impairment in executive abilities without memory deficits and is not aetiologically based.
It is noteworthy that memory impairment thought to be related to ageing, categorized by terms such as age-associated memory impairment [108], age-related cognitive decline and subjective memory impairment or cognitive decline in the elderly, has not been the focus of drug development. This may reflect a lack of economic incentive, as from a regulatory point of view such conditions are not considered to be illnesses, which is a prerequisite for approval of a drug. However, in the USA, food or dietary supplements have been promoted for such conditions. In this domain, disease treatment claims are not made, but rather ‘structure/function claims’ (21 CFR 101.93) are advanced. The emerging concept of preclinical Alzheimer’s disease, wherein there may or may not be evidence of cognitive impairment but biological evidence of Alzheimer’s disease pathology [95, 109] is present, partially address the poorly defined space between normal cognitive ageing and pathological cognitive impairment.
In summary, diagnostic criteria for clinical trials of Alzheimer’s disease have remained consistent for more than a quarter of a century, and those for MCI trials for more than a decade. The new research criteria mainly incorporate biomarkers to increase the likelihood of Alzheimer’s disease pathology in patients clinically diagnosed as having prodromal Alzheimer’s disease and MCI or to help define a preclinical or presymptomatic Alzheimer’s disease state.
Outcomes in clinical trials
A narrow set of outcomes for Alzheimer’s disease trials have been used since the mid-1980s. The primary outcomes of the trials can be described within cognitive, functional, global change and severity domains (Table 5). The ADAS-cog [110], ADCS-ADL scale [111], Disability Assessment for Dementia (DAD) [112], Clinician Interview Based Impression of Change with Caregiver Input (CIBIC+) or ADCS Clinical Global Impression of Change (ADCS-CGIC)) [113] and CDR [114] are among the most commonly used. The two ADL scales are often used interchangeably. The Progressive Deterioration Scale [115], however, is noteworthy for its use as an ADL rating in important regulatory trials for tacrine, galantamine and rivastigmine. A Severe Impairment Battery [116] is used as a primary outcome in moderate to severe Alzheimer’s disease trials with memantine and donepezil. The ADAS-cog was developed to assess cognitive areas commonly impaired in patients and is the standard cognitive outcome for the vast majority of mild and mild to moderate and prodromal or early Alzheimer’s disease trials. It includes tests of memory, praxis, orientation, language, reasoning and word-finding as well as, in extended versions, executive function and attention.
Table 5.
Outcomes used in clinical trials for Alzheimer’s disease and mild cognitive impairment
| Staging | Cognition | ADLs | Global |
|---|---|---|---|
| Mini-Mental State Examination (MMSE) | Alzheimer’s Disease Assessment Scale- cognitive subscale (ADAS-cog) | Alzheimer’s Disease Cooperative Study- Activities of Daily Living (ADCS-ADL) | Alzheimer’s Disease Cooperative Study- Clinical Global Impression of Change (ADCS-CGIC) |
| Clinical Dementia Rating Scale (CDR) | Neuropsychological Test Battery (NTB) | Disability in Alzheimer’s Disease (DAD) | Clinical Dementia Rating Scale-sum of boxes (CDR-SB) |
| Global Deterioration Scale (GDS) | Mini-Mental State Examination (MMSE) | Progressive Deterioration Scale (PDS) | Gottfries-Brane-Steen (GBS) |
| Gottfries-Brane-Steen (GBS) | Severe Impairment Battery (SIB) | Bristol activities of daily living (ADL) |
The Neuropsychological Test Battery (NTB) was introduced in early 2001 for a trial of the synthetic Aβ42 vaccine AN1792 [22]. It is an individualized selection of neuropsychological tests mainly from the Wechsler Memory Scale, the Rey Auditory Verbal Learning Test and verbal fluency tests that are combined into three batteries assessing immediate and delayed memory and executive function [117].
In theory, this composite score may be more sensitive to change than the ADAS-cog for patients with milder Alzheimer’s disease who retain episodic memory capacity or the ability to learn. The NTB has been used instead of or in addition to the ADAS-cog in a few phase 2 and 3 trials. The ADAS-cog, however, can be expanded to include tests of executive function and episodic memory [110].
The ADCS-CGIC [113] for 3- to 18-month trials is the most commonly used example of a global change rating originally required by the FDA in 1990 [14]. The CDR is the most commonly used severity rating in trials of 12 months and longer. The former is used to assess clinically meaningful change from baseline, and the latter to assess current severity of dementia, either on a staging scale of 0–3 (0, 0.5, 1, 2, 3) or of 0–18 by adding the scores for six cognitive and functional areas.
Since the first 6-month trials, co-primary outcomes assessing cognition and either function or clinical global change have been used in phase 2 and 3 trials that are intended as pivotal studies. For regulatory purposes the EMA and FDA have required two primary outcomes, the ADAS-Cog and either the ADCS-ADL or ADCS-CGIC. Recently, the FDA suggested in their draft guidance that the CDR-SB be used as a ‘composite’ and sole outcome in prodromal Alzheimer’s disease trials and a single neuropsychological assessment can be used as sole outcome for preclinical Alzheimer’s disease trials [96, 118]. The EMA has also endorsed the use of the CDR-SB as sole primary outcome as evidenced by ongoing 2-year registration trials conducted in patients with prodromal Alzheimer’s disease. However, a disease-modifying claim will be granted by the EMA only providing that there is a clinically relevant treatment effect supported by additional CDR sub-items exploratory analyses and results from key secondary endpoints as well as compelling evidence from the biomarker programme showing delay in neurodegeneration. The EMA requires a comprehensive assessment of efficacy to support the primary outcome that itself must be interpretable as clinically meaningful in addition to being statistically significant.
In summary, outcomes for phase 2 and 3 trials in patients with mild to moderate Alzheimer’s disease have been largely the same since 1988. Any variations have been in the use of a cognitive outcome in addition to the ADAS-cog or the choice of ADL rating, global change or severity scales. Recently, the CDR-SB has been used as sole clinical outcome in two prodromal Alzheimer’s disease trials and sanctioned by the EMA and FDA, although secondary outcomes will be important in the interpretation of any positive CDR-SB primary outcome.
The introduction of biomarkers in trials
The serial failures of drugs in 12- to 18-month trials, especially the anti-Aβ agents, led to a suggestion that the primary reasons for failure were that the patients selected for the trials with mild to moderate disease were both too far advanced into their illness and, simultaneously, some patients did not have the neuropathology of Alzheimer’s disease. Therefore, according to this reasoning, the drugs did not have enough opportunity to work as the brain contained too much amyloid substrate, the neurodegeneration was too advanced or the patients did not have the illness in the first place. This rationale combined with the wish to not just improve symptoms but to modify the progression of Alzheimer’s disease pathology encouraged focus on advancing potential biomarkers for diagnosis, sample enrichment, disease progression and for targeting patients in an earlier prodromal or mild dementia phase within a clinical trials context.
The advancement of the amyloid hypothesis, as well as advances in Aβ and tau assay technology, volumetric MRI and amyloid PET scans led to the use of putative CSF and imaging biomarkers in clinical trials. The original purpose of CSF Aβ and tau assays in trials was as diagnostic and prognostic markers of illness and later as potential therapeutic, predictive, pharmacodynamic or surrogate markers of the effects of a drug. Finally, the EMA in 2011 and FDA in 2013 [96] recognized their potential use as markers to enrich sample selection in early-stage Alzheimer’s disease. The EMA qualified both amyloid PET imaging and CSF Aβ42 and tau assays as enrichment biomarkers for enrolling participants with either prodromal or mild to moderate Alzheimer’s disease in regulatory clinical trials [89, 90].
Most current longer-term trials use a biomarker in a subset of participants, usually for exploratory and sometimes for disease-modification purposes. Thus far biomarkers have been used in an Aβ-centric way. Biomarkers that are affected by the drug might be used for enrichment and prediction, but are limited for use as a surrogate clinical outcome. For example, an Aβ marker that is affected by a test drug cannot be used as a surrogate clinical outcome marker, but could be used as a potential marker of pharmacodynamics.
It is useful to consider the types of biomarkers that have been used in recent Alzheimer’s disease trials: diagnostic – for determining diagnosis; enrichment – for enhancing entry criteria; prognostic – for determining the course of illness; predictive – for determining treatment outcomes; surrogate – to substitute for clinical outcomes. However, for the most part biomarkers still require validation for the particular purpose for which they are used and need more than just correlation with clinical change.
The biomarkers used in clinical trials were developed with the intention of reflecting disease and have been used extensively in observational studies. They were introduced into clinical trials as potential prognostic and predictive markers. Biomarker outcomes in trials have been counterintuitive and are often difficult to interpret; the Aβ vaccine AN1792 was associated with decreased brain volumes and decreases in amyloid plaques at autopsy, while xaliproden was associated with changes in MRI volumes and bapineuzumab with decreases in CSF tau but not Aβ. These changes, however, occurred in the absence of detectable clinical change. By comparison, another Aβ antibody, solanezumab, was associated with no change in biomarkers but the possibility of a clinical effect in a subgroup with mild Alzheimer’s disease.
One explanation for counter-intuitive outcomes is that analyses of biomarker subsets from individual trials lack statistical power to detect significant changes related to treatment effects. On the other hand, the trials including biomarkers are not designed to make a constructive comparison between biomarker-positive and biomarker-negative patients. Such trials could potentially validate biomarkers for diagnostic and labelling purposes, and possibly as supportive evidence of clinical outcomes, but would require either validation in a biomarker-negative population or neuropathological evidence. Another consideration is that biomarker determinations and cut-off points are set arbitrarily and vary across studies.
A method for validating a biomarker for a particular drug development programme is to document the change in biomarker in phase 1, validate by correlating with the clinical outcome in phase 2, and then use it in confirmatory phase 3 trials. The given drug however would have to show efficacy at the phase 2 proof of concept stage, which is something that has not been accomplished in Alzheimer’s disease trials for drugs other than cholinesterase inhibitors [5].
An example of both qualification of a biomarker and a companion biomarker is Takeda/Zinfandel seeking both to qualify a compound biomarker composed of a TOMM40 genotype, APOE genotype and age, as a prognostic biomarker for risk of MCI due to Alzheimer’s disease, and as a predictive biomarker of the efficacy of very low-dose pioglitazone to delay onset of MCI due to Alzheimer’s disease. This will involve a single phase 3 trial of about 5800 participants without cognitive impairment who will be followed for 5 years to investigate the onset of ‘MCI due to Alzheimer’s disease’ or Alzheimer’s disease dementia [119].
There is considerable hope that biomarkers can first accurately measure biological change that indicates disease progression and, secondly, be validated as surrogate markers of clinical effect and therefore, by virtue of being more precise than the clinical measures, increase the statistical power of clinical trials, thus requiring fewer participants for faster and more efficient trials. In such a scenario the standard clinical outcomes (considered to be insensitive to change especially in early illness) can be wholly replaced [92]. The outcomes of the recent bapineuzumab and solanezumab trials raised further questions regarding the usefulness of current biomarkers as outcomes for trials. Consideration regarding the use of biomarkers should be given to several factors including the type of therapeutic intervention, the clinical stage of illness and the time dependence of biomarker changes during illness.
Although a risk factor and not a biomarker per se, APOE genotype has been commonly treated as a diagnostic or prognostic biomarker. Particular development programmes and trials were designed to predict or assess drug response based on non-carriage of an APOE ε4 allele, such as studies with rosiglitazone and bapineuzumab and the ongoing prevention trial with pioglitazone.
Regulatory considerations for drug development in Alzheimer’s disease
The roles, responsibilities and actions of and guidelines developed by the regulatory authorities are generally not well understood even by experts in the field of drug development for Alzheimer’s disease. Among many other responsibilities, the FDA, EMA and other agencies worldwide regulate and assure the safety, effectiveness, quality, labelling and manufacturing standards of prescription and non-prescription drugs. Specifically they judge whether an investigational drug intended for marketing is safe and effective for its proposed use, the benefits of the drug outweigh the risks, the proposed labelling (i.e. package insert or prescribing information) is appropriate, and the manufacturing methods and controls used to maintain quality are adequate in order to approve the drug for marketing. In Europe, Regulation (EC) No. 726/2004 states that drugs for neurodegenerative disorders must be evaluated centrally by the EMA rather than separately by each individual member state.
As the science changes, regulatory positions evolve as well In general regulatory criteria for marketing of therapies for Alzheimer’s disease require a demonstration of cognitive efficacy and improvements in function, i.e. ADL, and/or evidence of overall clinical improvement (according to FDA draft guidelines [14, 118] and EMA formal guidelines [120]). Regulators have generally facilitated drug development by providing standards for later phase development. For example, the FDA provided unofficial guidelines for trial methods for demonstrating disease modification in 1996 [121], and more recently proposed guidelines for prodromal Alzheimer’s disease [96]. The EMA issued detailed guidelines for disease modification [120], and qualified novel methodologies, including the use of biomarkers for enrichment.
However, guidelines may constrain some development programmes by limiting how drugs with vastly different actions used in a pleomorphic disorder can be developed. Effectively, the guidelines encourage very similar programmes regardless of the different characteristics of the drugs or diagnostic subtlety. Pharmaceutical companies and academics, therefore, may restrict themselves to planning standard protocols that might not be the most appropriate for the drug under development.
For these reasons, regulatory agencies may be involved very early in drug development programmes encouraging companies to seek scientific advice meetings. The EMA, for instance, has established the Scientific Advice Working Party (SAWP) which consists of experts in areas including non-clinical quality and biostatistics, and connects with a network of external clinical experts and patient representatives from the EU. These processes serve to bring together the major stakeholders to develop common protocols for developing new therapeutics for Alzheimer’s disease.
Recent development programmes have tended to place greater emphasis on patients with mild or early Alzheimer’s disease, but it might be expected that efficacy of a treatment that delays disease progression would be demonstrated in trials at different stages of illness. Phase 3 trials for symptomatic drugs commonly have a duration of 6 or 12 months, compared to 18 and 24 months for drugs that could be considered as disease modifying in mild/mild to moderate and in prodromal Alzheimer’s disease, respectively; however, study length does not determine the intention for a disease-modifying trial.
According to the EMA, cholinesterase inhibitors are considered standard care (despite their controversial health effectiveness status) such that a drug being developed needs to be assessed also in a phase 3 add-on design wherein the new drug is used in a placebo-controlled trial in patients already maintained on cholinesterase inhibitors. However, this occurs de facto as it is very difficult to recruit to long-term clinical trials patients with Alzheimer’s disease who are not taking these drugs.
Disease-modification claims
The quest for a disease-modifying marketing claim to be endorsed by the EMA or FDA is driven by the pharmaceutical industry’s assessment that it would sell more drug and at a higher price with such a claim. By comparison, patients and physicians would welcome a drug that worked over a long period whether or not it is called a disease modifier. In the late 1990s, the FDA informally suggested two methods to demonstrate disease modification that would be sufficient for a health claim: a randomized-start and a randomized-withdrawal design [121].
Until recently only the EMA offered guidelines that suggested a method for disease modification. According to the EMA, biomarkers should correlate with the presumed mechanism of action of the drug and ultimately, it is hoped, with the clinical efficacy of the drug [89]. So far, however, no such relationship has been observed other than in small subsets of larger trials that showed null results in terms of the primary outcomes for the drug in question. For example, an Aβ vaccine, AN1792, may reduce CSF phosphorylated-tau (P-tau) protein [22] and amyloid plaque density on autopsy but with no apparent clinically advantageous effects [122]. A trial of bapineuzumab including only 29 patients showed a small decrease in fibrillar Aβ on PiB-PET imaging and a decrease in CSF P-tau after 18 months of treatment [123, 124]. There was a potential trend, depending on the statistical analysis, for tramiprosate to be associated with increased hippocampal volume in a subgroup [125]. In phase 3 bapineuzumab trials relative decreases in PiB-PET uptake and CSF P-tau were observed in subsets of patients [Janssen CTAD presentation, 2012]. All these observations, however, were unrelated to clinical improvement (which did not occur) with the experimental drugs, and do not provide evidence to support the biomarkers in question.
In early 2013 the FDA produced guidelines for a disease-modification marketing claim in the context of early-stage Alzheimer’s disease that are more compatible with the those of the EMA [96, 118]. By early stage the FDA means ‘symptomatic’, preclinical Alzheimer’s disease (so-called stage 3 preclinical) and MCI due to Alzheimer’s disease which it considers to be the same as prodromal disease. These draft guidelines allow for the possibility ‘that a claim of disease modification could be supported by evidence of a meaningful effect on a biomarker in combination with a clinical benefit’, or through the use of a randomized-start design trial in which patients who were initially receiving placebo and then assigned to active treatment would be expected to fail to catch up in order for the drug to receive a disease-modification claim. The FDA prefers the latter option and first proposed this clinical trial design two decades earlier [121]. It was noted, however, that for the former to be the case ‘there should be widespread evidence-based agreement in the research community that the chosen biomarker reflects a pathophysiologic entity that is fundamental to the underlying disease process. There is currently no consensus as to what particular biomarkers would be appropriate to support clinical findings in trials in early AD [Alzheimer’s disease]’.
Fig. 4 shows a schematic example of a randomized-start design, demonstrating the type of clinical effect that could be considered disease modifying. Indeed, in the late 1990s a 12-month trial of propentofylline built in both a randomized-start and randomized-withdrawal design by re-randomizing at 12 months and in a double-blind manner extending the trial by 6 months [54]. However, the drug was withdrawn from development.
Fig. 4.

Illustration of a randomized-start design for a regulatory clinical trial to demonstrate disease modification. Reproduced with permission from Leber [120].
In evaluating the development plans of mainly anti-amyloid drugs, the EMA has issued further scientific advice as to how to contextualize a disease-modification claim in light of the quest to treat patients with very early stage disease and to develop diagnostic companion biomarkers. Assuming that prodromal Alzheimer’s disease and dementia are part of a continuum, the efficacy of a treatment that delays disease progression should be demonstrated in two trials. The first trial should recruit patients with early stage disease with virtually no functional impairment at baseline and efficacy should be demonstrated on a composite endpoint, such as the CDR-SB, or possibly a single primary endpoint providing that it is validated in this population. The second trial should recruit patients with mild or mild to moderate disease, with efficacy demonstrated on two co-primary endpoints addressing both cognition and function. The evidence from both trials should be robust and supported by secondary endpoints including responder analyses and biomarkers that show delay in brain neurodegeneration.
The FDA has emphasized that a disease modification claim would be seen as an extraordinary major claim, implying that nearly all individuals at risk of Alzheimer’s disease would need to take the drug, putting the prospects for approval of a drug with a disease-modification claim in a realistic context. One challenge for disease-modifying drugs will be their costs especially if the majority of older people use them. Cost-effectiveness estimates in this area are controversial, although the findings of a Swedish simulation study suggested that the main costs were the price of the drug and the cost of an expected 1-year decrease in mortality. An optimistic, incremental cost-effectiveness ratio was estimated at $50,000 per Quality-Adjusted Life Year, with no cost savings, but below an expected willingness-to-pay range, at least in Sweden [126]. However, long-term data are needed to understand the progression of change beyond the trial period, and particularly with drugs intended to interfere with the amyloid cascade to assess for any worsening or rebound that may occur with prolonged therapy.
Discussion
The only drugs approved for the treatment of Alzheimer’s disease over the past 30 years have been cholinesterase inhibitors and memantine, and none has been approved since 2003. Nearly 200 drugs were advanced to at least the level of phase 2 development. Moreover, the approved drugs show limited clinical effect and there is controversy regarding whether they are therapeutically useful (Fig. 5. The most probable explanation for this failure, of course, is that the tested drugs simply were not effective for their intended indication. Alzheimer’s disease is a complex, multi-determined disease with a pleomorphic phenotype. There are many potential drug targets and so far no validated targets except perhaps for the cholinergic system.
Fig. 5.

A comparison of the same effects of donepezil with standard deviation bars showing the distribution of drug and placebo outcomes and standard error bars indicating the precision of the outcomes measures. Reproduced with permission from Lindner et al. [2].
Although very few of the many negative trials give clear information about why results were null or inconclusive, the methods used to develop the drugs should be viewed with caution and critically appraised. Important issues include whether development programmes and trial methods have gone wrong, and are there better approaches to late-stage trials that might enhance the likelihood of success?
Explaining after the fact why trials failed despite our best efforts is speculative and any rationalizations are likely to be incorrect. One current explanation is that patients should have been treated earlier, particularly those with milder illness and who have amyloid markers associated with Alzheimer’s disease pathology. However, although rational and based on observations that amyloid biomarkers in some cohorts precede symptoms, there is no supporting evidence that this approach will be effective. Many phase 2 and 3 trials now include a diagnosis of prodromal Alzheimer’s disease supported by amyloid PET scans or CSF Aβ despite the limited evidence about biomarkers validity.
The only consistency in drug development for Alzheimer’s disease that might constitute a successful approach is for drugs that affect the cholinergic system. The cholinesterase inhibitors, including those that were not marketed, showed consistent, albeit small, statistical efficacy over 6 and sometimes 12 months. Even so the drugs do not achieve the minimum clinically important drug–placebo difference of 4 points on the ADAS-cog that was sought by experts in the 1990s; in addition, they are not effective in MCI, i.e. in patients who are likely to have amyloid pathology and progress to Alzheimer’s disease. Thus, a ‘treat-early’ hypothesis is unlikely to be sustained with cholinesterase inhibitors. A model of clinical trials that was recently qualified by the FDA and EMA [127] demonstrated that cholinesterase inhibitors do not change illness course and have negligible long-term effects on the ADAS-cog.
Muscarinic M1 agonists showed slight but overall consistent effects. Furthermore, other drugs in development that affect the cholinergic system including α7 allosteric modulators and 5-HT6 antagonists have shown measurable cognitive effects across compounds and are in active development. Moreover, the phase 2 and 3 trials showed efficacy with from 400 to 700 patients, far fewer than the sizes of phase 2 and 3 trials for the anti-amyloid drugs that did not show significant effects. In retrospect this level of consistency demonstrates a robust proof of concept for the cholinergic hypothesis despite the small magnitude of the clinical effects. It is notable that there have been no formal a priori attempts to maximize the effects of cholinesterase inhibitors, for example to identify and target clinical subgroups that might be better responders.
By comparison, the results of trials of drugs for other targets have been disappointing. For example, although well-reasoned and based on preclinical studies, anti-inflammatory agents, conjugated oestrogens, secretase inhibitors and Aβ vaccines have had unexpected ‘opposite’ (i.e. cognition-impairing) effects, and failed in trials intended to be confirmatory. Phase 3 trials should be hypothesis confirming and such surprises should not be common.
A milestone for drug development is the progression from phase 2, a safety and proof of concept phase, to pivotal trials in phase 2b or 3. The regular failures in phase 3 suggest that decisions may not be based on the evidence from phase 2, that phase 2 is not informative, or that we may be acting on signals that are not signals; in particular, over-valuing or misinterpreting post hoc subset analyses. For example, the phase 2 trials of tarenflurbil and bapineuzumab did not show significant outcomes, phase 2 safety findings for semagacestat demonstrated major toxicity and phase 2 was avoided for solanezumab except to establish minimal safety for phase 3 and pharmacodynamic changes in Aβ. Under these circumstances, the negative outcomes of the subsequent phase 3 trials should not have been surprising.
No anti-amyloid therapy phase 3 trial has been preceded by a positive phase 2 proof of concept trial. Moreover, the fact that the protocols of some pivotal trials had to be changed during the study because of toxicity means that characteristics of the drugs, dosages and targets were not known before entering confirmatory trials. In summary, an unfortunate characteristic of development programmes for Alzheimer’s disease has been that phase 2 does not provide ‘proof of concept’ to predict phase 3, but rather is used as a bridge to the next stage. Thus the large phase 3 trials, instead of being confirmatory, are often exploratory.
The decision to advance a drug may be more dependent on how pharmaceutical companies make decisions than on an evidence-based likelihood of efficacy. A phase 3 trial may be undertaken without phase 2 support based on business considerations about the future sales and profits over the duration of the drug’s marketing life. Therefore, the sponsor may be willing to spend relatively large amounts on late-stage trials while recognizing the high probability of failure in pursuit of extremely large returns over the life of the drug. As the phase 2 trials are negative, the phase 3 trials are conducted without adequate prior information for planning, often with data gleaned from post hoc subgroup analyses, and methods duplicated from previous, unsuccessful trials. The new trial represents, in effect, a large call option for the pharmaceutical company.
Although many drugs should not have been brought into phase 2 or 3, for many more development was in fact stopped early and appropriately in phase 1 and 2a and these trials were not included is this review focused on late-stage development. Some drugs in phase 3 programmes do have significant efficacy results from phase 2 trials to support their advancement. These drugs are mainly small molecules, so-called ‘symptomatic’ often cholinergically acting compounds, and not considered disease-modifying agents.
Another characteristic of the methods used for trials in Alzheimer’s disease is that drugs with very different therapeutic activities and outcome expectations are developed by using essentially the same phase 2 and phase 3 clinical trial designs as were used with cholinesterase inhibitors in the early 1990s [5, 128]. There are few adaptations in trial design for the unique characteristics of different drugs, durations of use, mechanisms by which they exert their effects, or for any expected different outcomes. Late-phase development does not seem to be tailored to a drug’s unique characteristics or to the patients who might benefit.
The minimal changes in trial methods may be a consequence of having no successes to build on and the perceived risk aversion of companies and academics to making changes. Thus small changes occur: MMSE criteria ranges are expanded or contracted, trial durations are lengthened, similar outcomes are exchanged, sample sizes are increased, amyloid biomarkers are used to increase diagnostic confidence and smaller clinical effects are sought. Notably, trials now require drug–placebo differences of only 1 to 2 points on the ADAS-cog over 18 to 24 months, i.e. half the effect in three times the duration of treatment than had been sought with the cholinesterase inhibitors (Fig. 5).
More refined diagnostic and prognostic markers have advantages and disadvantages. The use of biomarkers may lead to sample enrichment of patients who might benefit from a particular drug, future identification of valid subtypes and drug-responsive genotypes and phenotypes. On the other hand, the current markers being advanced may be pseudo-specific to the illness and its progression, or may indicate more advanced illness.
In this non-systematic review, important drugs may have missed and others may have been over-emphasized. Yet it is apparent that phase 2 and 3 drug development has been over-represented by anti-amyloid approaches. It is beyond the scope of this review to comment on the validity of the amyloid cascade hypothesis. It is not possible to extrapolate from trial failures to refute the hypothesis; to do so it must be known or demonstrated that the drug entered the brain, had a clear effect on the target, and then resulted in no effect. Without such information it can only be concluded that these approaches have been tested inadequately.
Conclusions and future directions
The sustainability of and methods of conducting large clinical development programmes in Alzheimer’s disease are questionable. Many trials are including earlier-stage diagnoses or at-risk states, requiring greater numbers of participants, and longer follow-up periods now extending beyond 18 months. The prevention trials are longer still, possibly including a majority of participants who may not develop clinical Alzheimer’s disease during the follow-up period or their lives. Specifically, phase 2 and 3 development programmes have grown from trials with 400 to 700 participants and two 6-month trials, to 1200 to 2300 participants in trials extending to 24 months.
There is a need to better appreciate the pleomorphism of the illness, its competing risk factors and precipitants such as age, genetics, environmental factors and cerebrovascular and cardiovascular disease. The wide range of targets make it unlikely that affecting one alone will lead to a more than minimally effective therapeutic agent.
The goal of enrichment with biomarkers is targeted research. However, targeted clinical trials also need to be focused, based on the drug, patient characteristics and ideally a companion biomarker. Better, smarter phase 2 studies are needed to identify potentially effective drugs for phase 3. This could involve fewer patients, drug-specific outcomes and protocol designs that can be changed or adapted after randomization. Phase 3 trials may need to be more efficiently designed with clinically relevant and more precise outcomes.
Past failures and the dichotomies of symptomatic versus disease-modifying, early versus late and amyloid versus not suggest that there is still much to learn about late-phase development. Less emphasis should be placed on consensus, received wisdom, precedence and wishfulness and more on prior knowledge, as consensus does not mean that what is done is correct. The lack of progress despite substantial effort may rest, as stated by Leber, a former FDA director, ‘Not in our methods, but in our ignorance’ [129].
Fig. 3.
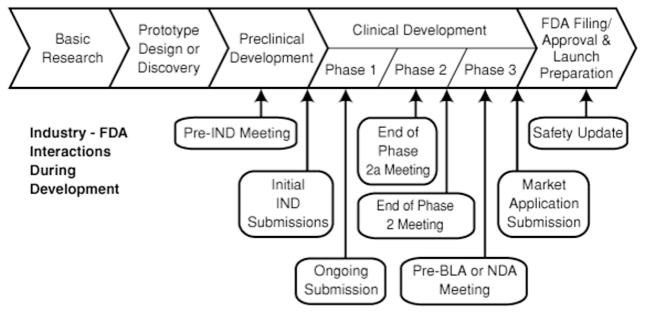
The drug development process from preclinical development to approval. The focus of this review is phase 2 to phase 3 development. In general, the trials discussed were planned as phase 2, 2a or 3. Some phase 2 trials discussed were pivotal and could have been the basis for regulatory approval if the results had been positive. Modified from the FDA publication, Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm
Abbreviations: BLA: biologics license application; FDA: Food and Drug Administration; IND: Investigational new drug application; NDA: New drug application.
Table 2.
Alzheimer’s disease (AD) treatment: recent therapeutic approaches in phase 2 or 3. Mainly symptomatics, targeting neurotransmission
| Main mechanism of action or class | RCTs completed (examples) | RCTs ongoing (examples) |
|---|---|---|
| Cholinergic agents | ||
| Acetylcholinesterase inhibitors, butrylcholinesterase inhibitors |
Donepezil, rivastigmine, galantamine: improve cognition in mild–moderate AD [39]; (>45 RCTs, >18,000 subjects, mainly 6-month duration) [31–33] MCI: no clinical effects, increased adverse events [74] |
|
|
| ||
| Muscarinic receptor agonists | Some compounds tested in phase 1 RCTs (e.g. talsaclidine, AF-102B, AF267B), were discontinued due to adverse effects due to peripheral cholinergic stimulation [21, 24] | |
|
| ||
| Nicotinic receptor agonists (modulators of the nicotinic receptors α7, α2β2, α4β2, α6β2): effects on several neurotransmitters (acetylcholine, dopamine, noradrenaline, GABA, glutamate) |
AZD-3480 (modulators of nicotinic receptors α4β2, α2β2): no evidence of benefit in a 3-month phase 2 RCT in 567 patients with mild–moderate AD [130] EVP-6124 (modulator α7 receptor): positive results in a 24-week phase 2b RCT in 409 cases of mild–moderate AD [131] |
MT-4666 (α7 modulator): 6 months, phase 2 RCT in ≈ 450 patients with mild–moderate AD AZD-3480: phase 2 RCT in mild–moderate AD (1 year, ≈ 300 participants) ABT-126: two phase 2, mild–moderate AD, 6 months ≈ 820 participants with and without stable ChEI treatment EVP-6124: two phase 3 trials in 1580 subjects with mild–moderate AD, 26 weeks. MK-7622 (α7 modulator): phase 2b, 12 to 24 weeks in 640 patients with mild–moderate AD (dose-ranging study) |
|
| ||
| Glutamatergic agents | ||
| NMDA receptor antagonist Modulate glutamatergic transmission |
Memantine: clinical benefits in moderate– severe AD, mainly 6 months [132]. Not effective in mild AD [133] DAOI-B (NMDA enhancer): 6 months, phase 2 RCT in 86 subjects with MCI or mild AD (not published) |
AVP-923 (dextromethorphan/quinidine): phase 2 RCTs in mild–moderate AD (main outcome: behavioural problems, 10 weeks, ≈ 200 participants) |
|
| ||
| Serotoninergic agents | ||
| 5-HT6 receptor antagonists: effects on cholinergic, glutamatergic and monoaminergic neurons |
SB742457: positive results in three phase 2 RCTs, 6 months, ≈ 1460 patietns with mild– moderate AD; 1 negative RCT [134] Lu AE58054: positive outcomes in one phase 2 RCT in mild–moderate AD, 6 months, 278 participants (NCT01019421) |
Lu AE58054: several phase 3 trials with about 2500 patients with mild–moderate AD on cholinesterase inhibitors |
|
| ||
|
Monoamine oxidase inhibitors: enhance serotoninergic, noradrenergic, dopaminergic function, neuroprotective |
Selegiline and vitamin E for moderate AD [61] Selegiline: meta-analysis of 17 RCTs including 1143 subjects with moderate– severe AD, durations from 3 to 105 weeks, some positive effects, but overall no significant clinically meaningful benefits | RO4602522 (EVT 302): phase 2 RCT in 495 patients with moderate AD, 1 year |
|
| ||
| Multi-target-directed ligands | ||
| Ladostigil (TV-3326): derivative of rasagiline and rivastigmine, acts as an AChEI and MAO-I, antioxidant properties, modulates APP processing | Phase 2 RCTs in MCI (3 years, ≈ 200 participants) and mild–moderate AD (26 weeks + 26 weeks open label, ≈ 188 participants) | |
RCT: randomized controlled trial;, GABA: γ-Aminobutyric acid; NMDA: N-methyl-D-aspartate AChEI: acetylcholinesterase inhibitor, APP: amyloid precursor protein
Table 4.
Alzheimer’s disease (AD) treatment: recent and current therapeutic approaches in phase 2 or 3. Other approaches
| Main mechanism of action | RCTs completed (examples) | RCTs ongoing (examples) |
|---|---|---|
|
Nutraceuticals and food supplements Macro/micronutrients, alone and in combination: antioxidants, omega-3-PUFAs, vitamins B |
PUFA: 6-month RCT in 174 subjects with mild– moderate AD did not report beneficial effect on cognition and CSF markers of AD [153, 154]. 18- month, phase 3 RCT in 285 participants with mild–moderate AD showed absence of benefits on cognition and functional status [81] | PUFA: several RCTs in which PUFAs are tested alone or combined with other supplements (e.g. lipoic acid) or lifestyle interventions. Subjects with different levels of cognitive impairment have been enrolled, duration 6 months to 3 years |
|
| ||
| Phosphodiesterase inhibitors | ||
| Regulate cGMP signalling pathways involved in synaptic plasticity and inflammation |
PF-04447943: selective PDE 9A inhibitor tested in a phase 2 RCT in ≈ 200 subjects with mild– moderate AD (completed 2009, not published) MK0952: PDE4 inhibitor, phase 2 RCT in mild– moderate AD (completed, results not published) Cilostazol: PDE3 inhibitor, approved for stroke prevention. Open trial in 10 patients with moderate AD: safety, possible cognitive benefit [155] |
|
|
| ||
| Tyrosine kinase inhibitor | Masitinib, phase 3, 6 months ≈ 400 participants with mild–moderate AD (NCT01872598) | |
|
| ||
| Cholesterol lowering | ||
| HMG-CoA reductase inhibitors have multiple effects: ↓ Aβ production, Aβ-mediated neurotoxicity, antioxidant, anti-inflammatory | Simvastatin and atorvastatin: two phase 3 RCTs, ≈ 1100 subjects, showed no benefits in mild– moderate AD after 18 months [78, 79] | Simvastatin: RCTs of effects on AD biomarkers, 1 year, ≈ 120 participants; and preventing/ delaying progression to dementia in aMCI, 2 years, ≈ 640 participants |
|
| ||
|
Insulin and glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonists Improve insulin-mediated signalling, brain glucose metabolism, neuro- transmission, possible effects on Aβ neurotoxicity |
Intranasal insulin: 4-month phase 2 RCT, 111 subjects with aMCI and mild AD; some cognitive benefits reported [156] |
Intranasal insulin: Phase 2/3 in aMCI and mild AD, 18 months, ≈ 240 participants Exendin-4 (GLP-1R): phase 2 RCT in 230 subjects with MCI or early AD, 3 years Liraglutide (GLP-1R): phase 2 RCT in 206 subjects with mild AD, 1 year |
cGMP, cyclic guanosine monophosphate; CSF, cerebrospinal fluid; GSK-3, glycogen-synthase-kinase-3; PDE, phosphodiesterase; PUFAs, polyunsaturated fatty acids; ARIA-E, amyloid-related imaging abnormalities with parenchymal oedema; ARIA-H, amyloid-related imaging abnormalities with intracerebral microhaemorrhages. RCT: randomized controlled trial; aMCI: amnestic mild cognitive impairment; HMG-CoA: 3-hydroxy-3-methyl-glutaryl-CoA reductase; ↓: reduction.
Acknowledgments
The authors received travel support from the Journal of Internal Medicine.
LSS was supported in part by the USC Alzheimer’s Disease Research Center, NIH P50 AG05142 and NIH R01 AG037561.
BW was supported in part by the Swedish Brain Power and the StratNeuro programmes, Karolinska Institutet.
Footnotes
Disclosures and conflict of interest statements
LSS has received grants from the NIH P50 AG05142, R01 AG033288 and R01 AG037561, the State of California, the Alzheimer’s Association for a registry for dementia and cognitive impairment trials and grants or research support from Baxter, Genentech, Johnson & Johnson, Eli Lilly, Novartis and Pfizer. He has served as a consultant for and received consulting fees from Abbott Laboratories, AC Immune, Allon, AstraZeneca, Baxter, Biogen Idec, Biotie, Bristol-Myers Squibb, Elan, Eli Lilly, EnVivo, GlaxoSmithKline, Johnson & Johnson, Lundbeck, MedAvante, Merck, Novartis, Piramal, Pfizer, Roche, Sanofi, Servier, Takeda, Tau Rx, Toyama and Zinfandel.
PM has received research support from Lundbeck, Novartis and Pfizer and served as a consultant for and received consulting fees from Baxter, Bristol-Myers Squibb, Eli Lilly, Lundbeck, Merck, Merz, Novartis, Pfizer, Roche, Sanofi and Servier.
HF has received grants from CIHR, Pacific Alzheimers Research Foundation, NIH, the Alz Society of Canada. He has received research support or performed clinical trials with Biogen, Elan, GE Health, Lundbeck, Myriad, Glaxo, ONO, Bayer, Roche, Servier, Janssen, Eisai, Pfizer, Lilly, Sanofi, Novartis, Neotherapeutics, Boehringer Ingelheim. He has served as consultant for and received consulting fees from Elan, Lundbeck, Servier, Sanofi, Pfizer, Eisai, Eli Lilly, Novartis, Roche, Bayer, Neurocrine, ONO, Glaxo, Janssen, Boehringer Ingelheim. While on leave from UBC between 2009–2011, he was a paid full time employee of Bristol-Myers Squibb for which he received salary, stock, and stock options.
RJ has received research support from Eli Lilly, Janssen Alzheimer Immunotherapy, Servier, Abbott and Genentech and personal fees for advisory board meetings and/or chairmanship or lecture fees at symposia from Eisai, Elan, Eli Lilly, Janssen Alzheimer Immunotherapy, Lundbeck, Merz, Novartis, Nutricia, Otsuka, Pfizer and Roche.
VM and LP have no competing interests to disclose and no specific funding was received for writing this article. The views expressed in this article are the personal views of VM and LP and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agency or any of the committees or working parties of the regulatory agencies for which the authors work.
BW has received research support from Dainippon Sumitomo Pharma Co. Ltd and has served as a consultant at advisory board meetings for AC Immune, Axon, Diagenic, Eli Lilly, Johnson & Johnson, Lundbeck, Merz, Novartis, Pfizer, Roche and Servier.
MK has received grants from the Alzheimer’s Association, Alzheimer’s Research and Prevention Foundation, Academy of Finland, EU 7th Framework and Swedish Research Council. She has served as a consultant at advisory board meetings for Elan, Johnson & Johnson, Merz, Novartis, Nutricia and Pfizer.
FM, NA, EG, and HF have no conflicts of interest to disclose.
References
- 1.Becker RE, Greig NH, Giacobini E. Why Do So Many Drugs for Alzheimer’s Disease Fail in Development? Time for New Methods and New Practices? Journal of Alzheimer’s Disease. 2008;15:303–25. doi: 10.3233/jad-2008-15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindner MD, McArthur RA, Deadwyler S, Hampson R, Tariot PN. Development, Optimization and Use of Preclinical Behavioral Models to Maximize the Productivity of Drug Discovery for Alzheimer’s Disease. In: McArthur RA, Borsini F, editors. Animal and Translational Models for CNS Drug Discovery. San Diego: Academic Press; 2008. pp. 93–157. [Google Scholar]
- 3.Pharmaceutical Research and Manufacturers of America. Researching Alzheimer’s Medicines: Setbacks and Stepping Stones. 2012. [Google Scholar]
- 4.Giacobini E. Cholinesterases in human brain: the effect of cholinesterase inhibitors on Alzheimer’s disease and related disorders. In: Giacobini E, Pepeu G, editors. The Brain Cholinergic System in Health and Disease. Oxon, UK: Informa Healthcare; 2006. pp. 235–64. [Google Scholar]
- 5.Schneider LS. Issues in Design and Conduct of Clinical Trials for Cognitive-Enhancing Drugs. In: McArthur RA, Borsini F, editors. Animal and Translational Models for CNS Drug Discovery. San Diego: Academic Press; 2008. pp. 21–76. [Google Scholar]
- 6.Tacrine as a Treatment for Alzheimer’s Dementia. New England Journal of Medicine. 1991;324:349–52. doi: 10.1056/NEJM199101313240525. [DOI] [PubMed] [Google Scholar]
- 7.Davis KL, Thal LJ, Gamzu ER, et al. A double-blind, placebo-controlled multicenter study of tacrine for Alzheimer’s disease. The Tacrine Collaborative Study Group [see comments] New England Journal of Medicine. 1992;327:1253–9. doi: 10.1056/NEJM199210293271801. [DOI] [PubMed] [Google Scholar]
- 8.Murphy MF, Hardiman ST, Nash RJ, Huff FJ, Demkovich JJ, Dobson C, Knappe UE. Evaluation of HP 029 (velnacrine maleate) in Alzheimer’s disease. Annals of the New York Academy of Sciences. 1991;640:253–62. doi: 10.1111/j.1749-6632.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 9.Thal LJ, Schwartz G, Sano M, et al. A multicenter double-blind study of controlled-release physostigmine for the treatment of symptoms secondary to Alzheimer’s disease. Physostigmine Study Group. Neurology. 1996;47:1389–95. doi: 10.1212/wnl.47.6.1389. [DOI] [PubMed] [Google Scholar]
- 10.Olin JT, Schneider LS, Doody RS, et al. Clinical evaluation of global change in Alzheimer’s disease: identifying consensus. Journal of Geriatric Psychiatry & Neurology. 1996;9:176–80. doi: 10.1177/089198879600900404. [DOI] [PubMed] [Google Scholar]
- 11.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–64. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- 12.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 13.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, D.C: American Psychiatric Association Press; 1987. [Google Scholar]
- 14.Leber P. Guidelines for the clinical evaluation of anti-dementia drugs, 1st draft. Rockville, MD: United States Food and Drug Amdinistration; 1990. [Google Scholar]
- 15.Drachman DA, Leavitt J. Human memory and the cholinergic system: A relationship to aging? Archives of Neurology. 1974;30:113–21. doi: 10.1001/archneur.1974.00490320001001. [DOI] [PubMed] [Google Scholar]
- 16.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease [letter] Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 17.Bowen DM, Smith CB, White P, Davison AN. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. 1976;99:459–96. doi: 10.1093/brain/99.3.459. [DOI] [PubMed] [Google Scholar]
- 18.Coyle J, Price D, DeLong M. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–90. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 19.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences. 1991;12:383–8. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 20.Golde TE, Schneider LS, Koo EH. Anti-Aβ Therapeutics in Alzheimer’s Disease: The Need for a Paradigm Shift. Neuron. 2011;69:203–13. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. The Lancet Neurology. 2010;9:702–16. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 22.Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 23.United States Food and Drug Administration Peripheral and Central Nervous System Drugs Advisory Committee Meeting; July 7, 1989; Rockville, MD: Department of Health and Human Services, Public Health Service, Food and Drug Administration; 1989. [Google Scholar]
- 24.McArthur RA, Gray J, Schreiber R. Cognitive effects of muscarinic M1 functional agonists in non-human primates and clinical trials. Current Opinion in Investigational Drugs. 2010;11:740– 60. [PubMed] [Google Scholar]
- 25.Farlow M, Gracon SI, Hershey LA, Lewis KW, Sadowsky CH, Dolan-Ureno J. A controlled trial of tacrine in Alzheimer’s disease. The Tacrine Study Group [see comments] Journal of the American Medical Association. 1992;268:2523–9. [PubMed] [Google Scholar]
- 26.Knapp MJ, Knopman DS, Solomon PR, Pendlebury WW, Davis CS, Gracon SI. A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer’s disease. The Tacrine Study Group [see comments] Journal of the American Medical Association. 1994;271:985–91. [PubMed] [Google Scholar]
- 27.Thal LJ, Ferguson JM, Mintzer J, Raskin A, Targum SD. A 24-week randomized trial of controlled-release physostigmine in patients with Alzheimer’s disease. Neurology. 1999;52:1146–52. doi: 10.1212/wnl.52.6.1146. [DOI] [PubMed] [Google Scholar]
- 28.Imbimbo BP, Martelli P, Troetel WM, Lucchelli F, Lucca U, Thal LJ. Efficacy and safety of eptastigmine for the treatment of patients with Alzheimer’s disease. Neurology. 1999;52:700–8. doi: 10.1212/wnl.52.4.700. [DOI] [PubMed] [Google Scholar]
- 29.Imbimbo BPTWM, Martelli P, Lucchelli F the Eptastigmine Study Group. A 6-Month Double-Blind, Placebo-Controlled Trial of Eptastigmine i Alzheimer’s Disease. Dement Geriatr Cogn Disord. 2000:17–24. doi: 10.1159/000017208. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Arrieta JM, Schneider L. Metrifonate for Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2006:CD003155. doi: 10.1002/14651858.CD003155.pub3. [DOI] [PubMed] [Google Scholar]
- 31.Birks J, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2006:CD001190. doi: 10.1002/14651858.CD001190.pub2. [DOI] [PubMed] [Google Scholar]
- 32.Birks J, Grimley Evans J, Iakovidou V, Tsolaki M. Rivastigmine for Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2009:CD001191. doi: 10.1002/14651858.CD001191.pub2. [DOI] [PubMed] [Google Scholar]
- 33.Loy C, Schneider L. Galantamine for Alzheimer’s disease and mild cognitive impairment. Cochrane Database of Systematic Reviews 2006, Issue 1. Cochrane Database of Systematic Reviews. 2006:CD001747. doi: 10.1002/14651858.CD001747.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatellier G, Lacomblez L. Tacrine (tetrahydroaminoacridine; THA) and lecithin in senile dementia of the Alzheimer type: a multicentre trial. Groupe Francais d’Etude de la Tetrahydroaminoacridine [see comments] BMJ (Clinical Research Ed) 1990;300:495–9. doi: 10.1136/bmj.300.6723.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eagger SA, Levy R, Sahakian BJ. Tacrine in Alzheimer’s disease [see comments] Lancet. 1991;337:989–92. doi: 10.1016/0140-6736(91)92656-m. [DOI] [PubMed] [Google Scholar]
- 36.Schneider LS, Olin JT. Overview of clinical trials of hydergine in dementia [see comments] Archives of Neurology. 1994;51:787–98. doi: 10.1001/archneur.1994.00540200063018. [DOI] [PubMed] [Google Scholar]
- 37.Jones RW, Laake K, Øksengård AR. D-cycloserine for Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2002;2:Art No: CD003153. doi: 10.1002/14651858.CD003153. DOI: 101002/14651858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birks J, Flicker L. Selegiline for Alzheimer’s diseaseArt. No. Cochrane Database of Systematic Reviews. 2003:1. doi: 10.1002/14651858.CD000442. [DOI] [PubMed] [Google Scholar]
- 39.Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2006:CD005593. doi: 10.1002/14651858.CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loveman E, Green C, Kirby J, Takeda A, Picot J, Payne E, Clegg A. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technology Assessment. 2006:10. doi: 10.3310/hta10010. [DOI] [PubMed] [Google Scholar]
- 41.Takeda A, Loveman E, Clegg A, Kirby J, Picot J, Payne E, Green C. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer’s disease. International Journal of Geriatric Psychiatry. 2006;21:17–28. doi: 10.1002/gps.1402. [DOI] [PubMed] [Google Scholar]
- 42.Schneider LS, Dagerman KS, Higgins JPT, McShane R. Lack of Evidence for the Efficacy of Memantine in Mild Alzheimer Disease. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.69. archneurol.2011.69. [DOI] [PubMed] [Google Scholar]
- 43.Aisen PS, Davis KL, Berg JD, et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study [see comment] Neurology. 2000;54:588–93. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 44.Mulnard RA, Cotman CW, Kawas C, et al. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer’s Disease Cooperative Study [Erratum appears in JAMA 2000 Nov 22–29;284(20):2597] JAMA. 2000;283:1007–15. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- 45.Hake A, Tariot P, Hochadel T, Schneider L, Lahiri DK, Farlow M. Apolipoprotein E genotype in a 48-week multicenter double-blind placebo-controlled transdermal selegiline trial in mild to moderate alzheimer disease. Journal of Neuroscience. 2001;187:S55. [Google Scholar]
- 46.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 47.Aisen P, Schafer K, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 48.Soininen H, West C, Robbins J, Niculescu L. Long-Term Efficacy and Safety of Celecoxib in Alzheimer’s Disease. Dementia and Geriatric Cognitive Disorders. 2007;23:8–21. doi: 10.1159/000096588. [DOI] [PubMed] [Google Scholar]
- 49.Thal LJ, Carta A, Clarke WR, et al. A 1-year multicenter placebo-controlled study of acetyl-L-carnitine in patients with Alzheimer’s disease. Neurology. 1996;47:705–11. doi: 10.1212/wnl.47.3.705. [DOI] [PubMed] [Google Scholar]
- 50.Thal LJ, Calvani M, Amato A, Carta A. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology. 2000;55:805–10. doi: 10.1212/wnl.55.6.805. [DOI] [PubMed] [Google Scholar]
- 51.Le Bars PL, Katz MM, Berman N, Itil TM, Freedman AM, Schatzberg AF. A Placebo-Controlled, Double-blind, Randomized Trial of an Extract of Ginkgo Biloba for Dementia. JAMA: The Journal of the American Medical Association. 1997;278:1327–32. doi: 10.1001/jama.278.16.1327. [DOI] [PubMed] [Google Scholar]
- 52.Thal LJ, Grundman M, Berg J, et al. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology. 2003;61:1498–502. doi: 10.1212/01.wnl.0000096376.03678.c1. [DOI] [PubMed] [Google Scholar]
- 53.Amaducci L, Maurer K, Winblad B, et al. A long-term, double-blind, placebo-controlled efficacy and safety study of nicergoline in patients with mild to moderate Alzheimer’s disease. Journal of the European College of Neuropsychopharmacology. 1999:9. [Google Scholar]
- 54.Kittner B, RÖSsner M, Rother M. Clinical Trials in Dementia with Propentofylline. Annals of the New York Academy of Sciences. 1997;826:307–16. doi: 10.1111/j.1749-6632.1997.tb48481.x. [DOI] [PubMed] [Google Scholar]
- 55.Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. The Lancet Neurology. 2008;7:483–93. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- 56.Sevigny JJ, Ryan JM, van Dyck CH, Peng Y, Lines CR, Nessly ML On behalf of the MK-677 Protocol 30 Study Group. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology. 2008;71:1702–8. doi: 10.1212/01.wnl.0000335163.88054.e7. [DOI] [PubMed] [Google Scholar]
- 57.Schneider LS, Farlow MR, Porsteinsson A, Shimakura A, Nakagawa M, Iwakami N. The neuroprotective and neurotrophic agent T-817MA for Alzheimer disease: randomized, double-blind, placebo-controlled proof of concept trial outcomes (O3-06-06) Alzheimer’s Association International Conference on Alzheimer’s Disease Alzheimer’s & Dementia. 2013:9. [Google Scholar]
- 58.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 59.Winblad B, Engedal K, Soininen H, et al. A 1-year, randomized, placebo-controlled study of donepezil in patients with mild to moderate AD. [see comment] Neurology. 2001;57:489–95. doi: 10.1212/wnl.57.3.489. [DOI] [PubMed] [Google Scholar]
- 60.Mohs RC, Doody RS, Morris JC, et al. A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology. 2001;57:481–8. doi: 10.1212/wnl.57.3.481. erratum appears in Neurology 2001 Nov 27;57(10):1942. [DOI] [PubMed] [Google Scholar]
- 61.Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study [see comment] New England Journal of Medicine. 1997;336:1216–22. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 62.Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry. 1982;139:1136–9. doi: 10.1176/ajp.139.9.1136. [DOI] [PubMed] [Google Scholar]
- 63.Morris JC, McKeel DW, Jr, Storandt M, et al. Very mild Alzheimer’s disease: Informant-based clinical, psychometric, and pathologic distinction from normal aging. Neurology. 1991;41:469. doi: 10.1212/wnl.41.4.469. [DOI] [PubMed] [Google Scholar]
- 64.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of Neurology. 1999;56:303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 65.Mild Cognitive Impairment; United States Food and Drug Administration Peripheral and Central Nervous System Drugs Advisory Committee; [March 13, 2001]. 2001. http://www.fda.gov/ohrms/dockets/ac/01/minutes/3724m1.html. [Google Scholar]
- 66.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. [see comment] New England Journal of Medicine. 2005;352:2379–88. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 67.Feldman HH, Ferris S, Winblad B, et al. Effect of rivastigmine on delay to diagnosis of Alzheimer’s disease from mild cognitive impairment: the InDDEx study. [see comment] Lancet Neurology. 2007;6:501–12. doi: 10.1016/S1474-4422(07)70109-6. [DOI] [PubMed] [Google Scholar]
- 68.Winblad B, Gauthier S, Scinto L, et al. Safety and efficacy of galantamine in subjects with mild cognitive impairment. Neurology. 2008;70:2024–35. doi: 10.1212/01.wnl.0000303815.69777.26. [DOI] [PubMed] [Google Scholar]
- 69.Thal LJ, Ferris SH, Kirby L, et al. A Randomized, Double-Blind, Study of Rofecoxib in Patients with Mild Cognitive Impairment. Neuropsychopharmacology. 2005;30:1204–15. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 70.Smith AD, Smith SM, de Jager CA, et al. Homocysteine-Lowering by B Vitamins Slows the Rate of Accelerated Brain Atrophy in Mild Cognitive Impairment: A Randomized Controlled Trial. PLoS ONE. 2010;5:e12244. doi: 10.1371/journal.pone.0012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jelic V, Kivipelto M, Winblad B. Clinical trials in mild cognitive impairment: lessons for the future. Journal of Neurology, Neurosurgery & Psychiatry. 2006;77:429–38. doi: 10.1136/jnnp.2005.072926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology. 2004;63:651–7. doi: 10.1212/01.wnl.0000134664.80320.92. [DOI] [PubMed] [Google Scholar]
- 73.Doody RS, Ferris SH, Salloway S, et al. Donepezil treatment of patients with MCI. Neurology. 2009;72:1555–61. doi: 10.1212/01.wnl.0000344650.95823.03. [DOI] [PubMed] [Google Scholar]
- 74.Raschetti R, Albanese E, Vanacore N, Maggini M. Cholinesterase Inhibitors in Mild Cognitive Impairment: A Systematic Review of Randomised Trials. PLoS Med. 2007;4:e338. doi: 10.1371/journal.pmed.0040338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.European Medicines Agency. Guideline on medicinal products for the treatment of Alzheimer’s disease and other dementias. 2008. Pre-Authorisation Evaluation of Medicines for Human Use. CPMP/EWP/553/95 Rev. 1. [Google Scholar]
- 76.Schneider LS, Sano M. Current Alzheimer’s disease clinical trials: Methods and placebo outcomes. Alzheimer’s and Dementia. 2009;5:388–97. doi: 10.1016/j.jalz.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ. Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001;358:455–60. doi: 10.1016/s0140-6736(01)05623-9. [DOI] [PubMed] [Google Scholar]
- 78.Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease. Neurology. 2010;74:956–64. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- 79.Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, Aisen PS. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77:556–63. doi: 10.1212/WNL.0b013e318228bf11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aisen PS, Schneider LS, Sano M, et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: a randomized controlled trial. JAMA. 2008;300:1774–83. doi: 10.1001/jama.300.15.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quinn JF, Raman R, Thomas RG, et al. Docosahexaenoic Acid Supplementation and Cognitive Decline in Alzheimer Disease: A Randomized Trial. JAMA. 2010;304:1903–11. doi: 10.1001/jama.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Douillet P, Orgogozo JM. What we have learned from the xaliproden Sanofi-aventis trials. JNHA - The Journal of Nutrition, Health and Aging. 2009 doi: 10.1007/s12603-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 83.Aisen PS, Gauthier S, Vellas B, Briand R, Saumier D, Laurin J, Garceau D. Alzhemed: A Potential Treatment for Alzheimers Disease. Current Alzheimer Research. 2007;4:473–8. doi: 10.2174/156720507781788882. [DOI] [PubMed] [Google Scholar]
- 84.Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients With Mild Alzheimer Disease. JAMA: The Journal of the American Medical Association. 2009;302:2557–64. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salloway S, Sperling R, Keren R, et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77:1253–62. doi: 10.1212/WNL.0b013e3182309fa5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doody RS, Raman R, Farlow M, et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. New England Journal of Medicine. 2013;369:341–50. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 87.Coric V, van Dyck CH, Salloway S, et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate alzheimer disease. Archives of Neurology. 2012;69:1–12. doi: 10.1001/archneurol.2012.2194. [DOI] [PubMed] [Google Scholar]
- 88.Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.European Medicines Agency. Qualification opinion of Alzheimer’s disease novel methodologies/biomarkers for PET amyloid imaging (positive/negative) as a biomarker for enrichment, for use in regulatory clinical trials in predementia Alzheimer’s disease. 2012. EMA/CHMP/SAWP/892998/2011. [Google Scholar]
- 90.European Medicines Agency. Qualification opinion of Alzheimer’s disease novel methodologies/biomarkers for the use of CSF AB 1–42 and t-tau and/or PET-amyloid imaging (positive/ negative) as biomarkers for enrichment, for use in regulatory clinical trials in mild and moderate Alzheimer’s disease. 2012. EMA/CHMP/SAWP/893622/2011. [Google Scholar]
- 91.Vellas B, Andrieu S, Sampaio C, Wilcock G. Disease-modifying trials in Alzheimer’s disease: a European task force consensus. The Lancet Neurology. 2007;6:56–62. doi: 10.1016/S1474-4422(06)70677-9. [DOI] [PubMed] [Google Scholar]
- 92.Vellas B, Andrieu S, Sampaio C, Coley N, Wilcock G. Endpoints for trials in Alzheimer’s disease: a European task force consensus. The Lancet Neurology. 2008;7:436–50. doi: 10.1016/S1474-4422(08)70087-5. [DOI] [PubMed] [Google Scholar]
- 93.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. [see comment] Lancet Neurology. 2007;6:734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 95.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. The Lancet Neurology. 2010;9:1118–27. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 96.Kozauer N, Katz R. Regulatory Innovation and Drug Development for Early-Stage Alzheimer’s Disease. New England Journal of Medicine. 2013;368:1169–71. doi: 10.1056/NEJMp1302513. [DOI] [PubMed] [Google Scholar]
- 97.Braak H, Del Tredici K. Where, when, and in what form does sporadic Alzheimer’s disease begin? Current Opinion in Neurology. 2012;25:708–14. doi: 10.1097/WCO.0b013e32835a3432. [DOI] [PubMed] [Google Scholar]
- 98.Schneider LS. Prevention therapeutics of dementia. Alzheimer’s & Dementia. 2008;4:S122–S30. doi: 10.1016/j.jalz.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 99.Chui H, Zheng L, Reed B, Vinters H, Mack W. Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimer’s Research & Therapy. 2011;4:1. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kryscio R, Abner E, Schmitt F, et al. A randomized controlled Alzheimer’s disease Prevention trial’s evolution into an exposure trial: The Preadvise trial. The Journal of Nutrition, Health & Aging. 2013;17:72–5. doi: 10.1007/s12603-013-0004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeKosky ST, Williamson JD, Fitzpatrick AL, et al. Ginkgo biloba for Prevention of Dementia: A Randomized Controlled Trial. JAMA. 2008;300:2253–62. doi: 10.1001/jama.2008.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vellas B, Coley N, Ousset P-J, et al. Long-term use of standardised ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): a randomised placebo-controlled trial. The Lancet Neurology. 2012;11:851–9. doi: 10.1016/S1474-4422(12)70206-5. [DOI] [PubMed] [Google Scholar]
- 103.ADAPT Research Group. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007;68:1800–8. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
- 104.Peters R, Beckett N, Forette F, et al. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): a double-blind, placebo controlled trial. The Lancet Neurology. 2008;7:683–9. doi: 10.1016/S1474-4422(08)70143-1. [DOI] [PubMed] [Google Scholar]
- 105.Solomon A, Mangialasche F, Richard E, et al. Advances in the prevention of Alzheimer’s disease and dementia. Journal of Internal Medicine. 2013 doi: 10.1111/joim.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wischik CM, Bentham P, Wischik DJ, Seng KM. O3-04-07: Tau aggregation inhibitor (TAI) therapy with rember™ arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. Alzheimer’s & Dementia. 2008;4:T167. [Google Scholar]
- 107.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crook T, Bartus RT, Ferris SH, Whitehouse P, Cohen GD, Gershon S. Age-associated memory impairment: Proposed diagnostic criteria and measures of clinical change — report of a national institute of mental health work group. Developmental Neuropsychology. 1986;2:261–76. [Google Scholar]
- 109.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. The Alzheimer’s Disease Cooperative Study. Alzheimer Disease & Associated Disorders. 1997;11:S13–21. [PubMed] [Google Scholar]
- 111.Galasko D, Bennett D, Sano M, Ernesto C, Thomas R, Grundman M, Ferris S. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. Alzheimer Disease & Associated Disorders. 1997;11:33–9. [PubMed] [Google Scholar]
- 112.Gelinas I, Gauthier L, McIntyre M, Gauthier S. Development of a functional measure for persons with Alzheimer’s disease: the disability assessment for dementia. American Journal of Occupational Therapy. 1999;53:471–81. doi: 10.5014/ajot.53.5.471. [DOI] [PubMed] [Google Scholar]
- 113.Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer’s Disease Cooperative Study. Alzheimer Disease & Associated Disorders. 1997;11:S22–32. doi: 10.1097/00002093-199700112-00004. [DOI] [PubMed] [Google Scholar]
- 114.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. [see comment] Neurology. 1993;43:2412–4. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 115.DeJong R, Osterlund OW, Roy GW. Measurement of quality-of-life changes in patients with Alzheimer’s disease. Clinical Therapeutics. 1989;11:545–54. [PubMed] [Google Scholar]
- 116.Schmitt FA, Ashford W, Ernesto C, et al. The Severe Impairment Battery: concurrent validity and the assessment of longitudinal change in Alzheimer’s disease. Alzheimer Disease & Associated Disorders. 1997;11:S51–6. [PubMed] [Google Scholar]
- 117.Harrison J, Minassian SL, Jenkins L, Black RS, Koller M, Grundman M. A Neuropsychological Test Battery for Use in Alzheimer Disease Clinical Trials. Arch Neurol. 2007;64:1323–9. doi: 10.1001/archneur.64.9.1323. [DOI] [PubMed] [Google Scholar]
- 118.United States Food and Drug Administration. Guidance for Industry Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease (FDA-2013-D-0077) DRAFT. US Food and Drug Administration, Center for Drug Evaluation and Research; 2013. [Google Scholar]
- 119.Roses AD, Welsh-Bohmer KA, Burns DK, et al. A pharmacogenetics-supported clinical trial to delay onset of mild cognitive impairment due to Alzheimer’s disease. The Journal of Nutrition, Health & Aging. 2012:841. [Google Scholar]
- 120.European Medicines Agency. Guideline on Medicinal Products for the Treatment of Alzheimer’s Disease and other Dementias. 2009. Committee for Medicinal Products for Human Use. CPMP/EWP/553/95 Rev. 1. [Google Scholar]
- 121.Leber P. Slowing the progression of Alzheimer disease: methodologic issues. Alzheimer Disease & Associated Disorders. 1997;11:S10–21. discussion S37–9. [PubMed] [Google Scholar]
- 122.Holmes C, Boche D, Wilkinson D, et al. Long-term effects of A[beta]42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. The Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 123.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-[beta] load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. The Lancet Neurology. 2010;9:363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 124.Blennow K, Zetterberg H, Rinne JO, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Archives of Neurology. 2012:1–9. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- 125.Gauthier S, Aisen P, Ferris S, et al. Effect of tramiprosate in patients with mild-to-moderate alzheimer’s disease: Exploratory analyses of the MRI sub-group of the alphase study. The Journal of Nutrition, Health & Aging. 2009;13:550–7. doi: 10.1007/s12603-009-0106-x. [DOI] [PubMed] [Google Scholar]
- 126.Sköldunger A, Johnell K, Winblad B, Wimo A. Mortality and treatment costs have a great impact on the cost-effectiveness of disease modifying treatment in Alzheimer’s disease – A simulation study. Current Alzheimer Research. 2013;10:207–16. doi: 10.2174/1567205011310020011. [DOI] [PubMed] [Google Scholar]
- 127.European Medicines Agency. Qualification opinion of a novel data driven model of disease progression and trial evaluation in mild and moderate Alzheimer’s disease. 2013 Jul 12; EMA/CHMP/SAWP/420174/2013. http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2013/07/WC500146179.pdf 2013.
- 128.Schneider LS. Prevention therapeutics of dementia. Alzheimer’s & Dementia. 2008;4:S122–30. doi: 10.1016/j.jalz.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 129.Leber P. Not in our methods, but in our ignorance. [comment] Archives of General Psychiatry. 2002;59:279–80. doi: 10.1001/archpsyc.59.3.279. [DOI] [PubMed] [Google Scholar]
- 130.Frolich L, Ashwood T, Nilsson J, Eckerwall G. Effects of AZD3480 on cognition in patients with mild-to-moderate Alzheimer’s disease: a phase IIb dose-finding study. J Alzheimers Dis. 2010;24:363–74. doi: 10.3233/JAD-2011-101554. [DOI] [PubMed] [Google Scholar]
- 131.Hilt D, Gawryl M, Koenig G, Dgetluck N, Harrison J, Moebius H, Lowen G. Positive effects on cognition and clinical function in mild to moderate Alzheimer’s disease patients with a selective alpha-7 nicotinic partial agonists: interpretation of effects based on a PK/PD model. 5th Conference Clinical Trials on Alzheimer’s Disease; Monte Carlo: The Journal of Nutrition, Health & Aging; 2012. p. 819. [Google Scholar]
- 132.McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia [update of Cochrane Database Syst Rev. 2005;(3):CD003154] Cochrane Database of Systematic Reviews. 2006:CD003154. doi: 10.1002/14651858.CD003154.pub5. [DOI] [PubMed] [Google Scholar]
- 133.Schneider LS, Dagerman KS, Higgins JPT, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch Neurol. 2011;68:991–8. doi: 10.1001/archneurol.2011.69. [DOI] [PubMed] [Google Scholar]
- 134.Schneider L. Pharmacological Treatment of Alzheimer’s Disease. In: Hampel HCM, editor. Alzheimer’s Disease – Modernizing Concept, Biological Diagnosis and Therapy. Basel: Karger; 2012. pp. 122–67. [Google Scholar]
- 135.Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A Randomized Pilot Clinical Trial of the Safety of Pioglitazone in Treatment of Patients With Alzheimer Disease. Arch Neurol. 2011;68:45–50. doi: 10.1001/archneurol.2010.229. [DOI] [PubMed] [Google Scholar]
- 136.Vellas B, Sol O, Snyder PJ, et al. EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Current Alzheimer research. 2011;8:203–12. doi: 10.2174/156720511795256053. [DOI] [PubMed] [Google Scholar]
- 137.Aisen PS, Gauthier S, Ferris SH, et al. Tramiprosate in mild-to-moderate Alzheimer’s disease – a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study) Archives of Medical Science. 2011;7:102–11. doi: 10.5114/aoms.2011.20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. The Lancet Neurology. 2008;7:779–86. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 139.Winblad B, Andreasen N, Minthon L, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. The Lancet Neurology. 2012;11:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- 140.Schneeberger A, Mandler M, Mattner F, Schmidt W. AFFITOME(R) technology in neurodegenerative diseases: the doubling advantage. Human vaccines. 2010;6:948–52. doi: 10.4161/hv.6.11.13217. [DOI] [PubMed] [Google Scholar]
- 141.Scheltens P, Sperling R, Salloway S, Fox N. Bapineuzumab IV phase 3 results. 5th Conference Clinical Trials on Alzheimer’s Disease; Monte Carlo: The Journal of Nutrition, Health & Aging; 2012. p. 797. [Google Scholar]
- 142.Doody R. Safety and efficacy of solanezumab in patient with mild to moderate Alzheimer’s disease: results from phase 3. 5th Conference Clinical Trials on Alzheimer’s Disease; Monte Carlo: The Journal of Nutrition, Health & Aging; 2012. pp. 801–2. [Google Scholar]
- 143.Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of Amyloid Removal in Patients With Alzheimer Disease Treated With Gantenerumab. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.1538. archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- 144.Dodel R, Neff F, Noelker C, Pul R, Du Y, Bacher M, Oertel W. Intravenous Immunoglobulins as a Treatment for Alzheimer’s Disease: Rationale and Current Evidence. Drugs. 2010;70:513–28. doi: 10.2165/11533070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 145.Dodel R, Rominger A, Bartenstein P, et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. The Lancet Neurology. 2013 doi: 10.1016/S1474-4422(13)70014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baxter. Deerfield. 2013. Baxter announces topline results of phase 3 study of immunoglobulin for Alzheimer’s disease. [Google Scholar]
- 147.Hampel H, Ewers M, Berger K, Annas P. Lithium Trial in Alzheimer’s Disease: A Randomized, Single-Blind, Placebo-Controlled, Multicenter 10-Week Study. Journal of Clinical Psychiatry. 2009;70:922–31. [PubMed] [Google Scholar]
- 148.Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. The British Journal of Psychiatry. 2011;198:351–6. doi: 10.1192/bjp.bp.110.080044. [DOI] [PubMed] [Google Scholar]
- 149.Tariot PN, Schneider LS, Cummings J, et al. Chronic Divalproex Sodium to Attenuate Agitation and Clinical Progression of Alzheimer Disease. Arch Gen Psychiatry. 2011;68:853–61. doi: 10.1001/archgenpsychiatry.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.del Ser T, Steinwachs KC, Gertz HJ, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis. 2012;33:205–15. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- 151.TauRX T. First UK patients enroll in Phase 3 clinical trials of LMTX™ for Alzheimer’s Disease. 2013. May 22, (press release) [Google Scholar]
- 152.Wischik DJ. Disease-modifying power estimation for TauRx global phase 3 trial program in AD with tau-aggregation inhibitor LMTX. 5th Conference Clinical Trials on Alzheimer’s Disease; Monte Carlo: The Journal of Nutrition, Health & Aging; 2012. p. 824. [Google Scholar]
- 153.Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, et al. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Archives of neurology. 2006;63:1402–8. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- 154.Freund-Levi Y, Hjorth E, Lindberg C, et al. Effects of omega-3 fatty acids on inflammatory markers in cerebrospinal fluid and plasma in Alzheimer’s disease: the OmegAD study. Dementia and geriatric cognitive disorders. 2009;27:481–90. doi: 10.1159/000218081. [DOI] [PubMed] [Google Scholar]
- 155.Arai H, Takahashi T. A combination therapy of donepezil and cilostazol for patients with moderate Alzheimer disease: pilot follow-up study. Am J Geriatr Psychiatry. 2009;17:353–4. doi: 10.1097/JGP.0b013e31819431ea. [DOI] [PubMed] [Google Scholar]
- 156.Craft S, Baker LD, Montine TJ, et al. Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment: A Pilot Clinical Trial. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.233. archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]