

Abstract
NMR spectroscopy of helical membrane proteins has been very challenging on multiple fronts. The expression and purification of these proteins while maintaining functionality has consumed countless graduate student hours. Sample preparations have depended on whether solution or solid-state NMR spectroscopy was to be performed – neither have been easy. In recent years it has become increasingly apparent that membrane mimic environments influence the structural result. Indeed, in these recent years we have rediscovered that Nobel laureate, Christian Anfinsen, did not say that protein structure was exclusively dictated by the amino acid sequence, but rather by the sequence in a given environment (Anfinsen, 1973) [106]. The environment matters, molecular interactions with the membrane environment are significant and many examples of distorted, non-native membrane protein structures have recently been documented in the literature. However, solid-state NMR structures of helical membrane proteins in proteoliposomes and bilayers are proving to be native structures that permit a high resolution characterization of their functional states. Indeed, solid-state NMR is uniquely able to characterize helical membrane protein structures in lipid environments without detergents. Recent progress in expression, purification, reconstitution, sample preparation and in the solid-state NMR spectroscopy of both oriented samples and magic angle spinning samples has demonstrated that helical membrane protein structures can be achieved in a timely fashion. Indeed, this is a spectacular opportunity for the NMR community to have a major impact on biomedical research through the solid-state NMR spectroscopy of these proteins.
Keywords: Solid state NMR, Oriented sample NMR, Magic angle spinning NMR, Helical membrane proteins, Membrane protein structure, Membrane influence on structure, Membrane protein environment, PISEMA, Structural validation
1. Introduction
No other technology besides solid-state NMR (ssNMR) can characterize membrane protein structure and dynamics in native-like lipid bilayer environments at atomic resolution. Other techniques can provide low resolution or specific site information, but only ssNMR has the demonstrated capability to characterize high resolution structures. Characterization in lipid bilayer environments has been demonstrated to be critical for achieving the native structure of helical membrane proteins. Protein structure is the result of the sum of the intra-protein interactions and the interactions between the protein and its environment [106]. ssNMR is currently the structural technique that is capable of providing the most native-like environment for atomic resolution structural characterizations of membrane proteins. It is also the only technology that can evaluate protein dynamics under native-like conditions, i.e. at physiological temperature in liquid crystalline lipid bilayers. These statements need justification and that is the goal of this perspectives article. Here, we present background information on membrane proteins and why this environment is both difficult to model and has such a significant impact on protein structure. Furthermore, we establish a justification for ssNMR characterizations of membrane protein structures through a discussion of two structures determined by ssNMR compared with characterizations by both solution NMR and X-ray crystallography.
2. Membrane proteins and their environment
Here, the focus is on helical transmembrane (TM) proteins and not on the relatively rare β-barrel proteins. The tertiary structural stability for these two classes of membrane proteins is very different. The β-strands that form a β-barrel have extensive arrays of hydrogen bonds to stabilize the tertiary structure in contrast to the weak van der Waals interactions that typically stabilize membrane proteins with multiple TM helices.
It has been reasoned for a long time that the surface of the protein facing the fatty acyl environment would be hydrophobic. More than a decade ago when the first structure of the voltage dependent KvaP K+ channel was obtained [1] this concept was revisited. Some publications supported the concept that arginine sidechains carrying a charge could be located in the low dielectric fatty acyl environment consistent with this original KvAP structure [2,3], others disagreed [4]. The voltage sensing domain has multiple Arg residues on TM helix, S4. In Fig. 1 the structures of tetrameric KcsA, a monomer of KvAP and a monomer of Kv1.2 are compared. In Fig. 1b none of the TM helices of the voltage sensing domain (green) span the membrane. In fact, two of the TM helices originate and terminate near the middle of the lipid bilayer. Today, it is again well appreciated that charged residues and highly hydrophilic sites are costly from an energetics perspective to place in the fatty acyl environment. Consequently, it is also well accepted that the KvAP structure is highly distorted. A more recent structure of the voltage sensing domain (Fig. 1c) from Kv1.2 [5] presents a credible structure for this domain. All 4 TM helices span the bilayer and TM 4 (labeled S4 in the figure) that carries the arginine residues is in a position packed against the conductance domain helices of an adjacent monomer (not shown in Fig. 1c, but like those in Fig. 1a). Not only are charged residues excluded from the protein surface facing the fatty acyl environment, but highly polar residues such as histidine, aspargine, and glutamine are virtually non-existent on such surfaces of membrane proteins. Aromatic hydrophilics (tryptophan and tyrosine) have long been known to form belts on the protein exterior near the hydrophilic interface that, in part, serve to orient the TM structure relative to its environment [6]. Serine and occasionally threonine are observed on the exterior (facing the acyl chains), but they have the ability to hydrogen bond back to the backbone in a helix. Glycine which has only a hydrogen atom for a sidechain would expose the hydrophilic backbone to the fatty acyl environment if it was located on the exterior. However, conserved glycine residues are very rare on the exterior of multi-helix TM proteins [7]. A final note is that often phenylalanine is found on the hydrophobic exterior, where these sidechains appear to cover up hydrophilic sites, such as exposed hydroxyl or amide backbone sites. These observations are useful in recognizing structural perturbations in membrane protein structures.
Fig. 1.
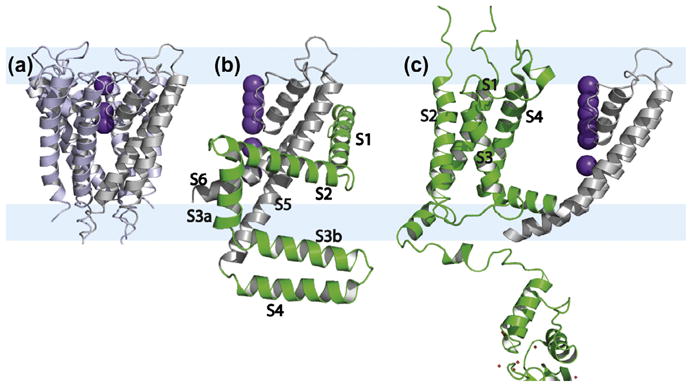
Crystal structures of (a) the tetrameric KcsA (PDB: 1BL8) [105] and the monomers of (b) KvAP (1ORQ) [1] and (c) Kv1.2 (PDB: 3LUT) [5] to simplify the view of these large structures. The conductance domain is in gray, the voltage sensing domain of KvAP and Kv1.2 with 4 TM helices is in green. The hydrophilic interfacial regions are pale blue bands underlying the three structures. The electron density associated with partial occupancy of the K+ ions is shown in purple. These ions and the conductance domain provide an accurate picture of the bilayer normal for orienting these structures.
The interior of the TM domain of multi-helix membrane proteins is considerably more hydrophobic than the water soluble counterparts [8]. During folding, helices appear to be inserted across the membrane as structural units [9] and once again exposed charges would need to be accommodated. In addition, by avoiding strong and specific electrostatic interactions it is easier to rearrange the structure without having to compensate for the exposure of these charges or highly hydrophilic sites to the hydrophobic environment. Furthermore, structural rearrangements appear to be a common strategy for achieving a variety of functional states in helical membrane proteins and therefore, strongly hydrophilic sites are integrated into the protein interior only when it is essential for protein function. Specifically, the formation of hydrogen bonds in a low dielectric environment could be a dangerous folding strategy, since once formed it is difficult to rearrange the hydrogen bond without water being present. Water is known to be a catalyst for hydrogen bond exchange [10] and in the membrane interior water is scarce and therefore hydrogen bond lifetimes are long and structural inter-conversions restricted. As a result it is not surprising that the TM interior has far fewer charged residues, a reduced number of highly hydrophilic residues and a normal content of weakly hydrophilic residues [8]. Surprisingly, proline and glycine, the most significant helix destabilizing residues, are common in TM helices. Proline is unique in that it does not have an amide hydrogen for hydrogen bonding creating a weak point in helices that is referred to as a pro-kink site [11]. In other words, this is a site where the helix may or may not be kinked. Glycine residues may also form pro-kink sites, due to the large expanse of ϕ, ψ torsional space that is uniquely accessible to this residue. Glycine and alanine residues are commonly used to facilitate helix-helix packing in the TM domain [12–14]. Since specific electrostatic interactions are scarce numerous van der Waals interactions are needed to stabilize tertiary structure. By using small residues on one face of the helix (i to i + 4 or i to i + 7 residues) it is possible to have not only increased van der Waals interactions with a neighboring helix, but also weak electrostatic interactions between the helical backbones such as CαH–O hydrogen bonds [15]. The overall result is that the glycine residues are used to stabilize tertiary structure while sacrificing some secondary structural stability [7].
In addition to matching the low dielectric of the membrane in the fatty acyl region with that in the protein, much has been written about hydrophobic mismatch between the hydrophobic length of the protein and the thickness of the membrane environment [16–19]. The lipid compositions of membranes vary and even the composition of membranes in which a given protein is fully functional varies over the lifecycle of a cell or during various stress conditions. Consequently, the protein needs to function under a variety of lipid conditions [20]. This is further complicated in that the inner and outer leaflets of membranes have different compositions. The most dramatic differences occur between the liquid crystalline domains and domains, known as rafts that are purported to have a high composition of sphingomyelin and cholesterol [21–23]. Sphingomyelin and cholesterol reduce the fluidity and increase the hydrophobic thickness of the membrane. Not surprisingly, these raft-like domains appear to solubilize different proteins than those that are soluble in liquid crystalline domains. One of the clear examples of these raft-like domains is observed in the budding of influenza viral particles from cellular membranes [24]. Neurominidase and haemagglutinin are tethered to these domains by their relatively long hydrophobic helices. The M2 protein is also part of the viral particle, but is thought to be trapped in a small region of liquid crystalline lipid [25] from the environment surrounding the raft-like domain. Most of the variations in lipid composition for membrane proteins do not result in such significant changes in the hydrophobic thickness of membranes. An important property of these lipid bilayers, in addition to the hydrophobic thickness, is the profile of lateral pressure, which stems from a dramatic hydrophilic/hydrophobic boundary in the glycerol backbone region of the bilayer [26]. Such a pressure profile may have significant structural implications for TM proteins [27,28].
The lipid backbone (glycerol moiety) and phosphorus-headgroup region of the lipids form the interfacial region between the bulk aqueous environment and the hydrophobic fatty acyl region of the membrane interior. The two interfacial regions have almost the same thickness as the hydrophobic domain of the membrane (see Fig. 1) [29]. This is a region that includes a hydrophilic domain with a dielectric constant that ranges upward to three times that of the bulk aqueous environment [30]. As a result electrostatic interactions will be weakened and their range shortened. The corresponding region in TM membrane proteins is often where ligands bind and therefore, an understanding of the structure and dynamics in this region is of great importance. However, this is the poorest understood structural domain of membrane proteins, since membrane mimetic models used for structural studies are weakest in the interfacial region and our ability to recognize native-like structure in this region is also very poor. Promising results from ssNMR have suggested that this will be another major strength for this spectroscopy [31,32] although few efforts have attempted to model the complex mixture of headgroups present in native membranes. Potentially, the studies of membrane proteins in cells or in cellular membranes [33–38] will lead to structural characterizations of proteins in a membrane that includes all of this complexity.
3. Implications for membrane protein structural biology
The complexity of the membrane environment generates an anisotropic environment for the protein in which these proteins are oriented with respect to this environment to carry out specific functions. For water soluble proteins the aqueous media is an isotropic environment for their functions, and this environment is also used in the preparation of crystals for X-ray diffraction or samples for solution NMR spectroscopy. In other words the environment for water soluble proteins has been mimicked very well for their structural studies and the resultant protein structures have been excellent representations of these water soluble proteins in their native environment. This success generated a reputation that has been assumed to hold for structural studies of membrane proteins. However, the environment for membrane proteins is extremely difficult to model without the use of lipids, their fatty acyl chains, their head-groups, and their extreme amphipathic character. In Fig. 2 the structure of the M2 protein embedded in a lipid bilayer of dioleoylphosphatidylcholine and dioleoylphosphatidylethanolamine is presented showing the match of the hydrophobic surface of M2 with the hydrophobic domain of the bilayer. In addition, a relatively thick interfacial region between the bulk aqueous environment and the fatty-acyl environments is clearly identified by the oxygen and nitrogen atoms of the headgroups. In aqueous preparations of lipids, monomers exist in equilibrium with aggregates that form lipid bilayers. The concentration of monomers in such preparations is on the order of nM. However, in modeling the membrane environment with detergents the concentration of monomeric detergents in the presence of micelles is often 6 orders of magnitude greater (i.e. in the mM range). Such high concentrations of monomeric organics can cause considerable problems by penetrating into membrane protein structures in ways that are not native-like [27]. Moreover, it is very difficult to match the hydrophobicity of the native membrane interior or the hydrophilicity of the head-group region of membranes with detergent based environments. In addition, the well-defined hydrophobic thickness of lipid bilayers is not well defined by detergent micelles that have variable hydrophobic dimensions [39].
Fig. 2.
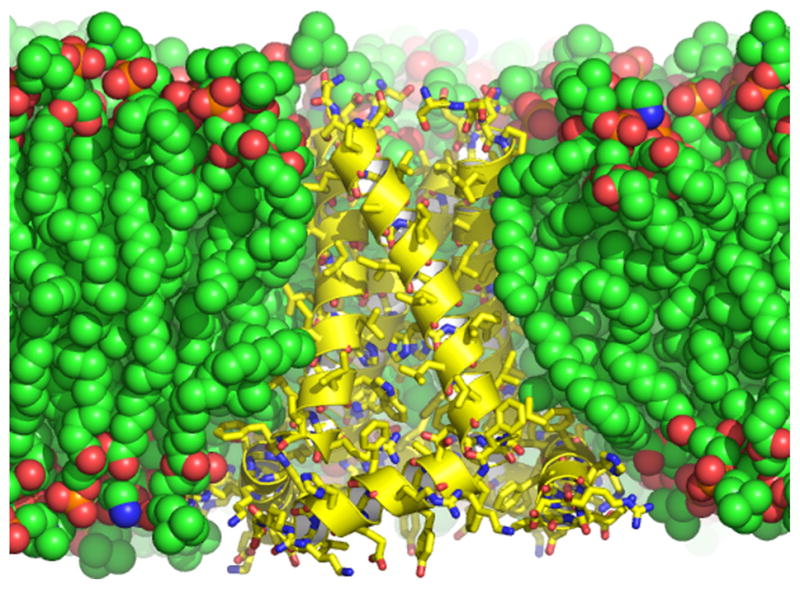
The conductance domain of the M2 proton channel from Influenza A shown in a lipid bilayer of dioleoylphosphatidylcholine and dioleoylphoshatidylethanolamine in which the structural data were obtained and in which the structure was refined using restrained molecular dynamics (PDB: 2L0J) [80]. The protein structure shown in yellow with a helical cartoon and sticks for the heavy atoms is a tetrameric structure composed of a TM and an amphipathic helix, the latter interacting with both the hydrophobic and hydrophilic regions of the bilayer. The heavy atoms of the lipids are shown as space filling spheres. Carbon is green or yellow, oxygen is red, phosphorous is orange, and nitrogen is blue.
When considering the influence of the membrane environment on the structure of membrane proteins, the size of the protein is significant. Very large membrane proteins or membrane protein complexes, such as electron transport or light harvesting complexes have a relatively small portion of their stabilizing interactions with the environment. In comparison monomeric proteins or small oligomeric structures, such as the four helix bundle of the M2 protein from Influenza A virus has a substantial surface area for each helix exposed to the fatty acyl environment. Consequently, crystal structures of large complexes may be more likely to be native-like while smaller proteins, such as M2 or protein domains or the voltage sensing domain of KvAP (Fig. 1) may be more sensitive to the environment and hence at greater risk for structural deformations from a detergent-based membrane mimetic. In addition, many membrane proteins have cofactors, such as heme groups that can stabilize complexes through their interactions with helical bundles. As a result, the structural perturbations induced by membrane mimetics are typically associated with those membrane proteins that are not stabilized with cofactors. However, even structures with 10 or more TM helices have been shown to be destabilized and distorted by membrane mimetic environments [27,28] and therefore there is a large class of proteins that need to be characterized in a lipid bilayer environment. Moreover, what has been established is a critique of the TM domains and not of the entire membrane protein structure. For the interfacial domains there is much less understanding of native vs. non-native structures.
For crystallography site specific interactions between unit cells are required to form well-diffracting crystals. Such interactions are not compatible with a liquid crystalline lipid bilayer and consequently the environment has to be mimicked using detergent solutions. Recently, however, an increasing number of crystals have been formed with an environment that includes some lipids. Such efforts appear to be improving the crystallographic results. Moreover, to crystallize membrane proteins from bicelles [40,41], representing a mixture of detergents and lipids, or a detergent based cubic phase [42–44] seem to hold promise for better structures. Unfortunately, these crystal environments all include crystal contacts between proteins that can lead to structural deformations [45,46]. These contacts are more damaging in membrane proteins, because the helix-helix interactions are relatively weak. For solution NMR the use of detergent micelles results in a single aqueous-detergent interface, more significant water penetration into the hydrophobic core, and a weak lateral pressure profile [27,28,47]. The use of amphipols to surround the hydrophobic core of membrane proteins [48,49] or nanodiscs [50–52] that trap lipids surrounding the protein in a small disk provide better membrane mimetic environments, but only limited results for helical membrane proteins have been obtained to date.
4. Understanding membrane protein biophysics from ssNMR structures
While relatively few ssNMR structures have been deposited in the Protein Data Bank there have been enough to clearly show that this technology is the structural technology for helical membrane proteins especially those, whose structure is significantly influenced by the membrane environment. Here, we present only two of these solid state NMR structures that provide important insights into the influence of the environment on the protein structure.
The first structure in a lipid bilayer – Gramicidin A
The first structural characterization in a lipid bilayer of a functioning ‘protein,’ was the monovalent cation selective channel, gramicidin A (gA). The structure and dynamics, including the backbone and sidechains were characterized in a liquid crystalline lipid environment at 30 °C (Fig. 3a and b) [53–55]. This 15 amino acid peptide has an alternating pattern of L and D amino acids leading to a β-strand in which all of the side-chains are on one side of the strand that wraps into a helix with 6.3 residues per turn. This unique structure, known as a “head to head” single stranded dimer (i.e. amino terminus to amino terminus dimer) was characterized with orientational restraints from 15N anisotropic chemical shifts, 15N–1H and 15N–13C dipolar interactions, as well as 2H quadrupolar interactions using uniformly aligned lipid bilayer samples. The dynamics in the backbone were characterized from powder pattern averaging [56] and from anisotropic 15N T1 relaxation measurements [57,58]. The dimeric nature of the structure was determined from REDOR distance measurements between the monomers [59,60]. Note that the backbone amides line an aqueous pore and that the indole N–H groups are all positioned so as to hydrogen bond with hydrophilic sites in the interfacial region of the lipid bilayer.
Fig. 3.

Dimeric gramicidin A structures viewed perpendicular (top) and parallel (bottom) to the pore axis (bottom). (a and b) The ‘head to head’ single stranded structure known to be a monovalent cation selective channel in membranes (PDB: 1MAG) [55]; (c and d) double helical antiparallel structure, not observed in lipid bilayers (PDB: 2XDC) [45]; and (e and f) another double helical antiparallel structure this time Cs+ bound in the pore, but again, not observed in lipid bilayers (PDB: 1GMK) [69].
Not only was this the first structure determined in a liquid crystalline lipid bilayer, but it was the first TM structure determined by ssNMR. The resolution of the structure was very high due to the nature of the orientational restraints. Today, such absolute restraints are more widely appreciated because of their use in solution NMR as residual dipolar restraints [61]. In ssNMR there is the added advantage that the alignment tensor is fixed by the glass slides used for alignment, such that the normal to the bilayer is parallel with the axis of the magnetic field. In gA the backbone torsion angles were determined within 3° [54,62]. Because of the accuracy of the restraints only four restraints per peptide plane were needed to achieve such high resolution. This illustrated the advantage of absolute restraints over the use of distance or torsional restraints alone that are classified as relative restraints. In other words each distance or torsional restraint, restricts one portion of the structure with respect to another portion of the structure, as opposed to absolute restraints that characterize one portion of the structure relative to a laboratory fixed axis frame. For gA, the torsion angles for the conformational states of the side chains were characterized to within 10° [54] and high resolution models for the side chain motions were also characterized [63,64]. While the single site-specific labeling strategy used for gA is not generally applicable to membrane proteins that cannot be economically synthesized via a chemical synthesis strategy, the gA structure demonstrated that these orientational restraints could be used to characterize high resolution structure.
Other structures of gA have been obtained, several by solution NMR spectroscopy in organic solvents and others in detergent micelles. Only these latter structures are consistent with the structure characterized by ssNMR [65–67]. However, none of the multiple X-ray structures have achieved the native ‘head to head’ conformation that forms the monovalent cation selective channel. While both 1ALX [68] and 2XDC [45] (Fig. 3c–f) have claimed to be the native structure in lipid bilayers we have shown that this conformation does not support a hydrated pore for conductance [59]. We were able to show this by replacing the four tryptophan residues with phenylalanine and in so doing the intertwined double helix (Fig. 3c and d) is the minimum energy conformation in the lipid bilayer. However, unlike the ’head to head’ structure (Fig. 3a and b) where the backbone amide protons exchange for deuterium – this intertwined double helical structure does not [59]. Indeed, the wild type (i.e. with tryptophan) intertwined double helical structures represent minimum energy conformations only in isotropic or nearly isotropic environments. The reason that these structures are not the minimum energy conformation in lipid bilayers is that the 4 tryptophan residues (in the C-terminal half of the sequence) are distributed over the entire length of an antiparallel double helix thereby burying some of the indoles near the bilayer center (Fig. 3c and e). The head to head dimer places all of these indoles within hydrogen bonding distance of the lipid interface (Fig. 3a). In other words, the membrane environment has dictated the structure of this peptide while the crystal lattice environment tolerates or stabilizes the hydrophilic indoles in the interstices of what would be a very low dielectric of fatty acyl native environment. The larger pore intertwined double helix structure [69] shown in Fig. 3e and f has not been observed in lipid bilayers [59].
Another critical lesson from the studies of gA was that the kinetics of the conformational interchange between various double stranded and single stranded structures was very slow in a lipid bilayer resulting in a half life for the antiparallel double helix of approximately 3 days at 69 °C in DMPC bilayers [70]. In other words, the ability to break and reform the set of hydrogen bonds in the backbone was severely compromised by the low dielectric of the lipid bilayer and the lack of an aqueous environment. In fact, the catalysis for this process in mixed organic solvents was shown to be dependent on the protic solvent concentration with a dramatic cooperativity index (a Hill coefficient) of 6.5. In the lipid bilayer the catalyst for hydrogen bond exchange would be the water molecules. Since water is at a very low concentration the process is indeed slow suggesting that in general, the rearrangement of hydrogen bonds is difficult in the TM environment. This explains why helices are not expected to form from a random coil in the bilayer interior, but rather are proposed to form in the interfacial region and are then inserted across the bilayer [9]. Consequently, one might expect to see the potential for forming inter-helix hydrogen bonds to be rare in the TM domains unless there is access to water through, for instance, a hydrophilic pore. Otherwise the formation of an unintended hydrogen bond could result in misfolding [71].
The proton channel of influenza A – The M2 protein
The TM helix of the influenza A M2 proton channel was first characterized in 2001 [72] and in 2002 a tetrameric structure of the M2 proton channel was published based on additional interhelical distance restraints [73]. What was striking about these data was the uniformity of the helical structures [74]. During this time it was discovered that images of the helical wheels were observed in the Separated Local Field spectra obtained with the PISEMA pulse sequence [75] where the 15N anisotropic chemical shift and 15N–1H dipolar spin interactions are correlated [76,77]. These patterns, known as PISA wheels, are remarkably sensitive to the local structure. Even though the chemical shift tensors vary somewhat from residue to residue, the positions of the resonances in the spectra (with the exception of glycine) are dominated by small structural perturbations and not so significantly by the variation in tensor element magnitudes or orientations. Calculations of the PISA wheels, assuming uniform tensors demonstrate that a variation of only ±6° in the backbone torsion angles virtually obliterates the PISA wheel pattern [11,78]. Consequently, the observation of such a pattern defines a relatively uniform set of helical torsion angles. These patterns have now been obtained from many different proteins, not always for the full length of the helix, because occasionally kinks in the structure occur. For instance, the M2 TM helices have kinked helices when the pore is blocked by amantadine [79] resulting in different PISA wheels for the N- and C-terminal helical segments.
This helix uniformity stems from the strengthening of the hydrogen bonds in the helices due to the low dielectric environment. As the crystal structures have improved in resolution so has the uniformity of the helices characterized by X-ray diffraction [78]. This helix uniformity generates a structural challenge for membrane proteins that are dependent upon a variety of conformational states for their functional activities. Glycine and proline residues, as mentioned earlier, reduce the secondary structural stability so that deformations of these helices can take place [7], such as the kink in the vicinity of Gly34 in the M2 proton channel, mentioned above [79], or in the M2 conductance domain without the inhibitor [80].
For gramicidin we needed four orientational restraints per residue to characterize the backbone torsion angles and to eliminate degenerate solutions. For these α-helical structures, the PISA wheel identifies a resonance pattern with 3.6 residues per repeat limiting the range of possible torsion angles to those in the vicinity of an α-helix. Consequently, only two specific-site restraints per residue are required to accurately and unambiguously define the orientation of the peptide plane excluding what would otherwise be a substantial range of degenerate solutions [81].
In 2008 the first crystal structures of the M2 TM domain were achieved with and without drug bound [82]. The structure without drug (Fig. 4a and b) was an asymmetric tetramer resulting from two non-native interactions. First, antiparallel tetramers were packed in the crystal lattice sharing an extensive van der Waals surface and a salt bridge between tetramers. Additionally, both polyethylene glycol and octylglucoside molecules were embedded in what would be the pore of this proton channel, presumably the result of a high concentration of monomeric organics in this environment [28,39]. In fact, one of the glucose residues (from octylglucoside) hydrogen bonds with one of the unique His37 residues that is responsible for the proton selectivity of this channel. The overall result is an asymmetric tetramer having splayed helices and fenestrations from the aqueous pore into the fatty acyl environment for nearly two-thirds of the membrane thickness. It is clear that the membrane mimetic used has not adequately modeled the native membrane environment for this crystal structure.
Fig. 4.

Structures of the M2 proton channel from Influenza A viewed perpendicular (top) and parallel (bottom) to the pore axis (bottom). The hydrophilic interfacial regions are pale yellow bands underlying the three structures and in between is the hydrophobic region of the bilayer. (a and b) An asymmetric X-ray crystal structure (PDB: 3BKD) [82] of the TM domain (residues 22–46) obtained from detergent based crystals; (c and d) a solution NMR structure of the conductance domain obtained from DHPC micelles (residues 18–60, PDB: 2RLF) [83] with bound rimantadine on the lipid facing surface (space filling view); (e and f) a ssNMR structure of the conductance domain (residues 22–62, PDB: 2L0J) [80] from liquid crystalline bilayers of DOPC and DOPE. The His37 residues shown in space filling view.
Also in 2008 a solution NMR structure of the conductance domain including the amphipathic helix on the C-terminal side of the TM helix was characterized (Fig. 4c and d) [83]. However, this amphipathic helix did not interact with the micelle surface, but instead, formed a four helix bundle in the bulk aqueous environment. This structure in DHPC micelles, did not bind drug in the pore, but on the external surface in a fourfold symmetric fashion. Importantly, the TM helix tilt was only 16° with respect to the axis of the pore [8], a much smaller angle than what had previously been observed for the tilt of these helices in the bilayer solubilized TM domain structures and for the full length protein [79,84]. This small tilt angle from the solution NMR structure suggested that the pore entrance was too narrow for the drug to enter and consequently, the drug bound to an external site [85]. This external site is in close proximity to the binding site for the drug in the lipid bilayer and the specific site on the protein has been recognized as a secondary low affinity site by REDOR experiments performed on the TM domain in lipid bilayers [86]. The variable hydrophobic thickness allowed by detergent micelles has resulted in a more tightly packed bundle of helices compared to the native structure that permits drug binding in the pore. Moreover, the surface of the micelle did not attract the amphipathic helix, which had been shown to bind to the lipid surface in a 2002 study of the full length protein [84]. Once again a poor membrane mimetic environment has resulted in a structure that is non-native and misleading.
In 2010 the conductance domain structure of wild type M2 was characterized in DOPC/DOPE lipids using OS ssNMR (Fig. 4e and f) [80]. Like the other α-helical structures characterized using orientational restraints, this structure has a high resolution backbone conformation from these restraints. The tilt of the N-terminal half of the TM helices is 32° generating an N-terminal access for drug binding similar to the values determined from the shorter TM domain constructs, but twice the value obtained from the solution NMR structure. The structure in lipid bilayers has no fenestrations to the fatty acyl environment and the amphipathic helix is embedded in the lipid bilayer interfacial region. In fact, the binding of the amphipathic helix to the lipid bilayer eliminates the secondary drug binding site, since two of the sidechains from the amphipathic helix pack into the hydrophobic pocket that generated this secondary binding site [80]. In addition, a small set of distance restraints from MAS ssNMR defined the quaternary structure. Uniquely, the His37 pKas (8.2, 8.2, 6.3 and <5.0) determined in 2006 [87] led to a detailed model for proton specificity and conductance in this channel through the formation of a dimer-of-dimers structure for these imidazole sidechains at neutral pH. This structure resulted in a shared charge between adjacent imidazoles thereby reducing charge repulsion in this low dielectric environment [87,88]. A great deal of biophysical data supports this model as do numerous interhelical distances from these imidazoles obtained using MAS ssNMR in the conductance domain and in the full length protein [89,90].
Even though these three wild type structures from X-ray diffraction, solution and solid-state NMR are all tetrameric with a TM helix spanning the membrane, it is clear that the helices have packed differently in these structures with important implications for the pore and the channel’s functional mechanism. Clearly, the structure obtained in liquid crystalline lipid bilayers has uniquely generated a native-like structure upon which biological understanding can be reliably based.
Initial studies of the full length M2 protein using MAS spectroscopy have been published [38,90,91], suggesting that the histidine tetrad functions similarly in the full length compared with the conductance domain, however a detailed pH titration and assignment of the resonances is needed. Recently, the MAS spectra of 13C labeled valine full length M2 obtained from isolated Escherichia coli membranes, where the M2 was expressed is the same as that following isolation, purification and reconstitution in synthetic lipid bilayers [38]. Once again, it is clear that the robust and native-like environment for membrane proteins is a lipid bilayer.
5. A central role for ssNMR of membrane protein structural biology
There may be no other major field of research where ssNMR spectroscopy is needed more than for the structures of helical membrane proteins. There is no other approach that can characterize these proteins at atomic resolution in their native-like lipid bilayer environment or even in situ environments [34,35,37,92,93]. Furthermore, these environments are a major contributor to the mix of molecular interactions that stabilize the tertiary and quaternary structures of, especially small helical membranes proteins, i.e. those with 10 or fewer TM helices. It is now clear from the considerable number of helical membrane protein structures in the Protein Data Bank that have been characterized by X-ray crystallography and solution NMR in detergent based environments that these environments often, do not provide the native set of molecular interactions that stabilize the native structure of these proteins [27,28].
Shown in Fig. 5 is a comparison of a few resonance frequencies observed by OS ssNMR from the WT M2 (22–62; 2L0J) in lipid bilayers (Fig. 4e and f) with the predicted OS ssNMR frequencies based on the coordinates from the 2RLF solution NMR structure of WT M2 (19–61) (Fig. 4c and d) and the 3BKD crystal structure of M2 (22–46) (Fig. 4a and b). This latter structure has an Ile to SelenoMet mutation. There are other crystal and solution NMR structures, but they have either thermo-stabilizing mutations or are drug resistant mutants of the wild type sequence. The solution NMR structure had a set of fourfold symmetric restraints, but was refined without symmetry imposed. While the anisotropic chemical shift and dipolar interaction predictions are quite similar for the four monomers, what is shown in Fig. 5 is the monomer that represented the best fit to the observed ssNMR data. Many of the predicted resonance frequencies are quite similar (W41, H37, L43 F55, and L59), however S31, L46, F47 are not close. The tilt of the TM helices in the N-terminus differ by ~16° accounting for the S31 deviation. In addition, the data for L46 and F47 show a dramatic kink between the TM and amphipathic helices in 2L0J, while in 2RLF there is loop between these helices. The data comparison of predicted and observed data for F55 and L59 suggest similar structures, but there is little chemical shift dispersion for these helices that are nearly parallel to the bilayer surface. The displacement of the amphipathic helix into the aqueous environment and away from the bilayer surface is a structural detail that is not picked up by the orientational restraints. A calculation of the anisotropic chemical shift root mean square deviation (RMSD) between the observed 2L0J data and the predicted date from 2RLF is 28 ppm and for the dipolar interaction, 2.4 kHz.
Fig. 5.

Comparison of a sample of observed and predicted OS ssNMR data from wild type constructs of the Influenza A M2 protein. (Red) Observed resonance frequencies for S31, H37, W41, L43, L46, F47, F55, and L59 from M2 (22–62) in liquid crystalline lipid bilayers used to define the 2L0J structure [80]. The letter designations are used only for the 2L0J resonances. (Purple) Predicted resonance frequencies from the solution NMR structure (2RLF) [83] in detergent micelles of M2 (19–61) for the same sites. (Green) Predicted resonance frequencies from the X-ray crystal structure (3BKD) [82] in detergent based crystals of M2 (22–46) for the same sites except for F47, F55, and L59 which were not in this construct. 3BKD is an asymmetric structure and hence the four helices give rise to different resonance frequencies. Different shades of green are used to color code the different helices in 3BKD. The data and predictions are plotted on an absolute scale for the dipolar coupling. Both the predications and data for F47, F55, and L59 are used in the structures as negative values.
To put these deviations in perspective calculations have also been performed on a recent X-ray crystal structure [44] of diacylglycerol kinase (DgkA) characterized in a lipidic cubic phase (Murray and Cross, to be published). This is a trimeric structure and the best fit monomer (one not involved in crystal contacts) resulted in an anisotropic chemical shift RMSD with previously published OS ssNMR data [94] of 5 ppm and 0.5 kHz RMSD for the dipolar interactions. Consequently, the RMSD calculated here between the observed data and that predicted for the M2 solution NMR structure shows a factor of 5 larger deviations than those observed for the DgkA crystal structure, where a good fit that is almost within the experimental error is obtained. This is not to say that lipidic cubic phase (actually a detergent phase) is the crystallographic answer for modeling the membrane environment. It was further shown that crystal contacts significantly distorted the other monomers in the structure and thermal stabilization mutations distorted the non-WT structures. Moreover, as alluded to above, the crystal structure of gramicidin A, 2XDC, was obtained from lipidic cubic phase that did not support the native gramicidin A structure [45,46].
The predicted resonances for the M2 crystal structure display dramatic asymmetry in the tetramer. None of the helices fit the experimental data well. Since L46 is the final residue in this construct we did not include it in a calculation of RMS deviations for the anisotropic chemical shift and dipolar interactions. The two helices that most closely fit have an RMSD for the chemical shift of 22 and 26 ppm, similar to that observed for the calculation from the solution NMR structure, however, the RMSD for the dipolar interactions is smaller ~0.7 kHz for the best helix and 1.5 kHz for the second best. The other two helices are much worse. The asymmetry in the X-ray structure has implications for discussions of proton conductance mechanisms and indeed, the authors when they published this structure chose to consider the structure to be a hybrid of two functional states [82]. Unfortunately, there is no justification for this, since the cause of the asymmetry is the presence of detergents wedged between the helices interacting with the functionally important H37 and W41 residues [39]. In addition, there are major crystal contacts that appear to further influence the structure [27].
Importantly, we have demonstrated here, a way in which OS ssNMR data can be used to validate structures that are not obtained in a lipid bilayer environment. For the DgkA validation we used data from two amino acid labeled samples – methionine and tryptophan that were published years before the crystal structure. It was not necessary to have the individual resonances assigned to make a quality assessment of the structure. There is also a solution NMR structure of DgkA [95], again published after the OS ssNMR data [94] and this structure displays much larger RMS deviations between predicted and observed OS data from the structure obtained in liquid crystalline lipid bilayers (Murray and Cross, to be published).
Impediments for the characterization of membrane protein structures by ssNMR spectroscopy are rapidly disappearing. Strategies for the heterologous expression of these proteins are well refined and described in the literature, as are strategies for the extraction of membrane proteins from the cellular membrane or from inclusion bodies. Affinity chromatography has revolutionized the purification of these proteins and reconstitution into lipid bilayers is becoming much better understood. We and others find that the final and complete removal of detergent is critical for generating a homogeneous preparation that provides good line widths [96]. Removal of detergents is also important for the stability of the sample. Multiple approaches can be used to characterize trace quantities of detergent, such as evaporative light scattering [97].
The preparation of oriented samples has long been viewed as a tedious process, and the uniformity of alignment has often been highly variable. By eliminating the detergent and by carefully controlling the hydration conditions our current protocol is robust and can be performed by multiple individuals with very similar results [96]. Along with improvements in the uniformity of alignment has come consequential improvements in both spectral resolution and sensitivity. Orientational restraints that were pioneered with single site labels can now be achieved from amino acid specific labels of proteins generating dozens of restraints per spectrum [98]. The samples are approximately 40–50% by weight water and the molar ratio of protein:lipid is between 1:50 and 1:100 resulting in approximately 5–10 mgs of protein per sample. Such quantities of protein are readily achieved by current expression systems for proteins under 25 kDa. It is also possible to obtain orientational restraints from MAS spectroscopy of liposome preparations above the lipid phase transition temperature, using a technique known as rotational alignment [99].
Importantly, this structural approach is also dependent on obtaining sparse distance restraints. The uniformity of the helical structures coupled with limited variation in amino acid composition has resulted in very limited dispersion for many of the MAS resonances in 13C–13C correlations spectra, especially for the hydrophobic amino acids that are so common. The quality of the MAS spectra have also been improving and importantly this is true for proteoliposome spectra, where the proteins are also in a native-like environment [36,90,100–104]. Indeed, the protocols that we use for MAS and OS sample preparations are identical up to the point where we spread proteoliposomes onto glass slides. From our perspective the careful and complete elimination of detergents from the sample has improved both the OS and MAS samples.
As importantly as sample preparation, the use of orientational restraints permits a great reduction in the number of distance restraints required for achieving a unique structural solution [98]. While the orientational restraints only provide secondary structural solutions they also orient the helices with respect to the bilayer normal both in terms of tilt and rotation angle. The result is an even further reduction in the number of distances required to achieve a membrane protein structure, at least for the structure of the TM domain. Consequently, a complete and unambiguous set of resonance assignments for the MAS spectra is not needed. Instead, unique assignments for the more unusual amino acid residues in the TM helices are needed. This greatly reduces the challenge for obtaining distance restraints. While many PhD dissertations were required for each of the structures of gramicidin A and M2, today the structure of membrane proteins with 2 and 3 TM helices can be achieved by a single PhD student, even from first expression through the development of purification and reconstitution to OS and MAS ssNMR data collection and analysis.
There is no substitute for the proven structural technology of ssNMR that can characterize the native structures of this very large and vitally important class of proteins in a native-like environment that clearly influences their structure. A major investment in biological ssNMR instrumentation is needed so that many researchers can have access to the tools required to characterize these structures in native-like lipid bilayer preparations to address this structural biology challenge.
Acknowledgments
This work was supported in part by NIH Grants AI-023007 and AI-074805. The NMR experiments were performed at the National High Magnetic Field Laboratory supported by cooperative agreement (DMR-1157490) from the National Science Foundation and the State of Florida.
References
- 1.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 2.Hessa T, White SH, von Heijne G. Membrane insertion of a potassium-channel voltage sensor. Science. 2005;307:1427. doi: 10.1126/science.1109176. [DOI] [PubMed] [Google Scholar]
- 3.Yoo J, Cui Q. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys J. 2008;94:L61–L63. doi: 10.1529/biophysj.107.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grabe M, Lecar H, Jan YN, Jan LY. A quantitative assessment of models for voltage-dependent gating of ion channels. Proc Natl Acad Sci USA. 2004;101:17640–17645. doi: 10.1073/pnas.0408116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Wang Q, Ni F, Ma J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc Natl Acad Sci USA. 2010;107:11352–11357. doi: 10.1073/pnas.1000142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Killian JA, von Heijne G. How proteins adapt to a membrane–water interface. Trends Biochem Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Sharma M, Zhou HX, Cross TA. Glycines: role in alpha-helical membrane protein structures and a potential indicator of native conformation. Biochemistry. 2012;51:4779–4789. doi: 10.1021/bi300090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou HX, Cross TA. Modeling the membrane environment has implications for membrane protein structure and function: influenza A M2 protein. Protein Sci: Publ Protein Soc. 2013;22:381–394. doi: 10.1002/pro.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popot JL, Engelman DM. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 1990;29:4037–4041. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 10.Arumugam S, Pascal S, North CL, Hu W, Lee KC, Cotten M, Ketchem RR, Xu F, Brenneman M, Kovacs F, Tian F, Wang A, Huo S, Cross TA. Conformational trapping in a membrane environment: a regulatory mechanism for protein activity? Proc Natl Acad Sci USA. 1996;93:5872–5876. doi: 10.1073/pnas.93.12.5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Page RC, Li C, Hu J, Gao FP, Cross TA. Lipid bilayers: an essential environment for the understanding of membrane proteins. Magn Reson Chem: MRC. 2007;45:S2–S11. doi: 10.1002/mrc.2077. [DOI] [PubMed] [Google Scholar]
- 12.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 13.Javadpour MM, Eilers M, Groesbeek M, Smith SO. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha —H···O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Killian JA. Hydrophobic mismatch between proteins and lipids in membranes. Biochim Biophys Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 17.Park SH, Opella SJ. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J Mol Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Fattal DR, Ben-Shaul A. A molecular model for lipid–protein interaction in membranes: the role of hydrophobic mismatch. Biophys J. 1993;65:1795–1809. doi: 10.1016/S0006-3495(93)81249-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs FA, Denny JK, Song Z, Quine JR, Cross TA. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J Mol Biol. 2000;295:117–125. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]
- 20.Sanders CR, Mittendorf KF. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry. 2011;50:7858–7867. doi: 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 22.Heberle FA, Petruzielo RS, Pan J, Drazba P, Kucerka N, Standaert RF, Feigenson GW, Katsaras J. Bilayer thickness mismatch controls domain size in model membranes. J Am Chem Soc. 2013;135:6853–6859. doi: 10.1021/ja3113615. [DOI] [PubMed] [Google Scholar]
- 23.Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harbor Perspect Biol. 2011;3:1–17. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantor RS. Solute modulation of conformational equilibria in intrinsic membrane proteins: apparent “Cooperativity” without binding. Biophys J. 1999;77:2643–2647. doi: 10.1016/S0006-3495(99)77098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou HX, Cross TA. Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cross TA, Murray DT, Watts A. Helical membrane protein conformations and their environment. Eur Biophys J: EBJ. 2013;42:731–755. doi: 10.1007/s00249-013-0925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiener MC, White SH. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of X-ray and neutron diffraction data. III. Complete structure. Biophys J. 1992;61:434–447. doi: 10.1016/S0006-3495(92)81849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nymeyer H, Zhou HX. A method to determine dielectric constants in nonhomogeneous systems: application to biological membranes. Biophys J. 2008;94:1185–1193. doi: 10.1529/biophysj.107.117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higman VA, Varga K, Aslimovska L, Judge PJ, Sperling LJ, Rienstra CM, Watts A. The conformation of bacteriorhodopsin loops in purple membranes resolved by solid-state MAS NMR spectroscopy. Angew Chem, Int Ed Engl. 2011;50:8432–8435. doi: 10.1002/anie.201100730. [DOI] [PubMed] [Google Scholar]
- 32.Park SH, Casagrande F, Cho L, Albrecht L, Opella SJ. Interactions of interleukin-8 with the human chemokine receptor CXCR1 in phospholipid bilayers by NMR spectroscopy. J Mol Biol. 2011;414:194–203. doi: 10.1016/j.jmb.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu R, Wang X, Li C, Santiago-Miranda AN, Pielak GJ, Tian F. In situ structural characterization of a recombinant protein in native Escherichia coli membranes with solid-state magic-angle-spinning NMR. J Am Chem Soc. 2011;133:12370–12373. doi: 10.1021/ja204062v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacso T, Franks WT, Rose H, Fink U, Broecker J, Keller S, Oschkinat H, Reif B. Characterization of membrane proteins in isolated native cellular membranes by dynamic nuclear polarization solid-state NMR spectroscopy without purification and reconstitution. Angew Chem, Int Ed Engl. 2012;51:432–435. doi: 10.1002/anie.201104987. [DOI] [PubMed] [Google Scholar]
- 35.Kamihira M, Vosegaard T, Mason AJ, Straus SK, Nielsen NC, Watts A. Structural and orientational constraints of bacteriorhodopsin in purple membranes determined by oriented-sample solid-state NMR spectroscopy. J Struct Biol. 2005;149:7–16. doi: 10.1016/j.jsb.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc Natl Acad Sci USA. 2012;109:4863–4868. doi: 10.1073/pnas.1116478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sivertsen AC, Bayro MJ, Belenky M, Griffin RG, Herzfeld J. Solid-state NMR evidence for inequivalent GvpA subunits in gas vesicles. J Mol Biol. 2009;387:1032–1039. doi: 10.1016/j.jmb.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew Chem, Int Ed Engl. 2012;51:8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 41.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 42.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Curr Opin Struct Biol. 2011;21:559–566. doi: 10.1016/j.sbi.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li D, Lyons JA, Pye VE, Vogeley L, Aragao D, Kenyon CP, Shah ST, Doherty C, Aherne M, Caffrey M. Crystal structure of the integral membrane diacylglycerol kinase. Nature. 2013;497:521–524. doi: 10.1038/nature12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofer N, Aragao D, Caffrey M. Crystallizing transmembrane peptides in lipidic mesophases. Biophys J. 2010;99:L23–25. doi: 10.1016/j.bpj.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Separovic F, Killian JA, Cotten M, Busath DD, Cross TA. Modeling the membrane environment for membrane proteins. Biophys J. 2011;100:2073–2074. doi: 10.1016/j.bpj.2011.02.058. author reply 2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoonens M, Comer J, Masscheleyn S, Pebay-Peyroula E, Chipot C, Miroux B, Dehez F. Dangerous liaisons between detergents and membrane proteins. The case of mitochondrial uncoupling protein 2. J Am Chem Soc. 2013;135:15174–15182. doi: 10.1021/ja407424v. [DOI] [PubMed] [Google Scholar]
- 48.Tribet C, Audebert R, Popot JL. Amphipols: polymers that keep membrane proteins soluble in aqueous solution. Proc Natl Acad Sci USA. 1996;93:15047–15050. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Popot JL, Althoff T, Bagnard D, Baneres JL, Bazzacco P, Billon-Denis E, Catoire LJ, Champeil P, Charvolin D, Cocco MJ, Cremel G, Dahmane T, de la Maza LM, Ebel C, Gabel F, Giusti F, Gohon Y, Goormaghtigh E, Guittet E, Kleinschmidt JH, Kuhlbrandt W, Le Bon C, Martinez KL, Picard M, Pucci B, Sachs JN, Tribet C, van Heijenoort C, Wien F, Zito F, Zoonens M. Amphipols from A to Z. Annu Rev Biophys. 2011;40:379–408. doi: 10.1146/annurev-biophys-042910-155219. [DOI] [PubMed] [Google Scholar]
- 50.Bayburt TH, Sligar SG. Membrane protein assembly into nanodiscs. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G. Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure. 2013;21:394–401. doi: 10.1016/j.str.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ketchem RR, Lee KC, Huo S, Cross TA. Macromolecular structural elucidation through solid state NMR-derived orientational constraints. J Biomol NMR. 1996;8:1–14. doi: 10.1007/BF00198135. [DOI] [PubMed] [Google Scholar]
- 54.Ketchem RR, Roux B, Cross TA. High-resolution polypeptide structure in a lamellar phase lipid environment from solid-state NMR derived orientational constraints. Structure. 1997;5:1655–1669. doi: 10.1016/s0969-2126(97)00312-2. [DOI] [PubMed] [Google Scholar]
- 55.Ketchem RR, Hu W, Cross TA. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 56.Nicholson LK, Teng Q, Cross TA. Solid-state nuclear magnetic resonance derived model for dynamics in the polypeptide backbone of the gramicidin A channel. J Mol Biol. 1991;218:621–637. doi: 10.1016/0022-2836(91)90706-c. [DOI] [PubMed] [Google Scholar]
- 57.North CL, Cross TA. Analysis of polypeptide backbone T1 relaxation data using an experimentally derived model. J Magn Reson. 1993;101B:35–43. [Google Scholar]
- 58.North CL, Cross TA. Correlations between function and dynamics: time scale coincidence for ion translocation and molecular dynamics in the gramicidin channel backbone. Biochemistry. 1995;34:5883–5895. doi: 10.1021/bi00017a018. [DOI] [PubMed] [Google Scholar]
- 59.Cotten M, Fu R, Cross TA. Solid-state NMR and hydrogen-deuterium exchange in a bilayer-solubilized peptide: structural and mechanistic implications. Biophys J. 1999;76:1179–1189. doi: 10.1016/S0006-3495(99)77282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu R, Cotten M, Cross TA. Inter- and intramolecular distance measurements by solid-state MAS NMR: determination of gramicidin A channel dimer structure in hydrated phospholipid bilayers. J Biomol NMR. 2000;16:261–268. doi: 10.1023/a:1008372508024. [DOI] [PubMed] [Google Scholar]
- 61.Tjandra N, Omichinski JG, Gronenborn AM, Clore GM, Bax A. Use of dipolar 1H–15N and 1H–13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat Struct Biol. 1997;4:732–738. doi: 10.1038/nsb0997-732. [DOI] [PubMed] [Google Scholar]
- 62.Ketchem RR, Hu W, Tian F, Cross TA. Structure and dynamics from solid state NMR spectroscopy. Structure. 1994;2:699–701. doi: 10.1016/s0969-2126(00)00070-8. [DOI] [PubMed] [Google Scholar]
- 63.Hu W, Lazo ND, Cross TA. Tryptophan dynamics and structural refinement in a lipid bilayer environment: solid state NMR of the gramicidin channel. Biochemistry. 1995;34:14138–14146. doi: 10.1021/bi00043a019. [DOI] [PubMed] [Google Scholar]
- 64.Lee KC, Huo S, Cross TA. Lipid–peptide interface: valine conformation and dynamics in the gramicidin channel. Biochemistry. 1995;34:857–867. doi: 10.1021/bi00003a020. [DOI] [PubMed] [Google Scholar]
- 65.Arseniev AS, Barsukov IL, Bystrov VF, Lomize AL, Ovchinnikov Yu A. 1H-NMR study of gramicidin A transmembrane ion channel. Head-to-head right-handed, single-stranded helices. FEBS Lett. 1985;186:168–174. doi: 10.1016/0014-5793(85)80702-x. [DOI] [PubMed] [Google Scholar]
- 66.Pascal SM, Cross TA. High-resolution structure and dynamic implications for a double-helical gramicidin A conformer. J Biomol NMR. 1993;3:495– 513. doi: 10.1007/BF00174606. [DOI] [PubMed] [Google Scholar]
- 67.Townsley LE, Tucker WA, Sham S, Hinton JF. Structures of gramicidins A, B, and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 2001;40:11676–11686. doi: 10.1021/bi010942w. [DOI] [PubMed] [Google Scholar]
- 68.Burkhart BM, Gassman RM, Langs DA, Pangborn WA, Duax WL. Heterodimer formation and crystal nucleation of gramicidin D. Biophys J. 1998;75:2135–2146. doi: 10.1016/S0006-3495(98)77656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doyle DA, Wallace BA. Crystal structure of the gramicidin/potassium thiocyanate complex. J Mol Biol. 1997;266:963–977. doi: 10.1006/jmbi.1996.0837. [DOI] [PubMed] [Google Scholar]
- 70.Xu F, Cross TA. Water: foldase activity in catalyzing polypeptide conformational rearrangements. Proc Natl Acad Sci USA. 1999;96:9057–9061. doi: 10.1073/pnas.96.16.9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanders CRM, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci: Publ Protein Soc. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishimura K, Kim S, Zhang L, Cross TA. The closed state of a H+ channel helical bundle: combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 74.Kim S, Cross TA. Uniformity, ideality, and hydrogen bonds in transmembrane alpha-helices. Biophys J. 2002;83:2084–2095. doi: 10.1016/S0006-3495(02)73969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu CH, Ramamoorthy A, Opella SJ. High resolution heteronuclear dipolar solid-state NMR specroscopy. J Magn Reson A. 1994;109:270–272. [Google Scholar]
- 76.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 78.Page RC, Kim S, Cross TA. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure. 2008;16:787–797. doi: 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quine JR, Cross TA. Protein structure in anisotropic environments: unique structural fold from orientational constraints. Concepts NMR. 2000;12:71–82. [Google Scholar]
- 82.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tian C, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci: Publ Protein Soc. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sharma M, Li C, Busath DD, Zhou HX, Cross TA. Drug sensitivity, drug-resistant mutations, and structures of three conductance domains of viral porins. Biochim Biophys Acta. 2011;1808:538–546. doi: 10.1016/j.bbamem.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dong H, Yi M, Cross TA, Zhou HX. Calculations and validation of the pH-dependent structures of the His37-Trp41 quartet, the heart of acid activation and proton conductance in the M2 protein of Influenza A virus. Chem Sci. 2013;4:2776–2787. doi: 10.1039/C3SC50293G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Can TV, Sharma M, Hung I, Gor’kov PL, Brey WW, Cross TA. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza A M2 protein functional insights. J Am Chem Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miao Y, Cross TA, Fu R. Identifying inter-residue resonances in crowded 2D C–C chemical shift correlation spectra of membrane proteins by solid-state MAS NMR difference spectroscopy. J Biomol NMR. 2013;56:265–273. doi: 10.1007/s10858-013-9745-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liao SY, Fritzsching KJ, Hong M. Conformational analysis of the full-length M2 protein of the influenza A virus using solid-state NMR. Protein Sci: Publ Protein Soc. 2013;22:1623–1638. doi: 10.1002/pro.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu R, Wang X, Li C, Miranda AN, Pielak GJ, Tian F. In situ detection of a recombinant human protein in native E. coli membranes by solid-state NMR. J Am Chem Soc. 2011;133:12370–12373. doi: 10.1021/ja204062v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Renault M, Cukkemane A, Baldus M. Solid-state NMR spectroscopy on complex biomolecules. Angew Chem, Int Ed Engl. 2010;49:8346–8357. doi: 10.1002/anie.201002823. [DOI] [PubMed] [Google Scholar]
- 94.Li C, Gao P, Qin H, Chase R, Gor’kov PL, Brey WW, Cross TA. Uniformly aligned full-length membrane proteins in liquid crystalline bilayers for structural characterization. J Am Chem Soc. 2007;129:5304–5305. doi: 10.1021/ja068402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Horn WD, Sanders CR. Prokaryotic diacylglycerol kinase and undecaprenol kinase. Annu Rev Biophys. 2012;41:81–101. doi: 10.1146/annurev-biophys-050511-102330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Das N, Murray DT, Cross TA. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat Protoc. 2013;8:2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park SH, Casagrande F, Chu M, Maier K, Kiefer H, Opella SJ. Optimization of purification and refolding of the human chemokine receptor CXCR1 improves the stability of proteoliposomes for structure determination. Biochim Biophys Acta. 2012;1818:584–591. doi: 10.1016/j.bbamem.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Murray DT, Das N, Cross TA. Solid state NMR strategy for characterizing native membrane protein structures. Acc Chem Res. 2013 doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, Marassi FM, Opella SJ. Structure determination of a membrane protein in proteoliposomes. J Am Chem Soc. 2012;134:2047–2056. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A. J Am Chem Soc. 2012;134:7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhate MP, McDermott AE. Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc Natl Acad Sci USA. 2012;109:15265–15270. doi: 10.1073/pnas.1211900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gustavsson M, Traaseth NJ, Veglia G. Probing ground and excited states of phospholamban in model and native lipid membranes by magic angle spinning NMR spectroscopy. Biochim Biophys Acta. 2012;1818:146–153. doi: 10.1016/j.bbamem.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shi L, Kawamura I, Jung KH, Brown LS, Ladizhansky V. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew Chem, Int Ed Engl. 2011;50:1302–1305. doi: 10.1002/anie.201004422. [DOI] [PubMed] [Google Scholar]
- 104.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 106.Anfinsen CB. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]