

Short abstract
A genome-wide computational screen for targets of the PmrA transcription factor in Salmonella typhimurium has identified novel target genes.
Abstract
Background
The PmrAB (BasSR) two-component regulatory system is required for Salmonella typhimurium virulence. PmrAB-controlled modifications of the lipopolysaccharide (LPS) layer confer resistance to cationic antibiotic polypeptides, which may allow bacteria to survive within macrophages. The PmrAB system also confers resistance to Fe3+-mediated killing. New targets of the system have recently been discovered that seem not to have a role in the well-described functions of PmrAB, suggesting that the PmrAB-dependent regulon might contain additional, unidentified targets.
Results
We performed an in silico analysis of possible targets of the PmrAB system. Using a motif model of the PmrA binding site in DNA, genome-wide screening was carried out to detect PmrAB target genes. To increase confidence in the predictions, all putative targets were subjected to a cross-species comparison (phylogenetic footprinting) using a Gibbs sampling-based motif-detection procedure. As well as the known targets, we detected additional targets with unknown functions. Four of these were experimentally validated (yibD, aroQ, mig-13 and sseJ). Site-directed mutagenesis of the PmrA-binding site (PmrA box) in yibD revealed specific sequence requirements.
Conclusions
We demonstrated the efficiency of our procedure by recovering most of the known PmrAB-dependent targets and by identifying unknown targets that we were able to validate experimentally. We also pinpointed directions for further research that could help elucidate the S. typhimurium virulence pathway.
Background
The PmrAB two-component regulatory system is part of a multicomponent feedback loop that acts as one of the key regulatory mechanisms of Salmonella typhimurium virulence [1-3]. The PmrAB regulatory system is itself responsive to Fe3+ and mild acid [4] and senses Mg2+ indirectly by communicating with the Mg2+-sensitive PhoPQ system [5-8] via PmrD [1,9]. PmrD is hypothesized to transduce the signal from the PhoPQ system to the PmrAB system via a posttranslational modification. The gene pmrD is transcriptionally activated by the PhoPQ system but repressed by the PmrAB system [1,9,10]. The PmrAB system is required for resistance to the cationic antibiotic polymyxin B [11] and to Fe3+-mediated killing [4]. The Mg2+-dependent regulation of PmrAB was shown to be important for gene expression in an intracellular environment [12]. Fe3+-dependent PmrAB regulation, on the other hand, has been hypothesized to be essential for survival in extracellular environments [13]. A region in DNA to which the PmrA protein binds has been identified by DNA footprinting analysis [14,15].
In contrast to pmrD, other known target genes of PmrAB in S. typhimurium are transcriptionally activated. One group of targets is involved in LPS modification. PmrAB-induced modifications include the addition of 4-amino-4-deoxy-L-arabinose (Ara4N) and phosphoethanolamine (pEtN) to lipid A [16]. Loci involved in the Ara4N modification of lipid A are ugd [6] and the pmrHFIJKLM loci, both of which are responsible for Ara4N biosynthesis [2,16-18] and incorporation of Ara4N into lipid A [19,20]. LPS modifications are hypothesized to allow bacterial survival within macrophages by lowering the affinity of the LPS for amphipathic cationic peptides with antimicrobial activity that are produced as a consequence of the innate immune response.
A second class of targets are directly dependent on PmrAB, but have as-yet-undefined functions. pmrC (co-transcribed with pmrAB [21]) and pmrG (located upstream of the pmrHFIJKLM operon) are both transcriptionally activated by PmrAB. Mutations in pmrG did not affect the resistance to polymyxin B [2]. Tamayo et al. recently identified two additional targets of PmrAB - yibD and dgoA. However, none of these was involved in resistance to polymyxin B or to high concentrations of Fe3+ [22]. These genes might therefore represent a group of as-yet-unidentified functions regulated by the PmrAB system [22]. Also, PmrAB-regulated genes involved in resistance to Fe3+ and pEtN addition to LPS remain to be identified [22]. Together with the recent indications of new PmrAB-dependent functions, this raises the possibility that not all PmrAB targets have yet been identified. Therefore, in this study we used an in silico approach to predict targets of the PmrAB regulatory system. Several methodologies exist for genome-wide screening using a motif model (or mathematical representation) of experimentally verified regulatory sites [23-27]. These assign to each possible motif position in the genome a score (the specifics of which depend on the methodology) that indicates how well the subsequence located at that position matches the motif model. Genome-wide screenings of this type have proved successful in detecting additional targets of the regulator being investigated. However, more reliable predictions for motifs in specific pathways have been obtained by incorporating cross-species comparisons (phylogenetic footprinting) [26,28-34]. Because evolutionary forces tend to preferentially retain functional DNA sequences, motifs that are conserved in the intergenic regions of orthologs derived from several related species are more likely to be biologically relevant [35,36].
In this study, we combine both approaches. Putative targets identified by a genome-wide screening were, whenever possible, analyzed by phylogenetic footprinting based on Gibbs sampling [33,34,37]. Four interesting targets were validated by wet lab experiments and the PmrA box of a representative target was subjected to site-directed mutagenesis.
Results
Genome-wide screening using a PmrA motif model
Gibbs sampling was used to detect PmrA-binding motifs in the intergenic regions of three experimentally verified PmrAB targets (ugd, pmrC, pmrG). The logo of the statistically overrepresented motif detected is represented in Figure 1. This motif corresponded to the PmrA-binding site experimentally identified by Aguirre et al. [15] and partially overlapped the PmrA-binding site delineated by Wosten et al. [14]. They detected this site upstream of the transcription start of pmrC, in the intergenic region between pmrG and pmrH, and upstream of ugd (on the plus strand) [14,15]. We used the obtained motif model in a genome-wide screening of the S. typhimurium intergenic sequences [38]. Table 1 summarizes the results of our screening, using a threshold as described in Materials and methods. From experimentally verified examples, it appears that the PmrA motif can be biologically functional not only when present on the plus strand (as in the case of pmrH), but also when located on the minus strand (for example in pmrG) [14]. Therefore, both strands of the genome sequence were screened.
Figure 1.
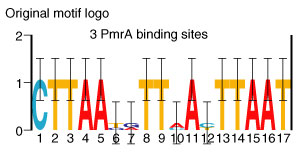
Consensus sequence of the PmrA box. Motif logo representing the initial motif model used to screen the S. typhimurium intergenic sequences.
Table 1.
List of the putative PmrAB targets in S. typhimurium
| Name | Description | Score | Instance | Alignment | Footprint | Distribution (COG) | Distribution [38] |
| Minus strand | |||||||
| STM1273 | Putative nitric oxide reductase | 0.848436 | CTTAATGTTTTCTTAAT | / | / | 1000 | All Salmonella only |
| STM2132 | Pseudogene; frameshift; putative RBS for STM2133 | 0.814252 | TTTTAGATTCACTTAAT | / | / | 1000 | Some or all Salmonella only |
| STM4596 | Paralog of E. coli ORF, hypothetical protein (AAC73478.1); BLAST hit to putative inner membrane protein | 0.806962 | TTTAATATTCACTTAAA | / | / | 1000 | Some Salmonella only |
| STM3131 | Putative cytoplasmic protein; putative RBS for STM3130; putative first gene of operon with STM3130 (putative hypothetical protein) | 0.801641 | CTTAATTTTTACTTATT | / | / | 1000 | All Salmonella only |
| STM1020 | Gifsy-2 prophage | 0.791616 | CTTATTGTTAAGTCAAT | / | / | 1000 | Other distributions |
| stdA | STM3029; paralog of E. coli putative fimbrial-like protein (AAC73813.1); BLAST hit to putative fimbrial-like protein | 0.788548 | CAAAACATTAACTTAAT | / | / | 1000 | Subspecies 1 only? |
| ugd | STM2080; S. typhimurium UDP-glucose 6-dehydrogenase | 0.781719 | CTCAGAATTAACTTAAT | m | + | 1100 | All nine genomes |
| sinR | STM0304; S. typhimurium SINR protein. (SW:SINR_SALTY) transcriptional regulator | 0.780204 | CTTGATATCATCTTAAT | / | / | Subspecies 1 only | |
| STM3131 | Putative cytoplasmic protein; putative RBS for STM3130; putative first gene of operon with STM3130; (putative hypothetical protein) | 0.772846 | CTTAATACTCACATTAT | / | / | 1000 | Other distributions |
| STM4413 | Putative imidazolonepropionase and related amidohydrolases; putative RBS for STM4412; first gene of operon with STM4412 (D-galactonate transport) | 0.771153 | GTGAATGTTAAATTAAT | / | / | 1000 | Some or all Salmonella only |
| ybdO | STM0606; ortholog of E. coli putative transcriptional regulator LYSR-type (AAC73704.1); BLAST hit to putative transcriptional regulator, LysR family | 0.769839 | CTTAATGTAGAGTTTAT | m | + | 1110 | All Salmonella only |
| oraA | STM2828; ortholog of E. coli regulator, OraA protein (AAC75740.1); BLAST hit to regulator | 0.766748 | CTTGATGGTAATTTAAC | m | - | 1110 | All nine genomes |
| sdhC | STM0732; Ortholog of E. coli succinate dehydrogenase, cytochrome b556 (AAC73815.1); Putative RBS for sdhD; first gene of putative operon encoding succinate dehydrogenase | 0.765950 | CTTATTATTCCCTTAAG | / | / | 1000 | All nine genomes |
| ycaR | STM0987; Ortholog of E. coli ORF, hypothetical protein (AAC74003.1); BLAST hit to putative inner membrane protein; Putative RBS for kdsB; first gene of a putative operon with ksdB (CMP-3-deoxy-D-manno-octulosanate transferase) | 0.765889 | TTCAATATTAACATAAT | / | / | 1000 | All nine genomes |
| lasT | STM4600; Ortholog of E. coli ORF, hypothetical protein (AAC77356.1); BLAST hit to putative tRNA*tRNA methyltransferase | 0.765754 | ATTTAGGATAATTTAAT | nd | / | 1110 | All nine genomes |
| STM2137 | Putative cytoplasmic protein | 0.764036 | TTTAACCTTAATTTAAT | nd | / | 1100 | Some Salmonella only |
| STM1672 | Putative cytoplasmic protein | 0.762904 | ATTAATAGTCACTTATT | / | / | 1000 | Subspecies 1 only? |
| gcvA | STM2982; Ortholog of E. coli positive regulator of gcv operon (AAC75850.1); first gene of putative operon (gcvA, ygdD, ygdE containing a SAM-dependent methyltransferase) | 0.761166 | CTTAATGTCGAATGAAT | m | + | 1111 | All nine genomes |
| ycgO | STM1801; Ortholog of E. coli ORF, hypothetical protein (AAC74275.1); BLAST hit to putative CPA1 family, Na:H transport protein | 0.760685 | TTTAACATTAACATAAT | m | + | 1110 | All nine genomes? |
| STM2287 | Paralog of E. coli putative sulfatase* phosphatase (AAC75329.1); BLAST hit to putative cytoplasmic protein | 0.759519 | CTTATTATTCACATAAC | / | / | 1000 | Some or all Salmonella only? |
| yebW | STM1852; Ortholog of E. coli ORF, hypothetical protein (AAC74907.1); BLAST hit to putative inner membrane lipoprotein | 0.754895 | CTCAATGTTAACTACTT | / | / | 1000 | All nine genomes? |
| STM0897 | Hypothetical protein Fels-1 prophage | 0.754468 | CGTAAGGCTCTTTTAAT | / | / | 1000 | Some Salmonella only |
| lpfA | STM3640; S. typhimurium long polar fimbrial protein A precursor; first gene of a putative fimbriae synthesis operon | 0.753228 | ATTAAGAATAAATTAAT | / | / | 1000 | Other distributions |
| Plus strand | |||||||
| yjdB* | STM4293; S. typhimurium hypothetical 61.6 kDa protein in basS*pmrA-adiY intergenic region. (SW:YJDB_SALTY) putative integral membrane protein; Putative RBS for basR; first gene of the putative operon (yjdB basR basS) | 0.930146 | CTTAAGGTTCACTTAAT | m | + | 1111 | All nine genomes |
| ugd | STM2080; S. typhimurium UDP-glucose 6-dehydrogenase | 0.913666 | CTTAATATTAACTTAAT | m | + | 1100 | All nine genomes |
| yfbE/ais | STM2297; Ortholog of E. coli putative enzyme (AAC75313.1); first gene of the yfbE operon; shared intergenic with ais | 0.912660 | CTTAATGTTAATTTAAT | m | + | 1111 | All nine genomes? |
| STM1269*/STM1268 | Putative chorismate mutase; intergenic shared with STM1268 | 0.888478 | CTTAATGTTATCTTAAT | / | / | 1000 | All Salmonella only |
| STM0692 | Paralog of E. coli nitrogen assimilation control protein (AAC75050.1); putative transcriptional regulator, LysR family | 0.814773 | CTTGATGTTGATTTAAT | / | / | 1000 | All Salmonella only |
| ybjG/mdfA* | STM0865; Ortholog of E. coli orf, hypothetical protein (AAC73928.1); putative permease; intergenic shared with mdfA (multidrug translocase) | 0.810981 | CTTTAAGGTTAATTTAA | m | + | 1111 | All nine genomes |
| STM2901 | Hypothetical protein putative cytoplasmic protein; located downstream of pathogenicity island 1 | 0.803712 | CTTAATATCAATATAAT | / | / | 1000 | Other distributions |
| yhjC/yhjB | STM3607; Ortholog of E. coli putative transcriptional regulator LysR-type (AAC76546.1); intergenic shared with yhjB (putative transcriptional regulator) | 0.796967 | TTGAATATTAATTTAAT | nd | / | 1110 | All nine genomes? |
| yjbE/pgi | STM4222; Ortholog of E. coli orf, hypothetical protein (AAC76996.1); BLAST hit to putative outer membrane protein; first gene of the putative operon (yjbE, yjbF, yjbG, yjbH) consisting of putative outer membrane (lipo)proteins; intergenic shared with pgi (glucosephosphate isomerase) | 0.791181 | TTTAATTTTAACTTATT | / | / | 1000 | All nine genomes? |
| yibD* | STM3707; Ortholog of E. coli putative regulator (AAC76639.1); BLAST hit to putative glycosyltransferase | 0.790879 | CTTAATAGTTTCTTAAT | m | + | 1100 | Other distributions |
| STM1926/flhC | Putative cytoplasmic protein Putative RBS for STM1926; first gene of a putative operon with yecG (putative universal stres protein); shared intergenic with flhC en flhD (flagellar transcriptional activator) | 0.790699 | CCTAATGTTCACTTTTT | / | / | 1000 | Some or all Salmonella only |
| STM0334/STM0335 | Putative cytoplasmic protein; shared intergenic with STM0335 | 0.789514 | TTTCATATTCATTTAAT | / | / | 1000 | Some Salmonella only |
| ybdN | STM0605; Ortholog of E. coli orf, hypothetical protein (AAC73703.1); BLAST hit to putative 3-phosphoadenosine 5-phosphosulfate sulfotransferase (PAPS reductase)*FAD synthetase Putative RBS for ybdM; first gene of a putative operon with ybdM (hypothetical transcriptional regulator) | 0.788778 | ATTAATATAAATTTAAT | nd | / | 1100 | All nine genomes? |
| glgB | STM3538; Ortholog of E. coli 1,4-alpha-glucan branching enzyme (AAC76457.1); BLAST hit to 1,4-alpha-glucan branching enzyme; Putative RBS for glgX; putative first gene of operon involved in glycogen synthesis | 0.779808 | TTTAAGGGTAGCTTAAT | m | - | 1111 | All nine genomes |
| leuO | STM0115; S. typhimurium probable activator protein in leuabcd operon. (SW:LEUO_SALTY) putative transcriptional regulator (LysR family) | 0.776490 | ATTAATGTTAACTTTTT | m | - | 1111 | All nine genomes |
| STM0343 | Paralog of E. coli orf, hypothetical protein (AAC75237.1); BLAST hit to AAC75237.1 identity in aa 10 - 512 putative Diguanylate cyclase*phosphodiesterase domain | 0.774271 | ATTAATGTTACTTTAGT | nd | / | 1100 | Subspecies 1 only |
| orf242 | STM1390 S. typhimurium ORF242 (gi|4456866) putative regulatory proteins, merR family | 0.773644 | CTTAGTCTTCATTTGAT | / | / | 1000 | Other distributions |
| STM1868A/mig-3 | Lytic enzyme; intergenic shared with mig-3 (phage assembly protein) | 0.773462 | CTTAATGATTATTTATT | / | / | 1000 | ? |
| STM2763/STM2726 | Paralog of E. coli prophage CP4-57 integrase (AAC75670.1); BLAST hit to putative integrase; intergenic shared with STM2726 (putative inner membrane) | 0.772053 | ATTAATGTCCATTTAGT | / | / | 1000 | S. typhimurium only |
| pntA | STM1479; Ortholog of E. coli pyridine nucleotide transhydrogenase, alpha subunit (AAC74675.1); Blast hit to AAC74675.1 pyridine nucleotide transhydrogenase (proton pump), alpha subunit; Putative RBS for pntB; first gene of the putative operon (pntA, pntB) | 0.770547 | TTTAATGTTAATTTCTT | m | - | 1111 | All nine genomes |
| STM0057/cit2 | Putative citrate-sodium symport; intergenic shared with citC2 (citrate lyase synthetase) | 0.767968 | CTCATGGTTCATTGAAT | nd | / | 1110 | Other distributions |
| yrbF | STM3313; Ortholog of E. coli putative ATP-binding component of a transport system (AAC76227.1); Blast hit to AAC76227.1 putative ABC superfamily (atp_bind) transport protein; Putative RBS for yrbE; RegulonDB:STMS1H003330; first gene of putative yrb operon (ABC transporter) | 0.766758 | CCTAATTTTGACTTTAT | m | + | 1111 | All nine genomes |
| yejG | STM2220; Paralog of E. coli orf, hypothetical protein (AAC75242.1); Blast hit to putative cytoplasmic protein | 0.767099 | CTTTATGTTTATTTTAT | m | + | 1111 | All nine genomes |
| slsA | STM3761; putative inner membrane protein | 0.765418 | CTTTATGTTATTTAAAT | nd | / | 1110 | Other distributions |
| yhcN | STM3361; Ortholog of E. coli orf, hypothetical protein (AAC76270.1); Blast hit to putative outer membrane protein | 0.764452 | ATTAGTGTATACTTAAT | m | + | 1111 | All nine genomes? |
| yceP | STM1161; Ortholog of E. coli orf, hypothetical protein (AAC74144.1); Blast hit to putative cytoplasmic protein | 0.764191 | TTTATTGTTCATATAAT | m | + | 1100 | All nine genomes |
| STM4098 | putative arylsulfate sulfotransferase | 0.763003 | TCTAATATTTATTTAAT | nd | / | 1100 | Subspecies 1 only? |
| stfA | STM0195; S. typhimurium major fimbrial subunit StfA | 0.762241 | ATCAATTTTAATTTAAT | / | / | 1000 | Some Salmonella only |
| atpF | STM3869; Ortholog of E. coli membrane-bound ATP synthase, F0 sector, subunit b (AAC76759.1); Blast hit to imembrane-bound ATP synthase, F0 sector, subunit b; Putative RBS for atpH; first gene of a putative operon encoding putative ATP synthase | 0.760841 | CAGAAGGTTAACTAGAT | m | + | 1111 | All nine genomes |
| yegH/wza | STM2119; Ortholog of E. coli putative transport protein (AAC75124.1); Blast hit to putative inner membrane protein; intergenic shared with wza (putative polysaccharide export protein) | 0.760004 | ATTAATATTAAATGAAT | m | - | 1111 | All nine genomes |
| yjgD/argI | STM4470; S. typhimurium hypothetical protein in argI-miaE intergenic region (ORF15.6). (SW:YJGD_SALTY) putative cytoplasmic protein; Putative binding site for ArgR; shared intergenic regions with argI (arginine ornithine transferase); first gene of a putative operon with miaE (tRNA hydroxylase) | 0.759514 | ATTAAAATTCACTTTAT | m | + | 1111 | All nine genomes |
| sseJ/STM1630* | STM1631; S. typhimurium secreted effector; regulated by SPI-2; shared intergenic with STM1630 (putative inner membrane protein) | 0.758303 | CTTAAGAAATATTTAAT | / | / | 1000 | Some Salmonella only |
| csrA | STM2826; S. typhimurium carbon storage regulator | 0.756990 | CTTAGGTTTAACAGAAT | m | + | 1111 | All nine genomes |
| dinP/yafK | STM0313; Ortholog of E. coli damage-inducible protein P; putative tRNA synthetase (AAC73335.1); Blast hit to AAC73335.1 DNA polymerase IV, devoid of proofreading, damage-inducible protein P; intergenic shared with yafKJ (periplasmic protein, putative amido transferase) | 0.756938 | CATACTGTACACTTAAA | m | + | 1111 | All nine genomes |
| STM0346 | Putative outer membrane protein; Homolog of ail and ompX | 0.756369 | CATTAGGTGCTCTTAAT | / | / | 1000 | Some Salmonella only |
| ybfA/STM0707 | STM0708; Ortholog of E. coli orf, hypothetical protein (AAC73793.1); Blast hit to putative periplasmic protein; intergenic shared with STM0707 (hypothetical protein) | 0.754265 | ATTAGTATTAATTTAAC | m | + | 1111 | All nine genomes? |
| yncD/STM1587 | STM1587; Ortholog of E. coli putative outer membrane receptor for iron transport (AAC74533.1); Blast hit to paral putative outer membrane receptor; intergenic shared with STM1586 (putative receptor) | 0.754063 | CATTTTCTTAACTTAAT | m | - | 1100 | All nine genomes |
| yafC/STM0275 | STM0256; Ortholog of E. coli putative transcriptional regulator LysR-type (AAC73313.1); Blast hit to putative transcriptional regulator, LysR family; intergenic shared STM0275 (drug efflux protein) | 0.753257 | CAAAATATCAATTTAAT | m | - | 1111 | Other distributions |
Name: name of the gene in the S. typhimurium genome (NC_003197). For genes that are divergently transcribed and have a shared intergenic region, the gene for which the motif is detected on the plus strand is indicated first and the gene for which the motif is on the minus strand is indicated after the slash. Description: annotation of the encoded proteins and genome location of the genes (derived from GenBank and Sanger annotation). Score: normalized score assigned to the respective motifs by MotifLocator. Site: instance of the motif as detected in the respective intergenic sequence. Distribution (COG): distribution of the protein as determined by our analysis. The distribution is indicated by a binary profile that indicates the presence 1 versus absence 0 of the protein in species (serovars) of, respectively, Salmonella, E. coli, Shigella and Yersinia (for example, 1111 indicates protein present in all four species; 1000: protein present in Salmonella species only). Distribution: distribution of the protein encoded by the corresponding gene in nine bacterial genomes as determined by McClelland et al. [38]. Proteins having close homologs in at least one Salmonella strain but not in E. coli or K. pneumoniae are indicated by 'some Salmonella only'. Genes that contain close homologs in all genomes are indicated by 'all nine genomes'. Other combinations are indicated by 'other distributions'. ? indicates that the authors were not certain about the statement. Differences between the distribution as determined by McClelland et al. and the one determined by our analysis is due to the difference in selection criteria used to identify close homologs (see Materials and methods). Alignment: indicates whether the intergenic regions in the dataset could be locally aligned (nd, no local alignment detected that contained the original sequence of S. typhimurium; m, local alignment detected. If the dataset only contained homologs from Salmonella species, local alignments were considered noninformative (indicated by /)). Footprint: denotes whether the PmrA motif is conserved in the close homologs. +, the retrieved putative PmrA motif is conserved; -, the intergenic sequences of the orthologs could be locally aligned but the PmrA motif was not part of the conserved regions. Most promising PmrAB targets that contained a PmrA motif matching the PmrA consensus (Figure 4) are in bold face. PmrA motifs that are experimentally validated in this study are indicated by an asterisk.
Identification of close homologs
We can expect to detect conserved biologically active PmrA motifs only in species that have a functional counterpart of the S. typhimurium PmrAB system. Of all the completely sequenced bacterial species, only the genomes of S. typhimurium, S. typhi, Escherichia coli, Shigella flexneri and Yersinia pestis contain the amino-acid motif that determines the specificity of the sensor protein PmrB (the amino acids suggested to be involved in binding Fe3+ [4]). Also, the protein domains involved in the binding of PmrA to DNA were almost perfectly conserved in the PmrA orthologs in the species above (PF00486 domain, see supplementary information on our website [39]). Therefore, these γ-proteobacterial species were used to perform phylogenetic footprinting analysis. For each gene containing a potential hit of the PmrA motif in the S. typhimurium genome sequence, close homologs were selected as described in Materials and methods.
Phylogenetic footprinting using Gibbs sampling
For each dataset we aimed at constructing a local multiple alignment. We used Gibbs sampling to generate motifs that can be used as alignment seeds. Alignments were subsequently constructed based on the positions of these motif seeds. Potential seeds were selected using a heuristic described in the supplementary information on [39]. Such multiple alignments summarize the motifs in the intergenic sequences that are conserved between species. We used the alignments to verify whether the putative PmrA motifs retrieved by the genome-wide screening were conserved in other species. Table 1 gives an overview of the results of the genome-wide screening and the phylogenetic footprinting approach (individual alignments are displayed in the supplementary information at [39]).
Detailed analysis of the putative PmrAB targets
Putative PmrA motifs were detected in the intergenic regions of genes encoding transcriptional regulators, outer-membrane and secreted proteins, proteins with functions involved in flagella and fimbria synthesis, proteins with a function related to the modification of cellular components, putative transport proteins, proteins involved in amino-acid synthesis and also in phage remnants. As mentioned above, if the putative PmrAB-regulated genes contained close homologs in other species, the intergenic sequences of these close homologs were locally aligned to check whether putative PmrA motifs were conserved in these other species as well. For some of the datasets, however, no local alignment could be identified (no motif detected). Closer inspection showed that most of these datasets contained highly homologous paralogs of the original sequence. The intergenic sequences of these paralogs showed an overall low degree of conservation (for example, STM0057) with the original intergenic sequence in S. typhimurium (data not shown). In some of the datasets, a local alignment of the respective intergenic regions could be detected, but the putative PmrA motif was not present within the conserved parts of the alignment (for example, leuO). For these putative PmrAB targets, phylogenetic footprinting could not strengthen the confidence in the prediction of the PmrA motif. If such putative motifs are biologically active, their activity will be restricted to Salmonella serovars or S. typhimurium.
Our analysis revealed that PmrA motifs, present in the intergenic sequences of known PmrAB-dependent S. typhimurium genes, were also conserved in the intergenic sequences of the orthologs of these genes in related species (Figure 2). An overview of the alignments of these known targets is given below.
Figure 2.
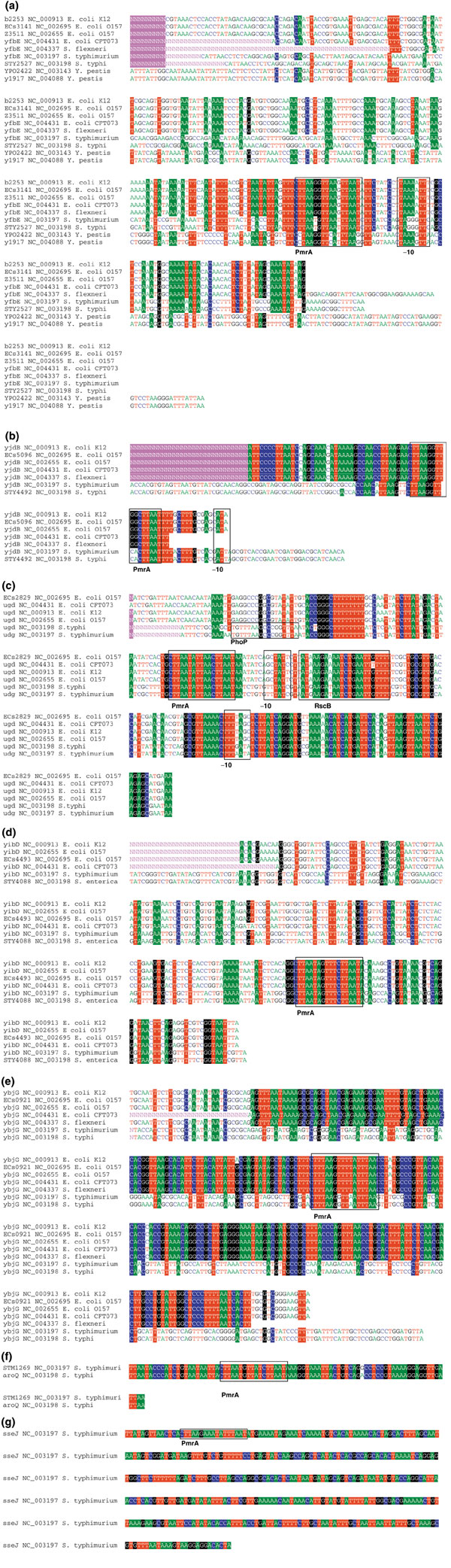
Local alignments of the most promising targets. Examples of local alignments obtained by phylogenetic footprinting of known PmrAB targets and of some promising potential targets. Known motifs or (putative) PmrA motifs are indicated by a box. (a) yfbE (pmrH); (b) yjdB (pmrC); (c) ugd; (d) yibD; (e) ybjG (mig-13); (f) STM1269 (aroQ); (g) sseJ.
pmrH (the first gene of an operon that contains the genes pmrHFIJKLM; Table 1) is the only known PmrAB-regulated gene for which the PmrA motif is conserved in all genome sequences analyzed (including that of Y. pestis). In pmrC, encoding a gene with unknown function [15,22], the PmrA motif is conserved in the intergenic regions of its orthologs in E. coli strains, Salmonella species and Shigella. ugd encodes a UDP-D-glucose dehydrogenase required for the synthesis of Ara4N. Three two-component systems are involved in its regulation (PmrAB, PhoPQ and RcsCB) [12,15] and this is reflected in the presence of the corresponding motifs: ugd contains PmrA, PhoP and RcsB motifs. The experimentally confirmed PmrA motif on the plus strand and part of the -10 sequence as determined by Aguirre et al. have been conserved in S. typhimurium, S. typhi and E. coli [15]. The promoter of ugd also has a hit of the PmrA motif on the minus strand. This was, however, not confirmed by DNA footprint analysis [15] and might represent a false positive. The PhoP motif on the plus strand in ugd of Salmonella, although occurring as a dyad, is not conserved in close orthologs and was recently demonstrated to be non-functional [12]. The recognition site for the RcsB protein [12] is also conserved in E. coli. Lastly, yibD encodes a putative glycosyltransferase. The PmrA motif is conserved in E. coli. yibD has recently been identified as a PmrAB target by a genome-wide mutagenesis study. Its actual function is still unknown [22].
Experimental validation by expression analysis
Our in silico predictions pointed towards putative targets of the PmrAB regulatory system. Some of these have functions that were previously not associated with the PmrAB system. To prove the strength of our in silico approach, four potential targets were selected for biological validation: yibD (novel at the time of our analysis), aroQ (STM1269), mig-13 and sseJ. aroQ and yibD were selected because a perfect repeat of the previously described PmrA half-site (CTTAAT [15]) was detected in their respective intergenic regions. mig-13 (Figure 2) was chosen because it has previously been reported as a gene selectively induced in macrophages, but with further unknown regulation [40]. sseJ (Figure 2) was further analyzed because although PmrAB-regulated genes have been implicated in animal virulence [2], no direct link between SPI-2 (Salmonella pathogenicity island 2) gene regulation and PmrAB has been demonstrated yet.
For each of these targets, green fluorescent protein (GFP) reporter fusions were constructed and their expression was determined by fluorescence-activated cell sorter (FACS) analysis in wild-type S. typhimurium and a pmrA::Tn10d mutant. Because the PmrAB system is sensitive to Mg2+ and Fe3+ concentration, we tested the effect of these signals on the expression of the fusions [22] (Table 2). All experiments were performed at pH 5.8 and pH 7.7. All fusions tested exhibited the same PmrAB-dependent expression behavior at both pH levels. In all experiments, pmrC was used as a positive control.
Table 2.
Expression analysis of the GFP reporter fusions
| Fusion | Strain | 10 mM MgCl2 | 10 μM MgCl2 | 100 μM FeCl3 | 10 mM MgCl2 100 μM FeCl3 | 10 μM MgCl2 100 μM FeCl3 |
| pmrC::GFP | WT | 6.06 (0.18) | 16.8 (1.42) | 70.53 (3.84) | 27.39 (4.41) | 83.2 (3.21) |
| pmrA - | 1.00 (0.01) | 1.02 (0.02) | 1.08 (0.03) | 1.03 (0.03) | 1.16 (0.12) | |
| mig-13::GFP | WT | 6.17 (1.55) | 13.50 (2.02) | 35.81 (4.67) | 17.86 (5.04) | 49.23 (5.43) |
| pmrA - | 2.69 (0.11) | 4.32 (0.48) | 5.2 (0.09) | 2.67 (0.16) | 9.64 (1.19) | |
| aroQ::GFP | WT | 2.32 (0.22) | 20.39 (1.54) | 19.39 (0.53) | 4.38 (0.19) | 19.48 (2.07) |
| pmrA - | 1.06 (0.02) | 1.09 (0.02) | 1.71 (0.09) | 1.02 (0.01) | 1.09 (0.03) | |
| yibD::GFP | WT | 1.25 (0.02) | 1.67 (0.26) | 33.35 (7.01) | 27.52 (5.64) | 52.46 (8.98) |
| pmrA - | 1.26 (0.02) | 1.21 (0.06) | 1.30 (0.02) | 1.14 (0.02) | 1.81 (0.44) | |
| sseJ::GFP | WT | 7.68 (1.55) | 11.25(1.46) | 22.58 (1.01) | 3.80 (1.13) | 8.03 (1.27) |
| pmrA - | 5.64 (0.72) | 8.72 (1.05) | 7.35 (1.55) | 2.99 (0.43) | 6.47 (1.36) |
All experiments were performed twice. Values indicate the average mean peak fluorescence measurements of at least three samples for the populations grown under the conditions indicated for one representative experiment. Values in parentheses represent standard deviations. All values are expressed in arbitrary units. Strains used: WT = ATCC14028s and pmrA- = pmrA::Tn10d. For pmrC, aroQ, mig-13 and yibD, values represented in the table correspond to experiments performed at pH 7.7. Similar results were obtained at pH 5.8 (data not shown). For sseJ, values correspond to experiments performed at pH 5.8 because at this pH the overall measured expression was higher. The constitutive gfp fusion (pFPV25.1) varied less than 10% between the conditions tested.
The pmrC fusion showed a clear induction by either Mg2+ deprivation or Fe3+ excess. The observed level of induction was higher for the Fe3+-dependent signal than for the Mg2+-dependent signal and the combination of both signals seemed to act synergistically. For both signals, induction was abrogated in a pmrA::Tn10d background, indicating that induction by Mg2+ and Fe3+ is solely PmrAB dependent. For the mig-13 fusion, similar observations were made, although induction by low Mg2+ and the synergistic effect of both signals were less pronounced. mig-13 also exhibited a considerable background expression level both in a pmrA::Tn10d mutant and in the uninduced state in a wild-type background. aroQ was strongly induced by low Mg2+ and induction was abrogated in a pmrA::Tn10d background. The influence of Fe3+ was less pronounced. In the case of yibD, the opposite was found: the yibD gene was barely induced by low Mg2+ but Fe3+ excess resulted in a large induction. For the yibD fusion, although Fe3+ excess, but not Mg2+ deprivation, seemed to be a sufficient signal to trigger expression, both signals acted synergistically. Also, induction of yibD was abrogated in a pmrA::Tn10d background. Compared to the other fusions, the observed expression levels of the sseJ fusion were rather low in the test conditions. Because sseJ showed a higher overall expression level at pH 5.8, these data were considered most representative (see Table 2). Results show an upregulation of sseJ expression in elevated Fe3+ concentrations that was absent in the pmrA::Tn10d background. As observed for mig-13, sseJ was expressed at a background level in the mutant pmrA::Tn10d. Interestingly, even at low concentrations, Mg2+ seemed to counteract the Fe3+-dependent induction.
Site-directed mutagenesis of the PmrA box
We constructed a set of mutant PmrA box sequences by site-directed mutagenesis of the PmrA box of yibD. AT → GC and GC → AT substitutions were introduced in the first half-site of the PmrA box (Figure 3a). We focused on the first half-site, as in the experimentally verified target pmrC, the second half-site overlaps with the -35 promoter site [14]. Expression was compared in different mutagenized fusions and the nonmutated fusion in the wild type and in the pmrA::Tn10d strain in all conditions mentioned above. For simplicity, only the expression values for two inducing conditions are displayed in Figure 3b. One is induction by the combined action of high Fe3+ and low Mg2+ concentrations and the other is the induction by raised Fe3+ levels in the presence of high Mg2+. Observations under all other conditions allowed us to draw similar conclusions. Substitutions in the third and fifth positions of the motif box completely abrogated PmrAB-dependent expression. Mutations of the first, second, fourth or sixth position reduced PmrAB-dependent induction. Note that for the mutation in the second position, expression was very low but not completely abrogated. Results from this site-directed mutagenesis experiment of one representative PmrAB target allowed us to demonstrate unequivocally that the PmrA box we identified was responsible for PmrAB-dependent transcriptional activation. It also allowed us to further delineate the sequence requirements of the PmrA consensus.
Figure 3.
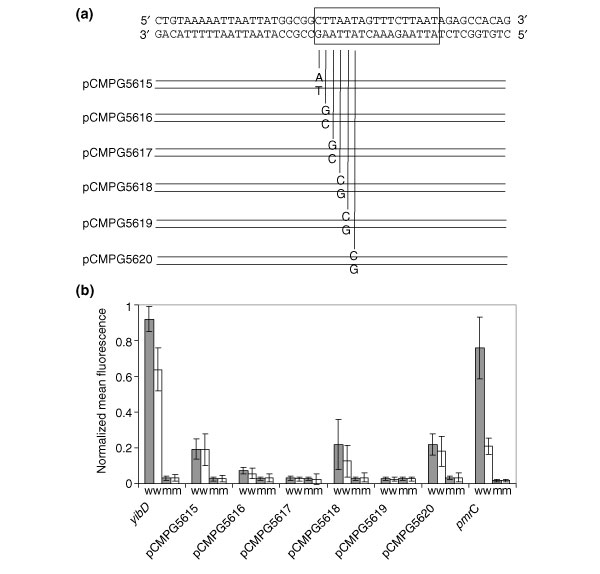
Site-directed mutagenesis of the PmrA box in yibD. (a) Construction of six species of the yibD promoter mutant, designated pCMPG5615 to pCMPG5620, each with a single base substitution (T → G or A → C) in the PmrA box. Promoters were fused to GFP and promoter activity was assessed by FACS analysis. (b) Plot of the normalized expression values of the six mutant fusions and the wild-type fusion measured in two distinct conditions in the wild type and pmrA::Tn10d mutant background. Gray bars represent condition 1 (pH 7.7, 100 μM FeCl3 + 10 μM MgCl2), white bars correspond to the expression values observed in condition 2 (pH 7.7, 100 μM FeCl3 + 10 mM MgCl2). w, wild-type background; m, pmrA::Tn10d mutant background. The pmrC::GFP fusion was included as a positive control. Bars represent the standard deviations of three independent measurements.
Other promising PmrAB targets
On the basis of the instances of the PmrA motif in experimentally verified PmrAB targets of Salmonella (verified previously or validated in this study), a PmrA consensus was built (Figure 4). The motif consensus of PmrA was converted into a regular expression (A/C)(C/T)T(A/T)A(T/G/A) N5NTT(A/T)A(T/A/G). To construct this regular expression we only considered the two conserved half-sites, because the PmrA motif is believed to be a dyad [15]. We preferred the part between the conserved half-sites of the regular expression to be represented as degenerate (that is, N5). Indeed, the observed degree of conservation in the intermediate part of the motif model (Figure 4b) is probably related to the restricted sample size of the training set rather than being an intrinsic property of the motif. Promising motifs (indicated in bold in Table 1) are, therefore, motifs that match this regular expression and thus contain nucleotides that occur in the conserved half-sites of one of the experimentally verified examples. Promising targets for which the putative PmrA motif was also conserved in species other than Salmonella were mig-13, yrbF, yjgD, ybdO, yejG, lasT and ybdN. Promising targets only present in S. typhimurium and/or S. typhi were STM1269 (aroQ), STM1273, sseJ and lpfA. Note that this listing is just based on an arbitrary selection criterion, that is, a preliminary PmrA motif consensus that will be improved as more PmrAB targets become experimentally validated. As well as the targets mentioned above, Table 1 contains other targets that are of interest because their annotation relates to the PmrAB system (such as yncD).
Figure 4.
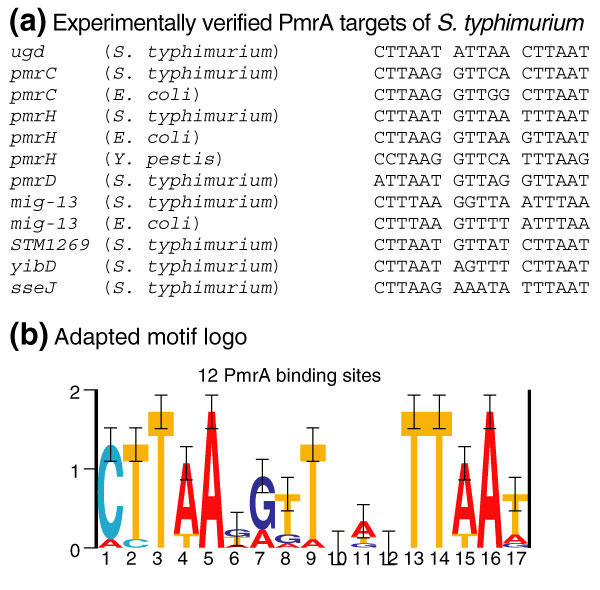
Refined consensus of the PmrA box. (a) Alignment of all experimentally verified PmrA sites ([15] or this work) in S. typhimurium [1]. PmrA sites in the orthologs of these respective experimentally verified genes are also displayed if these PmrA motif instances deviated from the PmrA motif in S. typhimurium. (b) An adapted motif model of the PmrA site was built (represented by its logo) on the basis of the sequences represented in (a).
Discussion
Putative PmrAB targets were detected by genome-wide screening of S. typhimurium intergenic sequences using a PmrA motif model. If possible, the confidence in the predicted motifs was strengthened by a cross-species comparison: we tested whether the PmrA motif was conserved in the intergenic regions of close homologs in related species. To this end, we developed a two-step procedure for phylogenetic footprinting. In the first step, a motif-detection procedure based on Gibbs sampling was performed to generate a list of motifs. In the second step, these motifs were used as seeds to generate local multiple alignments. Eventually, the biological relevance of the obtained alignments was assessed.
We used the alignments rather than a listing of the high-scoring motifs obtained by Gibbs sampling for the following reasons. First, we observed, as also reported by McCue et al., a high overall similarity in intergenic regions of the selected species [34]. In general, the overall degree in conservation between the intergenic sequences of close homologs is about 93.56% for the sequenced representatives of Escherichia and Shigella species, 69.21% for Shigella and Salmonella and 53.31% for Salmonella and Yersinia. As a result of this property (high correlation in the data), not only the motif itself turns out to be conserved, but also its local neighborhood. Moreover, the degree of conservation between the aligned sequences in a biologically relevant alignment will reflect, in most cases, the phylogenetic relatedness of the species from which the sequences are derived (see Figure 2 for examples). By selecting the most promising alignment seeds (based on the appropriate heuristics for the scores) and constructing a local alignment with these seeds, we could also evaluate the local neighborhood of the seed. If this one seemed to be conserved as well, we could be more confident in the obtained alignment and in the motifs contained within the conserved parts. Therefore, the use of local alignments allows a better judgment on the reliability of the motifs.
Second, Gibbs sampling is a stochastic procedure. The algorithm has to be run repeatedly on the same dataset, each time generating potentially different motifs. As a consequence, the output of a motif-detection approach can be simultaneously redundant and non-exhaustive: some statistically strong motifs are detected repeatedly in different runs. On the other hand, some motifs might never be detected. Indeed, because Gibbs sampling was originally designed for unrelated sequences and because of the high correlation in the data, the number of possible equally scoring motifs (local optima) might be so high that many runs have to be performed before all motifs have been covered. All these local optima coincide with motifs that, when used as seeds, will result in a similar alignment. The same alignment can thus be obtained by several motifs, but there is no guarantee that all possible motifs that result in the same alignment will be detected by Gibbs sampling. Therefore, an alignment is a better summary of the degree of conservation between the intergenic regions than a listing of the highest-scoring motifs.
Moreover, regulatory systems such as PmrAB might have acquired some very species-specific targets. For such highly specialized regulatory systems, motifs are likely to be present in the intergenic sequences of a selected subset of orthologs only. Because such motifs occur in a restricted number of sequences of the dataset, they will not necessarily correspond to the highest-scoring motifs. Thus, they might be overlooked when selecting on high-scoring motifs by, for instance, setting a threshold on the score. Once a reliable local alignment of a set of intergenic sequences is obtained, one can judge the degree of confidence to put on the prediction of the motif of interest not only by checking in which subset of species the motif is conserved, but also by taking into consideration other factors, such as the functional annotation of the putative target. The motifs that we select on the basis of our heuristic will result in a biologically relevant alignment that includes the maximal number of species. As such, our heuristic tries to overcome the fact that Gibbs sampling is intrinsically unable to cope with correlated data. Note that the motif of interest (PmrA motif) does not necessarily have to correspond to the motif used to produce the alignment.
We showed that our in silico phylogenetic footprinting approach can be used to confirm targets detected by genome-wide screening. So far, it can only be used for species that show a high degree of conservation in their intergenic regions, similar to the conservation observed in this study. As more complete genomic data become available, the approach might be extended to other species.
As suggested previously [34], the high observed similarity in intergenic sequences might be due to the small phylogenetic distance between the species we analyzed. However, it cannot be excluded that because of the small size of the intergenic regions in bacteria and the very similar habitat and mechanism of regulation among the γ-proteobacterial species used in this study, a large part of the complete intergenic region is functional and therefore conserved. This hypothesis was also put forward by Rajewsky et al. [41]. The alignment of the intergenic region of the well-characterized ugd indeed points in that direction. Large parts of the conserved regions of the alignments correspond to experimentally verified motifs.
Remarkably, most potential PmrAB-regulated genes exhibited a footprint of the PmrA motif in E. coli only, and several target genes had no counterpart at all in organisms other than Salmonella species. This indicates a high degree of specialization of the PmrAB two-component system in Salmonella. Such specialization could also explain the considerable differences between PmrAB-dependent regulons in related species. For instance, in both Y. pestis and S. typhimurium the attenuated virulence of phoP mutants is ascribed to a defect in LPS modification, a process shown to be PmrAB-dependent [42]. So far, two S. typhimurium loci have been postulated to be involved in this LPS remodeling: pmrHFIJKLM and ugd. Only for pmrH did we detect an ortholog in Y. pestis and a conserved footprint of the PmrA motif in the promoter region of this ortholog. The Ugd protein does not even have a functional counterpart in Y. pestis. This low similarity in PmrAB regulon composition indicates that a different network of genes must be responsible for a similar phenotype in distinct species. This is not completely unexpected in view of the very different LPS composition of Salmonella and Y. pestis [42].
For most of the known experimentally verified targets, clear phylogenetic footprints of the PmrA motif could be detected in the intergenic regions of close homologs. In the intergenic region of pmrD we could recover the consensus sequence only partially (that is, one half-site) because the second half-site overlaps with the coding region (data not shown) and this was not included in the current analysis. Another PhoPQ-dependent gene that contributes to resistance to antimicrobial peptides is mig-14 [10]. However, we could not find the presence of a clear PmrA consensus in the promoter of mig-14. Neither could we detect a PmrA motif in dgoA, which was previously shown to be regulated by PmrAB [22]. This would indicate that both targets are only indirectly dependent on PmrAB. It can, however, not be excluded that they represent false negatives of our screening.
As well as the known targets, several putative new predictions could be made. Some of these predictions are consistent with previously published observations. Indeed, the PmrAB system is part of a complex regulatory cascade acting downstream of the pleiotropic PhoPQ system. The PhoPQ regulon is responsible for intracellular survival of bacteria and genes dependent on PhoPQ are induced in bacteria inside macrophages. Part of the PhoPQ regulon has been discovered to be dependent only indirectly on PhoPQ, via PmrAB. This PmrAB-dependent subset is known to confer resistance to cationic peptides by encoding genes involved in LPS modification and genes contributing to resistance to raised Fe3+ concentration. Genes encoding proteins involved in modification of membrane components and outer-membrane proteins are therefore sensible additional putative PmrAB targets. Another target worth mentioning in view of the Fe3+-sensitivity of the PmrAB system is yncD, which encodes a putative outer-membrane receptor for iron transport.
Phage remnants, such as mig-3, have been described as macrophage inducible, PhoPQ-dependent genes [40], and thus can be PmrAB-dependent. This might explain the PmrA motif in the intergenic region between STM1868A and mig-3. Detweiler et al. showed that two genes, virK and somA, both coexpressed with the SPI-2 system, confer resistance to cationic peptides and their expression is PhoPQ-dependent. Also, four fimbrial operons had genes that were coexpressed with SPI-2 [43]. Predictions of the PmrAB-dependence of sseJ, which encodes a secreted effector of the SPI-2 system, or of genes that are involved in fimbrial synthesis (such as in lpfA, encoding the S. typhimurium long polar fimbria A precursor), could therefore be in agreement with these findings. Recently, the study of Kim et al. related PmrAB-dependent regulation to swarming motility functions in S. typhimurium [44]. This could explain why we detected a putative PmrA motif in the intergenic region of flhC, which encodes a master transcriptional activator of flagellar genes.
To confirm the predictive power of our methodology further, four putative PmrAB targets were validated biologically. Expression analysis clearly demonstrated PmrAB-dependence of yibD, which confirms the recent observations of Tamayo et al. [22]. The observed PmrA-dependence of mig-13 accords with its being upregulated in macrophages in in vivo conditions [40]. The clear PmrAB-dependence of aroQ, which encodes a periplasmic chorismate mutase, is striking. In general, chorismate mutases are involved in the synthesis of tyrosine and phenylalanine and are key to the synthesis of a plethora of secondary metabolites [45]. The function of periplasmic chorismate mutases, which differ from the cytoplasmic chorismate mutases in their long carboxy-terminal extension [46], is still unclear. Periplasmic AroQ proteins have also been detected in Y. pestis, Pseudomonas aeruginosa, Mycobacterium species, Erwinia herbicola and in the phytoparasitic nematode Meloidogyne javanica [46]. Strikingly, all these organisms containing AroQ interact with a eukaryotic host. This observation, together with the fact that AroQ is dependent on the key virulence regulator PmrAB in S. typhimurium, suggests that the as-yet-unknown function of AroQ might be involved in bacteria-host interactions.
Despite its low expression level in our in vitro conditions, the sseJ fusion showed a clear PmrAB-dependent induction by Fe3+ excess. SseJ is a secreted effector protein that is translocated across the membrane of the Salmonella-containing vacuole (SCV) by SPI-2. From recent evidence it was speculated that the putative acyltransferase activity of SseJ would be involved in modifying the lipid composition of the SCV [47,48], thereby interfering with the trafficking and maturation properties of the SCV in infected cells. The PmrA-dependence of sseJ would therefore link expression of genes involved in bacterial LPS modification with those involved in regulating the lipid composition of the SCV membrane.
Further experimental analysis will shed light on how these previously undescribed PmrAB-dependent proteins, with unknown functions, relate to the known part of the PmrAB-dependent regulon.
The extent to which each of the tested strains reacted to the signals Fe3+ or/and Mg2+ varied considerably. This is not surprising in view of the complex regulatory system that integrates both these signals. Indeed, both signals are transduced via the PhoPQ, PmrD, PmrAB multicomponent system, which includes a posttranslational signal transduction and a transcriptional feedback loop [1]. Depending on the affinities between the interacting components of such dynamic systems, small changes in the initial concentrations of the components might result in large differences in the observed expression levels [49]. A more detailed study of the dynamics of this system might reveal how such systems can integrate signals so differently.
Site-directed mutagenesis of the PmrA box in the yibD promoter indicated a crucial role for the T at position 3 and the A at position 5 of the first half-site of the motif. As can be deduced from the consensus site in Figure 4, no degeneracy is allowed at positions 3 and 5. This observation allows us to extrapolate to a certain extent the sequence requirements of the PmrA box in yibD to other PmrAB targets. Some positions seem essential, whereas it appears that the specific choice of nucleotide at the other positions affects the level of induction. By altering the nucleotides, the binding affinities of the regulatory protein to the box can be modified, allowing specific fine-tuning of gene expression in a cell.
Conclusions
We conclusively demonstrate that our in silico approach reliably identifies additional PmrAB-dependent targets. Although false positives will still be present among these predicted targets, the method offers an interesting approach for further elucidation of genetic networks involved in the expression of S. typhimurium virulence genes. We predicted the PmrAB-dependent regulation of four additional targets: yibD, aroQ, mig-13 and sseJ. Our approach might become extendable to other species when more genome sequences become available.
Materials and methods
Selection of intergenic sequences
Genome sequences were obtained from GenBank [50]. All intergenic regions used in this study were extracted using the modules of INCLUSive [51] to automatically parse GenBank entries [52]. Here, we define an intergenic sequence as a region that contains the noncoding sequence between two coding regions. No overlap with coding regions is allowed. Intergenic regions with lengths smaller than 10 base-pairs (bp) were discarded because of computational reasons.
Construction of motif models
A motif model (a probabilistic representation of the consensus DNA pattern that is recognized by the respective regulatory protein) for PmrA was constructed using MotifSampler [53]. The PmrA training set consisted of the promoter regions of three known PmrAB-regulated genes (ugd [7,15,17], pmrH [7,14,17,19] and pmrC [2,14]) for which the binding of the PmrA protein to the promoter regions was verified by DNA footprint analysis [14,15].
Genome-wide screening
The intergenic regions of the complete genome of S. typhimurium LT2 (NC_003197 [38]) were screened using MotifLocator [54,55]. The scoring scheme used by MotifLocator is extensively described in Thijs et al. [55] and uses an extension of the classical position-weight matrix scoring scheme [56]. Given the motif model θ and the background model Bm a score W(x) is computed for each window x of length l in the sequence S. W(x) compares the score of the subsequence within the window being generated by the motif model to the score of the subsequence within the window being generated by the background model. bj: nucleotide at position j in the segment.
![]()
Both the plus and minus strand were screened using a background model of order 3. The higher-order background allows implicit compensation for motifs that are located in a context highly resembling the global nucleotide composition of the genome. To apply a threshold on the scores, the scores of different motifs were normalized such that their values ranged between 0 and 1. The normalized scores 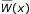 are displayed in Table 1.
are displayed in Table 1.
![]()
Hits with a score above 0.75 were retained (corresponding to a selection of the 0.003% top-scoring hits of the total number of possible motif positions in the genome. Possible positions are identified as overlapping windows of length l). To give a rough assessment of the number of hits with a score similar to the chosen threshold that could be expected from the specific nucleotide composition of the genome, we generated 100 random sets of intergenic sequences using a third-order background model. These random sets were scored with the same PmrA motif model. From these results it appeared that the true set contained three times more hits with a score above the threshold than an average random genome.
Identification of datasets
Highly similar homologs of the putative PmrAB-regulated genes were identified in the genome sequences of S. typhimurium (NC_003197), S. typhi (NC_003198), S. flexneri (NC_004337), E. coli O157:H7 (NC_002695), E. coli O157:H7 EDL933 (NC_002655), E. coli K12 (NC_000913), Y. pestis CO92 (NC_003143) and Y. pestis KIM (NC_004088). In general, only true orthologs are likely to have retained a similar function and therefore a similar mechanism of regulation [35]. However, ortholog identification is a difficult problem and discriminating between true orthologs and paralogs is not always straightforward. Because our motif-detection algorithm is, to some extent, robust against the presence of noise and allows for the presence of sequences that do not contain the motif [57], we did not make an a priori distinction between true orthologs and paralogs if both appeared highly similar to the original protein. This motivated us to use the principle of clusters of orthologs for dataset construction [58]. The pairwise BLAST scores obtained by mutually aligning the whole-genome sequences using BlastP [59] were used as input of the cluster program TRIBE-MCL [60]. Stringent criteria were applied to retain only closely related orthologs and paralogs (cut-off of the BLAST hit was an E-value of 1e-80). For those proteins that, when BLASTed against themselves, gave rise to an E-value higher than 1e-80 (yjbE, STM1926, STM0344, yhcN, STM1868A, yceP, atpF, yjgD, csrA, ybfA) the threshold was relaxed (E-value 1e-20). The choice of the stringent threshold was essential to maximally reduce the noise in the datasets.
Phylogenetic footprinting by Gibbs sampling
We used a two-step procedure for phylogenetic footprinting. In the first step, Gibbs sampling is performed to generate a list of motifs. Subsequently, local alignments are generated by selecting motifs that can be used as alignment seeds and by assessing the relevance of the alignments by a test statistic.
Motif detection by Gibbs sampling
An advanced Gibbs sampling procedure for motif detection was used (MotifSampler). MotifSampler allows us to search for overrepresented motifs in each dataset. The motif length, the maximal number of different motifs and the background model are user-defined parameter settings of the algorithm described in Thijs et al. [53]. A recent extension of the algorithm allows to automatically determine the number of instances of a certain motif per sequence [55] and requires a predefined indication on the prior probability of expecting at least one motif per sequence. For each dataset, 100 runs of the MotifSampler were performed under the following conditions: motif lengths varying from 6, 8, 10, 12; background order 0; prior probability default value 0.7. Because Gibbs sampling is a stochastic procedure, each run can give rise to different motifs. To summarize the results from the 100 runs, all detected motifs were mutually compared and similar motifs were grouped. The information content was used as a similarity measure to compare motifs. For each dataset, therefore, a list of different potential motifs was obtained. Motifs in this list were ranked according to their log-likelihood score (LL-score) [53].
Generating reliable local alignments using the detected motifs
From the obtained list, motifs that could be used as seeds to generate a biologically relevant alignment were selected using a heuristic [61]. Motifs with a high LL-score that occurred preferentially once in each sequence were chosen (starting with those motifs that had the highest consensus score). Moreover, we preferentially selected motifs that occurred in the maximal number of species. For each dataset, multiple alignments were constructed using the position of these retrieved motifs as alignment initializations (seeds) until a reliable alignment was obtained. The alignment was considered biologically relevant if within a window of 100 bp around the motif it exhibited a degree of conservation that reflected the overall observed homology between intergenic sequences of the selected species (interspecies homology).
To assign a more quantitative criterion to the alignment, a p-value was assigned to each alignment that was calculated as follows: for each window of 100 bp around the motif, the largest conserved block not overlapping with the core motif was identified (using the consensus score of minimal 0.7 as minimal similarity measure). This p-value expresses the probability of observing a conserved block of the same length in a randomly aligned dataset of similar composition. Distributions of conserved blocks in randomly aligned sequences were constructed [62]. These random datasets take into account the observed high pairwise sequence homology between intergenic sequences derived from similar species (serovars) (homology between E. coli sequences, homology between Salmonella sequences, homology between Y. pestis sequences), but not the interspecies homology between intergenic regions (for example, between E. coli and Salmonella). All alignments with a p-value less than 0.15 (the p-value of ugd) were considered as relevant (indicated in Table 1 with 'm'). Because the obtained alignments are local, they are gapless. In some cases, more than one alignment might be essential to cover the complete intergenic region.
Selected alignments are displayed in the supplementary information at [39]. Sequence editing was done in BioEdit [63].
Functional annotation
Functional annotation was derived from the National Center for Biotechnology Information (NCBI) [38,52] and from the Sanger annotation of S. typhi [64]. Specific genomic context was derived from NCBI [38,52]. The distribution of the putative targets (unique for Salmonella species versus more widely distributed), as derived from our clusters of orthologous groups (COGs), was verified by comparison to the analysis of McClelland et al. [38], who included, in addition to the species we used, several subspecies of Salmonella (six genomes), and species more distantly related to Salmonella (Klebsiella pneumoniae).
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 3. Bacteria were grown overnight at 37°C with aeration in Luria-Bertani (LB) broth or in the nitrogen minimal medium of Nelson and Kennedy [65] with modifications as previously described [66]. The pH of the medium was buffered with 100 mM Tris-HCl, adjusted to pH 7.4 or pH 5.8. MgCl2 was added at a final concentration of 10 μM or 10 mM. FeCl3 was used at a final concentration of 100 μM from a freshly prepared 10 mM stock. Antibiotics were used, when appropriate, at the following concentrations: tetracycline, 30 μg/ml; and ampicillin, 100 μg/ml.
Table 3.
Bacterial strains
| Strain or plasmid | Relevant genotype | Reference or source | |
| Salmonella | ATCC 14028s | Wild type | ATCC |
| JSG421 | pmrA::Tn10d | [21] kind gift of J.S. Gunn | |
| LB5010 | metA22 metE551 ilv-452 leu-3121 trpΔ2 xyl-404 galE856 hsdLT6 hsdSA29 hsdSB121 rpsL120 | [67] | |
| E. coli | DH5α | Fφ80ΔlacZM15 Δ(lacZYAargF)U169 deoP recA1 endA1 hsdR17 (rk-mk-) | Gibco BRL |
| Plasmids | pCRII-TOPO | Cloning vector, AmpR | Invitrogen |
| pFPV25 | ColE1 mob bla promoterless gfpmut3, AmpR | [40] kind gift of R. Valdivia and S. Falkow | |
| pFPV25.1 | Constitutive rpsM promoter in pFPV25, AmpR | [40] kind gift of R. Valdivia and S. Falkow | |
| pCMPG5611 | pFPV25 with yibD promoter | This work | |
| pCMPG5612 | pFPV25 with ybjG promoter | This work | |
| pCMPG5613 | pFPV25 with STM1269 promoter | This work | |
| pCMPG5614 | pFPV25 with yjdB promoter | This work | |
| pCMPG5621 | pFPV25 with sseJ promoter | This work | |
| pCMPG5615 | pCMPG5611 with point mutated putative PmrA motif C→A | This work | |
| pCMPG5616 | pCMPG5611 with point mutated putative PmrA motif T→G | This work | |
| pCMPG5617 | pCMPG5611 with point mutated putative PmrA motif T→G | This work | |
| pCMPG5618 | pCMPG5611 with point mutated putative PmrA motif A→C | This work | |
| pCMPG5619 | pCMPG5611 with point mutated putative PmrA motif A→C | This work | |
| pCMPG5620 | pCMPG5611 with point mutated putative PmrA motif T→C | This work | |
Molecular methods
Plasmid DNA, after passage through S. typhimurium LB5010 [67], was introduced into bacterial strains by electroporation. Polymerase chain reaction (PCR) was carried out in a Personal Mastercycler (Eppendorf, Hamburg, Germany) with Pfx DNA polymerase, using the manufacturer's instructions. The constructs containing the putative promoter regions of yibD, mig-13, aroQ, sseJ and pmrC and the site-directed mutated yibD promoter fragments were all verified by sequence analysis.
Construction of plasmids
To construct the gfp reporter fusions of yibD, mig-13, aroQ, sseJ and pmrC, the primers listed in Table 4 were used in a PCR reaction (as described above) to amplify the respective promoter regions from the ATCC14028s genome. Restriction sites in the primers are indicated in bold face in Table 4. The promoter fragments were digested with EcoRI and BamHI and cloned into pCRII-TOPO that had been digested with EcoRI and BamHI. The promoter fragments were subcloned as EcoRI/BamHI fragments into the corresponding sites of pFPV25 [40], resulting in the pCMPG plasmids listed in Table 3. These plasmids were electroporated into ATCC14028s and JSG421 [21] after propagation through LB5010, as described above. Cloning steps were carried out in E. coli DH5α.
Table 4.
Primers used to construct the GFP promoter fusions
| Name | Sequence 5' to 3' | Description |
| Amplification of promoter regions | ||
| Pro-115 | CCGAATTCTAATTCGAGTTGCTTAAAGGCGGC | Amplification of yibD promoter region |
| Pro-116 | CCGGATCCGCTCCCGCATTATATAACGGG | Amplification of yibD promoter region |
| Pro-117 | CCGAATTCGCCAATAAAAACCGCGCAGAGTG | Amplification of mig-13 promoter region |
| Pro-118 | CCGGATCCAGCGAGTTGTTAAGGTTTTCCAGC | Amplification of mig-13promoter region |
| Pro-119 | CCGAATTCGAAGATTCCGCAGAATCAACGGCC | Amplification of aroQ promoter region |
| Pro-120 | CCGGATCCGGTGCTGCACATCAATAAAGAACAAAG | Amplification of aroQ promoter region |
| Pro-121 | CCGAATTCGTATTGCATCTGGGCGGTCATCG | Amplification of pmrC promoter region |
| Pro-122 | CCGGATCCAGGCGATTTGCCCAAGAACAGG | Amplification of pmrC promoter region |
| Pro-224 | ATGAATTCGCTTCCCCATCCCAAACCACC | Amplification of sseJ promoter region |
| Pro-225 | ATGGATCCGGAAGGCGTGCGCTTTCTTTTATC | Amplification of sseJ promoter region |
Restriction sites in the primers are indicated in bold type.
The single base-pair substitutions in the putative PmrA motif occurring in the yibD promoter sequence were introduced via a PCR approach using the QuickChange Site-Directed mutagenesis kit (Stratagene, La Jolla, CA), according to the manufacturer's instructions. The yibD promoter sequence introduced into pCRII-TOPO was used as the parent plasmid and appropriate primers were applied (sequences not shown). The site-directed mutated yibD promoter fragments were subcloned into pFPV25 (EcoRI/BamHI), resulting in plasmids pCMPG5615 → pCMPG5620, as listed in Table 3. These reporter plasmids were electroporated into ATCC14028s and JSG421.
Fluorescence-activated cell sorter-based expression analysis
Bacterial strains harboring the reporter constructs were grown overnight in nitrogen (N)-minimal medium pH 7.4 plus 10 mM MgCl2, harvested, washed in N-minimal medium pH 7.4 without MgCl2, and diluted 1:100 in N-minimal medium pH 7.4 plus 10 mM MgCl2. Mid-log-phase bacteria were then inoculated into the indicated media and grown for 3 h to allow expression of GFP. Bacteria were diluted into PBS and analyzed by flow cytometry with a Becton Dickinson FACSCalibur and CellQuest acquisition and analysis software [40] with gates set to forward and side scatters characteristic of the bacteria.
Nomenclature
As the gene names used in the annotation of the S. typhimurium genome sequence do not always match the 'common' names used in the PmrAB literature, we give a summary of the synonyms below. STM1269 (aroQ); ybjG (mig-13); pmrAB (basSR); ugd (udg, pagA, pmrE); pmrHFIJKLM (yfbE, pmrF, yfbG, STM2300, pqa, STM2302, STM2303); pmrC (yjdB); pmrG (ais); pbgP (yfbE,pmrH).
Availability of data
All additional information of our analysis is available on our supplementary website [39].
Acknowledgments
Acknowledgements
This work is partially supported by Instituut voor de aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (IWT) projects STWW-00162, STWW-Genprom, GBOU-SQUAD-20160; Research Council KULeuven GOA Mefisto-666, IDO genetic networks; and FWO: Fund for Scientific Research-Flanders (Belgium) projects G.0115.01 and G.0413.03; IUAP V-22 (2002-2006). K.M. is a postdoctoral researcher of the FWO: Fund for Scientific Research-Flanders (Belgium). We thank John Gunn and Stanley Falkow and their respective co-workers for the kind gift of the strains and four anonymous reviewers for valuable comments on the manuscript.
References
- Kato A, Latifi T, Groisman EA. Closing the loop: the PmrA/PmrB two-component system negatively controls expression of its posttranscriptional activator PmrD. Proc Natl Acad Sci USA. 2003;100:4706–4711. doi: 10.1073/pnas.0836837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun. 2000;68:6139–6146. doi: 10.1128/IAI.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jansen R, Gaastra W, Arkesteijn G, van der Zeijst BA, van Putten JP. Identification of genes affecting Salmonella enterica serovar enteritidis infection of chicken macrophages. Infect Immun. 2002;70:5319–5321. doi: 10.1128/IAI.70.9.5319-5321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. A signal transduction system that responds to extracellular iron. Cell. 2000;103:113–125. doi: 10.1016/s0092-8674(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Garcia Vescovi E, Soncini FC, Groisman EA. Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell. 1996;84:165–174. doi: 10.1016/s0092-8674(00)81003-x. [DOI] [PubMed] [Google Scholar]
- Soncini FC, Groisman EA. Two-component regulatory systems can interact to process multiple environmental signals. J Bacteriol. 1996;178:6796–6801. doi: 10.1128/jb.178.23.6796-6801.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini FC, Garcia Vescovi E, Solomon F, Groisman EA. Molecular basis of the magnesium deprivation response in Salmonella typhimurium: identification of PhoP-regulated genes. J Bacteriol. 1996;178:5092–5099. doi: 10.1128/jb.178.17.5092-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox LF, Wosten MM, Groisman EA. A small protein that mediates the activation of a two-component system by another two-component system. EMBO J. 2000;19:1861–1872. doi: 10.1093/emboj/19.8.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky IE, Ernst RK, Miller SI, Falkow S. mig-14 is a Salmonella gene that plays a role in bacterial resistance to antimicrobial peptides. J Bacteriol. 2002;184:3203–3213. doi: 10.1128/JB.184.12.3203-3213.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland KL, Martin LE, Esther CR, Spitznagel JK. Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J Bacteriol. 1993;175:4154–4164. doi: 10.1128/jb.175.13.4154-4164.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouslim C, Groisman EA. Control of the Salmonella ugd gene by three two-component regulatory systems. Mol Microbiol. 2003;47:335–344. doi: 10.1046/j.1365-2958.2003.03318.x. [DOI] [PubMed] [Google Scholar]
- Chamnongpol S, Dodson W, Cromie MJ, Harris ZL, Groisman EA. Fe(III)-mediated cellular toxicity. Mol Microbiol. 2002;45:711–719. doi: 10.1046/j.1365-2958.2002.03041.x. [DOI] [PubMed] [Google Scholar]
- Wosten MM, Groisman EA. Molecular characterization of the PmrA regulon. J Biol Chem. 1999;274:27185–27190. doi: 10.1074/jbc.274.38.27185. [DOI] [PubMed] [Google Scholar]
- Aguirre A, Lejona S, Vescovi EG, Soncini FC. Phosphorylated PmrA interacts with the promoter region of ugd in Salmonella enterica serovar typhimurium. J Bacteriol. 2000;182:3874–3876. doi: 10.1128/JB.182.13.3874-3876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CR. Lipid A modifications in polymyxin-resistant Salmonella typhimurium: PmrA-dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J Biol Chem. 2001;276:43111–43121. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol. 1998;27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- Breazeale SD, Ribeiro AA, Raetz CR. Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. Origin of lipid A species modified with 4-amino-4-deoxy-L-arabinose. J Biol Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CR. Accumulation of a polyisoprene-linked amino sugar in polymyxin-resistant Salmonella typhimurium and Escherichia coli: structural characterization and transfer to lipid A in the periplasm. J Biol Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- Gunn JS, Miller SI. PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J Bacteriol. 1996;178:6857–6864. doi: 10.1128/jb.178.23.6857-6864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo R, Ryan SS, McCoy AJ, Gunn JS. Identification and genetic characterization of PmrA-regulated genes and genes involved in polymyxin B resistance in Salmonella enterica serovar Typhimurium. Infect Immun. 2002;70:6770–6778. doi: 10.1128/IAI.70.12.6770-6778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. Methods for calculating the probabilities of finding patterns in sequences. Comput Appl Biosci. 1989;5:89–96. doi: 10.1093/bioinformatics/5.2.89. [DOI] [PubMed] [Google Scholar]
- van Helden J, Andre B, Collado-Vides J. A web site for the computational analysis of yeast regulatory sequences. Yeast. 2000;16:177–187. doi: 10.1002/(SICI)1097-0061(20000130)16:2<177::AID-YEA516>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Panina EM, Mironov AA, Gelfand MS. Comparative analysis of FUR regulons in gamma-proteobacteria. Nucleic Acids Res. 2001;29:5195–5206. doi: 10.1093/nar/29.24.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, De Moor B. Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 2003;31:1753–1764. doi: 10.1093/nar/gkg268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MS, Koonin EV, Mironov AA. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res. 2000;28:695–705. doi: 10.1093/nar/28.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laikova ON, Mironov AA, Gelfand MS. Computational analysis of the transcriptional regulation of pentose utilization systems in the gamma subdivision of Proteobacteria. FEMS Microbiol Lett. 2001;205:315–322. doi: 10.1016/S0378-1097(01)00495-5. [DOI] [PubMed] [Google Scholar]
- Rodionov DA, Gelfand MS, Mironov AA, Rakhmaninova AB. Comparative approach to analysis of regulation in complete genomes: multidrug resistance systems in gamma-proteobacteria. J Mol Microbiol Biotechnol. 2001;3:319–324. [PubMed] [Google Scholar]
- Tan K, Moreno-Hagelsieb G, Collado-Vides J, Stormo GD. A comparative genomics approach to prediction of new members of regulons. Genome Res. 2001;11:566–584. doi: 10.1101/gr.149301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrecht B, Marchal K, Vanderleyden J, Michiels J. Prediction and overview of the RpoN-regulon in closely related species of the Rhizobiales. Genome Biol. 2002;3:research0076.1–0076.11. doi: 10.1186/gb-2002-3-12-research0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, Derbyshire V, Lawrence CE. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue LA, Thompson W, Carmack CS, Lawrence CE. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 2002;12:1523–1532. doi: 10.1101/gr.323602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Elnitski L, Church DM, Dubchak I, Hardison RC. Cross-species sequence comparisons: a review of methods and available resources. Genome Res. 2003;13:1–12. doi: 10.1101/gr.222003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MS. Recognition of regulatory sites by genomic comparison. Res Microbiol. 1999;150:755–771. doi: 10.1016/S0923-2508(99)00117-5. [DOI] [PubMed] [Google Scholar]
- McGuire AM, Hughes JD, Church GM. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 2000;10:744–757. doi: 10.1101/gr.10.6.744. [DOI] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- Supplementary information http://www.esat.kuleuven.ac.be/~kmarchal/Supplementary_Information_GenBiol_2004/PmrAwebsite.html
- Valdivia RH, Falkow S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science. 1997;277:2007–2011. doi: 10.1126/science.277.5334.2007. [DOI] [PubMed] [Google Scholar]
- Rajewsky N, Socci ND, Zapotocky M, Siggia ED. The evolution of DNA regulatory regions for proteo-gamma bacteria by interspecies comparisons. Genome Res. 2002;12:298–308. doi: 10.1101/gr.207502. 10.1101/gr.207502. Article published online before print in January 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchen PG, Prior JL, Oyston PC, Panico M, Wren BW, Titball RW, Morris HR, Dell A. Structural characterization of lipo-oligosaccharide (LOS) from Yersinia pestis: regulation of LOS structure by the PhoPQ system. Mol Microbiol. 2002;44:1637–1650. doi: 10.1046/j.1365-2958.2002.02990.x. [DOI] [PubMed] [Google Scholar]
- Detweiler CS, Monack DM, Brodsky IE, Mathew H, Falkow S. virK, somA and rcsC are important for systemic Salmonella enterica serovar Typhimurium infection and cationic peptide resistance. Mol Microbiol. 2003;48:385–400. doi: 10.1046/j.1365-2958.2003.03455.x. [DOI] [PubMed] [Google Scholar]
- Kim W, Killam T, Sood V, Surette MG. Swarm-cell differentiation in Salmonella enterica serovar Typhimurium results in elevated resistance to multiple antibiotics. J Bacteriol. 2003;185:3111–3117. doi: 10.1128/JB.185.10.3111-3117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosselaere F, Vanderleyden J. A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Crit Rev Microbiol. 2001;27:75–131. doi: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- Calhoun DH, Bonner CA, Gu W, Xie G, Jensen RA. The emerging periplasm-localized subclass of AroQ chorismate mutases, exemplified by those from Salmonella typhimurium and Pseudomonas aeruginosa. Genome Biol. 2001;2:research0030.1–0030.16. doi: 10.1186/gb-2001-2-8-research0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol Microbiol. 2002;44:645–661. doi: 10.1046/j.1365-2958.2002.02912.x. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Ohl ME, Miller SI. The Salmonella enterica serovar typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella-containing vacuole. Infect Immun. 2003;71:418–427. doi: 10.1128/IAI.71.1.418-427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- GenBank http://www.ncbi.nih.gov/genomes/MICROBES/Complete.html
- Thijs G, Moreau Y, De Smet F, Mathys J, Lescot M, Rombauts S, Rouze P, De Moor B, Marchal K. INCLUSive: INtegrated Clustering, Upstream sequence retrieval and motif Sampling. Bioinformatics. 2002;18:331–332. doi: 10.1093/bioinformatics/18.2.331. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2002;30:17–20. doi: 10.1093/nar/30.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs G, Marchal K, Lescot M, Rombauts S, De Moor B, Rouze P, Moreau Y. A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J Comput Biol. 2002;9:447–464. doi: 10.1089/10665270252935566. [DOI] [PubMed] [Google Scholar]
- BioI@Sista Software: MotifLocator http://www.esat.kuleuven.ac.be/~dna/BioI/Software.html
- Thijs G. PhD thesis. Katholieke Universiteit Leuven, Faculty of Applied Sciences; 2003. Probabilistic methods to search for regulatory elements in sets of coregulated genes. [Google Scholar]
- Schneider T, Stormo G, Gold L, Ehrenfeucht J. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Marchal K, Thijs G, De Keersmaecker S, Monsieurs P, De Moor B, Vanderleyden J. Genome-specific higher-order background models to improve motif detection. Trends Microbiol. 2003;11:61–66. doi: 10.1016/S0966-842X(02)00030-6. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Development of a heuristic for the selection of alignment seeds from a list of motifs generated by Gibbs sampling ftp://www.esat.kuleuven.ac.be/pub/sista/marchal/Supplementary_information_GenBiol_2004/website/reports/heuristic.pdf
- Methodology to identify conserved sequence blocks in an alignment of intergenic sequences ftp://www.esat.kuleuven.ac.be/pub/sista/marchal/Supplementary_information_GenBiol_2004/website/reports/Validation_Alignments_221003.pdf
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nuceicl Acids Symp. 1999;41:95–98. [Google Scholar]
- The Sanger Institute http://www.sanger.ac.uk/Info/Intro
- Nelson DL, Kennedy EP. Magnesium transport in Escherichia coli. Inhibition by cobaltous ion. J Biol Chem. 1971;246:3042–3049. [PubMed] [Google Scholar]
- Snavely MD, Miller CG, Maguire ME. The mgtB Mg2+ transport locus of Salmonella typhimurium encodes a P-type ATPase. J Biol Chem. 1991;266:815–823. [PubMed] [Google Scholar]
- Bullas LR, Ryu JI. Salmonella typhimurium LT2 strains which are r- m+ for all three chromosomally located systems of DNA restriction and modification. J Bacteriol. 1983;156:471–474. doi: 10.1128/jb.156.1.471-474.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]