

Abstract
A novel fungal gene encoding the Rhizomucor miehei l-asparaginase (RmAsnase) was cloned and expressed in Escherichia coli. Its deduced amino acid sequence shared only 57% identity with the amino acid sequences of other reported l-asparaginases. The purified l-asparaginase homodimer had a molecular mass of 133.7 kDa, a high specific activity of 1,985 U/mg, and very low glutaminase activity. RmAsnase was optimally active at pH 7.0 and 45°C and was stable at this temperature for 30 min. The final level of acrylamide in biscuits and bread was decreased by about 81.6% and 94.2%, respectively, upon treatment with 10 U RmAsnase per mg flour. Moreover, this l-asparaginase was found to potentiate a lectin's induction of leukemic K562 cell apoptosis, allowing lowering of the drug dosage and shortening of the incubation time. Overall, our findings suggest that RmAsnase possesses a remarkable potential for the food industry and in chemotherapeutics for leukemia.
INTRODUCTION
The enzyme l-aspariginase (l-asparagine amidohydrolase; EC 3.5.1.1) catalyzes the hydrolysis of l-asparagine to l-aspartic acid and ammonia. The enzyme is widely distributed among plants, animals, and microorganisms (1–3). It can be used to reduce the formation of acrylamide in baked goods (4–6), and its antileukemic capacity has garnered much attention for its application in the treatment of malignancies of the lymphoid system, acute lymphoblastic leukemia, and non-Hodgkin's lymphoma (7).
As acrylamide is carcinogenic to humans, much effort has been invested in identifying possible ways of reducing its levels in foods and, thus, consumer exposure. The predominant acrylamide formation pathway is via a Maillard reaction between the amino acid asparagine and reducing sugars (8). However, most acrylamide-reducing methods limit not only acrylamide formation but also the formation of desirable Maillard products, which can have a negative impact on product taste and appearance (5). Application of l-asparaginase provides a possible alternative method for the mitigation of acrylamide formation that has little effect on the general formation of Maillard products in baked goods (5, 6).
To date, the best-known bacterial l-asparaginases are from Escherichia coli and Erwinia carotovora, and these have been employed for several decades as effective drugs for the treatment of acute lymphoblastic leukemia and leukemia lymphosarcoma (7, 9). However, these l-asparaginases can also have undesirable side effects, including anaphylaxis, pancreatitis, diabetes, leucopoenia, neurological seizures, and coagulation abnormalities that may lead to intracranial thrombosis or hemorrhage (7, 10). These side effects are considered to arise from allergic responses, and the use of l-asparaginases from different organisms might alleviate the problem (11). Hence, attempts are being made to find novel l-asparaginases or ways of enhancing the chemotherapeutic effect of this drug for the treatment of leukemia.
Though many l-asparaginases have been cloned from bacteria, very few studies have focused on fungal l-asparaginases, which may have fewer adverse side effects when used for the treatment of leukemia or lymphosarcoma (12–14). In the present work, a novel gene encoding the l-asparaginase from Rhizomucor miehei CAU432, a strain of thermophilic fungus that thrives at an optimum temperature of 50°C (15), was cloned. The heterologous expression, purification, and characterization of this recombinant l-asparaginase (RmAsnase) are described. We further investigate the application of RmAsnase for the degradation of acrylamide in baked goods and for synergistic treatment of leukemia when combined with Astragalus membranaceus lectin (AML), reported in our previous work (16).
MATERIALS AND METHODS
Strains, vectors, and reagents.
E. coli strains DH5α and BL21 were used for plasmid propagation and as a host for expression of the l-asparaginase gene, respectively. Vector pET-28a(+) was obtained from Novagen (Madison, WI). PrimeSTAR HS DNA polymerase and restriction endonucleases were purchased from TaKaRa (Tokyo, Japan). T4 DNA ligase was from New England BioLabs (Ipswich, MA). A multifunctional DNA purification kit was purchased from BioTeke (Beijing, China). 3-(4,5-Dimethylthiazo-l-2-yl)-2,5-diphenyltetrazolium bromide (MTT), d-acrylamide, and the substrates l-asparagine, d-asparagine, and l-glutamine were purchased from Sigma Chemical Company (St. Louis, MO). All other chemicals used were of analytical grade, unless otherwise stated.
Microorganism and cultivation.
The R. miehei strain used in this study, strain CAU432, has been deposited in the China General Microbiological Culture Collection Center (CGMCC; http://www.cgmcc.net/) under CGMCC accession no. 4967. For isolation of genomic DNA, this strain was cultivated at 50°C for 2 days in medium containing the following (g/liter): oat flour, 10; tryptone, 10; yeast extract, 10; MgSO4·7H2O, 0.3; FeSO4, 0.3; and CaCl2, 0.3. The mycelia were collected and ground to a fine powder under liquid nitrogen.
Cloning of the l-asparaginase gene and sequence analysis.
DNA manipulations were performed as described by Sambrook and Russell (17). Genomic DNA was isolated from R. miehei CAU432 using the cetyltrimethylammonium bromide method (18). To obtain RNA for reverse transcription-PCR (RT-PCR), cells were grown, collected, and ground as described above. Total RNA was isolated using a TRIzol kit (Invitrogen, Carlsbad, CA), and mRNA was purified using an Oligotex mRNA midikit (Qiagen, Düsseldorf, Germany).
R. miehei CAU432 genomic DNA was used as the template for subsequent PCR amplification. To clone the l-asparaginase gene, degenerate primers AsnDF and AsnDR (Table 1) were designed on the basis of the conserved blocks of amino acid residues (FVVLHGTDTM and ETFGAGNAP) of known l-asparaginases using the CODEHOP (Consensus Degenerate Hybrid Oligonucleotide Primers) algorithm (http://blocks.fhcrc.org/codehop.html). A putative homologous consensus region of the l-asparaginase gene was amplified using the degenerate primers and analyzed by sequencing the PCR products. PCR conditions were as follows: a hot start at 94°C for 5 min, followed by 10 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, with a 0.5°C decrease in annealing temperature per cycle, and then 20 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min. The PCR product was purified, ligated to the pMD18-T vector, and transformed into competent E. coli DH5α cells.
TABLE 1.
Primers used in this study
| Primer | Primer sequencea (5′ → 3′) |
|---|---|
| AsnDF | TCGTCCTGCACGGCacngayacnatg |
| AsnDR | CGTTGCCGGCGccaaangtytc |
| Asn5′GSP | AAGACTACCCCTTGCAAAGGAGGTG |
| Asn3′GSP | TGCAAGTGCGCTTAGCTTCATG |
| Asn5′NGSP | AAGGAGGTGCGAGAAAAGCG |
| Asn3′NGSP | GCGCTTAGCTTCATGTTGGAGGA |
| AsnDNAF | ATGGATTCGAGAACGACTGCTCAT |
| AsnDNAR | TTATTCTTTTCCTAGAAGTTGTGCTATCTC |
| RmAsnAF | GGGTTTCATATGGATTCGAGAACGACTGCT |
| RmAsnAR | ATTCCGCTCGAGTTCTTTTCCTAGAAGTTGTGC |
N = A, T, C, or G; Y = C or T. Restriction enzyme sites incorporated into primers are underlined. Lowercase nucleotides represent degenerate cores in CODEHOP primers.
The full-length cDNA sequence of l-asparaginase was obtained by 5′ and 3′ rapid amplification of cDNA ends (RACE) using a BD SMART RACE cDNA amplification kit (Clontech, Palo Alto, CA). The PCR conditions for RACE were a hot start at 94°C for 5 min, followed by 30 cycles of 30 s at 94°C, 30 s at 68°C, and 1 min at 72°C and a final step of 10 min at 72°C. PCR was performed with the following primer pairs (Table 1): Asn5′GSP and universal primer A mix for the first PCR, followed by a nested PCR with primers Asn5′NGSP and nested universal primer A (BD Biosciences, Franklin, NJ) for 5′ RACE. Similarly, 3′ RACE was performed with primer Asn3′GSP and universal primer A mix, followed by nested PCR with Asn3′NGSP and nested universal primer A. The obtained PCR product was purified, cloned, and sequenced. The 5′ and 3′ flanking sequences obtained by 5′ and 3′ RACE were assembled with those of the consensus region to form the full-length cDNA sequence containing the open reading frame (ORF) of the l-asparaginase gene. This l-asparaginase cDNA sequence was subjected to BLAST analysis. To amplify this region from the genomic DNA of R. miehei CAU432, the same PCR conditions were used with the specific primers AsnDNAF and AsnDNAR (Table 1). The purified PCR products were ligated with the pMD18-T vector and transformed into E. coli DH5α cells for sequencing.
Nucleotide and deduced amino acid sequences were analyzed using DNAMAN software (Lynnon Biosoft). BLAST analysis was performed at the NCBI server (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The amino acid sequences were aligned using the ClustalW program (ftp://ftp-igbmc.u-strasbg.fr/pub/ClustalW/). The signal peptide was analyzed by the SignalP (version 4.0) server (http://www.cbs.dtu.dk/services/SignalP/). Search analysis of conserved domain and signature sequences was carried out using the ScanProsite program (http://prosite.expasy.org/scanprosite/).
Heterologous expression of the gene in E. coli.
The ORF encoding RmAsnase was amplified from the cDNA of R. miehei CAU432 by PCR with two primers: RmAsnAF and RmAsnAR (Table 1). NdeI and XhoI sites (Table 1, underlined) were added to the forward and reverse primers, respectively. PCR conditions were as follows: a hot start at 94°C for 5 min and 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min, followed by 1 cycle of 72°C for 10 min. The purified PCR product was digested with NdeI and XhoI; subcloned into the pET-28a(+) vector, which had been digested with the same restriction enzymes; and transformed into competent E. coli BL21(DE3) cells for protein expression. A single colony of E. coli BL21 harboring the RmAsnase gene in pET-28a(+) was inoculated into terrific broth (TB) medium containing kanamycin (50 μg/ml) and incubated on a rotary shaker (200 rpm, 37°C) to an optical density at 600 nm of about 3.0. E. coli BL21 with an empty pET-28a(+) vector was used as a native control. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce enzyme expression, and the culture was then grown at 30°C for 12 h on a rotary shaker at 200 rpm.
Purification of recombinant l-asparaginase.
E. coli cells were harvested by centrifugation and resuspended in Tris-HCl buffer (50 mM, pH 8.5, 150 mM NaCl). Then, the cells were sonicated and centrifuged at 10,000 × g. The clear supernatant was collected and applied to an Ni-IDA (nickel-iminodiacetic acid) column (1 cm by 5 cm; GE Life Sciences) preequilibrated with buffer A (50 mM Tris-HCl, pH 8.5, 150 mM NaCl, 20 mM imidazole). Nonadsorbed protein was washed off with 15 column volumes (CV) of buffer A, followed by 5 CV of buffer B (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 500 mM NaCl, 50 mM imidazole) at a flow rate of 1.0 ml/min. Bound proteins were eluted with buffer C (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 200 mM imidazole). The protein eluted as a single peak, and the fractions showing l-asparaginase activity were pooled, concentrated, and buffer exchanged in 50 mM Tris-HCl (pH 8.0) using a 10-kDa-molecular-mass-cutoff ultrafiltration membrane. A sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) assay was carried out using a 12.5% running gel according to the method of Laemmli (19). After denaturation, the purified enzyme was digested with trypsin and then identified using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS).
Enzyme activity assay and protein determination.
To determine l-asparaginase activity, 50 μl of suitably diluted enzyme was incubated with 40 mM l-asparagine (or l-glutamine for l-glutaminase activity) in 200 μl of 50 mM phosphate buffer (pH 7.0) at 45°C for 10 min. The reaction was terminated by adding 50 ml 1.5 M trichloroacetic acid solution. The amount of ammonia liberated was determined by addition of Nessler's reagent (20) and spectrophotometric measurement at 450 nm. One unit of enzyme activity was defined as the amount of enzyme that liberated 1 μmol ammonia per minute under the described conditions. The protein concentration was determined by the method of Lowry et al. (21) using bovine serum albumin as the standard. The specific activity was expressed as U/mg protein.
Molecular mass determination.
The denatured RmAsnase was analyzed by SDS-PAGE in a 12.5% gel. The native molecular mass was estimated by use of a Superdex-300 gel filtration column (1 cm by 40 cm) in 50 mM phosphate buffer (pH 7.4) containing 150 mM NaCl at a flow rate of 0.3 ml/min. The molecular mass standards were alcohol dehydrogenase (150 kDa), phosphorylase b (97.2 kDa), chicken egg albumin (44.3 kDa), and α-chymotrypsinogen (25.6 kDa).
Biochemical characterization.
The optimal pH of RmAsnase was assayed at 30°C with 40 mM l-asparagine in 50 mM the following buffers: citrate buffer (pH 3.0 to 6.0), morpholineethanesulfonic acid (MES) buffer (pH 5.5 to 6.5), phosphate buffer (pH 6.0 to 8.0), Tris-HCl buffer (pH 7.0 to 9.0), and 2-(cyclohexylamino)ethanesulfonic acid (CHES) buffer (pH 8.5 to 10.0). The effect of pH on l-asparaginase stability was examined by incubating the purified enzyme in the different buffers at 30°C for 30 min. The residual activity of the treated samples was determined by the standard assay described above. The optimal temperature of the recombinant l-asparaginase was determined by performing the standard assay at temperatures of 20 to 80°C in 50 mM phosphate buffer (pH 7.0). Thermal stability was measured by assessing the residual enzyme activity after incubation of the enzyme at different temperatures for 30 min. The influence of metal ions (2 mM), EDTA, β-mercaptoethanol, and SDS in phosphate buffer (pH 7.0) on the l-asparaginase activity of purified RmAsnase was studied at 30°C for 30 min. The residual activity was assayed by using l-asparagine and comparison with the activity of the control (the enzyme without the addition of reagents).
Substrate specificity was determined using different compounds containing an amide bond. l-Asparaginase activity was determined using 40 mM substrate in 50 mM phosphate buffer (pH 7.0) at 45°C for 10 min and measuring the amount of ammonium ion using Nessler's reagent (20). One unit was defined as the amount of enzyme producing 1 μmol of ammonium ion per minute under these conditions. The kinetic parameters were determined by measuring the enzyme activities with six different substrate concentrations in 50 mM phosphate buffer (pH 7.0) at 45°C for 10 min. Vmax, Km, and kcat values were calculated using the Grafit program.
Reduction of acrylamide in baked goods.
Acrylamide formation was evaluated in biscuits as described by Anese et al. (22). Briefly, short-dough samples were prepared using cake flour (50 g), tap water (12.5 ml), sucrose (12.5 g), shortening (8 g), salt (0.6 g), glucose (0.65 g), sodium bicarbonate (0.25 g), and ammonium bicarbonate (0.5 g). The glucose, sodium bicarbonate, and ammonium bicarbonate were dissolved in water, and different concentrations of RmAsnase (in 50 mM phosphate buffer, pH 7.0) were added. The rest of the ingredients were mixed in, and the dough was kneaded and allowed to rest for 30 min at 4°C. The dough was rolled out to a 0.3-cm thickness, and 7-cm-diameter circles were cut out and left in a thermostatic cell at 45°C for 30 min. The samples were baked in a 260°C oven for 10 min.
Bread was prepared following the procedure described by Jiang et al. (23). The bread dough consisted of wheat flour (150 g), dry Saccharomyces cerevisiae yeast (3.0 g), salt (2.25 g), sugar (7.5 g), vegetable oil (4.5 g), and water (90 ml). The purified RmAsnase (in 50 mM phosphate buffer, pH 7.0) was incorporated into the flour at levels of 0.5 to 100 U/g flour. Then, the dough was mixed for 4 min, allowed to rest for 10 min, divided (into 50-g portions), kneaded, and allowed to rest again (15 min). Each dough portion was rolled out and proofed to three times its initial volume (at 40°C, 80% relative humidity). Loaves were baked at 200°C for 15 min.
All samples were homogenized and defatted with hexane. d-Acrylamide was added as an internal standard to 2 g of the defatted samples. Acrylamide was extracted from each sample by acetonitrile and concentrated by vacuum evaporation. This extract was then dissolved in deionized water and analyzed by gas chromatography (GC) for acrylamide using a C18 column with a flame ionization detector and helium gas as the carrier.
Cell culture, viability, and apoptosis assay.
Human leukemia cell lines Jurkat, U937, and K562 were obtained from the Chinese Academy of Medical Sciences (Beijing, China). Cells were maintained in RPMI 1640 medium containing 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell cultures were incubated at 37°C in a humidified atmosphere with 5% CO2. Cell viability was measured by the MTT method as described previously, with some modifications (16). Briefly, cells were dispensed in 96-well flat-bottom microtiter plates at a density of 1 × 105 cells/ml. After 24 h of incubation, they were treated with different concentrations of RmAsnase alone or in combination with AML for the indicated time periods. Cell apoptosis was analyzed using annexin V labeling and propidium iodide (PI) staining, followed by flow cytometry. After incubation, cells were treated ahead of the flow cytometric studies (Becton, Dickinson, Franklin Lakes, NJ) following a previously described method (16).
Nucleotide sequence accession number.
The l-asparaginase cDNA sequence was deposited in the GenBank nucleotide sequence database under accession no. KF290772.
RESULTS
Cloning and sequence analysis of an l-asparaginase gene from R. miehei.
On the basis of the conserved amino acid sequences of known l-asparaginase genes, a 622-bp fragment was amplified with degenerate primers AsnDF and AsnDR using genomic DNA from R. miehei CAU432 as the template. The 5′ and 3′ flanking regions of the fragment, which were approximately 1,000 and 1,600 bp, respectively, were obtained by RACE. The two flanking regions were then assembled with the core fragment to generate a 2,156-bp cDNA sequence containing a putative full-length ORF of 2,049 bp. The translated protein contained 682 amino acid residues with a predicted molecular mass of 75,316 Da and a theoretical pI of 6.76. The nucleotide and deduced amino acid sequences of the full-length cDNA and flanking regions of the gene encoding RmAsnase are shown in Fig. 1. Comparison of the cDNA sequence with the genomic sequences revealed six introns in the coding region. The N-terminal region contained no predicted signal peptide.
FIG 1.

Nucleotide and deduced amino acid sequences of the full-length cDNA and flanking regions of the RmAsnase gene. The translational initiation codon (ATG) and termination codon (TAA) are boxed. Six intron sequences are shown in lowercase letters with dotted underlining. A poly(A)+ sequence is double underlined. Conceptual translation of the ORF to the amino acids is shown in one-letter code below the respective codon. *, the stop codon.
Expression and purification of the recombinant l-asparaginase.
RmAsnase was expressed in E. coli BL21 as a soluble intracellular enzyme. The results of comparative analysis by SDS-PAGE and analysis of asparaginase activity are shown in Table S1 and Fig. S1 in the supplemental material, respectively. The specificity activity of the crude enzyme from E. coli with an empty vector was found to be only 8.8 U/mg. The activity of asparaginase from E. coli with an empty vector was negligible compared to that of the crude enzyme from E. coli BL21 harboring the RmAsnase gene in the pET-28a(+) vector (780 U/mg). RmAsnase was purified 2.6-fold by Ni-IDA chromatography with an overall yield of 48.8% and a specific activity of 1,985 U/mg (Table 2). The purified enzyme migrated on the SDS-polyacrylamide gel as a single, homogeneous band of 72 kDa, which matches its predicted molecular mass of 75,316 Da (Fig. 2). The native molecular mass of RmAsnase was determined by gel filtration to be 133.5 kDa, indicating that the enzyme is a homodimer (data not shown). Eleven internal peptides from MALDI-TOF MS were randomly selected from RmAsnase and were as follows: TTAHVPIYDNAAVEHQLRADRDEMLAPDFSR, HTPEHGYIPLPNYLAQSLAR, FHDPSHGFLSRSSSQENHADGVTLKEDFTR, TVIITGSQVPLTEVR, WPLVLRPTHIAK, LFPGINESTVRAFLAPPLQGVVLETYGAGNAPAR, EACDRGVVIVNCTQCRKGLVTDSYATGR, SQLLLDIFANISAR, DGHIAIVEYLLLHGASVHVRDRWGHTPLFVAVVGKHAQVVSMLRRAGAHLSVNEQSDMGPAWLK, IALEAGWPVNWAEPVEGR, and WGFTIIDK. These peptide sequences matched the deduced amino acid sequence of RmAsnase, confirming that the purified enzyme was indeed RmAsnase. The quantitative results and characterization of RmAsnase were carried out with electrophoresis-grade enzyme.
TABLE 2.
Summary of RmAsnase purification stepsa
| Purification step | Total activity (U) | Amt of protein (mg) | Sp act (U/mg) | Purification factor (fold) | Recovery (%) |
|---|---|---|---|---|---|
| Crude enzyme | 43,506.4 | 35.8 | 779.7 | 1 | 100 |
| Ni-IDA | 21,237.4 | 10.7 | 1,984.8 | 2.6 | 48.8 |
Enzymatic reactions were carried out for 10 min at 45°C in 50 mM phosphate buffer (pH 7.0), as described in the text.
FIG 2.
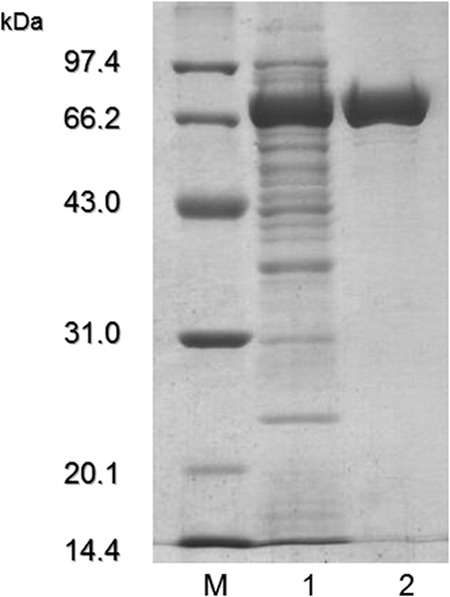
SDS-PAGE analysis of RmAsnase expressed in E. coli. Lane M, low-molecular-mass protein markers; lane 1, crude lysate; lane 2, purified RmAsnase.
Biochemical characterization of RmAsnase.
RmAsnase displayed maximal l-asparaginase activity at pH 7.0 in 50 mM sodium phosphate buffer (Fig. 3a). It was fairly stable over a pH range of 4.0 to 8.0, retaining more than 80% of its activity (Fig. 3b). The enzyme showed optimal activity at 45°C (Fig. 3c), and no activity loss was detected after incubation of the enzyme at 45°C for 30 min (Fig. 3d). Sn2+, K+, Ni2+, and Na+ had only slight inhibitory effects on RmAsnase activity (reducing it to 87, 92, 85, and 91% of its activity without a metal ion, respectively), whereas Mg2+ (28%), Ba2+ (30%), Cr2+ (37%), Fe3+ (41%), Ag+ (42%), Co2+ (69%), and, especially, Cu2+ (13%), Zn2+ (17%), and Hg2+ (24%) strongly inhibited the l-asparaginase activity. Ca2+ and Mn2+ significantly enhanced RmAsnase activity (to 144% and 183% of its activity without a metal ion, respectively), in contrast to their inhibition of many other l-asparaginases (11, 24). Both SDS (56%) and β-mercaptoethanol (55%) were strong inhibitors, as was EDTA (24%), suggesting that the enzyme is dependent on metal ions or disulfide bonds for its activity.
FIG 3.

Optimum pH (a), pH stability (b), optimum temperature (c), and thermostability (d) of RmAsnase. The influence of pH on l-asparaginase activity was determined at 45°C using different buffers: citrate (♢), MES (■), phosphate (▲), Tris-HCl (×), and CHES (○). The temperature profile was measured at different temperatures in 50 mM phosphate buffer (pH 7.0). Data represent means ± SDs (n = 3).
To demonstrate the substrate specificity of RmAsnase, its enzymatic activity was determined in terms of the amount of ammonium liberated from several relevant amide compounds. The enzyme showed the highest specificity for l-asparagine (1,985 U/mg), but it showed negligible activity with both d-asparagine (22 U/mg) and l-glutamine (36 U/mg) as the substrates. The Vmax, Km, and kcat values of RmAsnase for l-asparagine were 3,380.0 ± 133.4 μmol/min mg, 0.0253 ± 0.0024 mg/ml, and 676 ± 26.7 s−1, respectively (data not shown).
Reduction of acrylamide formation in baked goods by RmAsnase.
Applied trials were performed in biscuits and bread to evaluate RmAsnase's potential as an additive for general acrylamide mitigation in baked goods (Fig. 4). A clear, dose-dependent reduction in acrylamide levels was observed in both final products following addition of RmAsnase. More than 40% of the acrylamide in bread was eliminated when a low concentration of RmAsnase was added (0.5 U/g flour). Over 90% acrylamide formation was eliminated when the concentration of RmAsnase was increased to 10 U/g flour. In the biscuits, samples treated with the same amounts of RmAsnase showed only approximately 15% and 80% decreases in acrylamide, respectively.
FIG 4.
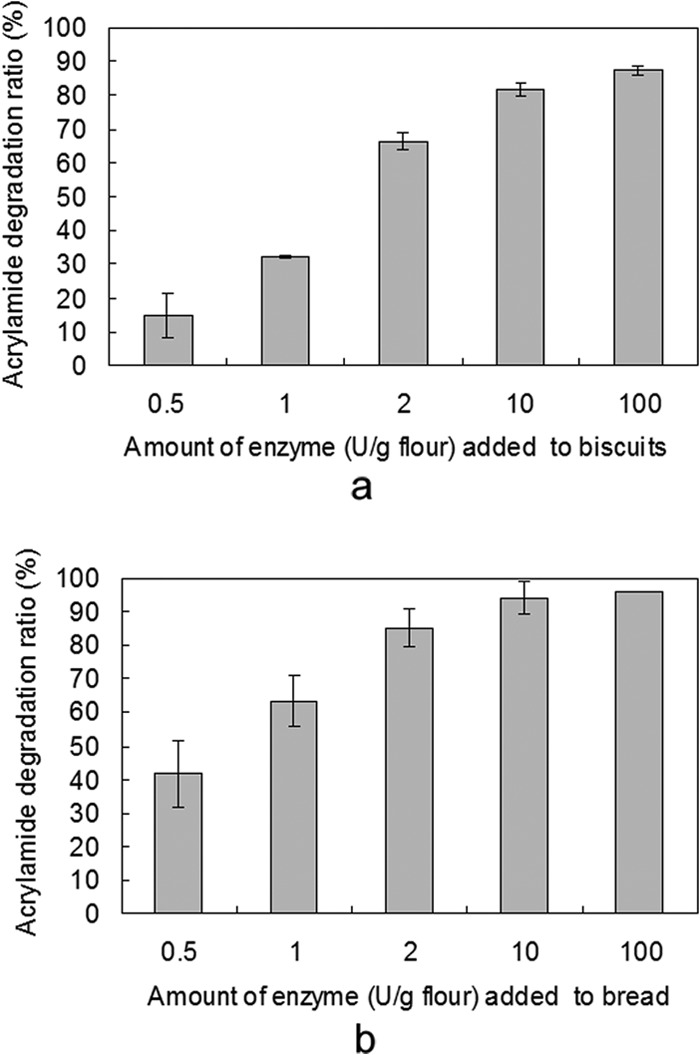
Reduction of acrylamide content by RmAsnase in baked biscuits (a) and bread (b). Biscuits or bread dough was pretreated with different amounts of RmAsnase or equal amounts of phosphate buffer (50 mM, pH 7.0) as a control. Data represent means ± SDs (n = 3).
Inhibition of leukemia cell proliferation by RmAsnase alone or combined with AML.
The cytotoxicity of RmAsnase to several human leukemia cell lines was demonstrated by MTT assay. Of the three cell types tested, the antiproliferative activity of RmAsnase, after 48 h of incubation, was the most efficient against Jurkat cells (more than 60%) (Fig. 5a). Clinically, the high dosages and long periods involved in l-asparaginase treatments eventually cause hypersensitivity, anaphylaxis, allergic reactions, and other toxic reactions (25, 26). We therefore performed a multidrug trial with AML, a plant lectin from Astragalus membranaceus, in this study. As shown in Fig. 5b, AML maintained its significant antiproliferative activity in K562 cells when combined with RmAsnase. In fact, these two proteins showed a synergistic effect, with stronger proliferation inhibition against Jurkat and U937 cells being obtained with the combination than with either drug alone.
FIG 5.

Antiproliferative activity of RmAsnase toward leukemia cells and synergistic treatment with RmAsnase and AML. Cells were incubated with different concentrations of RmAsnase for 48 h (a) or treated with 40 mU/ml RmAsnase alone or combined with 40 μg/ml AML for 48 h (b). Cell viability was checked by MTT assay. Results are representative of those from experiments performed in triplicate. In panel a, the leukemia cell lines were K562 (⧫), U937 (■), and Jurkat (▲).
Apoptosis induced by synergistic treatment with RmAsnase and AML.
To delineate the mechanism governing the synergistic antiproliferative activity of RmAsnase and AML, apoptosis of K562 cells treated by both of these compounds was determined by flow cytometry. As shown in Fig. 6a, incubation with 5 and 20 μg/ml AML resulted in 9.3% and 28.9% apoptotic cells, respectively, suggesting a dose-dependent effect. Although there were very few apoptotic cells when only RmAsnase was added, the apoptotic cell ratio was 20.8% in the cells treated with 5 μg/ml AML and 10 mU/ml RmAsnase. Moreover, most of the apoptotic cells in the early stages of the coincubation emerged 24 h earlier than those in the treatment with AML alone (Fig. 6b). It is therefore suggested that RmAsnase not only allows the dosage of AML to be decreased but also accelerates the induction of K562 cell apoptosis. The effect of RmAsnase on caspase-dependent AML-induced apoptosis was further investigated using a pan-caspase inhibitor, z-VAD-fmk (Medical and Biological Laboratories Company, Nagoya, Japan). The levels of inhibition of apoptosis induced by AML only or by AML combined with RmAsnase were very similar, suggesting that RmAsnase does not change the mechanism of intracellular apoptosis, which is mainly induced by AML (Fig. 6c).
FIG 6.
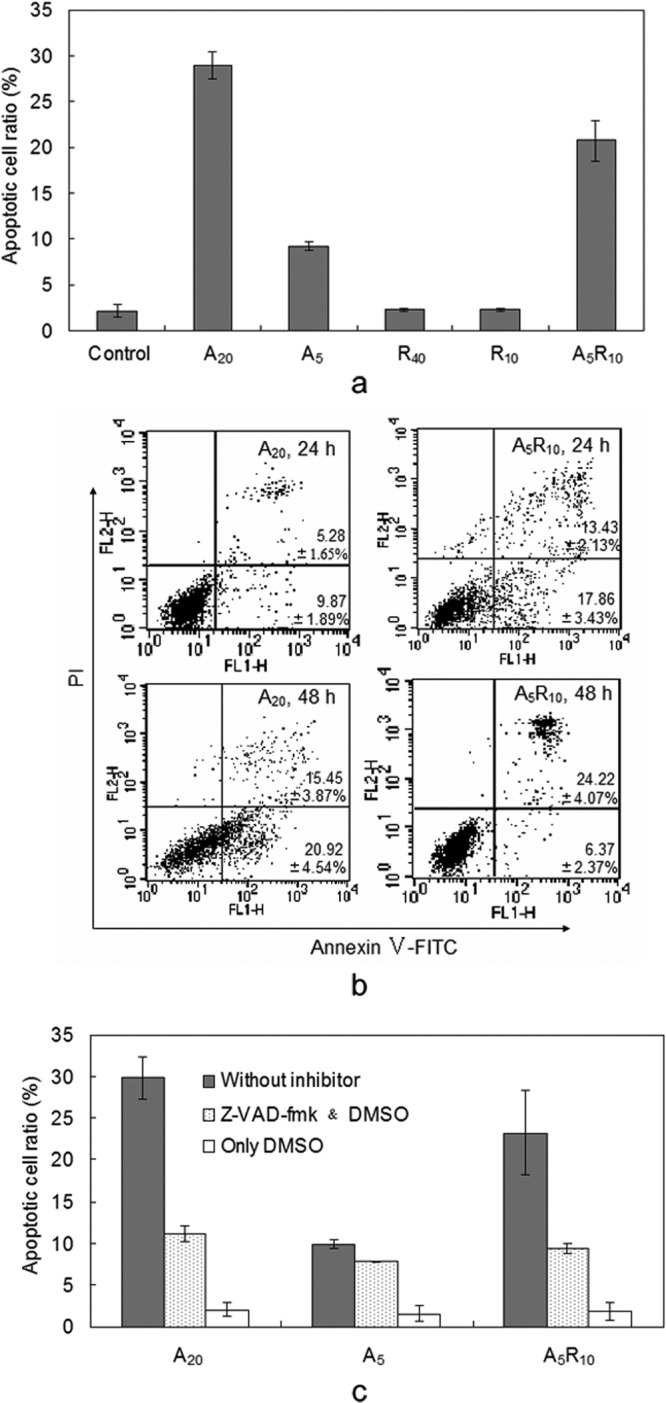
Mechanism of enhancement of antiproliferative activity of AML by RmAsnase in K562 cells. (a) Apoptosis of K562 cells was induced by a lower dose of AML when combined with RmAsnase; (b) apoptosis of K562 cells induced by AML was accelerated by RmAsnase; (c) effect of a caspase inhibitor on the synergistic induction of apoptosis of K562 cells by AML or AML combined with RmAsnase. A20 and A5, treatment with AML at 20 and 5 μg/ml, respectively; R40 and R10, treatment with RmAsnase at 40 and 10 mU/ml, respectively; A5R10, cotreatment with AML at 5 μg/ml and RmAsnase at 10 mU/ml; FITC, fluorescein isothiocyanate; DMSO, dimethyl sulfoxide. Apoptosis was measured using a flow cytometer. Results are representative of those from experiments performed in triplicate.
DISCUSSION
In the present study, the hypothesized gene encoding the l-asparaginase RmAsnase was successfully cloned from Rhizomucor miehei. On the basis of a BLAST homology search, the deduced amino acid sequence of RmAsnase showed 57% identity to the l-asparaginase from Vibrio cholerae (PDB accession no. 2OCD) and 56% identity to the l-asparaginases from E. coli (PDB accession no. 2P2D) (27) and Yersinia pestis (PDB accession no. 3NTX), all of which are type I bacterial l-asparaginases. The putative conserved domains of the amino acid sequence of RmAsnase could be divided into two parts including l-asparaginase and ankyrin (ANK) repeats, which showed domain architectures similar to those of an asparagine amidohydrolase from the filarial parasite Dirofilaria immitis (Q9U518) (28) and an l-asparaginase from the filamentous fungus Neurospora crassa (XP_961114).
According to the previous studies, two isozymes of l-asparaginase have been produced by Escherichia coli. Type I l-asparaginase from E. coli (EcAI) is located in the cytosol, whereas type II l-asparaginase from E. coli (EcAII) is located in the periplasmic region of the bacterium (29). Actually, l-asparaginase type I (EcAI) is a dimer of two intimate dimers, and l-asparaginase type II (EcAII) is a tetramer of identical subunits of 35.6 kDa (27, 29). Both of the two l-asparaginases were inactive as a homodimer of 135 kDa. Unlike bacterial l-asparaginases, which are found as tetramers of identical subunits with molecular masses in the range of 140 to 160 kDa, the purified RmAsnase homodimer's molecular mass was higher than that of some other fungal l-asparaginases (1, 5). The specific activity of RmAsnase was similar to that of l-asparaginase from Pectobacterium carotovorum (2,020.9 U/mg) (11) but much higher than the specific activities of most bacterial and fungal l-asparaginases (14, 30–32). The optimum pH and temperature of RmAsnase are similar to those reported for many l-asparaginases (2, 31, 33). Moreover, the enzyme showed negligible activity with l-glutamine as the substrate. Contamination with glutaminase activity is one of the causes of l-asparaginase toxicity to leukemia patients (6, 7), with only a few reported l-asparaginases showing little or no such activity (6, 11, 24). In view of the biochemical characterizations, RmAsnase could potentially serve for acrylamide reduction when added to baked goods or as an alternative drug for the treatment of leukemia (14, 31).
A clear, dose-dependent reduction in acrylamide levels was observed in the final products of bread and biscuits following addition of RmAsnase. However, it might have been due to the different processing protocols, particularly the different incubation times for these two baked goods, resulting in RmAsnase being more efficient at reducing the acrylamide content in bread than biscuits. Nevertheless, RmAsnase's ability to reduce acrylamide formation in biscuits was equivalent to that of commercial l-asparaginase (22) or l-asparaginase from Aspergillus oryzae (5).
Unlike normal cells, malignant cells are unable to synthesize endogenous l-asparagine, and a lack of this amino acid leads to their death (9). The general medical approach to leukemia therapy is therefore based on this metabolic defect in l-asparagine synthesis in some malignant cells (34). Thus, l-asparaginase has garnered interest for its beneficial pharmacological effect in the treatment of some types of human leukemias. In particular, l-asparaginases obtained from bacteria such as E. coli and Erwinia chrysanthemi have been used clinically to treat leukemia for several decades now (7, 9). However, the l-asparaginases from these two bacterial species have side effects, which are attributed in part to the presence of l-glutaminase activity or allergic responses to l-asparaginases from these sources (6, 11). Thus, l-asparaginases with low l-glutaminase activity or from different sources are of interest (7, 35). RmAsnase, the fungal l-asparaginase from R. miehei, showed antiproliferative activity against cells from several human leukemia cell lines and displayed very low l-glutaminase activity. Thus, RmAsnase may also be useful as an alternative to bacterial l-asparaginases in leukemia treatment (2, 6, 11, 32, 35).
Used alone, l-asparaginase is an effective drug for leukemia, especially acute lymphoblastic leukemia. However, resistance to the drug and side effects are observed in patients with relapses (9). Thus, agents that enhance the effect of l-asparaginase or overcome chemoresistance to the drug are needed for clinical treatment. In our previous work, a plant lectin from Astragalus membranaceus (AML) was shown to induce caspase-dependent apoptosis in human K562 cells (16). Here, we show that use of the fungus-originated RmAsnase not only allows a decrease in the AML dosage but also accelerates apoptosis of K562 cells. This result is similar to that obtained with another natural anticancer ingredient, curcumin, which also induces caspase-dependent apoptosis in leukemia cells and potentiates the antitumor activity of l-asparaginase (26). Some other drugs studied in multiagent regimens with l-asparaginases include vincristine, prednisone, and lomustine (25, 36, 37). To the best of our knowledge, this is the first trial to evaluate the antileukemia capacity of l-asparaginase combined with a lectin in vitro. Our results may help develop a new type of drug for application in combination chemotherapy for leukemia.
In conclusion, a novel fungal gene encoding the l-asparaginase from R. miehei (RmAsnase) was cloned and expressed in E. coli. The homodimer enzyme exhibited high specific activity and thermal and pH properties conducive to its use to reduce acrylamide levels in baked goods. RmAsnase displayed very low l-glutaminase activity and showed significant antiproliferative activity against several types of human leukemia cells. This l-asparaginase was further found to lessen the drug dosage and accelerate the apoptosis induced by a lectin in K562 cells when the two were combined. Thus, RmAsnase is suggested to have great potential for the food industry and for pharmaceutical exploitation in the treatment of leukemia.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the National Science Fund for Distinguished Young Scholars (no. 31325021) and the Chinese Universities Scientific Fund (no. 2012YJ107).
Footnotes
Published ahead of print 20 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03523-13.
REFERENCES
- 1.Eisele N, Linke D, Bitzer K, Na'amnieh S, Nimtz M, Berger RG. 2011. The first characterized asparaginase from a basidiomycete, Flammulina velutipes. Bioresour. Technol. 102:3316–3321. 10.1016/j.biortech.2010.10.098 [DOI] [PubMed] [Google Scholar]
- 2.Oza VP, Parmar PP, Patel DH, Subramanian RB. 2011. Cloning, expression and characterization of l-asparaginase from Withania somnifera L. for large scale production. 3 Biotech 1:21–26. 10.1007/s13205-011-0003-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahu MK, Sivakumar K, Poorani E, Thangaradjou T, Kannan L. 2007. Studies on l-asparaginase enzyme of actinomycetes isolated from estuarine fishes. J. Environ. Biol. 28:465–474 [PubMed] [Google Scholar]
- 4.Anese M, Quarta B, Peloux L, Calligaris S. 2011. Effect of formulation on the capacity of l-asparaginase to minimize acrylamide formation in short dough biscuits. Food Res. Int. 44:2837–2842. 10.1016/j.foodres.2011.06.025 [DOI] [Google Scholar]
- 5.Hendriksen HV, Kornbrust BA, Ostergaard PR, Stringer MA. 2009. Evaluating the potential for enzymatic acrylamide mitigation in a range of food products using an asparaginase from Aspergillus oryzae. J. Agric. Food Chem. 57:4168–4176. 10.1021/jf900174q [DOI] [PubMed] [Google Scholar]
- 6.Mahajan RV, Saran S, Kameswaran K, Kumar V, Saxena RK. 2012. Efficient production of l-asparaginase from Bacillus licheniformis with low-glutaminase activity: optimization, scale up and acrylamide degradation studies. Bioresour. Technol. 125:11–16. 10.1016/j.biortech.2012.08.086 [DOI] [PubMed] [Google Scholar]
- 7.Duval M, Suciu S, Ferster A, Rialland X, Nelken B, Lutz P, Benoit Y, Robert A, Manel AM, Vilmer E, Otten J, Philippe N. 2002. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children's Leukemia Group phase 3 trial. Blood 99:2734–2739. 10.1182/blood.V99.8.2734 [DOI] [PubMed] [Google Scholar]
- 8.Mottram DS, Wedzicha BL, Dodson AT. 2002. Acrylamide is formed in the Maillard reaction. Nature 419:448–449. 10.1038/419448a [DOI] [PubMed] [Google Scholar]
- 9.Narta UK, Kanwar SS, Azmi W. 2007. Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. Hematol. 61:208–221. 10.1016/j.critrevonc.2006.07.009 [DOI] [PubMed] [Google Scholar]
- 10.Geuenich S, Haberl C, Egger D, Kaspers U, Hültner L, Wilmanns W, Denzlinger C. 1998. Induction of leukotriene production by bleomycin and asparaginase in mast cells in vitro and in patients in vivo. Biochem. Pharmacol. 55:447–453. 10.1016/S0006-2952(97)00481-4 [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Venkata Dasu V, Pakshirajan K. 2011. Purification and characterization of glutaminase-free l-asparaginase from Pectobacterium carotovorum MTCC 1428. Bioresour. Technol. 102:2077–2082. 10.1016/j.biortech.2010.07.114 [DOI] [PubMed] [Google Scholar]
- 12.Ferrara MA, Severino NMB, Valente RH, Perales J, Bon EPS. 2010. High-yield extraction of periplasmic asparaginase produced by recombinant Pichia pastoris harbouring the Saccharomyces cerevisiae ASP3 gene. Enzyme Microb. Technol. 47:71–76. 10.1016/j.enzmictec.2010.05.001 [DOI] [Google Scholar]
- 13.Sarquis MI, Oliveira EM, Santos AS, Costa GL. 2004. Production of l-asparaginase by filamentous fungi. Mem. Inst. Oswaldo Cruz 99:489–492. 10.1590/S0074-02762004000500005 [DOI] [PubMed] [Google Scholar]
- 14.Shrivastava A, Khan AA, Shrivastav A, Jain SK, Singhal PK. 2012. Kinetic studies of l-asparaginase from Penicillium digitatum. Prep. Biochem. Biotechnol. 42:574–581. 10.1080/10826068.2012.672943 [DOI] [PubMed] [Google Scholar]
- 15.Katrolia P, Jia HY, Yan QJ, Song S, Jiang ZQ, Xu HB. 2012. Characterization of a protease-resistant alpha-galactosidase from the thermophilic fungus Rhizomucor miehei and its application in removal of raffinose family oligosaccharides. Bioresour. Technol. 110:578–586. 10.1016/j.biortech.2012.01.144 [DOI] [PubMed] [Google Scholar]
- 16.Huang LH, Yan QJ, Kopparapu NK, Jiang ZQ, Sun Y. 2012. Astragalus membranaceus lectin (AML) induces caspase-dependent apoptosis in human leukemia cells. Cell Prolif. 45:15–21. 10.1111/j.1365-2184.2011.00800.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 18.Lodhi MA, Ye GN, Weeden NF, Reisch BI. 1994. A simple and efficient method for DNA extractions from grape vine cultivars and Vitis species. Plant Mol. Biol. Rep. 12:6–13. 10.1007/BF02668658 [DOI] [Google Scholar]
- 19.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 20.Wade HE, Phillips BP. 1971. Automated determination of bacterial asparaginase and glutaminase. Anal. Biochem. 44:189–199. 10.1016/0003-2697(71)90360-5 [DOI] [PubMed] [Google Scholar]
- 21.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 22.Anese M, Quarta B, Frias J. 2011. Modelling the effect of asparaginase in reducing acrylamide formation in biscuits. Food Chem. 126:435–440. 10.1016/j.foodchem.2010.11.007 [DOI] [Google Scholar]
- 23.Jiang ZQ, Li XT, Yang SQ, Li LT, Tan SS. 2005. Improvement of the breadmaking quality of wheat flour by the hyperthermophilic xylanase B from Thermotoga maritima. Food Res. Int. 38:37–43. 10.1016/j.foodres.2004.07.007 [DOI] [Google Scholar]
- 24.Moreno-Enriquez A, Evangelista-Martinez Z, Gonzalez-Mondragon EG, Calderon-Flores A, Arreguin R, Perez-Rueda E, Huerta-Saquero A. 2012. Biochemical characterization of recombinant l-asparaginase (AnsA) from Rhizobium etli, a member of an increasing rhizobial-type family of l-asparaginases. J. Microbiol. Biotechnol. 22:292–300. 10.4014/jmb.1107.07047 [DOI] [PubMed] [Google Scholar]
- 25.Tallman MS, Appelbaum FR, Amos D, Goldberg RS, Livingston RB, Mortimer J, Weiden PL, Thomas ED. 1987. Evaluation of intensive postremission chemotherapy for adults with acute nonlymphocytic leukemia using high-dose cytosine arabinoside with l-asparaginase and amsacrine with etoposide. J. Clin. Oncol. 5:918–926 [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Geng QR, Wang L, Lu Y. 2012. Curcumin potentiates antitumor activity of l-asparaginase via inhibition of the AKT signaling pathway in acute lymphoblastic leukemia. Leukemia Lymphoma 53:1376–1382. 10.3109/10428194.2011.649478 [DOI] [PubMed] [Google Scholar]
- 27.Yun MK, Nourse A, White SW, Rock CO, Heath RJ. 2007. Crystal structure and allosteric regulation of the cytoplasmic Escherichia coli l-asparaginase I. J. Mol. Biol. 369:794–811. 10.1016/j.jmb.2007.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuji N, Morales TH, Ozols VV, Carmody AB, Chandrashekar R. 1999. Identification of an asparagine amidohydrolase from the filarial parasite Dirofilaria immitis. Int. J. Parasitol. 29:1451–1455. 10.1016/S0020-7519(99)00087-9 [DOI] [PubMed] [Google Scholar]
- 29.Swain AL, Jaskolski M, Housset D, Rao JK, Wlodawer A. 1993. Crystal structure of Escherichia coli l-asparaginase, an enzyme used in cancer therapy. Proc. Natl. Acad. Sci. U. S. A. 90:1474–1478. 10.1073/pnas.90.4.1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito K, Matsushima K, Koyama Y. 2012. Gene cloning, purification, and characterization of a novel peptidoglutaminase-asparaginase from Aspergillus sojae. Appl. Environ. Microbiol. 78:5182–5188. 10.1128/AEM.00765-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotzia GA, Labrou NE. 2007. l-Asparaginase from Erwinia chrysanthemi 3937: cloning, expression and characterization. J. Biotechnol. 127:657–669. 10.1016/j.jbiotec.2006.07.037 [DOI] [PubMed] [Google Scholar]
- 32.Pokrovskaya MV, Aleksandrova SS, Pokrovsky VS, Omeljanjuk NM, Borisova AA, Anisimova NY, Sokolov NN. 2012. Cloning, expression and characterization of the recombinant Yersinia pseudotuberculosis l-asparaginase. Protein Expr. Purif. 82:150–154. 10.1016/j.pep.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 33.Mohan Kumar N, Manonmani HK. 2013. Purification, characterization and kinetic properties of extracellular l-asparaginase produced by Cladosporium sp. World J. Microbiol. Biotechnol. 29:577–587. 10.1007/s11274-012-1213-0 [DOI] [PubMed] [Google Scholar]
- 34.Broome JD. 1981. l-Asparaginase: discovery and development as a tumor-inhibitory agent. Cancer Treat. Rep. 65:111–114 [PubMed] [Google Scholar]
- 35.Ramya LN, Doble M, Rekha VP, Pulicherla KK. 2012. l-Asparaginase as potent anti-leukemic agent and its significance of having reduced glutaminase side activity for better treatment of acute lymphoblastic leukaemia. Appl. Biochem. Biotechnol. 167:2144–2159. 10.1007/s12010-012-9755-z [DOI] [PubMed] [Google Scholar]
- 36.Saba CF, Hafeman SD, Vail DM, Thamm DH. 2009. Combination chemotherapy with continuous l-asparaginase, lomustine, and prednisone for relapsed canine lymphoma. J. Vet. Intern. Med. 23:1058–1063. 10.1111/j.1939-1676.2009.0357.x [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Wang ZH, Chen XQ, Li YJ, Wang KF, Xia YF, Xia ZJ. 2013. First-line combination of gemcitabine, oxaliplatin, and l-asparaginase (GELOX) followed by involved-field radiation therapy for patients with stage IE/IIE extranodal natural killer/T-cell lymphoma. Cancer 119:348–355. 10.1002/cncr.27752 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.