

Abstract
A prerequisite for DNA-based microbial community analysis is even and effective cell disruption for DNA extraction. With a commonly used DNA extraction kit, roughly two-thirds of subseafloor sediment microbial cells remain intact on average (i.e., the cells are not disrupted), indicating that microbial community analyses may be biased at the DNA extraction step, prior to subsequent molecular analyses. To address this issue, we standardized a new DNA extraction method using alkaline treatment and heating. Upon treatment with 1 M NaOH at 98°C for 20 min, over 98% of microbial cells in subseafloor sediment samples collected at different depths were disrupted. However, DNA integrity tests showed that such strong alkaline and heat treatment also cleaved DNA molecules into short fragments that could not be amplified by PCR. Subsequently, we optimized the alkaline and temperature conditions to minimize DNA fragmentation and retain high cell disruption efficiency. The best conditions produced a cell disruption rate of 50 to 80% in subseafloor sediment samples from various depths and retained sufficient DNA integrity for amplification of the complete 16S rRNA gene (i.e., ∼1,500 bp). The optimized method also yielded higher DNA concentrations in all samples tested compared with extractions using a conventional kit-based approach. Comparative molecular analysis using real-time PCR and pyrosequencing of bacterial and archaeal 16S rRNA genes showed that the new method produced an increase in archaeal DNA and its diversity, suggesting that it provides better analytical coverage of subseafloor microbial communities than conventional methods.
INTRODUCTION
Numerous molecular ecological studies have demonstrated that microorganisms are widely distributed in natural environments, where they play significant ecological roles in global elemental cycles, including in the deep, low-energy sedimentary habitat beneath the seafloor (1–6). In general, the activity of such subseafloor microbial communities is extremely low because of limited availability of energy sources (7–11), whereas phylogenetically diverse microbial life is present in living or necromass form (12–19).
To understand the biomass, diversity, and metabolic functions of naturally occurring microbial communities in deep-subseafloor sedimentary habitats, molecular ecological approaches (i.e., analyses of DNA, RNA, lipids, etc.) are the most powerful tools for analyses at community to single-cell levels. In fact, previous molecular ecological surveys of such habitats have revealed that the deeply buried ocean microbial ecosystem is distinct from those of all terrestrial ecosystems (20–27). However, those molecular analyses relied heavily upon available techniques and databases, most of which were not customized for analysis of deep-sedimentary life forms that may have survived for hundreds to thousands of years.
In this regard, an important issue that we should carefully consider is the potential of extraction bias; i.e., if significant biases occur during the experimental and analytical processes, we merely see the biased community. Does the result obtained under a certain condition represent the overall picture of the indigenous community? Numerous previous molecular ecological studies have used various DNA extraction procedures (24, 28–31), and hence, cautions have been raised that the use of different DNA extraction protocols may result in different microbial community structures (31). Others have pointed out that PCR-based molecular approaches might overlook some evolutionarily distinct deep subseafloor microbial populations because of bias introduced by PCR. For example, Teske and Sørensen clearly illustrated the possible bias in the amplification of archaeal sequences introduced by the use of conventional PCR primer sequences, which often produce mismatches to sequences of predominantly sedimentary archaea in deep sediments (21, 24, 32, 33). The potential for such bias may also have a critical impact on quantitative molecular analyses (e.g., measurement of gene quantity or copy number largely relies on stable amplification of the gene, as well as the DNA coverage of the primers or probe used). In fact, the presence of PCR inhibitors, such as humic acids in organic-rich marine sediments, may significantly diminish the amplification efficiency and cycle threshold (CT) values in quantitative real-time PCR analyses, and hence, inhibition-tolerant gene quantification using digital PCR is a useful alternative (33).
To address these methodological issues, the amount of target biomolecule extracted from the sample must first be determined. Extraction of DNA is the first step in many molecular analytical techniques and, thus, has a significant impact on the results obtained. We have often observed significant inconsistencies between cell abundance and molecular quantification using techniques such as conventional quantitative PCR (qPCR), indicating that the results must be understood to represent only the DNA-extractable and PCR-amplifiable fraction. Even with commonly used kits and protocols employing newly developed cell separation and enumeration techniques (34–36), we found that ∼75% of sedimentary microbial cells remained intact in the residue of DNA extraction, indicating that large fractions of many deep-subseafloor microbial communities have been missed in previous molecular ecological surveys.
In this study, we focused on improving the DNA extraction methodology for microbial communities in deep-subseafloor sediments. We demonstrate that by standardizing the chemical (alkaline) and physical (temperature) conditions, our newly established protocol increases (i) the cell disruption efficiency (up to ∼80%), (ii) the DNA extraction yield (2.7- to 52-times-higher yield than conventional kit-based methods), and (iii) the molecular quantity and diversity richness of archaeal genes.
MATERIALS AND METHODS
Sample description and subsampling procedure.
Marine subsurface sediment core samples were collected off the Shimokita Peninsula of Japan in 2006 (site C9001 hole C) during the D/V Chikyu shakedown cruise CK06-06 (37–40) and from the Nankai Trough plate subduction zone (site C0006 hole A) during Integrated Ocean Drilling Program (IODP) expedition 316 in 2008 (41) (Table 1). After core recovery, whole round cores (10 cm in length) were immediately placed in a freezer and maintained at −80°C until laboratory use. For microbiological analyses, the innermost part of the frozen whole round core was aseptically sampled using an electric band saw system in a clean booth and without sample thawing (42). Escherichia coli strain JM109 grown overnight in Luria-Bertani medium served as the positive control in experiments examining genome fragmentation in the alkaline treatment solution.
TABLE 1.
Sample description, microbial cell abundance, and TOCa content
| Expedition | Drillling site | Core | Depth (mbsf) | TOC (wt%) | Sulfateb (mM) | Methaneb (mM) | Microbial cell abundance (cells/g sediment)c | DNA extraction method | DNA recovery (ng/g sediment) | qPCR (copies/g sediment)d | qPCR/cell abundance ratio | Microbial abundance estimated from quantity of DNA with assumed avg genome size (kb) of: |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2,000 |
3,800 |
||||||||||||||
| CCN (cells/g sediment) | Biomass coverage (%) | CCN (cells/g sediment) | Biomass coverage (%) | ||||||||||||
| Chikyu shakedown cruise CK06-06 | C9001C | 2H-2 | 9.5 | 1.3 | 0.16 | 3.01 | 2.77 × 108 | Kit | 183.4 | 4.30 × 107 | 0.16 | 8.36 × 107 | 30.2 | 4.85 × 107 | 17.5 |
| Hot alkaline | 489.3 | 4.09 × 107 | 0.15 | 2.23 × 108 | 80.6 | 1.29 × 108 | 46.7 | ||||||||
| 5H-1 | 35.4 | 0.81 | NDe | 1.68 | 2.41 × 107 | Kit | 21.0 | 4.98 × 106 | 0.21 | 9.60 × 106 | 39.8 | 5.56 × 106 | 23.1 | ||
| Hot alkaline | 160.3 | 8.18 × 106 | 0.34 | 7.31 × 107 | 303 | 4.24 × 107 | 176 | ||||||||
| 10H-1 | 84.0 | 1.26 | ND | 1.15 | 2.91 × 107 | Kit | 2.2 | 1.96 × 106 | 0.07 | 9.96 × 105 | 3.4 | 5.77 × 105 | 2.0 | ||
| Hot alkaline | 113.3 | 1.80 × 106 | 0.06 | 5.17 × 107 | 177 | 2.99 × 107 | 103 | ||||||||
| 20H-3 | 177.4 | 1.2 | ND | 1.01 | 8.70 × 106 | Kit | BDL | 1.22 × 106 | 0.14 | NAf | NA | NA | NA | ||
| Hot alkaline | 71.7 | 1.08 × 106 | 0.12 | 3.27 × 107 | 376 | 1.90 × 107 | 218 | ||||||||
| 40H-10 | 364.0 | 0.82 | 0.04 | 1.99 | 1.21 × 105 | Kit | BDL | 3.76 × 104 | 0.31 | NA | NA | NA | NA | ||
| Hot alkaline | 2.3 | 1.48 × 105 | 1.23 | 1.07 × 106 | 886 | 6.21 × 105 | 513 | ||||||||
| IODP Expedition 316 | C0008A | 3H-6 | 22.0 | 0.37 | ND | 8.20 | 8.79 × 104 | Kit | BDL | 8.64 × 104 | 0.98 | NA | NA | NA | NA |
| Hot alkaline | 19.4 | 1.25 × 105 | 1.42 | 8.85 × 106 | 10,067 | 5.13 × 106 | 5,836 | ||||||||
DNA extraction using a commercial kit.
A total of 10 g of subsampled sediment was placed in a 15-ml conical tube. Extracellular DNA and other soluble molecules were removed by washing the sediment in 2 ml of 3% NaCl with rotation for 60 min at room temperature, followed by centrifugation at 3,000 × g for 10 min at 20°C and removal of the supernatant. Potential cell disruption during washing to remove extracellular DNA was evaluated by enumerating microbial cells in washed and unwashed (instantly fixed) sediment samples, which revealed that washing had a negligible effect (see Table S1 in the supplemental material).
An overnight culture of E. coli JM109 was centrifuged at 5,000 × g for 5 min at 4°C, and the supernatant was removed. Intracellular DNA was extracted from washed sediment or E. coli cells using either a PowerMax soil DNA isolation kit (MoBio Laboratories, Inc., Carlsbad, CA, USA) or FastDNA spin kit for soil (MP Biomedicals, LLC) in accordance with the manufacturer's instructions, with the exception of a slight modification to the shaking conditions. For the PowerMax soil DNA isolation kit, the sediment was vigorously shaken with autoclaved metal beads (5 mm in diameter) using a Shake Master auto (Bio Medical Science, Tokyo, Japan) operated at 1,500 rpm for 10 min. Since we obtained similar cell disruption efficiency with the two commercial kits, we used the DNA extract from the PowerMax soil DNA isolation kit for all of the following DNA-based analyses.
DNA extraction with alkaline lysis solution.
A prewarmed (50, 70, or 90°C) 2-ml volume of alkaline lysis solution consisting of 1 M NaOH, 5 mM EDTA (pH 8.0), and 1% SDS was added to either 2 g of washed sediment sample or pelleted E. coli in a 15-ml conical tube and heated at the respective temperature for 20 min. The heat-treated samples were then centrifuged at 10,000 × g for 1 min at 25°C, after which the supernatants were transferred to new tubes with 1.5 ml of neutralization solution consisting of 1 M HCl and 0.3 M Tris-HCl (pH 8.0). The remaining sediment was washed with 2 ml of prewarmed distilled water and centrifuged at 10,000 × g for 1 min at 25°C, and the supernatant was recovered into the same sample tube. The extract was treated with equal volumes (∼5.5 ml) of phenol-chloroform-isoamyl alcohol (25:24:1) and chloroform-isoamyl alcohol (24:1) and then precipitated by adding a 1/10 volume (∼0.5 ml) of 3 M sodium acetate, 3 μl of ethachinmate (Nippon Gene, Tokyo), and a 2.5-times-greater volume (∼10 ml) of ice-cold ethanol. The sample was then centrifuged at 15,000 × g for 30 min at 4°C, after which the pellet was washed with 70% ethanol, dried, and dissolved in 100 μl of 10 mM Tris-HCl (pH 8.0). The solution of DNA extracted from sediment was further purified using a spin column filled with polyvinylpolypyrrolidone (PVPP) to remove PCR inhibitors (e.g., humic acids). Acid-washed PVPP was prepared according to Holben et al. (1988) and resuspended in Tris-EDTA (TE) buffer at 6% (wt/vol). A 1-ml volume of the PVPP suspension was placed onto a microspin column (Suprec-EZ; TaKaRa Bio, Inc., Shiga, Japan) for DNA purification. The liquid was removed by centrifugation at 3,000 × g for 15 min at 25°C, followed by washing with 500 μl of distilled water. Then, 75 μl of the extracted DNA was placed into the column and the column was centrifuged at 3,000 × g for 15 min at 25°C. To gain higher recovery of the applied DNA, another 75 μl of 10 mM Tris-HCl (pH 8.0) was added to the same column, which was centrifuged again at 3,000 × g for 15 min at 25°C. The whole hot-alkaline DNA extraction scheme is summarized in Fig. S1 in the supplemental material.
Enumeration of microbial cells in sediment samples.
Prior to DNA extraction, frozen sediment samples (1 cm3 of each) were fixed with 9 ml of 2% paraformaldehyde for about 2 h at room temperature. The fixed slurry samples were washed twice with phosphate-buffered saline (PBS), resuspended in 10 ml of PBS-ethanol (1:1) solution, and stored at −20°C. The residue of the sediment after DNA extraction was resuspended in 3% NaCl (10% [vol/vol]). Enumeration of microbial cells in the slurry samples was performed according to a previously described protocol (34), with slight modifications as follows. Briefly, 50 μl of sediment slurry was mixed with 850 μl of 3% NaCl (filtered through a 0.22-μm membrane) and sonicated on ice for 1 min using an ultrasonic homogenizer (model UH-50; SMT Co. Ltd., Tokyo, Japan) set at 20 W, after which 100 μl of 10% hydrofluoric acid was added and the sample was incubated for 20 min at room temperature. After the acid treatment, 500 μl of the mixture was filtered using a 0.22-μm-pore-size black polycarbonate membrane (Millipore, Billerica, MA, USA). About 5 ml of 2.5% NaCl solution was placed into the filter tower prior to the addition of the supernatant to ensure even distribution of the cells on the filter. The membrane was then washed with 2.5 ml of TE buffer, and roughly 2 × 108 fluorescent microsphere beads (Fluoresbrite bright blue carboxylate microspheres [BB beads], 0.5 μm; Polysciences, Inc., Warrington, PA, USA) were added for use in focus adjustment (34). After air drying, one-fourth of the membrane was placed on the filtration device again and stained with SYBR green I (1/40 [vol/vol] SYBR green I in TE buffer). The stained filter was finally mounted on a glass microscope slide with 3 to 5 μl of mounting solution (2:1 mixture of Vectashield mounting medium H-1000 and TE buffer). Acquisition of microscopic fluorescence images (at 525/36 nm [center wavelength/bandwidth] and 605/52 nm by 490-nm excitation) was performed automatically using a fluorescence microscope system equipped with an automatic slide handler (35). The resulting images were analyzed using a macro in Metamorph software (Molecular Devices, Sunnyvale, CA, USA) to eliminate background signals and discriminatively enumerate cells on the membrane.
DNA quantification with HPLC.
Because alkaline treatment denatures DNA into single strands and the DNA extracts had high optical absorbance by humic substances at around the UV range, we digested extracted DNA molecules for quantification. To avoid enzyme inhibition by humic substances that still remained after DNA purification, 30 μl of each DNA extract was diluted 50-fold (i.e., to 1.5 ml) and then digested with 170 units of S1 nuclease in reaction buffer (TaKaRa Bio, Inc., Shiga, Japan) at 65°C for 12 h. The enzyme was inactivated after the reaction by treatment with 1.5 ml of chloroform-isoamyl alcohol (24:1). After reduction of the volume to less than 100 μl under vacuum centrifugation (DNA SpeedVac; Thermo Fisher Scientific, Inc., Waltham, MA, USA), the nucleotide solution was injected onto an LC-10A high-performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) equipped with a TSKgel ODS-100V column (5 μm, 4.6-mm inner diameter by 150 mm; Tosoh, Tokyo, Japan) and guard column cartridge (TSKgel Guardgel ODS-100V, 5 μm, 3.2-mm inner diameter by 15 mm; Tosoh). Standards for each nucleotide were obtained from a DNA GC kit (number 7160; Yamasa Co., Chiba, Japan). Each nucleotide was quantified relative to the standard peak area. The total amount of DNA was calculated by summation of the nucleotides measured.
PCR amplification and pyrosequencing.
To estimate the copy numbers of archaeal and bacterial 16S rRNA genes, qPCR was conducted with SYBR premix DimerEraser PCR master mix (TaKaRa Bio, Inc.) using a StepOnePlus real-time PCR system (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. The 16S rRNA gene primers EUB27F (43), EUB338R mix (I, 5′-GCTGCCTCCCGTAGGAGT-3′; II, 5′-GCAGCCACCCGTAGGTGT-3′; and III, 5′-GCTGCCACCCGTAGGTGT-3′) (44), EUB338F mix (I, 5′-ACTCCTACGGGAGGCAGC-3′; II, 5′-ACACCTACGGGTGGCTGC-3′; and III, 5′-ACACCTACGGGTGGCAGC-3′), and UNIV515R (5′-TTACCGCGGCKGCTGRCA-3′) (45) were used for bacteria, while the ARC806F (5′-GGACTACVSGGGTATCTAAT-3′) (46) and ARC958R (5′-YCCGGCGTTGAMTCCAATT-3′) (47) primers were used for archaea, as previously described (24). Due to the presence of PCR-inhibiting substances in the DNA extracts, we diluted the extracted DNA 100-fold, and 1 μl was used for qPCR. The lower detection limit of the qPCR analysis was determined from the amplification of at least three negative-control reactions. To examine the integrity of the E. coli genome, we used the primer pairs EUB27F and 1490R (45), EUB338F and 1490R, and EUB27F and EUB338R.
For pyrosequencing, amplification of 16S RNA gene fragments was conducted using the primers EUB27F and EUB338R mix for bacteria and ARC21F (5′-TTCCGGTTGATCCYGCCGGA-5′) (61) and ARC912R (I, 5′-CCCCCGCCAATTCCTTTAA-3′; II, 5′-CCCCCGTCAATTCCTTCAA-3′; and III, 5′-CCCCCGCCAATTTCTTTAA-3′) (62) for archaea, with some modifications relative to the sequences in the original publication. For sequencing using a GS FLX pyrosequencer (454 Life Sciences, Branford, CT, USA), another amplification (i.e., six additional cycles) was performed using the primers EUB27F and EUB338R mix for bacteria and UNIV530F (I, 5′-GTGCCAGCMGCCGCGG-3′, and II, 5′-GTGTCAGCCGCCGCGG-3′) and ARC912R for archaea, with 454 FLX Titanium adapters A and B and a 6-base sample identifier tag. Purification of the amplified products, quality checks, and sequencing using the GS FLX pyrosequencer were conducted by TaKaRa Bio, Inc.
Sequence data processing and statistical analyses of microbial community structures.
All of the sequence reads, including sample identifier tags and primer sequences, were first processed using the Pipeline Initial Process (http://pyro.cme.msu.edu/init/form.spr), which is part of the Ribosomal Database Project (48). The parameters for the Pipeline Initial Process were as follows: forward primer maximum edit distance, 2; maximum number of N′s, 0; minimum average exponential quality score, 20; reverse primer maximum edit distance, 0; and minimum sequence length, 150. Reads that did not match the tags and primer sequences were also eliminated during the process. A BLAST+ analysis with a customized computer script using the ARB SILVA sequence package (49) as the database (23) was then conducted to clean up the processed sequences such that nontarget sequences (e.g., bacterial sequences in the archaeal analysis and vice versa) were excluded in each pyrolibrary. The resulting sequences were screened for chimeras and then analyzed using the mothur utility package (50). Chao1 estimates (51) and Shannon diversity index (52) values were calculated using 97 and 99% sequence similarity cutoffs, respectively.
Sequence accession number.
The sequence data obtained in this study have been submitted to the DDBJ database under accession no. DRA001030.
RESULTS AND DISCUSSION
Cell disruption efficiency.
We first evaluated the cell disruption efficiency of the DNA extraction methods for subseafloor sediment samples (Fig. 1). Using two different commercial DNA extraction kits (i.e., the PowerMax soil DNA isolation kit [MoBio Laboratories, Inc.] and the FastDNA spin kit for soil [MP Biomedicals, LLC]), the average proportion of disrupted cells was 30.7% (range, 21.3 to 44.2%). These two DNA extraction kits, which employed mechanical cell disruption processes, yielded similar cell disruption efficiencies. The results indicated that approximately two-thirds of the total microbial cell numbers remained intact in residues after the DNA extraction (see Fig. S2 in the supplemental material). We then disrupted cells in subseafloor sediment by treatment with alkaline solution and heat. Lysis under alkaline conditions is widely used as a molecular approach to extract plasmids from E. coli cells (53). We tested the effects of harsher conditions involving heating. The cell disruption efficiencies after hot-alkaline treatment at 50 to 98°C ranged from 29.1 to 95.8%, which was 0.9 to 3.8 times greater than the efficiency of commercial kit-based mechanical disruption. The disruption efficiency for the shallowest sample (i.e., site C0008 core 2H-2, 9.5 m below the seafloor [mbsf]) (Fig. 1A) was consistently higher than 88% across all treatment temperatures tested. We also observed a tendency toward lower disruption efficiencies with increasing sample depth (Fig. 1A to E). However, the cell disruption efficiency was still greater than 47% even in the deepest sample we examined (i.e., core 40H-10, 364.0 mbsf) (Fig. 1E). The increase in disruption efficiency with increasing temperature indicates that loosening of the cell membrane at high temperature enhances the exposure of the hydrophilic part of membrane lipid molecules to the alkaline solution. In addition, the higher temperature also promotes saponification of the membrane lipids during the alkaline treatment, and those effects were found to be key for obtaining highly efficient cell disruption.
FIG 1.

Efficiency of microbial cell lysis in subseafloor sediments after DNA extraction. Sediments from 2H-2 (9.5 mbsf [meters below the seafloor]) (A), 5H-1 (35.4 mbsf) (B), 10H-1 (84.0 mbsf) (C), 20H-3 (177.4 mbsf) (D), and 40H-10 (364.0 mbsf) (E) from offshore of the Shimokita Peninsula and 3H-6 (22.0 mbsf) (F) from the Nankai Trough in Japan were treated with alkaline lysis solution at 50, 70, 90, or 98°C for 20 min, and the lysis efficiencies were determined with bead beating using a commercial kit (PowerMax soil DNA isolation kit [PM] or FastDNA spin kit for soil [FP]). The lysis percentage was calculated by enumerating cells before and after extraction. Error bars show standard deviations of at least triplicate measurements of triplicate extractions.
Degree of DNA fragmentation by hot-alkaline treatment.
Although we could obtain a higher percentage of disrupted cells when using the hot-alkaline treatment, the DNA extracted into the alkaline solution was denatured into the more labile single-stranded form. Therefore, we examined the degree of fragmentation of the extracted DNA using E. coli genomic DNA. The same hot-alkaline treatment was applied to a pellet of E. coli cells, and the extracted DNA was then analyzed using electrophoresis (Fig. 2A). Analysis of the gel image showed that the E. coli genome was broken into fragments shorter than 5 kb with the hot-alkaline treatment at 90°C and into fragments shorter than 1 kb at 98°C. Another examination with the nearly full-length PCR-amplified E. coli 16S rRNA gene showed that the fragmentation was critical for subsequent molecular analysis (e.g., 16S rRNA gene survey) when E. coli cells were disrupted at temperatures ranging from 90 to 98°C, as no amplifications were observed by PCR using the 27F-1490R primer pair (Fig. 2B). This result suggested that at least one strand break occurred in almost all of the 16S rRNA genes and resulted in null amplification with treatment at 90 and 98°C. In contrast, there was no significant difference in amplification of the 16S rRNA gene with treatment at 50 and 70°C (Fig. 2B). Given these gel-based observations, we concluded that heating at 70°C would provide for optimal preservation of DNA quality for subsequent molecular analyses.
FIG 2.
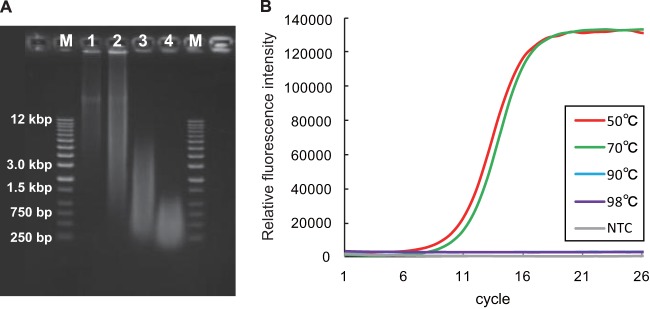
Degree of fragmentation of the E. coli genome by hot-alkaline treatment. (A) Electrophoretic image of the genome of E. coli extracted using the hot-alkaline method at 50°C (lane 1), 70°C (lane 2), 90°C (lane 3), and 98°C (lane 4) along with DNA size markers (kb DNA ladder; Agilent Technologies) (lanes M). (B) Real-time monitoring of nearly full-length 16S rRNA gene amplification with primers EUB27F and 1490R after extraction of E. coli genomic DNA using hot-alkaline extraction at various temperatures. NTC, no-template control.
Quantification of extracted DNA.
The DNA extracted using the hot-alkaline treatment was single stranded, and the DNA solution still contained humic substances. Although we could remove a large fraction of the humic substances by PVPP purification and obtain clear DNA extracts without significant loss of the applied DNA (i.e., more than 95% recovery of applied DNA [data not shown]), there was a substantial background in optical absorbance in the UV region around 260 nm. This was likely caused by remnant humic substances, which impeded subsequent spectrometric quantification of the extracted single-stranded DNA (data not shown). Therefore, to determine and compare the total quantity of DNA extracted using the hot-alkaline treatment with that obtained using a kit-based approach, we digested the DNA extracted using both approaches into nucleotides and quantified them using HPLC (Fig. 3A). Although the DNA concentration in samples from deeper sediments was below the detection limit (no nucleotide peak observed in HPLC traces) in the kit-extracted samples, DNAs prepared from all sampling depths using the hot-alkaline extraction method were measurable by HPLC. A comparison of the total amounts of DNA extracted using the hot-alkaline and kit-based methods showed that the DNA yield using hot-alkaline extraction was 2.7 to 52 times higher than that obtained using kit extraction (Fig. 3A).
FIG 3.
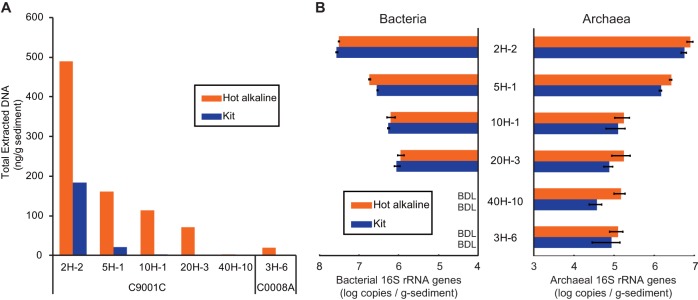
Quantification of extracted DNA. (A) Total extracted DNA quantified using HPLC. (B) qPCR-based quantification of bacterial and archaeal 16S rRNA genes. BDL, below quantification limit (6.37 × 105 copies/g of sediment) for bacteria; NTC, no-template control.
Figure 3B shows the copy numbers of bacterial and archaeal 16S rRNA genes, as estimated by qPCR analysis. Interestingly, the estimated copy numbers of archaeal 16S rRNA genes recovered using hot-alkaline DNA extraction were generally higher than those of genes recovered using a commercial kit. The most probable explanation is that the strong chemical and physical lysis conditions of the hot-alkaline method enhanced disruption of the subseafloor archaeal cells. Since archaeal cells in deep-subseafloor sediments might have a more rigid cell wall than bacteria (54), we infer that previous molecular ecological surveys might have caused biases for the archaeal community (24). These results are also consistent with the deep sequencing analysis, which showed an increase in archaeal diversity richness (described below).
The total copy numbers of 16S rRNA genes were generally inconsistent despite the increased amounts of total DNA extracted using the hot-alkaline method. The numbers of bacterial 16S rRNA gene copies as determined by qPCR analysis were lower in some samples extracted using the hot-alkaline method than in samples extracted using a commercial kit (Fig. 3B), probably due to the excess fragmentation of DNA released from the cells (especially for easily lysed bacteria) in the hot-alkaline solution. In our standardization of the optimum hot-alkaline treatment conditions using the E. coli genome, extracted genomic DNA retained sufficient integrity for amplification of nearly full-length 16S rRNA gene fragments (i.e., ∼1,500 bp). However, the stability of deep-subseafloor microbial DNA remains largely unknown. That is, the degree to which DNA of the extremely inactive subseafloor cells is damaged by the severe conditions of deep and ancient marine sediments has not been determined. If a large fraction of subseafloor microbial DNA has been damaged prior to extraction and is then further fragmented upon extraction, the amount of single-stranded DNA determined by HPLC as nucleotides may disagree with the qPCR results. Alternatively, the addition of DNA sequences that do not match to the primer sequences used (32, 33) potentially increased undetectable fraction of sequences over the kit extraction.
We performed a quantitative comparison between the quantity of DNA extracted and the microbial cell abundance. The total copy numbers of 16S rRNA genes were generally lower than the microbial cell abundances in organic-rich sediment samples down to 177.4 mbsf (Table 1). However, we could see an interesting trend in which the copy number of 16S rRNA genes got close to the microbial abundance in deep (364 mbsf) and organic-poor sediments (C9001C 40H-10 and C0008A 3H-6, respectively). In the DNA recovered using hot-alkaline DNA extraction, the total copy number of 16S rRNA genes was found to be higher than the microbial cell abundance. Considering the multiplicity of 16S rRNA genes in a microbial genome (55), the copy numbers detected are still not enough to cover all the microbial biomass. However, the increased qPCR copy numbers of archaeal 16S rRNA genes should contribute to higher coverage of the microbial community. We also estimated calculated cell numbers (CCN) based on the quantity of DNA extracted by assuming average genome sizes of microbes (2,000 kb and 3,800 kb). The CCN of DNA extracted from a shallow-subseafloor sample (C9001C 2H-2, 9.5 mbsf) appeared to fit well with the lysis efficiency (Fig. 1) for both of the extraction procedures. However, in deeper sediment (10H-1, 84.0 mbsf), the CCN for kit-extracted DNA was 10 times smaller than the fraction of cells lysed, whereas that for hot-alkaline-extracted DNA was 1.77 times greater than the total microbial cell abundance. We could not calculate the CCN for kit-extracted DNA in deeper sediment because of low DNA recovery. Although the CCN for DNA recovered using the hot-alkaline extraction method decreased with increasing depth, the depth-wise tendency was for the cell abundance to be greater than the CCN. The CCN was 8.9 times greater in the deepest sample (C9001C 40H-10, 364 mbsf) and 100 times greater in organic-poor sediments (C0008A 3H-6, 22.0 mbsf) than the cell abundance. The decreasing extractability by kit-based extraction and inversely increasing extractability using the hot-alkaline method is difficult to explain; however, it may possibly be due to the existence of DNA extraction-resistant cells in the subseafloor sediment, which might be derived from (endo)spore-related biomass. Recently Lomstein et al. found necromass to be greater than living biomass in marine subsurface sediments (16). Because of the impermeability of spore walls, it has been discussed that spores are generally missed in microbial cell detection by nucleic acid staining.
Comparison of the microbial community structures of samples prepared using hot-alkaline and kit-based DNA extraction methods.
Using pyrosequencing of the bacterial and archaeal 16S rRNA genes, we compared the microbial community structures (i.e., composition and diversity) based on DNA extracted with our new hot-alkaline method and a commercial kit (Fig. 4). The average number of sequence reads per sample was 15,517 (see Table S2 in the supplemental material). Phylogenetic analysis of the community composition in samples prepared using the two DNA extraction protocols showed significant differences in archaeal 16S rRNA genes (Fig. 4A). For example, the relative fraction of marine benthic group B (MBG-B, also referred to as the deep-sea archaeal group [DSAG]) generally increased (8.1 to 2,800 times) in samples prepared using the hot-alkaline extraction method, except for sample C9001C 40H-10 (364 mbsf), in which only one MBG-B sequence was detected with both extraction methods. In addition, the increased percentage of MBG-B archaea was generally consistent with the results of qPCR analysis, which showed increased recovery of archaeal 16S rRNA genes. This result was also consistent with the increase in total DNA yield with the hot-alkaline extraction method. The MBG-B are the predominant archaeal components detected in shallow- to deep-subseafloor sediments on the continental margins (15, 46, 56, 57), which harbor a phylogenetically diverse array of subseafloor archaea (58). We hypothesize that following burial in deep-sedimentary horizons, the MBG-B cell wall might become rigid upon starvation, and therefore, some of the DNA of these populations could be recovered by hot-alkaline treatment of the cells. On the other hand, the fraction of anaerobic methane-oxidizing Archaea (ANME-1) generally decreased in samples prepared using the hot-alkaline extraction method, except for sample C9001C 40H-10 (364 mbsf). The apparent disappearance of ANME-1 sequences for the hot-alkaline-extracted DNAs shown in Fig. 4 is most likely due to the other sequences that outnumbered ANME-1 sequences in the community reads.
FIG 4.

Characterization of extracted DNA. (A) Phylogenetic composition of archaeal 16S rRNA genes as determined by pyrosequencing of extracted DNAs. SAGMEG, South African gold mine euryarchaeotic group. (B and C) Archaeal diversity estimated by Chao1 species richness and Shannon-Wiener (H′) indices, calculated using pyrosequence data with similarity cutoffs of 97% (B) and 99% (C).
We observed a clear difference in the archaeal composition in samples from site C9001 hole 40H-10 prepared with each extraction method; members of the Halobacteriales and other groups (miscellaneous crenarcheaotic group [MCG], ANME-1, Methanosarcinales, and Thermoplasmatales) were the predominant archaeal components in samples extracted using the hot-alkaline method, whereas MCG archaea predominated in kit-extracted samples (>85%) (Fig. 4A). The relatively diverse archaea identified by analysis of the hot-alkaline-extracted DNA from site 40H-10 sediment were most likely detected as a result of the increased cell disruption efficiency provided by the hot-alkaline treatment, which is consistent with the largest increase (3.94 times) in the qPCR-detected archaeal population among the samples tested. These organisms therefore could remain undetected in samples analyzed using conventional standard molecular techniques.
In contrast to the archaeal communities, the composition of bacterial communities in samples prepared using each DNA extraction method was similar throughout the depths examined (see Fig. S3A in the supplemental material). These results were consistent with the qPCR results, which showed similar abundances of bacterial 16S rRNA genes between the two DNA extraction methods (Fig. 3B). This consistency provides another indication that archaeal cells are the major fraction of microbes that remain undisrupted by a conventional kit-based DNA extraction.
We then compared the α diversity of the two DNA extraction methods. Figure 4B and C show the Chao1 estimates (i.e., species richness) and Shannon-Wiener (H′) diversity indices calculated based on pyrosequencing of the archaeal a 16S rRNA gene fragments. For the archaeal communities, both the species richness (i.e., Chao1) and diversity (H′) of samples prepared by hot-alkaline DNA extraction were generally higher than those of samples prepared using a commercial kit (Fig. 4B). This is consistent with the amounts of DNA extracted, qPCR results, and community composition determined based upon analysis of the archaeal 16S rRNA genes, which were all impacted by the high cell disruption efficiency of the hot-alkaline method. One exception to the trend toward higher Chao1 and Shannon scores was site C9001 10H-1. In this sample horizon, the proportion of MBG-B sequences was increased by the hot-alkaline treatment and constituted 96.5% of the total sequence read (Fig. 4). Because of the increased MBG-B sequence recovery, we can infer that the relative abundance of other phylogenetic groups decreased. In addition, when we extended the similarity cutoff to 99%, the diversity indices of hot-alkaline-extracted DNA from site C9001 10H-1 were higher than those of kit-extracted DNA (Fig. 4C). Hot-alkaline extraction thus brought to light the microdiversity present within the MBG-B archaeal community.
For the bacterial communities, we observed increased diversity scores (both Chao1 and H′) for samples of hot-alkaline-extracted DNA from the deep subseafloor at site C9001 40H-10 and the old accretionary prism of site C0008 3H-6 (see Fig. S3B in the supplemental material). These two samples represented the lowest recovery of DNA using both extraction methods due to the low cell abundance (∼104 cells/cm3) (Table 1). Under the conditions characteristic of these deep-subseafloor sedimentary biospheres, the physiological state of the bacteria might be slightly different than that of bacteria in relatively shallow and more recently formed sedimentary habitats (2, 16, 59). Similar to the case with archaea, bacteria in these habitats may be more difficult to disrupt using standard kit-based DNA extraction methods. In fact, the diversity scores of bacterial communities from other horizons were similar or slightly lower for samples prepared using hot-alkaline extraction, which agreed well with other observations (i.e., qPCR and community composition). When we extended the similarity cutoff to 99%, both the species richness and diversity scores of all samples prepared by the hot-alkaline DNA extraction method increased (see Fig. S3C). As was the case with archaeal communities, this result indicated that there are some lysis-resistant bacterial components present that can only be explored using the hot-alkaline DNA extraction method.
Conclusion.
The improved DNA extraction efficiency provided by the hot-alkaline treatment method described here enabled us to demonstrate that the deep-subseafloor sedimentary biosphere harbors a large fraction of lysis-resistant cells that might have been missed in previous molecular ecological surveys based on standard DNA extraction protocols. In this study, we observed that microbial communities in the deep-subseafloor biosphere are more phylogenetically and physiologically diverse than previously expected. Our results do not mean that the DNA extraction method we standardized here could solve all the issues of bias, however. For example, there are still gaps between the amount of extracted DNA and the abundance of 16S rRNA genes, suggesting the presence of another factor(s) that may bias molecular detection and quantification of deep-subseafloor microbes. Nevertheless, the results of this study not only expand the molecular view of deep-subseafloor microbial communities, they also illustrate that there is much more that remains to be explored. More effective cell lysis and DNA retrieval must be key components of future molecular ecological studies at the community and single-cell levels if we are to obtain a better understanding of deep-subseafloor microbial ecosystems.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Tanaka and S. Fukunaga for technical assistance with the analytical work and Y. Suzuki and M. A. Lever for helpful discussions. We also thank the crew, drilling team, and onboard scientific parties of Chikyu shakedown expedition CK06-06 and Integrated Ocean Drilling Program (IODP) expedition 316.
This study was supported by the Japan Society for the Promotion of Science (JSPS) Strategic Fund for Strengthening Leading-Edge Research and Development (to JAMSTEC), the JSPS Funding Program for Next Generation World-Leading Researchers (NEXT Program, to F.I.), and JSPS Grants-in-Aid for Scientific Research (no. 24651018 and 24687004 to Y.M. and no. 24770033 to T.H.).
Footnotes
Published ahead of print 17 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04150-13.
REFERENCES
- 1.Edwards KJ, Becker K, Colwell F. 2012. The deep, dark energy biosphere: intraterrestrial life on earth. Annu. Rev. Earth Planet. Sci. 40:551–568. 10.1146/annurev-earth-042711-105500 [DOI] [Google Scholar]
- 2.Hoehler T, Jørgensen B. 2013. Microbial life under extreme energy limitation. Nat. Rev. Microbiol. 11:83–94. 10.1038/nrmicro2939 [DOI] [PubMed] [Google Scholar]
- 3.Jørgensen BB, Boetius A. 2007. Feast and famine—microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5:770–781. 10.1038/nrmicro1745 [DOI] [PubMed] [Google Scholar]
- 4.Kallmeyer J, Pockalny R, Adhikari R, Smith D, D'Hondt S. 2012. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. U. S. A. 109:16213–16216. 10.1073/pnas.1203849109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ. 2011. Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75:361–422. 10.1128/MMBR.00039-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkes RJ, Cragg BA, Wellsbury P. 2000. Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol. J. 8:11–28. 10.1007/PL00010971 [DOI] [Google Scholar]
- 7.D'Hondt S, Jørgensen BB, Miller DJ, Batzke A, Blake R, Cragg BA, Cypionka H, Dickens GR, Ferdelman T, Hinrichs KU, Holm NG, Mitterer R, Spivack A, Wang G, Bekins B, Engelen B, Ford K, Gettemy G, Rutherford SD, Sass H, Skilbeck CG, Aiello IW, Guerin G, House CH, Inagaki F, Meister P, Naehr T, Niitsuma S, Parkes RJ, Schippers A, Smith DC, Teske A, Wiegel J, Padilla CN, Acosta JL. 2004. Distributions of microbial activities in deep subseafloor sediments. Science 306:2216–2221. 10.1126/science.1101155 [DOI] [PubMed] [Google Scholar]
- 8.D'Hondt S, Spivack AJ, Pockalny R, Ferdelman TG, Fischer JP, Kallmeyer J, Abrams LJ, Smith DC, Graham D, Hasiuk F, Schrum H, Stancin AM. 2009. Subseafloor sedimentary life in the South Pacific Gyre. Proc. Natl. Acad. Sci. U. S. A. 106:11651–11656. 10.1073/pnas.0811793106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkes RJ, Webster G, Cragg BA, Weightman AJ, Newberry CJ, Ferdelman TG, Kallmeyer J, Jørgensen BB, Aiello IW, Fry JC. 2005. Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436:390–394. 10.1038/nature03796 [DOI] [PubMed] [Google Scholar]
- 10.Price PB, Sowers T. 2004. Temperature dependence of metabolic rates for microbial growth, maintenance, and survival. Proc. Natl. Acad. Sci. U. S. A. 101:4631–4636. 10.1073/pnas.0400522101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Røy H, Kallmeyer J, Adhikari R, Pockalny R, Jørgensen B, D'Hondt S. 2012. Aerobic microbial respiration in 86-million-year-old deep-sea red clay. Science 336:922–925. 10.1126/science.1219424 [DOI] [PubMed] [Google Scholar]
- 12.Futagami T, Morono Y, Terada T, Kaksonen AH, Inagaki F. 2009. Dehalogenation activities and distribution of reductive dehalogenase homologous genes in marine subsurface sediments. Appl. Environ. Microbiol. 75:6905–6909. 10.1128/AEM.01124-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imachi H, Aoi K, Tasumi E, Saito Y, Yamanaka Y, Saito Y, Yamaguchi T, Tomaru H, Takeuchi R, Morono Y. 2011. Cultivation of methanogenic community from subseafloor sediments using a continuous-flow bioreactor. ISME J. 5:1913–1925. 10.1038/ismej.2011.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inagaki F, Sakihama Y, Inoue A, Kato C, Horikoshi K. 2002. Molecular phylogenetic analyses of reverse-transcribed bacterial rRNA obtained from deep-sea cold seep sediments. Environ. Microbiol. 4:277–286. 10.1046/j.1462-2920.2002.00294.x [DOI] [PubMed] [Google Scholar]
- 15.Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A, Suzuki M, Takai K, Delwiche M, Colwell FS, Nealson KH, Horikoshi K, D'Hondt S, Jorgensen BB. 2006. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U. S. A. 103:2815–2820. 10.1073/pnas.0511033103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomstein B, Langerhuus A, D'Hondt S, Jørgensen B, Spivack A. 2012. Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature 484:101–104. 10.1038/nature10905 [DOI] [PubMed] [Google Scholar]
- 17.Schippers A, Neretin LN, Kallmeyer J, Ferdelman TG, Cragg BA, Parkes RJ, Jorgensen BB. 2005. Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433:861–864. 10.1038/nature03302 [DOI] [PubMed] [Google Scholar]
- 18.Langerhuus AT, Røy H, Lever MA, Morono Y, Inagaki F, Jørgensen BB, Lomstein BA. 2012. Endospore abundance and d:l-amino acid modeling of bacterial turnover in holocene marine sediment (Aarhus Bay). Geochim. Cosmochim. Acta 99:87–99. 10.1016/j.gca.2012.09.023 [DOI] [Google Scholar]
- 19.Teske AP. 2006. Microbial communities of deep marine subsurface sediments: molecular and cultivation surveys. Geomicrobiol. J. 23:357–368. 10.1080/01490450600875613 [DOI] [Google Scholar]
- 20.Biddle JF, Lipp JS, Lever MA, Lloyd KG, Sørensen KB, Anderson R, Fredricks HF, Elvert M, Kelly TJ, Schrag DP, Sogin ML, Brenchley JE, Teske A, House CH, Hinrichs KU. 2006. Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc. Natl. Acad. Sci. U. S. A. 103:3846–3851. 10.1073/pnas.0600035103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biddle J, Fitz-Gibbon S, Schuster S, Brenchley J, House C. 2008. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U. S. A. 105:10583–10588. 10.1073/pnas.0709942105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fry JC, Parkes RJ, Cragg BA, Weightman AJ, Webster G. 2008. Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 66:181–196. 10.1111/j.1574-6941.2008.00566.x [DOI] [PubMed] [Google Scholar]
- 23.Hoshino T, Morono Y, Terada T, Imachi H, Ferdelman T, Inagaki F. 2011. Comparative study of subseafloor microbial community structures in deeply buried coral fossils and sediment matrices from the Challenger Mound in the Porcupine Seabight. Front. Microbiol. 2:231. 10.3389/fmicb.2011.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipp JS, Morono Y, Inagaki F, Hinrichs K-U. 2008. Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454:991–994. 10.1038/nature07174 [DOI] [PubMed] [Google Scholar]
- 25.Lloyd K, Schreiber L, Petersen D, Kjeldsen K, Lever M, Steen A, Stepanauskas R, Richter M, Kleindienst S, Lenk S, Schramm A, Jørgensen B. 2013. Predominant archaea in marine sediments degrade detrital proteins. Nature 496:215–218. 10.1038/nature12033 [DOI] [PubMed] [Google Scholar]
- 26.Mills H, Reese B, Shepard A, Riedinger N, Dowd S, Morono Y, Inagaki F. 2012. Characterization of metabolically active bacterial populations in subseafloor Nankai Trough sediments above, within, and below the sulfate-methane transition zone. Front. Microbiol. 3:113. 10.3389/fmicb.2012.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morono Y, Terada T, Nishizawa M, Ito M, Hillion F, Takahata N, Sano Y, Inagaki F. 2011. Carbon and nitrogen assimilation in deep subseafloor microbial cells. Proc. Natl. Acad. Sci. U. S. A. 108:18295–18300. 10.1073/pnas.1107763108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roose-Amsaleg CL, Garnier-Sillam E, Harry M. 2001. Extraction and purification of microbial DNA from soil and sediment samples. Appl. Soil Ecol. 18:47–60. 10.1016/S0929-1393(01)00149-4 [DOI] [Google Scholar]
- 29.Luna G, Dell'Anno A, Danovaro R. 2006. DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 8:308–320. 10.1111/j.1462-2920.2005.00896.x [DOI] [PubMed] [Google Scholar]
- 30.Alain K, Callac N, Ciobanu M-C, Reynaud Y, Duthoit F, Jebbar M. 2011. DNA extractions from deep subseafloor sediments: novel cryogenic-mill-based procedure and comparison to existing protocols. J. Microbiol. Methods 87:355–362. 10.1016/j.mimet.2011.09.015 [DOI] [PubMed] [Google Scholar]
- 31.Webster G, Newberry CJ, Fry J, Weightman AJ. 2003. Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55:155–164. 10.1016/S0167-7012(03)00140-4 [DOI] [PubMed] [Google Scholar]
- 32.Teske A, Sørensen KB. 2008. Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J. 2:3–18. 10.1038/ismej.2007.90 [DOI] [PubMed] [Google Scholar]
- 33.Hoshino T, Inagaki F. 2012. Molecular quantification of environmental DNA using microfluidics and digital PCR. Syst. Appl. Microbiol. 35:390–395. 10.1016/j.syapm.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 34.Morono Y, Terada T, Masui N, Inagaki F. 2009. Discriminative detection and enumeration of microbial life in marine subsurface sediments. ISME J. 3:503–511. 10.1038/ismej.2009.1 [DOI] [PubMed] [Google Scholar]
- 35.Morono Y, Inagaki F. 2010. Automatic slide-loader fluorescence microscope for discriminative enumeration of subseafloor life. Sci. Drill. 9:32–36. 10.2204/iodp.sd.9.05.2010 [DOI] [Google Scholar]
- 36.Morono Y, Terada T, Kallmeyer J, Inagaki F. 2013. An improved cell separation technique for marine subsurface sediments: applications for high-throughput analysis using flow cytometry and cell sorting. Environ. Microbiol. 15:2841–2849. 10.1111/1462-2920.12153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi T, Koide O, Mori K, Shimamura S, Matsuura T, Miura T, Takaki Y, Morono Y, Nunoura T, Imachi H, Inagaki F, Takai K, Horikoshi K. 2008. Phylogenetic and enzymatic diversity of deep subseafloor aerobic microorganisms in organics- and methane-rich sediments off Shimokita Peninsula. Extremophiles 12:519–527. 10.1007/s00792-008-0157-7 [DOI] [PubMed] [Google Scholar]
- 38.Masui N, Morono Y, Inagaki F. 2008. Microbiological assessment of circulation mud fluids during the first operation of riser drilling by the deep-earth research vessel Chikyu. Geomicrobiol. J. 25:274–282. 10.1080/01490450802258154 [DOI] [Google Scholar]
- 39.Aoike KE. 2007. CK06-06 D/V Chikyu shakedown cruise offshore Shimokita, laboratory operation report. CDEX-JAMSTEC, Yokohama, Japan: http://www.godac.jamstec.go.jp/catalog/data/doc_catalog/media/CK06-06_902_all.pdf [Google Scholar]
- 40.Tomaru H, Fehn U, Lu Z, Takeuchi R, Inagaki F, Imachi H, Kotani R, Matsumoto R, Aoike K. 2009. Dating of dissolved iodine in pore waters from the gas hydrate occurrence offshore Shimokita Peninsula, Japan: 129I results from the D/V Chikyu shakedown cruise. Resour. Geol. 59:359–373. 10.1111/j.1751-3928.2009.00103.x [DOI] [Google Scholar]
- 41.Kimura G, Screaton E, Curewitz D, Expedition 316 Scientists 2008. Integrated Ocean Drilling Program Expedition 316 preliminary report NanTroSEIZE stage 1A: NanTroSEIZE shallow megasplay and frontal thrusts. IODP, La Jolla, CA. 10.2204/iodp.pr.316.2008 [DOI] [Google Scholar]
- 42.Masui N, Morono Y, Inagaki F. 2009. Bio-archive core storage and subsampling procedure for subseafloor molecular biological research. Sci. Drill. 8:35–37 [Google Scholar]
- 43.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74:2461–2470. 10.1128/AEM.02272-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56:1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow ME. (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Ltd., Chichester, United Kingdom [Google Scholar]
- 46.Takai K, Horikoshi K. 2000. Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl. Environ. Microbiol. 66:5066–5072. 10.1128/AEM.66.11.5066-5072.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, DeLong EF. 2008. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U. S. A. 105:3805–3810. 10.1073/pnas.0708897105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao A. 1987. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43:783–791. 10.2307/2531532 [DOI] [PubMed] [Google Scholar]
- 52.Krebs C. 1998. Ecological methodology, 2nd ed. Addison Wesley Longman, Inc., Menlo Park, CA [Google Scholar]
- 53.Ciccolini L, Shamlou P, Titchener-Hooker N, Ward J, Dunnill P. 1998. Time course of SDS-alkaline lysis of recombinant bacterial cells for plasmid release. Biotechnol. Bioeng. 60:768–770. [DOI] [PubMed] [Google Scholar]
- 54.Valentine DL. 2007. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat. Rev. Microbiol. 5:316–323. 10.1038/nrmicro1619 [DOI] [PubMed] [Google Scholar]
- 55.Lee ZM-P, Bussema C, Schmidt TM. 2009. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 37:D489–D493. 10.1093/nar/gkn689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inagaki F, Suzuki M, Takai K, Oida H, Sakamoto T, Aoki K, Nealson KH, Horikoshi K. 2003. Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Appl. Environ. Microbiol. 69:7224–7235. 10.1128/AEM.69.12.7224-7235.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vetriani C, Jannasch H, MacGregor B, Stahl D, Reysenbach A. 1999. Population structure and phylogenetic characterization of marine benthic archaea in deep-sea sediments. Appl. Environ. Microbiol. 65:4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inagaki F, Nakagawa S. 2008. Spatial distribution of the subseafloor life: diversity and biogeography, p 135–158 In Dilek Y, Furnes H, Muehlenbachs K. (ed), Links between geological processes, microbial activities and evolution of life, vol 4 Springer, Dordrecht, Netherlands [Google Scholar]
- 59.Finkel SE. 2006. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat. Rev. Microbiol. 4:113–120. 10.1038/nrmicro1340 [DOI] [PubMed] [Google Scholar]
- 60.Holben WE, Jansson JK, Chelm BK, Tiedje JM. 1988. DNA probe method for the detection of specific microorganisms in the soil bacterial community. Appl. Environ. Microbiol. 54:703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeLong EF. 1992. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U. S. A. 89:5685–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyashita A, Mochimaru H, Kazama H, Ohashi A, Yamaguchi T, Nunoura T, Horikoshi K, Takai K, Imachi H. 2009. Development of 16S rRNA gene-targeted primers for detection of archaeal anaerobic methanotrophs (ANMEs). FEMS Microbiol. Lett. 297:31–37 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.