

Short abstract
The size of a given gene family is remarkably similar in strains of the same species and in closely related species, suggesting that homologous gene families are vertically transmitted and depend little on horizontal gene transfer.
Abstract
Background
The wealth of genomic data in bacteria is helping microbiologists understand the factors involved in gene innovation. Among these, the expansion and reduction of gene families appears to have a fundamental role in this, but the factors influencing gene family size are unclear.
Results
The relative content of paralogous genes in bacterial genomes increases with genome size, largely due to the expansion of gene family size in large genomes. Bacteria undergoing genome reduction display a parallel process of redundancy elimination, by which gene families are reduced to one or a few members. Gene family size is also influenced by sequence divergence and physiological function. Large gene families show wider sequence divergence, suggesting they are probably older, and certain functions (such as metabolite transport mechanisms) are overrepresented in large families. The size of a given gene family is remarkably similar in strains of the same species and in closely related species, suggesting that homologous gene families are vertically transmitted and depend little on horizontal gene transfer (HGT).
Conclusions
The remarkable preservation of copy numbers in widely different ecotypes indicates a functional role for the different copies rather than simply a back-up role. When different genera are compared, the increase in phylogenetic distance and/or ecological specialization disrupts this preservation, albeit in a gradual manner and maintaining an overall similarity, which also supports this view. HGT can have an important role, however, in nonhomologous gene families, as exemplified by a comparison between saprophytic and enterohemorrhagic strains of Escherichia coli.
Background
One of the unexpected revelations of prokaryotic genomes has been the existence of significant gene redundancy. The existence of multiple gene copies in eukaryotes has been known for a long time and is considered an important element in their molecular evolution [1,2]. In pre-genomic times, however, bacteria were considered to be streamlined cells that carried very little, if any, redundant information in their genomes. It therefore came as a surprise when the genome of Escherichia coli K12 showed that nearly 30% of the coding sequences could be grouped into gene families that were similar enough to be assigned similar functions [3,4]. They were described as 'paralog' gene families, with the implicit assumption that their similarity reflected similar evolutionary descent, but actual or potential functional divergence. Since then, the presence of gene families typically containing between two and 30 copies has been described for nearly every prokaryotic genome sequenced. The number of paralogous genes and families appears to correlate well with an increase in genome size [5,6]. The relative contribution of these genes in each genome seems to be independent of phylogenetic affiliation and, for a limited dataset, appears to depend on genome size [7].
These gene families of diverse size and degree of similarity remain an important and little explored feature of prokaryotes. In eukaryotic genomes they are generally taken as the result of gene duplication. This would either supply the required gene dosage or the raw material for adaptation by mutation and selection acting on one of the copies that diverges in properties or function [1,8]. In E. coli, a model organism in which traditional genetics and physiology have already allowed the unequivocal identification of more than half of the coding genes, the role of paralog families (whatever their origin) seems much more operational than in eukaryotes [4]. For example, the different members of a gene family contribute the proper gene dosage or, most often, provide different specificities for similar chemical reactions or for other processes such as transport of different molecules. Regarding origin, duplication is not necessarily the only source for new members of a gene family in prokaryotes. The gene pools are known to vary enormously from one strain to another [9,10], and horizontal gene transfer (HGT) acts as a powerful source of innovation [11]. Therefore, HGT could provide gene families with members already divergent in sequence and function [12]. In prokaryotes, gene families could be the result of incomplete xenologous gene replacement by which a gene from another genome gets incorporated into a gene family with which it shares some sequence similarity. This process would provide additional physiological plasticity, and studies on the DNA composition of paralogous genes suggest that its contribution might be substantial [13]. The divergence of some of the members of the gene families or their DNA composition could be taken as evidence for a HGT origin [4]. It is unclear at the moment the extent to which each of these genomic forces (gene duplication and HGT) contributes to genome expansion and variability [5,14-16].
To address these issues we have compared the size of gene families across bacterial taxa. To try to shed light on the evolutionary origin of these initially redundant genes we have studied the distribution of gene family size among completed genomes of strains within the same bacterial species and over larger taxonomic distances. If the different family members were acquired by HGT their numbers will vary widely among different strains, as already detected for single genes in adaptive islands [17] or for whole families predicted to have been transferred as a whole [18]. On the other hand, if the family numbers are similar in different strains, vertical descent or a very old HGT will be a more likely origin. We have also determined the contribution of paralogous families to genome size for all 127 available eubacterial genomes, updating earlier work on a more limited dataset [7]. We have also tried to identify other factors affecting the number of members in a family, besides genome size, particularly sequence divergence, gene function and species lifestyle.
Results and discussion
Gene family size in bacterial genomes
Previous work on a more reduced set of sequenced genomes had determined that large genomes contain more paralogs and more gene families than smaller genomes [7]. Jordan and collaborators also found a correlation between the fraction of the genome occupied by gene families and the genome size; that is, larger genomes had a larger proportion of redundant genes. However, at the time of that analysis, the sequences of genomes larger than 5 million base pairs (5 Mbp) were not available. Now, the inclusion of genomes nearly twice as large confirms both trends (Figure 1): for example, nearly 50% of the genome is occupied by paralogous genes in Streptomyces coelicolor. A closer look at these data shows that larger genomes have larger gene families, as the average family size also increases with genome size (Figure 1, inset). Thus, the higher percentage of paralogs in large genomes is partly due to the expansion of existing gene families, together with a larger number of new families. The large-genomed species at one end of the distribution, such as Streptomyces, have gene families of up to 85 members, whereas the largest gene families in middle-sized genomes such as those of E. coli or Salmonella have more moderate numbers (40-45). This is reminiscent of the situation in eukaryotes, where the number of gene families increases with the number of genes in the genome at a lower rate than in prokaryotes [6], indicating that gene families have many more members in the larger eukaryotic genomes. Also consistent with this trend, some reconstructions of prokaryotic genome evolution based on gene content conclude that gene duplication has a critical role in the expansion of genome size [15].
Figure 1.
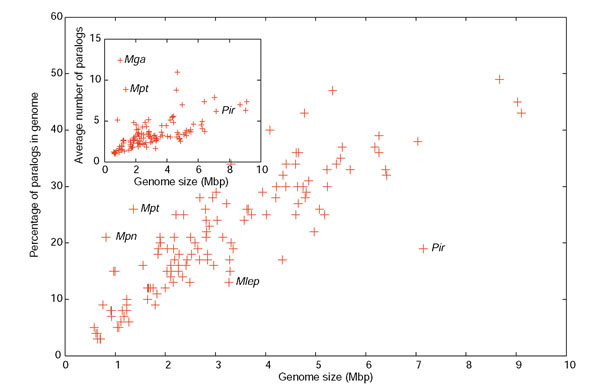
Relationship between percentage of genes belonging to paralogous families plotted versus genome size in 127 eubacterial genomes. Inset shows the average gene family size versus genome size for the same genomes, except Shigella flexneri, Bordetella pertusis, B. parapertussis and B. bronchiseptica, which contain a high number of IS elements. Some genomes with atypical values are identified: Mpn, Mycoplasma pneumoniae; Mpt, Mycoplasma penetrans; Mga, Mycoplasma gallisepticum; Mlp, Mycobacterium leprae; Pir, Pirellula sp.
Exceptions to the linear correlation in this graph are interesting to consider. On one hand, Pirellula (marked as Pir in Figure 1) has an enormous genome with a surprisingly low relative number of paralogs. This is due to an overrepresentation of small gene families and the absence of large ones (the largest gene family contains 57 members; see Additional data file 1). Pirellula is a marine bacterium and the reason for the reduced gene family size might be the homogeneity of the marine environment, in contrast to other large-genomed bacteria included in the graph which have the ability to survive in many different niches or in much more heterogeneous habitats, such as soil. In agreement with this, Pirellula has a greatly reduced number of transcriptional regulators, which again might reflect a relatively constant environment [19]. At the other end of the distribution, exceptions occur for three species that have small genomes with a larger-than-expected percentage of paralogs. All these species are mycoplasmas, and the high percentage of paralogs is due to a few gene families that are greatly expanded, including more than 25 members. In Mycoplasma penetrans, for example, these families include surface-exposed lipoproteins involved in antigenic variation [20], which are critical to the success of microbes exposed to the immune system of their hosts. On the other hand, the small genomes of other pathogenic bacteria correspond to intracellular parasites that do not need to evade the immune system [21], and these species show the smallest portion of paralogs. Finally, the largest gene families that we detected were those involving mobile genetic elements such as the IS elements of Shigella flexneri, where families surpassed 100 members (not included in the inset of Figure 1).
The data in Figure 1 cannot be viewed as a continuum, because small genomes are not ancestral to bigger ones. Instead, small genomes have been shown to be the result of reductive evolution, a process by which a larger-sized ancestor changes niche and undergoes a dramatic loss of DNA [22,23]. Both small and large genome fragments can be eliminated but the outcome of this process for gene families has not been documented. We have compared the number of members per gene family in two genomes that are undergoing rapid reductive evolution - Shigella flexneri 2a and Mycobacterium leprae TN - with larger-genomed close relatives (Figure 2). Shigella is a close relative of E. coli that has specialized in living as a human pathogen [24,25]. As a result of the expansion of the human population from Neolithic times a number of more generalistic or opportunistic pathogens found a new niche; Salmonella typhi might be a similar example [26]. In both cases there is a clear tendency to genome reduction accompanied by expansion of IS families (314 and 46 IS elements, respectively).
Figure 2.
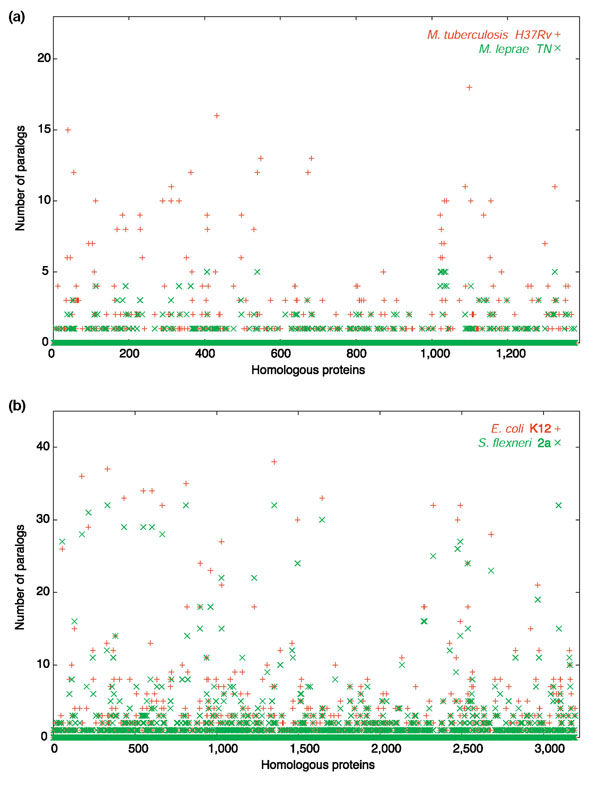
Gene family sizes in genomes undergoing reductive evolution compared to a phylogenetically related larger sequenced genome. (a) Mycobacterium leprae (reductive) vs Mycobacterium tuberculosis H37Rv; (b) Shigella flexneri (reductive) vs Escherichia coli K12. Orthologous genes in the genome pairs (identified by amino-acid sequence similarity) are displayed in arbitrary order and plotted against the number of homologs in their own genome (that is, paralogs). Only protein-coding genes are included. IS elements from S. flexneri 2a are excluded.
In Shigella there is a clear reduction in gene family copy number (Figure 2), which seems to be higher than would be expected from the random location of IS elements, suggesting that they might insert preferentially in gene family members. Something similar is found in the case of M. leprae (Figure 3), although in this case the main mechanism for gene inactivation is the generation of pseudogenes by mutation [27]. M. leprae is closely related to M. tuberculosis, with which it shares many homologous sequences. However, most gene families have been simplified in the short time period in which the leprosy bacillus has adopted its mainly intracellular lifestyle. This also illustrates the fact that, as described above, an early step in genome reduction allowed by intracellular parasitism or a narrower range of hosts is the shrinkage of gene families. It shows that the smaller percentage of paralogs in reduced genomes is probably due to simplification of existing gene families. A similar pattern was found in the small-genomed intracellular species Rickettsia and Buchnera when compared with free-living species of the same taxonomic group (see Additional data file 2). Thus, both genome expansion and reduction can be partly explained by the parallel growth or simplification, respectively, of gene families.
Figure 3.

The number of members in E. coli K12 gene families plotted versus mean sequence identity of pairwise comparisons among the members of each family.
Another feature we could detect in the evolution of gene families was that large families were more divergent (Figure 3). This could partly be due to a side-effect of the higher variability of a larger sample size or to misidentification of family members at low sequence identity levels. However, given the observed similarity of functions in these large families ([4,28] and R.P., A.M. and F.R-V., unpublished results), a substantial proportion must be true paralogous genes. Thus, this relationship can be interpreted as older (more divergent) families containing more members. Smaller families range from those with very similar members to those in which the members are very different. The latter probably represent either old families in which new members have not evolved because new duplications do not confer a selective advantage, or more recent incomplete xenologous replacements.
Gene family size in intraspecific and interspecific comparisons
The sequencing of several strains of a single species is now common in bacterial genomics. One of the most remarkable findings has been the different gene pools carried by strains that are highly similar if their housekeeping genes only are compared. For example, different virotypes of E. coli were shown to contain very different gene complements, with large pools of genes characteristic of each virotype [10]. Obvious candidates to vary would be multigene families. Thus, the comparison of the numbers of members within a single species might shed light in their origin. If the members of a gene family are frequently acquired by HGT from outside, the numbers should be expected to vary broadly in different lineages of the species (as a result of different acquisitions). On the other hand, if the numbers are similar, that would indicate that the families were already present in the common ancestor and represent a relatively stable feature of the genome.
We selected distinct prokaryotic taxa in which three or more strains have been fully sequenced (Escherichia coli, Streptococcus pyogenes, Staphylococcus aureus and Chlamydophila pneumoniae) and for each taxon established a list of homologous genes common to all strains. The gene family to which each homolog belonged was determined for each strain, and the number of family members compared for equivalent families (Figure 4). In all four species considered, the different strains showed a remarkably similar pattern of gene family size distributions: large gene families in one strain were also expanded in the others; small families were small, regardless of strain or virotype. Caution has to be exercised when examining these plots, as a gene can be a member of more than one gene family. However, although some of the gene families in Figure 4 are redundant, the parallel size pattern of gene families across strains is remarkably clear and seems to reflect a stable feature of the genome. Thus, the majority of gene families were most likely to have been formed by ancestral gene duplications or ancient gene transfers common to all strains. In addition, the preservation of gene family size in different strains strongly suggests that most family members have a high value for survival; redundant copies would otherwise be quickly eliminated.
Figure 4.

Gene family sizes for homologous genes in groups of strains belonging to the same species, represented as in Figure 2. (a) Chlamydophila pneumoniae strains; (b) Streptococcus pyogenes strains; (c) Escherichia coli strains; (d) Staphylococcus aureus strains. Strain denomination and graph code displayed in the top right-hand corner. Only protein-coding genes are included. Zero on the y-axis indicates single-copy genes; 1 indicates a gene family formed of two members.
We have obviously not excluded the possibility that nonhomologous gene families add to the differences among the compared genomes. For example, in a pairwise comparison between E. coli K12 and E. coli O157:H7, 186 genes belonging to paralog families were unique to K12 and 788 to O157:H7, versus 403 singletons (single-copy genes not belonging to families) unique to K12 and 883 to O157:H7. Thus, K12 keeps the same standard proportion of 30% paralogs for the differential gene pool. In O157:H7, on the other hand, paralogs account for 47% of the set of unique genes. The interpretation might be that the large islands that characterize the genome of the enterohemorrhagic virotype tend to carry a bigger proportion of families than the rest of the genome. Thus, it is possible that in some strains, HGT may contribute to expand and generate gene families that do not appear as homologs in closely related genomes. For example, 146 genes belonging to families of 10 or more members were detected in the O157:H7 differential pool, including three whole families of 14, 17 and 20 members with a G+C content of 57, 54 and 53%, respectively (the average G+C content in E. coli O157:H7 is 50.6%). The largest differential family in K12 had 11 members, which were not present in the enterohemorrhagic strain, and had a G+C content of 54.1% (the average G+C content of E. coli K12 is 50.5%).
To investigate whether the conservation in the size of homologous families was maintained across more divergent genomes, gene family plots were performed between species. A representative case for a Gram-negative (Pseudomonas) and a Gram-positive (Bacillus) comparison is illustrated in Figure 5. The preservation of family size was still remarkable, although, in the case of Pseudomonas, the number of orthologous genes is considerable smaller. The overall pattern of family sizes is preserved across these species. The two Bacillus species considered have the same genome size and one species contains larger numbers in some families but fewer in others (Figure 5b). The same trend was found in comparisons between species of Staphylococcus, Streptococcus, Salmonella and Mycoplasma (data not shown). It is also interesting to analyze the variation detected. Part of it can be attributed to differences in genome size. Pseudomonas syringae is approximately 200 kb larger than its other sequenced partners, which have mostly smaller gene families. However, part of the variability is also due to intrinsic differences between the species. For example, P. syringae contains some large gene families involved in invasion of the plant host and in pathogenesis [29]. One way to examine whether this variation can underlie the phenotypic/ecological characteristics of a given species is to visualize the size difference of each paralog group for some representative cases.
Figure 5.

Gene family sizes for homologous protein-coding genes in different species of the same genus. (a) Pseudomonas spp; (b) Bacillus spp. (c) Difference in the size of equivalent gene families between E. coli K12 and S. typhimurium LT2. Positive values indicate larger families in E. coli; negative values indicate larger families in S. typhymurium. The potG gene family is indicated.
Figure 5c shows the difference in gene family size in the interspecific comparison of E. coli K12 and S. typhimurium LT2. Both strains have similarly sized genomes (S. typhimurium is 218 kb larger) and a relatively high level of homology (3,026 orthologous genes). Of these, there are 572 homologs belonging to families that differ in size between the two genomes, and 435 belonging to families having the same number of members in both species. The rest are single-copy genes in both genomes. Forty-eight families were significantly larger (two or more extra copies) in E. coli, while 53 were larger in Salmonella. These differences can be taken as an example of the evolution of gene families in two diverging groups. Although the natural history of these model bacteria is not as well known as might be expected, it is generally believed that both Salmonella and Escherichia are mostly saprophytic facultative anaerobes that inhabit the intestine of vertebrates. The divergence between these two microbes arose after the origin of mammals around 120 million years ago. E. coli specialized as a commensal and an opportunistic pathogen of mammals, as witnessed, for example, by its ability to degrade lactose. On the other hand, Salmonella remains as a commensal in reptiles, with some serotypes colonizing mammals, but as a pathogen rather than a commensal and after developing strategies for intracellular invasion of the host [30,31]. Accepting this scenario, the fact that many gene families (and the number of members of each family) are preserved reflects a significant involvement in the saprophytic intestinal lifestyle, preserved over many millions of years. On the other hand, significant differences are starting to arise between the two species, perhaps reflecting their specialization in different hosts and lifestyles [32]. A dramatic example is the potG gene family, which has 13 more members in S. typhimurium than in E. coli (Figure 5). This is an ATP-binding component of spermidine/putrescine transport and for some reason its amplification has been selected in this species. Proteins involved in the transport of spermidine and putrescine have been shown to be involved in attachment to host cells and virulence [33]. Therefore, the size of this gene family might reflect the more pathogenic lifestyle of Salmonella.
Functional classification of gene families
Do certain functions predispose genes to form families? Do single genes that do not form families belong to a different category? To address these questions, extended gene families were identified, where a gene was not allowed to belong to more than one family. Thus, if gene A matched gene B, and gene B matched C, but A did not match C, all three were considered part of the same family, as it is likely that they are all evolutionarily derived from each other [34]. This method of transitive assembly of paralogs has been confirmed to include, in most cases, genes with related functions [4]. We found that, for all 127 sequenced species, singletons (genes in a single copy in a given genome) were massively overrepresented by genes with an unknown or hypothetical function. When only genes with a known or predicted function were included, these single genes without paralogs appeared equally distributed among the different functional categories. However, when genes belonging to families, especially large ones, were considered, a significant fraction had particular functions, such as transport of metabolites (data not shown). These data are, however, probably unrealistic because they represent the distribution of genes in sequenced genomes only, and certain species are overrepresented. In addition, larger genomes will also weigh more in this comparison than small genomes, as will species with several sequenced strains. We did, however, find relatively uniform results for individual genomes. Figure 6 shows such a distribution for two species, one Gram-negative (E. coli K12) and the other Gram-positive (B. subtilis). For E. coli, in which a large proportion of genes has been allocated a function, families with more than five members contain fewer unknown or hypothetical genes than do smaller families, and the distribution of functions among categories is unequal, with certain categories being overrepresented. Among these, genes involved in transport of different metabolites predominate (39% of the total), followed by those with transcription and replication/repair functions. In genes that do not belong to a family, however, most functional categories are equally represented and a large proportion of these singletons have an unknown function. The overrepresentation of unknown or hypothetical open reading frames (ORFs) could, in part, be due to many of these singletons not being real genes, as supported by their shorter length when compared to genes belonging to families. In the gamma-proteobacteria, for example, average singleton length is 127 nucleotides less than in genes belonging to families. It is also interesting to note that the phylogenetic distribution of these unknown singletons is not different from that of unknown paralogs (see Additional data file 4). In conclusion, some functions do appear to be more prone to develop families, although the functions overrepresented in a particular species may depend on its lifestyle.
Figure 6.

Proportions of assigned functions among genes belonging to families and singletons in B. subtilis and E. coli K12. Gene functions were assigned according to the Cluster of Orthologous Genes (COGs) classification [41]. Extended gene families are considered, in which a gene belongs to a single family only (see Materials and methods).
Conclusions
In eukaryotic genomes, a cornerstone of gene creation is extension of paralogous families by gene duplication [2]. This is reflected in the slow increase of new gene families with genome size, which does correlate with an increase in the size of the families [6]. The importance of DNA duplication in eukaryotes is probably also favored by the limitations of HGT in this group [35]. Despite the pervasiveness of HGT in prokaryotes, the increase in gene families with genome size is also robust (Figure 1). One obvious fact contributing to this situation might be that the pool of essential genes that have to be present for basic cell biology represents a larger percentage of a smaller genome, restricting the contribution of redundant genes with related functions and thus more expendable. However, this does not explain the high level of correlation maintained at the larger end of the range.
Of course, with the number of genomes available presently there is a certain representation bias, with a large input from human pathogens. Among these, small genomes often correspond to intracellular forms that are protected from the immune system of the host. Variability of antigen specificity is one paradigmatic case that justifies gene familes in extracellular pathogens of vertebrates, for example the PPE genes of Mycobacterium [28] and the Pap adhesins in E. coli [36]. The exceptional case of the mycoplasmas points in this direction as they possess small genomes but are extracellular mucosa-associated pathogens, and hence subjected to the host immune system [37]. At the other end of the genome size range there are many more free-living, saprophytic or opportunistic pathogens, a lifestyle that requires a highly versatile gene complement in order to survive, for example, both inside and outside a host. Again, the one exception is a single large-genome species from a relatively stable environment (Pirellula, which lives in the open ocean). Here, the possibility to carry out many different physiological activities is probably more advantageous than the ability to adapt the same activity to a wider range of conditions. Thus, as with other aspects of biology, the genomic properties of bacteria appear to be greatly conditioned by their specialist or generalist lifestyle.
The comparison of gene family size among strains from a single species shows a remarkable level of conservation, even when genome sizes are very different. This conservation indicates that gene family size is probably an ancestral feature rather than reflecting the acquisition of paralogs by HGT. This is consistent with evolutionary models based on bacterial gene content, which concluded that most protein gene families are transmitted by vertical inheritance [16]. The conservation that is detected even among more distantly related taxa strengthens this view, as in mostly free-living and very niche-diversified species such as Pseudomonas, there is a remarkable degree of conservation. This might reflect involvement of the gene families in more fundamental (less environment-dependent) processes of cell biology.
Genomic evolution simulations concluded that the amount of gene duplication is independent of HGT levels [15]. On the basis of these simulations, an upper limit of 20% was estimated for paralogs of xenologous origin. Assuming that the extra members of a gene family from our paralog plots represent an upper limit of HGT for established families, we calculate that gene transfer accounts for a maximum of 11% of a given family in E. coli (Figure 4c). However, this does not take into account families that are unique to a given strain and that may have a xenologous origin. The fact that these families are not included in the paralog plots (which display only homolog pairs between strains) suggests that they can represent transfers to a given strain. Thus, the paralog plots present a picture of stability and limited xenologous genes for already established families, but this is not inconsistent with the transfer of families that appear to be unique to a given strain or species. It could, theoretically, be more probable that gene families expand by horizontal transfers than by gene duplication [12]. This way, xenologous genes would already confer a functionally distinct role and would avoid the neutrality period in which redundant gene copies coexist and can be eliminated [38]. The results shown here suggest that the overrepresentation of duplications among transferred genes found by Hooper and Berg [13] might be a feature of these specific families but not of more ancient, homologous ones.
Materials and methods
The protein sequences of the 127 completely sequenced eubacterial genomes at the time this paper was submitted for publication were retrieved from the Genome division, Entrez retrieval system of the National Center for Biotechnology Information (NCBI; [39]). Table 1 shows a list with all the genomes used, with their genome size and accession numbers. To detect potentially homologous genes we started by carrying out an all-against-all BLASTP [40] search of every protein sequence in one genome against every protein sequence in all the other genomes. We then recorded the best reciprocal hit for each protein sequence with an E-value lower than 10-5 and sequence identity higher than 50% over more than 60% of the length. To validate the results, we performed some representative comparisons by studying the distribution of the ratio of bit score to the maximal bit score [41]. This method would separate probable homology from random similarity. We obtained almost identical results, with only a reduced set of the respective homologous genes being different in the two lists. For example, out of 3,026 homolog pairs between E. coli K12 and S. typhimurium detected by the reciprocal hit method, only one pair was found to differ with the bit score method. In addition, only three genes were detected with the reciprocal best-hit method that were not selected as homologs using the bit score method (using a cut-off value of 0.4). Finally, the bit-score ratio method identified 165 additional homologs that were not selected using reciprocal best-hits because they did not satisfy the length and/or sequence-identity requirements. Therefore, the list of homologous genes obtained by reciprocal best-hits was used for all the analyses.
Table 1.
Species used in the current work and their accession numbers
| Species | Accession number | Genome size (bp) |
| Agrobacterium tumefaciens str. C58 (Cereon) | NC_003062 | 2,841,581 |
| Agrobacterium tumefaciens str. C58 (U. Washington) | NC_003304 | 2,841,490 |
| Aquifex aeolicus VF5 | NC_000918 | 1,551,335 |
| Bacillus anthracis str. Ames | NC_003997 | 5,227,293 |
| Bacillus cereus ATCC 14579 | NC_004722 | 5,411,809 |
| Bacillus halodurans | NC_002570 | 4,202,353 |
| Bacillus subtilis subsp. subtilis str. 168 | NC_000964 | 4,214,814 |
| Bacteroides thetaiotaomicron VPI-5482 | NC_004663 | 6,260,361 |
| Bifidobacterium longum NCC2705 | NC_004307 | 2,256,646 |
| Bordetella bronchiseptica | NC_002927 | 5,339,179 |
| Bordetella parapertussis | NC_002928 | 4,773,551 |
| Bordetella pertussis | NC_002929 | 4,086,189 |
| Borrelia burgdorferi B31 | NC_001318 | 910,724 |
| Bradyrhizobium japonicum USDA 110 | NC_004463 | 9,105,828 |
| Brucella melitensis 16M | NC_003317 | 2,117,144 |
| Brucella suis 1330 | NC_004310 | 2,107,792 |
| Buchnera aphidicola str. APS (Acyrthosiphon pisum) | NC_002528 | 640,681 |
| Buchnera aphidicola str. Bp (Baizongia pistaciae) | NC_004545 | 615,980 |
| Buchnera aphidicola str. Sg (Schizaphis graminum) | NC_004061 | 641,454 |
| Campylobacter jejuni subsp. jejuni NCTC 11168 | NC_002163 | 1,641,481 |
| Candidatus Blochmannia floridanus | NC_005061 | 705,557 |
| Caulobacter crescentus CB15 | NC_002696 | 4,016,947 |
| Chlamydia muridarum | NC_002620 | 1,072,950 |
| Chlamydia trachomatis | NC_000117 | 1,042,519 |
| Chlamydophila caviae GPIC | NC_003361 | 1,173,390 |
| Chlamydophila pneumoniae AR39 | NC_002179 | 1,229,858 |
| Chlamydophila pneumoniae CWL029 | NC_000922 | 1,230,230 |
| Chlamydophila pneumoniae J138 | NC_002491 | 1,226,565 |
| Chlamydophila pneumoniae TW-183 | NC_005043 | 1,225,935 |
| Chlorobium tepidum TLS | NC_002932 | 2,154,946 |
| Chromobacterium violaceum ATCC 12472 | NC_005085 | 4,751,080 |
| Clostridium acetobutylicum | NC_003030 | 3,940,880 |
| Clostridium perfringens str. 13 | NC_003366 | 3,031,430 |
| Clostridium tetani E88 | NC_004557 | 2,799,251 |
| Corynebacterium diphtheriae | NC_002935 | 2,488,635 |
| Corynebacterium efficiens YS-314 | NC_004369 | 3,147,090 |
| Corynebacterium glutamicum ATCC 13032 | NC_003450 | 3,309,401 |
| Coxiella burnetii RSA 493 | NC_002971 | 1,995,275 |
| Deinococcus radiodurans | NC_001263 | 2,648,638 |
| Enterococcus faecalis V583 | NC_004668 | 3,218,031 |
| Escherichia coli CFT073 | NC_004431 | 5,231,428 |
| Escherichia coli K12 | NC_000913 | 4,639,221 |
| Escherichia coli O157:H7 | NC_002695 | 5,498,450 |
| Escherichia coli O157:H7 EDL933 | NC_002655 | 5,528,445 |
| Fusobacterium nucleatum subsp. nucleatum ATCC 25586 | NC_003454 | 2,174,500 |
| Gloeobacter violaceus | NC_005125 | 4,659,019 |
| Haemophilus ducreyi 35000HP | NC_002940 | 1,698,955 |
| Haemophilus influenzae Rd | NC_000907 | 1,830,138 |
| Helicobacter hepaticus ATCC 51449 | NC_004917 | 1,799,146 |
| Helicobacter pylori 26695 | NC_000915 | 1,667,867 |
| Helicobacter pylori J99 | NC_000921 | 1,643,831 |
| Lactobacillus plantarum WCFS1 | NC_004567 | 3,308,274 |
| Lactococcus lactis subsp. lactis | NC_002662 | 2,365,589 |
| Leptospira interrogans serovar lai str. 56601 | NC_004342 | 4,332,241 |
| Listeria innocua | NC_003212 | 3,011,208 |
| Listeria monocytogenes EGD-e | NC_003210 | 2,944,528 |
| Mesorhizobium loti | NC_002678 | 7,036,074 |
| Mycobacterium bovis subsp. bovis AF2122/97 | NC_002945 | 4,345,492 |
| Mycobacterium leprae | NC_002677 | 3,268,203 |
| Mycobacterium tuberculosis CDC1551 | NC_002755 | 4,403,836 |
| Mycobacterium tuberculosis H37Rv | NC_000962 | 4,411,529 |
| Mycoplasma gallisepticum R | NC_004829 | 996,422 |
| Mycoplasma genitalium | NC_000908 | 580,074 |
| Mycoplasma penetrans | NC_004432 | 1,358,633 |
| Mycoplasma pneumoniae | NC_000912 | 816,394 |
| Mycoplasma pulmonis | NC_002771 | 963,879 |
| Neisseria meningitidis MC58 | NC_003112 | 2,272,351 |
| Neisseria meningitidis Z2491 | NC_003116 | 2,184,406 |
| Nitrosomonas europaea ATCC 19718 | NC_004757 | 2,812,094 |
| Nostoc sp. PCC 7120 | NC_003272 | 6,413,771 |
| Oceanobacillus iheyensis HTE831 | NC_004193 | 3,630,528 |
| Pasteurella multocida | NC_002663 | 2,257,487 |
| Photorhabdus luminescens subsp. laumondii TTO1 | NC_005126 | 5,688,987 |
| Pirellula sp. | NC_005027 | 7,145,576 |
| Porphyromonas gingivalis W83 | NC_002950 | 2,343,476 |
| Prochlorococcus marinus str. MIT 9313 | NC_005071 | 2,410,873 |
| Prochlorococcus marinus subsp. marinus str. CCMP1375 | NC_005042 | 1,751,080 |
| Prochlorococcus marinus subsp. pastoris str. CCMP1378 | NC_005072 | 1,657,990 |
| Pseudomonas aeruginosa PA01 | NC_002516 | 6,264,403 |
| Pseudomonas putida KT2440 | NC_002947 | 6,181,863 |
| Pseudomonas syringae pv. tomato str. DC3000 | NC_004578 | 6,397,126 |
| Ralstonia solanacearum | NC_003295 | 3,716,413 |
| Rickettsia conorii | NC_003103 | 1,268,755 |
| Rickettsia prowazekii | NC_000963 | 1,111,523 |
| Salmonella enterica subsp. enterica serovar Typhi | NC_003198 | 4,809,037 |
| Salmonella enterica subsp. enterica serovar Typhi Ty2 | NC_004631 | 4,791,961 |
| Salmonella typhimurium LT2 | NC_003197 | 4,857,432 |
| Shewanella oneidensis MR-1 | NC_004347 | 4,969,803 |
| Shigella flexneri 2a str. 2457T | NC_004741 | 4,599,354 |
| Shigella flexneri 2a str. 301 | NC_004337 | 4,607,203 |
| Sinorhizobium meliloti | NC_003047 | 3,654,135 |
| Staphylococcus aureus subsp. aureus MW2 | NC_003923 | 2,820,462 |
| Staphylococcus aureus subsp. aureus Mu50 | NC_002758 | 2,878,040 |
| Staphylococcus aureus subsp. aureus N315 | NC_002745 | 2,814,816 |
| Staphylococcus epidermidis ATCC 12228 | NC_004461 | 2,499,279 |
| Streptococcus agalactiae 2603V/R | NC_004116 | 2,160,267 |
| Streptococcus agalactiae NEM316 | NC_004368 | 211,485 |
| Streptococcus mutans UA159 | NC_004350 | 203,0921 |
| Streptococcus pneumoniae R6 | NC_003098 | 2,038,615 |
| Streptococcus pneumoniae TIGR4 | NC_003028 | 2,160,837 |
| Streptococcus pyogenes M1 GAS | NC_002737 | 1,852,441 |
| Streptococcus pyogenes MGAS315 | NC_004070 | 1,900,521 |
| Streptococcus pyogenes MGAS8232 | NC_003485 | 1,895,017 |
| Streptococcus pyogenes SSI-1 | NC_004606 | 1,894,275 |
| Streptomyces avermitilis MA-4680 | NC_003155 | 9,025,608 |
| Streptomyces coelicolor A3(2) | NC_003888 | 8,667,507 |
| Synechococcus sp. WH 8102 | NC_005070 | 2,434,428 |
| Synechocystis sp. PCC 6803 | NC_000911 | 3,573,470 |
| Thermoanaerobacter tengcongensis | NC_003869 | 2,689,445 |
| Thermosynechococcus elongates BP-1 | NC_004113 | 2,593,857 |
| Thermotoga maritima | NC_000853 | 1,860,725 |
| Treponema pallidum | NC_000919 | 1,138,011 |
| Tropheryma whipplei TW08/27 | NC_004551 | 925,938 |
| Tropheryma whipplei str. Twist | NC_004572 | 927,303 |
| Ureaplasma urealyticum | NC_002162 | 751,719 |
| Vibrio cholerae | NC_002505 | 2,961,149 |
| Vibrio parahaemolyticus RIMD 2210633 | NC_004603 | 3,288,558 |
| Vibrio vulnificus CMCP6 | NC_004459 | 3,281,945 |
| Vibrio vulnificus YJ016 | NC_005139 | 3,354,505 |
| Wigglesworthia glossinidia (from Glossina brevipalpis) | NC_004344 | 697,724 |
| Wolinella succinogenes | NC_005090 | 2,110,355 |
| Xanthomonas axonopodis pv. citri str. 306 | NC_003919 | 5,175,554 |
| Xanthomonas campestris pv. campestris str. ATCC 33913 | NC_003902 | 5,076,188 |
| Xylella fastidiosa 9a5c | NC_002488 | 2,679,306 |
| Xylella fastidiosa Temecula1 | NC_004556 | 2,519,802 |
| Yersinia pestis CO92 | NC_003143 | 4,653,728 |
| Yersinia pestis KIM | NC_004088 | 4,600,755 |
To detect potential paralogous genes, we carried out an all-against-all BLASTP [40] search of every protein sequence in a genome against every protein sequence in the same genome. We define paralogs as protein sequences satisfying an E-value threshold of 10-5 in BLASTP [40] search and having at least 30% sequence identity over more than 60% of their lengths [3].
When comparing paralogs between two species, a gene family was created for each homologous gene detected in both genomes. This gave rise to some redundant families but ensured that the comparison between species was done between equivalent gene families. To describe the functional assignment of paralogous genes, extended gene families were created [3] that contained all genes that were interrelated by hits among any of their members. This is based on the transitive nature of sequence homology [34] and is supported by the findings on well-studied genomes of species with a relatively well-known metabolism. In these cases, extended gene families seem to be formed by genes involved in similar functions [4]. To minimize the incorporation of multidomain proteins in a family together with unrelated members [2], length cut-offs were kept at 60%. The assignment of a function to a gene was based on the Clusters of Orthologous Groups (COGs) classification [42].
Additional data files
Additional data file 1 is a PDF file of a figure showing the number of paralogs and the percentage of paralogous genes in the different-sized gene families in Pirelulla sp. compared to other large-sized genomes. Additional data file 2 is a PDF file of a figure showing gene family sizes in intracellular genomes that have undergone reductive evolution compared to related free-living organisms. Additional data file 3 contains legends to the figures in Additional data files 1 and 2. Additional data file 4 is a zip file containing the data from which the figures in the manuscript were made. The files are ordered following the figures as they appear in the text, and a readme text file explains the content of each file.
Supplementary Material
A figure showing the number of paralogs and the percentage of paralogous genes in the different-sized gene families in Pirelulla sp. compared to other large-sized genomes
A figure showing gene family sizes in intracellular genomes that have undergone reductive evolution compared to related free-living organisms
The legends to the figures in Additional data files 1 and 2
A zip file containing the data from which the figures in the manuscript were made
Acknowledgments
Acknowledgements
A.M. is the recipient of a 'Ramón y Cajal' research contract from the Spanish Ministry of Science and Technology (MCyT). Support from European Commission Project GEMINI (QLK3-CT-2002-02056) and MCYT project PM1999-0078 is also acknowledged. We thank Stuart Ingham for help with the graphics.
References
- Ohno S. Evolution by Gene Duplication. New York: Springer; 1970. [Google Scholar]
- Gogarten JP, Olendzenski L. Orthologs, paralogs and genome comparisons. Curr Opin Genet Dev. 1999;9:630–636. doi: 10.1016/S0959-437X(99)00029-5. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Liang P, Labedan B, Riley M. Physiological genomics of Escherichia coli protein families. Physiol Genomics. 2002;9:15–26. doi: 10.1152/physiolgenomics.00086.2001. [DOI] [PubMed] [Google Scholar]
- Hooper SD, Berg OG. Duplication is more common among laterally transferred genes than among indigenous genes. Genome Biol. 2003;4:R48. doi: 10.1186/gb-2003-4-8-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Kunin V, Ouzounis CA. Protein families and TRIBES in genome sequence space. Nucleic Acids Res. 2003;31:4632–4638. doi: 10.1093/nar/gkg495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Makarova KS, Spouge JL, Wolf YI, Koonin EV. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001;11:555–565. doi: 10.1101/gr.GR-1660R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and cosequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci. 2000;97:14668–14673. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RA, Burland V, Plunkett G, 3rd, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Hendrickson H. Lateral gene transfer: when will adolescence end? Mol Microbiol. 2003;50:739–749. doi: 10.1046/j.1365-2958.2003.03778.x. [DOI] [PubMed] [Google Scholar]
- Hooper SD, Berg OG. On the nature of gene innovation: duplication patterns in microbial genomes. Mol Biol Evol. 2003;20:945–54. doi: 10.1093/molbev/msg101. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen M. Genome evolution. Gene fusion versus gene fission. Trends Genet. 2000;16:9–11. doi: 10.1016/S0168-9525(99)01924-1. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen M. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002;12:17–25. doi: 10.1101/gr.176501. [DOI] [PubMed] [Google Scholar]
- Kunin V, Ouzounis CA. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003;13:1589–1594. doi: 10.1101/gr.1092603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschäpe H, Hacker J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- Glöckner FO, Kube M, Bauer M, Teeling H, Lombardot T, Ludwig W, Gade D, Beck A, Borzym K, Heitmann K, et al. Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc Natl Acad Sci USA. 2003;100:8298–8303. doi: 10.1073/pnas.1431443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Ishikawa J, Yamashita A, Oshima K, Kenri T, Furuya K, Yoshino C, Horino A, Shiba T, Sasaki T, Hattori M. The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acids Res. 2002;30:5293–5300. doi: 10.1093/nar/gkf667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T. Genome analysis of pathogenic bacteria - a review. Nippon Rinsho. 2000;58:1315–1325. [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/S0966-842X(98)01312-2. [DOI] [PubMed] [Google Scholar]
- Moran NA, Mira A. The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol. 2001;2:research0054.1–0054.12. doi: 10.1186/gb-2001-2-12-research0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, et al. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002;30:4432–4441. doi: 10.1093/nar/gkf566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G, 3rd, Rose DJ, Darling A, et al. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun. 2003;71:2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Brennan MJ, Delogu G. The PE multigene family: a 'molecular mantra' for mycobacteria. Trends Microbiol. 2002;10:246–249. doi: 10.1016/S0966-842X(02)02335-1. [DOI] [PubMed] [Google Scholar]
- Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, Gwinn ML, Dodson RJ, Deboy RT, Durkin AS, Kolonay JF, et al. Complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci USA. 2003;100:10181–10186. doi: 10.1073/pnas.1731982100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Wilson AC. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol. 1987;26:74–86. doi: 10.1007/BF02111283. Erratum in: J Mol Evol 26: 377. [DOI] [PubMed] [Google Scholar]
- Selander RK. DNA sequence analysis of the genetic structure and evolution of Salmonella enterica. In: van der Zeijst BAM, Hoekstra WPM, van Embden JDA, van Alphen AJW, editor. In Ecology of Pathogenic Bacteria Molecular and Evolutionary Aspects. Amsterdam, The Netherlands: Royal Netherlands Academy of Arts and Sciences; 1997. pp. 191–214. [Google Scholar]
- Winfield MD, Groisman EA. Role of nonhost environments in the lifestyles of Salmonella and Escherichia coli. Appl Environ Microbiol. 2003;69:3687–3694. doi: 10.1128/AEM.69.7.3687-3694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthysse AG, Yarnall HA, Young N. Requirement for genes with homology to ABC transport systems for attachment and virulence of Agrobacterium tumefaciens. J Bacteriol. 1996;178:5302–5308. doi: 10.1128/jb.178.17.5302-5308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SM. Bioinformatics: A Biologist's Guide to Biocomputing and the Internet. Natick, MA: Eaton Publishing; 2000. [Google Scholar]
- Andersson JO, Doolittle WF, Nesbo CL. Genomics. Are there bugs in our genome? Science. 2001;292:1848–50. doi: 10.1126/science.1062241. [DOI] [PubMed] [Google Scholar]
- Blomfield IC. The regulation of pap and type 1 fimbriation in Escherichia coli. Adv Microb Physiol. 2001;45:1–49. doi: 10.1016/s0065-2911(01)45001-6. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Park J, Chothia C. Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc Natl Acad Sci USA. 1998;95:14658–14663. doi: 10.1073/pnas.95.25.14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG. Catalyzing bacterial speciation: correlating lateral transfer with genetic headroom. Syst Biol. 2001;50:479–496. doi: 10.1080/10635150120286. [DOI] [PubMed] [Google Scholar]
- NCBI genomes. 2001. ftp://ftp.ncbi.nih.gov/genomes/Bacteria
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E, Daubin V, Moran NA. From gene trees to organismal phylogeny in prokaryotes: the case of the γ-proteobacteria. PLoS Biol. 2003;1:E19. doi: 10.1371/journal.pbio.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A figure showing the number of paralogs and the percentage of paralogous genes in the different-sized gene families in Pirelulla sp. compared to other large-sized genomes
A figure showing gene family sizes in intracellular genomes that have undergone reductive evolution compared to related free-living organisms
The legends to the figures in Additional data files 1 and 2
A zip file containing the data from which the figures in the manuscript were made