

ABSTRACT
In a genome-wide small interfering RNA (siRNA) screen, we recently identified the interferon (IFN)-inducible protein 35 (IFI35; also known as IFP35) as a factor required for vesicular stomatitis virus (VSV) infection. Studies reported here were conducted to further understand the role and requirement of IFI35 in VSV infection. Consistent with the siRNA screening data, we found that depletion of IFI35 led to reduced VSV replication at the level of viral gene expression. Although no direct interaction of IFI35 with the viral replication machinery was observed, we found that IFI35 negatively regulated the host innate immune response and rescued poly(I·C)-induced inhibition of VSV replication. Promoter-driven reporter gene assays demonstrated that IFI35 overexpression suppressed the activation of IFN-β and ISG56 promoters, whereas its depletion had the opposite effect. Further investigation revealed that IFI35 specifically interacted with retinoic acid-inducible gene I (RIG-I) and negatively regulated its activation through mechanisms that included (i) suppression of dephosphorylation (activation) of RIG-I and (ii) proteasome-mediated degradation of RIG-I via K48-linked ubiquitination. Overall, the results presented here suggest a novel role for IFI35 in negative regulation of RIG-I-mediated antiviral signaling, which will have implications for diseases associated with excessive immune signaling.
IMPORTANCE Mammalian cells employ a variety of mechanisms, including production of interferons (IFNs), to counteract invading pathogens. In this study, we identified a novel role for a cellular protein, IFN-inducible protein 35 (IFP35/IFI35), in negatively regulating the host IFN response during vesicular stomatitis virus (VSV) infection. Specifically, we found that IFI35 inhibited activation of the RNA sensor, the retinoic acid-inducible gene I (RIG-I), leading to inhibition of IFN production and thus resulting in better replication of VSV. The identification of a cellular factor that attenuates the IFN response will have implications toward understanding inflammatory diseases in humans that have been found to be associated with defects in the regulation of host IFN production, such as systemic lupus erythematosus and psoriasis.
INTRODUCTION
Negative-strand RNA viruses employ diverse strategies to counter the host innate immune response (1). Vesicular stomatitis virus (VSV), a prototypic member of the Rhabdoviridae family with a nonsegmented negative-strand RNA genome, replicates exclusively in the cytoplasm of host cells. Among the five proteins encoded by VSV, the nucleocapsid protein (N) and the matrix protein (M) are crucial for evading as well as impairing cellular antiviral responses (2). VSV N protein binds to newly synthesized viral genomic RNA during replication, which prevents the formation of double-stranded RNA (dsRNA) intermediates and thereby helps to avoid recognition by the viral RNA sensors to mount innate immune responses (1). On the other hand, VSV M causes a global inhibition of host gene expression by abrogating the nucleocytoplasmic export of host mRNAs (3–6). This leads to downregulation of the overall antiviral response during VSV infection. Although VSV is highly efficient in invading a wide range of cell types, its growth is attenuated in cells with a preexisting antiviral state (1). This suggests that VSV lacks an inherent mechanism(s) to counteract an active innate immune response immediately after entry into cells. In this context, VSV may need to depend on host cell factors to counter the antiviral response to allow efficient replication.
Our recent genomewide screen for host factors identified the interferon (IFN)-inducible gene IFI35 (also known as IFP35) as a factor required for VSV infection (7). It was intriguing to find the requirement for an interferon-stimulated gene (ISG) in virus infection, since the majority of ISGs are known to exert antiviral functions to facilitate clearance of viral infection. IFI35 is a 35-kDa protein that was first identified by screening of cDNA libraries of HeLa cells stimulated with IFN-γ (8). It contains an atypical leucine zipper domain that lacks the basic region essential for DNA binding but can homo- and heterodimerize with its binding partners through the N-myc-interacting domains (NIDs) (9, 10). It also interacts with another ISG, N-myc interacting protein (Nmi), to form a 200- to 400-kDa high-molecular-mass complex (HMMC) in response to IFN-α treatment (10). The interaction of IFI35 with Nmi prevents IFI35 from proteasomal degradation (11). However, the functional consequences of HMMC formation and/or the proteasomal degradation of IFI35 in the context of antiviral signaling are not yet understood.
In contrast to the classical role of ISGs in antagonizing virus infections, several ISGs are known to play regulatory functions to control excessive antiviral signaling. Recently, ISGs such as ISG56, Optineurin, gC1qR, LGP2, and SARM were shown to negatively regulate the antiviral response by interfering with interactions between signaling components (12–16), whereas another group of ISGs, including ISG15, A20, RNF125, TRIAD3a, PCBP2, and Ro52, induce degradation of signaling factors via the ubiquitin-proteasome system (17–22). Importantly, the physiological relevance of negative regulators is highlighted by the fact that many inflammatory diseases in humans, such as systemic lupus erythematosus and psoriasis, have been found to be associated with defects in the regulation of host IFN production (23). The observation that IFI35, an ISG, is required for VSV infection and does not interact directly with any of the viral components led us to consider the possibility that IFI35 may support VSV infection by functioning as a negative regulator of the innate antiviral response in infected cells.
Retinoic acid-inducible gene I (RIG-I) is a well-known pathogen recognition receptor (PRR) for negative-strand viruses such as VSV, Sendai virus, and influenza virus and some positive-strand viruses, such as hepatitis C virus and dengue virus (24). RIG-I recognizes short dsRNAs and single-stranded RNAs (ssRNAs) with 5′-triphosphate ends through a conserved DExD/H RNA helicase domain. The RNA helicase domain is essential for viral RNA recognition and activation, whereas the CARD domains are essential for downstream signaling (25). Many recent studies have focused on understanding the regulatory mechanisms of RIG-I activation mediated through posttranslational modifications such as phosphorylation and ubiquitination (26, 27). This is particularly important because downregulation of PRRs, in general, is crucial to prevent excessive signaling during virus infection and also after the infection is cleared. Studies have shown that dephosphorylation (of Ser8 and Thr170 residues) of RIG-I by the phosphatases PP1α/γ (28) and subsequent K63-linked ubiquitination are two major modifications that promote RIG-I activation (29–31). On the other hand, K48-linked ubiquitination and ISG15 conjugation (ISGylation) are the prominent mechanisms to downregulate RIG-I activation (17, 19, 28). Thus, the array of regulatory mechanisms employed by host cells underscores the importance of fine-tuning of the PRR response during virus infection.
In this report, we investigate the supportive role of IFI35 in VSV infection. Our studies revealed that IFI35 functions as a negative regulator of the host antiviral response. Specifically, we found that IFI35 negatively regulates the RIG-I-mediated antiviral signaling pathway. Our data suggest that IFI35 suppresses RIG-I activation and also mediates its proteasomal degradation through ubiquitination. Since RIG-I is the primary sensor during VSV infection (32), its downregulation may help the virus to replicate more efficiently. Overall, the key findings of this study uncover a new role for IFI35 in negatively regulating virus-triggered RIG-I antiviral signaling.
MATERIALS AND METHODS
Cells and reagents.
HeLa and HEK293 cells were obtained from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and the antibiotics penicillin (100 units/ml), kanamycin (20 units/ml), and streptomycin (20 units/ml) (PKS). Baby hamster kidney (BHK-21) cells were maintained in minimal essential medium (MEM) supplemented with 5% FBS and 1× PKS. We generated HEK293 cells stably expressing 3× Flag-tagged IFI35 (3xF-IFI35) by transfection of cells with a plasmid encoding IFI35 tagged with the 3× Flag sequence at its amino terminus and continuously passaging the cells in DMEM supplemented with G418 (1 mg/ml). A similar stable cell line (EV cells) transfected with an empty vector was also generated and used in control experiments. Following isolation, the stable cells were maintained in complete medium containing G418 (250 μg/ml). G418 was purchased from Invitrogen, and MG132, poly(I·C), and calyculin A were purchased from Sigma-Aldrich.
Viruses, infection, and titration.
Sendai virus (SeV) (Cantell strain) was obtained from Charles River Laboratories. Stocks of wild-type (wt) VSV and VSV-eGFP were prepared as described earlier (33, 34). VSVΔG virus and VSV nucleocapsids (NCs or RNPs) were prepared as described previously (35). VSV titers were determined by standard plaque assay on HeLa and BHK-21 cells. SeV infections were performed using 50 hemagglutinin units per ml (HA units/ml) for 16 h. VSV infections were performed at a multiplicity of infection (MOI) of 1 or 0.1 PFU per cell, unless otherwise indicated.
Antibodies.
VSV anti-M (23H12) antibody and anti-phospho RIG-I (Ser8) antibody were generously provided by D. Lyles (Wake Forest School of Medicine) and M. U. Gack (Harvard Medical School), respectively. Anti-ISG56 (rabbit polyclonal) antibody has been described before (36). A mouse monoclonal antibody (MAb) against IFI35 (Sc-100769; Santa Cruz), which detects a closely migrating doublet of endogenous IFI35 and a single band in plasmid-transfected cells, was used. IRF-3 (Sc-33641) and actin (Sc-47778) antibodies were from Santa Cruz Biotechnology Inc., anti-phospho IRF-3 (Ser-396) antibody (4947) was from Cell Signaling Technology Inc., anti-RIG-I (ab-65588) antibody was from Abcam, anti-HA (mouse MAb, HA.11 clone) antibody was from Covance, and anti-Flag (mouse MAb, clone M2) antibody and horseradish peroxidase (HRP)-conjugated goat anti-mouse (A4416) and goat anti-rabbit (A6154) antibodies were from Sigma-Aldrich. Alexa 594-conjugated goat anti-mouse IgG (A11032), Alexa 488-conjugated donkey anti-mouse IgG (A21202), and Alexa 594-conjugated goat anti-rabbit (A11037) antibodies were from Invitrogen.
Plasmid constructs.
The cDNA encoding the full-length open reading frame (ORF) of IFI35 was obtained from Origene and cloned into pcDNA 3.1(+) Neo vector (Invitrogen) at the KpnI and NotI sites to obtain the plasmid pcDNA-IFI35. N-terminally HA-tagged IFI35 (HA-IFI35) was constructed by cloning the IFI35 ORF into a modified pHyg vector (Clontech). N-terminally tagged 3xF-IFI35 was also constructed by inserting the IFI35 ORF from pcDNA-IFI35 into a modified pcDNA-3xFlag Neo vector. All the constructs were verified by sequencing, and all primers used are described in Table 1. The 5x-NF-κB luciferase (37), IFNβ-luciferase and ISG56-luciferase (38), myc-TBK-1 (39), and pEFBos-RIG-I and pEFBos-N-RIG-I (37) constructs have been described previously. myc-MAVS was a kind gift from L. Zhang (University of Nebraska-Lincoln). N-terminally tagged GFP-IRF-3 plasmid was a generous gift from M. U. Gack. pRK5-HA-Ubiquitin-K48 (17605) was purchased from Addgene (40).
TABLE 1.
Primers used in this study
| Primer | Sequence (5′ to 3′)a | Purpose |
|---|---|---|
| IFI35 KpnI-F | ATATATGGTACCGCCGCCACCATGTCAGCCCCACTGGATG | Amplification and cloning of IFI35 in pcDNA vector |
| IFI35 NotI-R | ATATATGCGGCCGCCTAGCCTGACTCAGAGGTG | Amplification and cloning of IFI35 in pcDNA vector |
| HAIFI35 KpnI-F | ATATATGGTACCGCCGCCACCATGTACCCATACGATGTTCCAGATTACGCTTCAGCCCCACTGGATGCC | Amplification and cloning of HA-tagged IFI35 |
| IFI35 NotI-R | ATATATGCGGCCGCCTAGCCTGACTCAGAGGTG | Amplification and cloning of HA-tagged IFI35 |
| VSV P-F | GTGACGGACGAATGTCTCATAA | Amplification of VSV P mRNA |
| VSV P-R | TTTGACTCTCGCCTGATTGTAC | Amplification of VSV P mRNA |
| VSV 2795-F | GTGACGGACGAATGTCTCATAA | Amplification of VSV antigenomic RNA |
| VSV2955-R | TGATGAATGGATTGGGATAACA | Amplification of VSV antigenomic RNA |
| Actin-F | CAAGTACTCCGTGTGTGGAT | Amplification of actin mRNA |
| Actin-R | CATACTCCTGCTTGCTGAT | Amplification of actin mRNA |
| IFN-β-F | ATGACCAACAAGTGTCTCCTCC | Amplification of IFN-β mRNA |
| IFN-β-R | GGAATCCAAGCAAGTTGTAGCTC | Amplification of IFN-β mRNA |
| IRF7-F | CCCACGCTATACCATCTACCT | Amplification of IRF-7 mRNA |
| IRF7-R | GATGTCGTCATAGAGGCTGTTG | Amplification of IRF-7 mRNA |
| RIG-I-F | CTGGACCCTACCTACATCCTG | Amplification of RIG-I mRNA |
| RIG-I-R | GGCATCCAAAAAGCCACGG | Amplification of RIG-I mRNA |
| GAPDH-F | GCAAATTCCATGGCACCGT | Amplification of GAPDH mRNA |
| GAPDH-R | TCGCCCCACTTGATTTTGG | Amplification of GAPDH mRNA |
| VSV DI | ACGAAGACCACAAAACCAGATAAAAA | Amplification of DI RNA |
| RPL32-F | GCCAGATCTTATGCCCCAAC | Amplification of RPL32 mRNA |
| RPL32-R | CGTGCACATGAGCTGCCTAC | Amplification of RPL32 mRNA |
| IFI35-F | AACAAAAGGAGCACACGATCA | Amplification of IFI35 mRNA |
| IFI35-R | CTCCGTTCCTAGTCTTGCCAA | Amplification of IFI35 mRNA |
Underlined sequences indicate restriction sites.
siRNA-mediated silencing.
For depletion of endogenous IFI35, we used a combination of two ORF-targeting small interfering RNA (siRNA) duplexes from Qiagen (Hs_IFI35_1, catalog no. SI00445760, and Hs_IFI35_2, catalog no. SI00445767). A nontargeting (NT) siRNA (catalog no. 1027281; Qiagen) was used as a negative control. All siRNAs were used at a final concentration of 15 nM, unless otherwise indicated. siRNA transfections were performed following a previously described protocol (7).
Promoter-driven reporter gene assays.
HEK293 cells were used for IFI35 knockdown by use of siRNAs as mentioned above, and 3xF-IFI35 stable cells were used in IFI35 overexpression experiments. Both types of cells were transfected with 0.5 μg of IFNβ-luc, NF-κB-luc, or ISG56-luc plasmid along with 50 ng of pRL-TK plasmid (transfection control) for 32 h. Subsequently, cells were infected with 50 HA units of SeV for 16 h before harvesting. Cell lysates were subjected to luciferase assay using a dual-luciferase assay kit (Promega). Luciferase activity was measured using a 20/20n luminometer (Turner Biosystems). Luciferase activity was expressed as the relative fold induction (n-fold) over the level of activity in the NT siRNA-transfected or stable EV cells after normalization to the Renilla luciferase activity.
qRT-PCR.
Total cellular RNA was extracted using TRIzol (Invitrogen) according to the manufacturer's protocol. First-strand cDNA was synthesized with 2 μg of total RNA by use of Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Oligo(dT) primers were used for synthesis of all cDNAs except the VSV antigenome, for which the VSV2955R primer was used. All primers were used at a final concentration of 200 nM. The cDNA was diluted 2-fold, and 200 ng of cDNA was used for each quantitative real-time PCR (qRT-PCR). In each reaction mixture, iTaq Universal SYBR green supermix (Bio-Rad) was used according to the manufacturer's protocol, using a Step One Plus real-time PCR system (Applied Biosystems) following the standard protocol provided by the manufacturer. Relative fold changes were automatically calculated by the Step One Plus real-time PCR system software (Applied Biosystems), following the ΔΔCT method. All primer sequences are listed in Table 1.
Plasmid transfections and Western blotting.
Transfection of plasmids was performed using Lipofectamine 2000 (Invitrogen) per the manufacturer's instructions. Usually, 4 to 6 h after transfection, fresh growth medium was added and incubated for 44 to 48 h. Transfection of viral RNPs was performed as described above, but with incubation for 6 to 8 h or until enhanced green fluorescent protein (EGFP) expression was observed by fluorescence microscopy. Western blotting and quantification of bands were performed as described before (35). Concentrations of primary antibodies used were as follows, unless otherwise indicated: anti-VSV M, 1:2,000; anti-actin, 1:10,000; anti-ISG56, 1:10,000; anti-IFI35, 1:2,000; anti-IRF-3, 1:2,000; anti-p-IRF-3, 1:1,000; anti-RIG-I, 1:1,000; anti-p-RIG-I, 1:1,000; anti-Flag MAb, 1:2,000; and anti-HA MAb, 1:2,000. The corresponding secondary antibodies were used at dilutions of 1:2,000 to 1:5,000.
Immunofluorescence (IF) microscopy.
Stable EV cells or 3xF-IFI35 cells were transfected with IRF-3-GFP plasmid and subsequently infected with 50 HA units/ml SeV for 16 h and fixed with 4% paraformaldehyde. IRF-3 was visualized from the fused GFP fluorescence, whereas 3× Flag-IFI35 was immunostained with anti-Flag MAb (1:600) and the secondary antibody Alexa 594–goat anti-mouse (1:1,000). In other experiments, HEK293 cells were mock infected or infected with SeV, fixed, and stained with antibodies for endogenous RIG-I (1:200) and IFI35 (1:200). The secondary antibodies used were Alexa 594–goat anti-rabbit (1:400) and Alexa 488–donkey anti-mouse (1:400), respectively. Nuclei were stained with 10 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). All images were acquired at a magnification of ×60, using an Olympus FV500/IX81 inverted laser scanning confocal microscope.
Co-IP assay.
Six-well plates containing 50 to 60% confluent HEK293 cells were used for coimmunoprecipitation (co-IP) experiments. For the interaction studies, cells were transfected with 1 μg of EV, HA-IFI35, or Flag-RIG-I plasmid for 30 h. One set of cells was then infected with SeV (50 HA units/ml) or left uninfected for another 16 h. After 46 h of transfection/infection, cells were lysed in lysis buffer as described before (35). For ubiquitination experiments, 0.5 μg (each) of the Flag-RIG-I and HA-Ubiquitin-K48 (HA-Ub-K48) plasmids along with increasing amounts of IFI35 plasmid (0.5, 1, and 2 μg) were transfected for 36 h and then treated with MG132 or its vehicle (dimethyl sulfoxide [DMSO]) for another 12 h. Antibodies used for immunoprecipitation were as follows: anti-Flag MAb (1:125) and anti-HA MAb (1:250). Following overnight incubation with the antibodies, 4 mg of protein A-Sepharose beads (GE Healthcare Bioscience AB) washed 3 times in lysis buffer was added to each sample and incubated for 6 to 8 h. Beads with bound immune complexes were thoroughly washed with lysis buffer, 30 to 40 μl 2× SDS-polyacrylamide gel electrophoresis (PAGE) sample preparation buffer was added, and the proteins were resolved in a 10% SDS-polyacrylamide gel. Subsequently, immunoblotting was carried out as described in the Western blotting section.
Statistical analysis.
The statistical significance of differences between groups was determined by two-tailed paired Student's t test. P values of <0.05 were considered statistically significant.
RESULTS
Depletion of IFI35 inhibits VSV replication.
Our recent genomewide RNA interference (RNAi) screen identified IFI35 as a factor required for VSV infection (7). Since IFI35 is an ISG, it was intriguing to observe its requirement for virus infection. To understand the involvement of IFI35 in VSV infection, we initially reconfirmed the high-throughput screen results by using siRNA duplexes targeting the open reading frame of IFI35 mRNA and examined the levels of IFI35 protein as well as the extent of virus replication. With increasing amounts of siRNA, we observed 8- to 10-fold reductions in the overall levels of IFI35 protein (Fig. 1A). Although IFI35 depletion appeared to have reached its peak with 5 nM siRNA, use of lower (1 nM or less) concentrations resulted in IFI35 depletion in a dose-dependent manner (data not shown). A 4- to 6-fold reduction in VSV M protein levels (measure of virus replication) was observed in cells depleted of IFI35 (Fig. 1A). Depletion of IFI35 also reduced VSV progeny production significantly (Fig. 1B). These data reconfirmed our genomewide screening results and suggested that IFI35 is required for VSV replication and growth.
FIG 1.

Depletion of IFI35 inhibits VSV replication. (A) HeLa cells were transfected with 15 nM NT siRNA or increasing amounts (5, 10, and 15 nM) of a combination of two different siRNAs targeting IFI35. At 60 h posttransfection, cells were infected with VSV (MOI = 1) for 4 h. Levels of IFI35 and VSV M were determined by immunoblotting with specific antibodies. Actin served as the loading control. (B) HeLa cells were transfected with 15 nM NT or IFI35 siRNA for 60 h and infected with VSV (MOI = 0.1) for 18 h. Supernatants were harvested, and virus titers were determined by plaque assay and expressed as PFU/ml. Average titers of viruses from NT- and siIFI35-treated cells were 2.7 × 105 and 3.9 × 104 PFU/ml, respectively. (C and D) IFI35 depletion inhibits VSV infection at the levels of virus transcription and replication. HeLa cells were transfected with 15 nM NT or IFI35 targeting siRNA for 60 h. Subsequently, the cells were infected with VSV (MOI = 0.1) for 12 h. VSV P mRNA (C) and VSV antigenomic RNA (D) levels in IFI35-depleted HeLa cells were determined by qRT-PCR. Values were normalized to the internal control β-actin level and expressed as relative changes over the NT sample value, which was set at 100. (E) IFI35 knockdown inhibits VSV DI RNA replication. NPeGFPL stable cells were transfected with 15 nM NT or IFI35 targeting siRNA for 60 h. The cells were then infected with DI particles (DI inf.) for 14 h, and the RNA replication products (both genomic and antigenomic) were quantitated by semiquantitative RT-PCR as described previously (41). IFI35 and RPL32 (internal control) mRNA levels were also examined. Data presented in panels B to D are from three independent experiments, and values represent means and standard deviations (SD) for duplicates. *, P < 0.05; **, P < 0.01.
To identify the step(s) of the VSV infection cycle where IFI35 might be involved, we determined the levels of viral M protein expression in cells depleted of IFI35 and transfected with viral nucleocapsids, infected with VSVΔG virus, or infected with VSV under single-cycle (MOI = 3) or multicycle (MOI = 0.1) infection conditions, as described previously (7, 35), and found that IFI35 primarily affected virus replication at the level of viral gene expression (data not shown). This observation was further confirmed by examining the levels of viral P mRNA (measure of transcription) and the viral antigenome (measure of replication) by quantitative real-time PCR. Results showed 4- to 5-fold reductions in P mRNA (Fig. 1C) and the antigenome (Fig. 1D) in cells depleted of IFI35 compared to the levels in NT siRNA-treated control cells. Since the observed reduction in genome replication could be due to inhibition of transcription, we examined VSV replication independent of transcription. This was performed by measuring replication of VSV defective interfering (DI) particle genomes in HEK293 cells (41) constitutively expressing the replication proteins of the virus (N, PeGFP, and L). We observed that DI particle RNA replication in the cells depleted of IFI35 was significantly (4- to 5-fold) reduced (Fig. 1E). Importantly, the levels of the viral N, PeGFP, and L proteins were not affected (data not shown), indicating that IFI35 depletion was responsible for the observed reduction of VSV DI RNA replication. Collectively, these data suggest a role for IFI35 in VSV infection at the level of viral RNA transcription and replication.
IFI35 rescues VSV infection by negatively regulating the host antiviral response.
Since our results demonstrated that IFI35 depletion led to reduced VSV RNA transcription and replication, we investigated whether IFI35 colocalizes or interacts with the VSV transcription and replication machinery. We did not observe any colocalization or specific interaction of IFI35 with the viral replication proteins N, P, and L (data not shown), suggesting that IFI35 is not directly involved in VSV replication but may be involved indirectly in modulating host cell pathways required for viral replication. IFI35 is an ISG and is induced in response to IFN-α/γ treatment (8, 10). In contrast to the classical role of ISGs in combating virus infection, many ISGs are known to regulate the antiviral response through negative-feedback mechanisms (23, 42). We considered the possibility that IFI35 is a negative regulator of antiviral responses and thereby may support VSV replication. Thus, we first examined the IFN-β mRNA production in HEK293 cells stably expressing 3× Flag-tagged IFI35 (3xF-IFI35) by real-time PCR. Stable cells (EV) transfected with an empty vector were used as a control. Because VSV is known to efficiently block host gene expression, we used SeV infection, which more potently activates an antiviral pathway similar to that induced by VSV (1). In comparison to the level in EV cells, we observed a 4-fold reduction in IFN-β mRNA levels in the 3xF-IFI35 stable cells infected with SeV (Fig. 2A). Additionally, we observed that when HEK293 cells were depleted of IFI35 by use of siRNAs, IFN-β mRNA levels were increased significantly (Fig. 2B). The effects of IFI35 on IFN-β mRNA production were similar whether SeV infection or poly(I·C) transfection was used (data not shown). To further investigate if IFI35 affects the production of ISGs, we examined the levels of endogenous ISG56 in EV or 3xF-IFI35 cells infected with SeV. Our results show that the ISG56 protein levels were significantly reduced in 3xF-IFI35 cells compared to EV cells (Fig. 2C). Collectively, the results suggest that IFI35 downregulates the host antiviral response.
FIG 2.
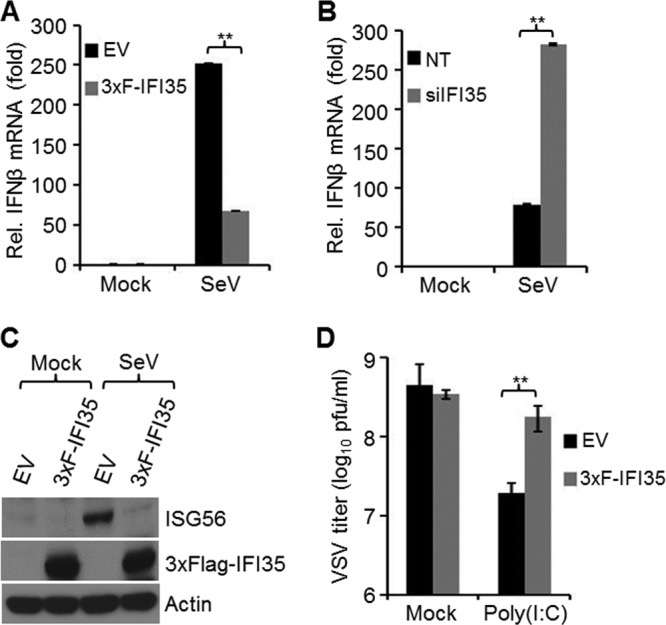
IFI35 rescues VSV infection by negatively regulating host antiviral response. (A and B) IFI35 downregulates IFN-β mRNA synthesis. EV and 3xF-IFI35 stable cells (A) or HEK293 cells transfected with NT or IFI35 siRNA (B) were infected with SeV for 16 h. Total RNA was isolated, and IFN-β mRNA was quantified by qRT-PCR. Values were normalized to the internal control (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) and expressed as relative fold changes over the mock-infected NT or EV sample value, which was set at 1. Values represent means and SD for duplicate reactions from two independent experiments. **, P < 0.01. (C) IFI35 overexpression suppresses ISG56 induction. EV and 3xF-IFI35 stable cells were infected with SeV for 16 h. Cell lysates were used for immunoblotting using ISG56, Flag M2 (detects 3xF-IFI35), and actin antibodies. (D) IFI35 overexpression rescues poly(I·C)-induced suppression of VSV infection. EV and 3xF-IFI35 cells were transfected with 2 μg poly(I·C) for 16 h and infected with VSV (MOI = 0.1) for 18 h. The supernatants were harvested, and virus titers were determined by plaque assay on BHK-21 cells. Virus titers are expressed as log10 PFU/ml. Values represent means and SD for three independent experiments. **, P < 0.01.
The observation that depletion of IFI35 potentiated the production of IFN-β and also resulted in reduced VSV infection prompted us to examine if overexpression of IFI35 could rescue VSV infection from suppression induced by the host antiviral response. Indeed, VSV growth was enhanced (∼10-fold) in poly(I·C)-transfected 3xF-IFI35 stable cells compared to the EV cells (Fig. 2D). Overall, these results indicate that IFI35 negatively regulates the host antiviral response and supports efficient replication of VSV.
IFI35 attenuates IFN-β activation.
To further probe into the IFN signaling pathway that is negatively regulated by IFI35, we examined the effects of IFI35 depletion and overexpression on the induction of various promoters, including those of IFN-β, NF-κB, and ISG56, all which are prominently activated in response to most virus infections (43). First, we tested the effect of IFI35 depletion on IFN-β promoter induction. To this end, HEK293 cells treated with NT or IFI35 siRNA were transfected with a plasmid (IFNβ-Luc) encoding firefly luciferase under the control of an IFN-β promoter. Subsequently, these cells were infected with SeV to induce the IFN-β promoter. Under these conditions, we observed a 4-fold increase in luciferase activity in IFI35-depleted cells compared to control cells (Fig. 3A). In SeV-infected 3xF-IFI35 stable cells, a significant (5-fold) reduction in luciferase activity was observed compared to that in SeV-infected EV cells (Fig. 3B). These data suggest that IFI35 downregulates IFN-β promoter induction, providing further support for our earlier observation of reduced IFN-β mRNA levels (Fig. 2A and B). Next, we examined the effect of IFI35 on induction of the NF-κB promoter. We found only a 2-fold increase or decrease in NF-κB-dependent promoter activity when IFI35 was depleted or overexpressed, respectively, in SeV-infected cells (Fig. 3C and D), suggesting that the NF-κB pathway may not play a major role in IFI35-mediated downregulation of IFN-β activation. Since we observed that IFI35 overexpression suppressed ISG56 protein induction (Fig. 2C), we then examined if this suppression was due to downregulation of ISG56 promoter activity. We found an approximately 5-fold enhancement in ISG56 promoter induction in cells depleted of IFI35 (Fig. 3E), and a 6-fold reduction in reporter activity was observed in cells overexpressing IFI35 compared to the control (EV) cells (Fig. 3F). Taken together, these results suggest that IFI35 downregulates the IFN-β activation pathway.
FIG 3.
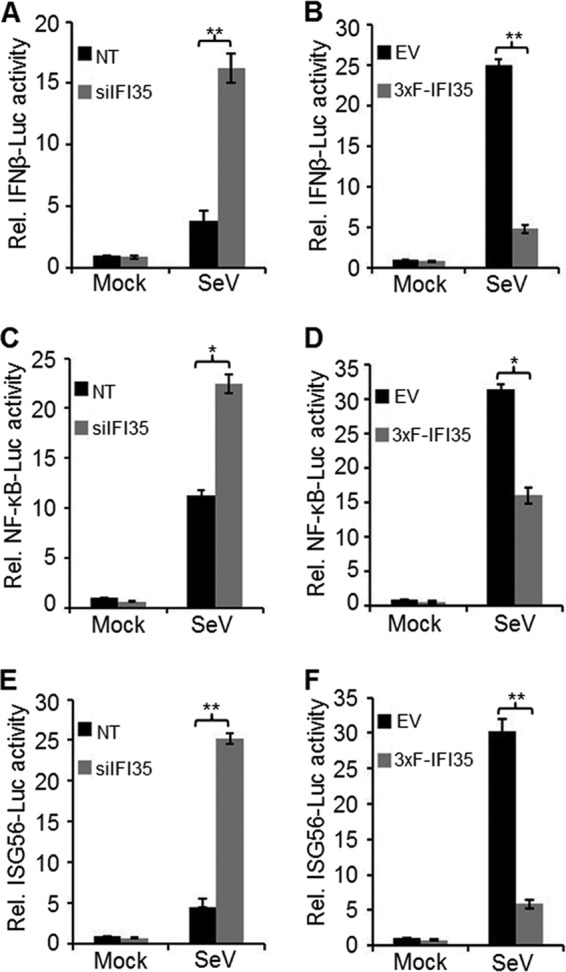
IFI35 attenuates IFN-β activation and signaling. (A and B) Effect of IFI35 on IFN-β promoter activation. (A) HEK293 cells were transfected with 15 nM NT or IFI35 targeting siRNA for a total of 76 h. After the first 24 h, cells were transfected with 500 ng of IFN-β luciferase reporter plasmid (IFNβ-Luc) along with 50 ng of pRL-TK plasmid and were incubated for another 36 h. For the final 16 h prior to harvesting, cells were either mock infected or infected with SeV. Cell lysates were used for dual-luciferase assay. (B) EV and 3xF-IFI35 stable cells were transfected with 500 ng of IFN-β luciferase reporter plasmid along with 50 ng of pRL-TK plasmid for 32 h. Subsequently, cells were infected with SeV for 16 h, and cell lysates were used for dual-luciferase assay. (C and D) Effect of IFI35 on NF-κB promoter activation. Experimental conditions were similar to those for panels A and B, except that 500 ng of NF-κB luciferase construct (NF-κB-Luc) was transfected in place of IFNβ-Luc. (E and F) Effect of IFI35 on ISG56 promoter activation. Experimental conditions were similar to those for panels A and B, except that 500 ng of ISG56 luciferase construct (ISG56-Luc) was transfected in place of the IFNβ-Luc construct. Values presented in all the promoter reporter assays are normalized to NT or EV control cells and expressed as relative fold changes over the mock-infected NT or EV sample value, which was set at 1. Values represent means and SD for three independent experiments. *, P < 0.05; **, P < 0.01.
IFI35 downregulates IRF-3 and IRF-7 activation.
Downregulation of IFN-β induction by IFI35 could occur either by direct interaction of IFI35 with the promoter elements or by interfering with activation of upstream signaling molecules. Since IFI35 lacks DNA binding activity (9) and therefore may not interact directly with the promoter elements, we investigated whether IFI35 suppresses activation of signaling factors that function upstream of IFN-β production, such as NF-κB, IRF-3, and IRF-7. We focused our investigation on IRF-3 and IRF-7 activation, as IFI35 showed relatively lower effects on NF-κB promoter activation (Fig. 3C and D). We first examined whether IRF-3 phosphorylation and nuclear translocation, both of which are hallmarks of IRF-3 activation (44), are affected by IFI35 depletion or overexpression. Depletion of IFI35 in HEK293 cells infected with SeV resulted in increased IRF-3 phosphorylation compared to that in NT siRNA-treated cells infected with SeV (Fig. 4A). Furthermore, IRF-3 phosphorylation was strongly suppressed in 3xF-IFI35 stable cells infected with SeV (Fig. 4B). Since phosphorylation of IRF-3 leads to its dimerization and nuclear translocation (45), we examined whether IFI35 inhibits nuclear translocation of IRF-3. To this end, HEK293 cells stably expressing 3xF-IFI35 or control (EV) cells were transfected with an N-terminally GFP-tagged IRF-3 construct (GFP-IRF-3). Infection of EV cells with SeV led to translocation of IRF-3-GFP from the cytoplasm into the nucleus, whereas the nuclear translocation was abolished in 3xF-IFI35 stable cells (Fig. 4C). These results indicate that IFI35 inhibits the activation of IRF-3, which likely accounts for the downregulation of IFN-β promoter induction.
FIG 4.

IFI35 downregulates activation of IRF-3 and IRF-7. (A and B) IFI35 downregulates IRF-3 phosphorylation. (A) HEK293 cells were transfected with 15 nM NT or IFI35 siRNA for 60 h and infected with SeV for another 16 h. Cell lysates were analyzed by immunoblotting using the indicated antibodies. (B) EV and 3xF-IFI35 stable cells were infected with SeV for 16 h, and cell lysates were analyzed by immunoblotting using the indicated antibodies. (C) IFI35 overexpression inhibits nuclear translocation of IRF-3. EV and 3xF-IFI35 stable cells were grown on coverslips, transfected with 1 μg GFP-IRF-3 plasmid for 32 h, and then infected with SeV for 16 h. 3xF-IFI35 was immunostained using anti-Flag antibody. Nuclei were stained with DAPI. (D and E) IFI35 downregulates IRF-7 induction. (D) HEK293 cells were transfected with 15 nM NT or IFI35 siRNA for 60 h and infected with SeV for another 16 h. Total RNA was isolated and subjected to qRT-PCR using IRF-7-specific primers. (E) IRF-7 mRNA was quantified as described for panel D, using EV and 3xF-IFI35 stable cells infected with SeV for 16 h. Values were normalized to the internal control GAPDH value and expressed as relative fold changes over the mock-infected EV or NT sample value, which was set at 1. Values represent means and SD for duplicate reactions from two independent experiments. *, P < 0.05.
Since IRF-7 in most cells other than plasmacytoid dendritic cells is induced upon virus infection and activated through positive feedback from IFN-α/β signaling (46), we examined whether IFI35 negatively regulates IRF-7 induction in the HEK293 cells used in our studies. Our results show that in SeV-infected cells, IRF-7 mRNA levels were upregulated when IFI35 was depleted (Fig. 4D) and reduced when IFI35 was overexpressed (Fig. 4E). Taken together, these results suggest that IFI35 negatively regulates activation of IRF-3 and IRF-7. Thus, the downregulation of the IFN-β promoter is likely due to the combined effect of suppression of IRF-3 and IRF-7 activation.
IFI35 negatively regulates RIG-I pathway activation.
To investigate if IFI35 downregulates the antiviral signaling pathway upstream of IRF-3 and IRF-7, we examined the activation of the IFN-β promoter by its signaling components, including TBK1, MAVS/IPS-1, and RIG-I, all of which are known to be activated during VSV or SeV infection (43). In 3xF-IFI35 stable cells, we observed significant reductions (3- to 5-fold) in IFN-β promoter activation induced by overexpression of RIG-I or the constitutively active form of RIG-I (N-RIG-I), but the activation was not significantly altered by overexpression of the downstream signaling molecules, MAVS or TBK1 (Fig. 5A). Consistent with these data, we also found significant enhancements in IFN-β promoter activation by overexpression of RIG-I or N-RIG-I in HEK293 cells depleted of IFI35 (Fig. 5B), whereas no significant effect was observed with MAVS or TBK1 overexpression in these cells (Fig. 5B). These data suggest that IFI35 negatively regulates the host antiviral pathway by exerting its effect at the level of RIG-I.
FIG 5.

IFI35 negatively regulates RIG-I pathway activation. (A) EV and 3xF-IFI35 stable cells were transfected with a plasmid encoding the RIG-I pathway components (0.5 μg each) MAVS, TBK1, RIG-I, or N-RIG-I, along with 0.25 μg IFNβ-Luc and 0.025 μg pRL-TK plasmid, for 48 h. The cells were lysed and used for dual-luciferase assays. (B) HEK293 cells were transfected with 15 nM NT or IFI35 siRNA for 24 h and then transfected for another 48 h with plasmids as described for panel A. Cells were lysed and used for dual-luciferase assays. Luciferase values were normalized to the EV or NT control cells and expressed as relative fold changes over the mock-infected EV or NT sample value, which was set at 1. Values represent means and SD for two independent experiments performed in duplicate. *, P < 0.05; **, P < 0.01; ns, not significant. (C and D) IFI35 negatively regulates RIG-I activation. (C) HEK293 cells were transfected with 15 nM NT or IFI35 siRNA for 60 h and infected with SeV for another 16 h. The cells were treated with 100 nM calyculin A for 30 min before harvesting. Cell lysates were analyzed by immunoblotting using the indicated antibodies. Actin served as a loading control. (D) EV and 3xF-IFI35 stable cells were infected with SeV for 16 h and processed as described for panel C. (E and F) Knockdown or overexpression of IFI35 enhances or suppresses RIG-I transcription, respectively. (E) HEK293 cells were transfected with 15 nM NT or IFI35 siRNA for 60 h and infected with SeV for another 16 h. Total RNA was isolated and used for quantification of RIG-I mRNA levels by qRT-PCR. (F) EV or 3xF-IFI35 stable cells were infected with SeV for 16 h and processed as described for panel E. Values were normalized to the internal control GAPDH level and expressed as relative fold changes over the mock-infected EV or NT sample value, which was set at 1. **, P < 0.01.
To understand how IFI35 regulates the activity of RIG-I, we examined the phosphorylation status of RIG-I. Dephosphorylation of Ser8 is a necessary prerequisite step in the RIG-I activation process (30). In SeV-infected HEK293 cells depleted of IFI35, we observed a significantly reduced level of the phosphorylated form of RIG-I by using a Ser8 phospho-specific RIG-I antibody (Fig. 5C). The total RIG-I levels were also slightly increased. In contrast, the level of RIG-I phosphorylation was partially restored in 3xF-IFI35 stable cells infected with SeV, and the total amount of RIG-I was also reduced, indicating that IFI35 overexpression led to suppression of RIG-I activation (Fig. 5D). Since we observed changes in total RIG-I levels by depletion or overexpression of IFI35, we examined whether IFI35 affected RIG-I induction at the level of transcription. The results show that RIG-I mRNA was upregulated in cells depleted of IFI35 (Fig. 5E), whereas in 3xF-IFI35 stable cells, it was reduced (Fig. 5F). Since RIG-I is an ISG, the reduced levels of RIG-I mRNA may have been due to overall downregulation of IFN-β activation. These results indicate that IFI35 downregulates the activation of RIG-I, leading to overall suppression of the antiviral response.
IFI35 interacts with and promotes RIG-I degradation via K48-linked ubiquitination.
To further understand the mechanism by which IFI35 negatively regulates RIG-I activation, we examined whether IFI35 and RIG-I colocalize and/or interact in cells. We observed enhanced colocalization of endogenous IFI35 and RIG-I in SeV-infected cells compared to mock-infected cells (Fig. 6A). To determine if the observed colocalization was due to a physical interaction between IFI35 and RIG-I, we performed an IP assay using HEK293 cells. Cells were cotransfected with plasmids encoding Flag-RIG-I and HA-IFI35 and subsequently were mock infected or infected with SeV. The cell extracts were subjected to co-IP with Flag (Fig. 6B, left panel) or HA (Fig. 6B, right panel) antibody and immunoblotted with HA or Flag antibody, respectively. The results show that RIG-I and IFI35 interact with each other in transfected cells in the presence or absence of SeV infection (Fig. 6B). Overall, both the colocalization and interaction studies suggest that IFI35 interacts specifically with RIG-I.
FIG 6.

IFI35 interacts with and promotes degradation of RIG-I via K48-linked ubiquitination. (A) SeV infection enhances colocalization of IFI35 and RIG-I. HEK293 cells were grown on coverslips and mock infected or infected with SeV for 16 h. The cells were fixed and immunostained with the indicated antibodies. Nuclei were stained with DAPI, and images were collected at a magnification of ×60. (B) IFI35 interacts with RIG-I in transfected cells. HEK293 cells were transfected with 0.5 μg (each) of the indicated plasmids for 32 h. One set of cells was mock infected, while the other set was infected with SeV for another 16 h. Co-IP and immunoblotting (IB) were performed with the indicated antibodies. Expression of proteins from the transfected plasmids was analyzed in the whole-cell lysates (WCL) by using the indicated antibodies. (C) IFI35 promotes proteasomal degradation of RIG-I. Sets of HEK293 cells were transfected with 0.5 μg of Flag-RIG-I along with increasing amounts of IFI35 (0.5 and 1 μg) for 36 h. One set of cells was treated with 10 μM MG132 and the other set with DMSO for 12 h. Cell lysates were analyzed by immunoblotting with the indicated antibodies. (D) IFI35 overexpression enhances K48-linked ubiquitination of RIG-I. Sets of HEK293 cells were transfected with Flag-RIG-I and HA-Ub-K48 constructs (0.5 μg [each]) along with increasing amounts of plasmid expressing IFI35 (0.5, 1, and 2 μg) for 36 h. One set of cells was treated with MG132 (10 μM) for 12 h and subsequently lysed and used for co-IP and immunoblotting with the indicated antibodies. Another set of cells was treated with DMSO for 12 h, and the whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (E) IFI35 knockdown reduces K48-linked ubiquitination of RIG-I. Sets of HEK293 cells were transfected with NT siRNA or increasing amounts of IFI35 siRNA (5, 10, and 20 nM) for 24 h. The cells were transfected with Flag-RIG-I and HA-Ubiquitin-K48 constructs (0.5 μg [each]) for another 36 h. Cells were subsequently processed as described for panel D.
Since negative regulators such as RNF125 and Siglec-G induce proteasomal degradation of RIG-I (19, 47), we examined whether IFI35 promotes degradation of RIG-I. Indeed, when HEK293 cells were transfected with Flag-RIG-I along with increasing amounts of IFI35, we observed dose-dependent reductions in the levels of Flag-RIG-I, but this effect was reversed when cells were treated with the proteasome inhibitor MG132 (Fig. 6C). This suggests that IFI35 promotes degradation of RIG-I through the proteasome machinery. It is well known that proteins degraded through the proteasome pathway are conjugated to ubiquitin chains via lysine 48 (K48) linkage (48). Thus, we examined whether IFI35 promotes K48-linked ubiquitination of RIG-I. As shown in Fig. 6D, dose-dependent increases in K48-linked ubiquitination of RIG-I were observed with increasing amounts of IFI35. The levels of free HA-Ub-K48 in whole-cell lysates decreased, indicating that the expressed HA-Ub-K48 was conjugated to RIG-I. Concomitant reduction of RIG-I levels was observed in the whole-cell lysates (Fig. 6D). Furthermore, we observed reductions in K48-linked ubiquitination of RIG-I when IFI35 was depleted using siRNAs (Fig. 6E). These data suggest that IFI35 interacts with RIG-I and negatively regulates its activation by promoting K48-linked ubiquitination and degradation by the proteasome pathway.
DISCUSSION
In the present study, we uncovered a new role for IFI35 as a negative regulator of the host antiviral response during VSV infection. Consistent with our recent high-throughput genome-wide siRNA screening study (7) revealing a requirement for IFI35 in VSV infection, we found that depletion of IFI35 led to a significant reduction in VSV replication. Conversely, overexpression of IFI35 could rescue VSV growth from the poly(I·C)-induced antiviral response. The observation that IFI35 did not interact with any of the viral components suggested that IFI35 likely exerted its effect by influencing cellular pathways. The results presented here indicate that IFI35 downregulates the host antiviral response mediated by RIG-I. IFI35 appears to downregulate RIG-I activity by at least two modes: (i) by keeping RIG-I in its phosphorylated (inactive) form and (ii) by mediating proteasomal degradation of RIG-I though ubiquitination. Whether the requirement of IFI35 is specific to VSV infection is not known at this time, although we observed a moderate requirement of IFI35 during lymphocytic choriomeningitis virus infection (7).
The current understanding of the role of IFI35 in virus infections is limiting. IFI35 is an ISG that is induced in response to treatment of cells with IFN-α/γ (8, 10). We have found that it is also induced by transfection of cells with poly(I·C) or by infection with SeV (data not shown). The only study reported so far suggests that IFI35 interacts with the bovine foamy virus (BFV) trans-activator protein Tas and that this interaction blocks the ability of Tas to activate viral gene transcription, resulting in inhibition of BFV replication (49). Thus, in this context, IFI35 exhibits an antiviral role and suppresses virus replication. Since infection of HeLa or HEK293 cells (which are of a nonhematopoietic origin) by foamy viruses does not induce a type I IFN response (50, 51), it is possible that the negative regulatory role of IFI35 in innate immune signaling was not observed readily in these studies. However, our data clearly demonstrate the supportive role of IFI35 in VSV infection through negative regulation of innate immune signaling. Because viruses differ significantly in their ability to induce or suppress the host innate antiviral responses, additional studies will be needed to evaluate the role and requirement of IFI35 during infection with diverse families of viruses.
Although host innate antiviral responses are of paramount significance for controlling viral infections, unregulated or excessive responses may be detrimental to the host. Therefore, identification of host cell factors and an understanding of the mechanisms that negatively regulate these processes are critical to prevent damage to the infected and neighboring, uninfected cells. In this regard, the role of ISGs that function as negative regulators of antiviral signaling is increasingly being appreciated. Previous studies have shown that several ISGs, such as ISG56, Optineurin, RNF125, and A20, downregulate the antiviral response through diverse mechanisms (12, 13, 19, 52) that include interference with interactions between various signaling components (12–16) and degradation of signaling factors via the ubiquitin-proteasome pathway. Our studies presented here show that IFI35 negatively regulates the RIG-I-mediated antiviral pathway through multiple mechanisms. Previous studies have shown that activation of RIG-I is controlled through ubiquitination or deubiquitination of the CARD domains (26). K48-linked ubiquitination of RIG-I is promoted primarily by the E3 ubiquitin ligase RNF125 (19) and the recently identified c-Cbl ubiquitin ligase (47), which results in degradation of RIG-I through the proteasome machinery. On the other hand, removal of K63-linked ubiquitin chains is mediated by the tumor suppressor CYLD, which leads to inactivation of RIG-I (53). The results presented here show that IFI35 downregulates the host antiviral response by promoting proteasomal degradation of RIG-I. IFI35 interacts with RIG-I and promotes its degradation through the proteasome pathway via K48-linked ubiquitination (Fig. 6). We did not observe any significant changes in K63-linked ubiquitination of RIG-I under conditions of IFI35 overexpression (data not shown), suggesting that negative regulation of RIG-I mediated by IFI35 occurs primarily through K48-linked ubiquitination and proteasomal degradation and may not involve deubiquitination. At this time, it is not known whether IFI35 directly mediates the K48-linked ubiquitination or facilitates the recruitment of an E3 ubiquitin ligase. Since IFI35 does not contain any typical E3 ubiquitin ligase domains, such as HECT, RING, or F-box motifs (54), the latter possibility of recruiting an E3 ubiquitin ligase seems more likely. It will be interesting to examine if the K48-linked ubiquitination of RIG-I in the IFI35–RIG-I complex is mediated by known E3 ubiquitin ligases of RIG-I, such as RNF125 and c-Cbl, or by an as yet unidentified E3 ubiquitin ligase (Fig. 7).
FIG 7.
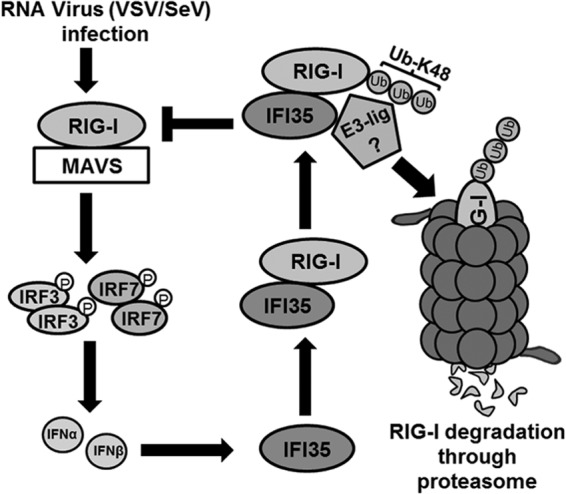
Proposed model depicting the role of IFI35 in negative regulation of RIG-I signaling. For the sake of simplicity, we show only the ubiquitin-mediated degradation pathway. Viruses such as VSV and SeV are recognized by RIG-I, which signals through downstream factors (IRF-3/IRF-7) leading to production of type I IFNs (IFN-α/β). IFN-α/β in turn induce the synthesis of ISGs, including IFI35, which interacts with RIG-I and inhibits its activation by promoting K48-linked ubiquitination and proteasomal degradation. The identity of the associated E3 ubiquitin ligase is unknown at this time. The negative-feedback loop mediated by IFI35 leads to downregulation of the host antiviral response.
Another mechanism of RIG-I inactivation by IFI35 appears to be through suppressing the dephosphorylation of RIG-I. Previously, it was shown that RIG-I is maintained in an inactive state by phosphorylation mediated by protein kinase C α/β (PKC-α/β) (55). The initial step of RIG-I activation involves dephosphorylation of the CARD domains, mediated by the phosphatases PP1α/γ (28). We observed that IFI35 overexpression led to enhancement of RIG-I phosphorylation (Fig. 5D), suggesting that IFI35 also regulates the activation of RIG-I through direct involvement in the phosphorylation event or through an indirect mechanism. Interestingly, our preliminary bioinformatic analysis uncovered several potential phosphorylation sites on IFI35 which are predicted to be phosphorylated by the PKC family of kinases (data not shown). Although the role of phosphorylation/dephosphorylation in modulating IFI35 function is not clear at this time, it is possible that phosphorylation of both IFI35 and RIG-I by a common family of kinases may represent an intricate mechanism for IFI35-mediated negative regulation of the RIG-I-mediated antiviral response.
Recently, it was reported that Nmi negatively regulates host antiviral signaling by promoting the degradation of IRF-7 (56). Since Nmi is known to interact with IFI35 and enhance its stability (11), it is possible that both proteins may act in a synergistic or additive fashion to negatively regulate a common antiviral signaling pathway, albeit through different targets. Since the formation of a high-molecular-mass IFI35-Nmi complex has been shown to be an IFN-α-stimulated event (10), it will be interesting to investigate if the interaction of Nmi and IFI35 plays any role in augmenting the negative regulatory effects during virus infections.
IFI35 belongs to a group of atypical helix-loop-helix (HLH) proteins, such as Id, which lack the basic DNA binding region and thus function in a dominant negative fashion to negatively regulate the function of other basic HLH (b-HLH) transcription factors (57, 58). In this study, we have uncovered a novel role of IFI35 in negatively regulating the host antiviral response by targeting the RIG-I signaling pathway. Mechanistically, we have demonstrated that IFI35 downregulates RIG-I activation by promoting its proteasomal degradation as well as keeping it in its phosphorylated (inactive) form. Further studies are required to understand the mechanistic details of this process and also to identify other factors involved in the IFI35-mediated negative regulatory loop. In conclusion, the identification of IFI35 introduces a new player to the existing group of molecules that regulate the RIG-I antiviral signaling pathway and also highlights the importance of negative regulation of cellular antiviral responses.
ACKNOWLEDGMENTS
We thank Y. Zhou and Terry Fangman (UNL Microscopy Core Facility) for help with fluorescence microscopy. We thank Deborah Brown (University of Nebraska-Lincoln) for providing help with quantitative real-time PCR and D. S. Lyles, M. U. Gack, and L. Zhang for reagents. We appreciate Z. H. Gill for excellent assistance in the laboratory.
Footnotes
Published ahead of print 26 December 2013
REFERENCES
- 1.Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol. Mol. Biol. Rev. 75:468–490. 10.1128/MMBR.00007-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rieder M, Conzelmann KK. 2009. Rhabdovirus evasion of the interferon system. J. Interferon Cytokine Res. 29:499–509. 10.1089/jir.2009.0068 [DOI] [PubMed] [Google Scholar]
- 3.von Kobbe C, van Deursen JM, Rodrigues JP, Sitterlin D, Bachi A, Wu X, Wilm M, Carmo-Fonseca M, Izaurralde E. 2000. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol. Cell 6:1243–1252. 10.1016/S1097-2765(00)00120-9 [DOI] [PubMed] [Google Scholar]
- 4.Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, Fontoura BM. 2005. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 17:93–102. 10.1016/j.molcel.2004.11.023 [DOI] [PubMed] [Google Scholar]
- 5.Her LS, Lund E, Dahlberg JE. 1997. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 276:1845–1848. 10.1126/science.276.5320.1845 [DOI] [PubMed] [Google Scholar]
- 6.Lyles DS. 2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 64:709–724. 10.1128/MMBR.64.4.709-724.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panda D, Das A, Dinh PX, Subramaniam S, Nayak D, Barrows NJ, Pearson JL, Thompson J, Kelly DL, Ladunga I, Pattnaik AK. 2011. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 108:19036–19041. 10.1073/pnas.1113643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bange FC, Vogel U, Flohr T, Kiekenbeck M, Denecke B, Bottger EC. 1994. IFP 35 is an interferon-induced leucine zipper protein that undergoes interferon-regulated cellular redistribution. J. Biol. Chem. 269:1091–1098 [PubMed] [Google Scholar]
- 9.Wang X, Johansen LM, Tae HJ, Taparowsky EJ. 1996. IFP 35 forms complexes with B-ATF, a member of the AP1 family of transcription factors. Biochem. Biophys. Res. Commun. 229:316–322. 10.1006/bbrc.1996.1799 [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Liao J, Meyerdierks A, Feng L, Naumovski L, Bottger EC, Omary MB. 2000. Interferon-alpha induces nmi-IFP35 heterodimeric complex formation that is affected by the phosphorylation of IFP35. J. Biol. Chem. 275:21364–21371. 10.1074/jbc.M003177200 [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Shpall RL, Meyerdierks A, Hagemeier M, Bottger EC, Naumovski L. 2000. Interferon-inducible Myc/STAT-interacting protein Nmi associates with IFP 35 into a high molecular mass complex and inhibits proteasome-mediated degradation of IFP 35. J. Biol. Chem. 275:36278–36284. 10.1074/jbc.M006975200 [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, Shu HB. 2009. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. U. S. A. 106:7945–7950. 10.1073/pnas.0900818106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mankouri J, Fragkoudis R, Richards KH, Wetherill LF, Harris M, Kohl A, Elliott RM, Macdonald A. 2010. Optineurin negatively regulates the induction of IFNbeta in response to RNA virus infection. PLoS Pathog. 6:e1000778. 10.1371/journal.ppat.1000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. 2005. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175:5260–5268 [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Xiao N, Liu F, Ren H, Gu J. 2009. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. U. S. A. 106:1530–1535. 10.1073/pnas.0811029106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. 2006. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7:1074–1081. 10.1038/ni1382 [DOI] [PubMed] [Google Scholar]
- 17.Kim MJ, Hwang SY, Imaizumi T, Yoo JY. 2008. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 82:1474–1483. 10.1128/JVI.01650-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430:694–699. 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 19.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. 2007. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. U. S. A. 104:7500–7505. 10.1073/pnas.0611551104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z. 2009. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 10:1300–1308. 10.1038/ni.1815 [DOI] [PubMed] [Google Scholar]
- 21.Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. 2008. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 181:1780–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. 2009. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 5:e1000650. 10.1371/journal.ppat.1000650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards KH, Macdonald A. 2011. Putting the brakes on the anti-viral response: negative regulators of type I interferon (IFN) production. Microbes Infect. 13:291–302. 10.1016/j.micinf.2010.12.007 [DOI] [PubMed] [Google Scholar]
- 24.Wilkins C, Gale M., Jr 2010. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22:41–47. 10.1016/j.coi.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 26.Maelfait J, Beyaert R. 2012. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 76:33–45. 10.1128/MMBR.05012-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixit E, Kagan JC. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv. Immunol. 117:99–125. 10.1016/B978-0-12-410524-9.00004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU. 2013. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38:437–449. 10.1016/j.immuni.2012.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 30.Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. 2010. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J. Biol. Chem. 285:20252–20261. 10.1074/jbc.M109.089912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. 2010. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 84:3220–3229. 10.1128/JVI.02241-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610. 10.1084/jem.20080091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das SC, Nayak D, Zhou Y, Pattnaik AK. 2006. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 80:6368–6377. 10.1128/JVI.00211-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das SC, Pattnaik AK. 2005. Role of the hypervariable hinge region of phosphoprotein P of vesicular stomatitis virus in viral RNA synthesis and assembly of infectious virus particles. J. Virol. 79:8101–8112. 10.1128/JVI.79.13.8101-8112.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinh PX, Beura LK, Panda D, Das A, Pattnaik AK. 2011. Antagonistic effects of cellular poly(C) binding proteins on vesicular stomatitis virus gene expression. J. Virol. 85:9459–9471. 10.1128/JVI.05179-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo J, Peters KL, Sen GC. 2000. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 267:209–219. 10.1006/viro.1999.0135 [DOI] [PubMed] [Google Scholar]
- 37.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699. 10.1128/JVI.79.5.2689-2699.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. 2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 84:1574–1584. 10.1128/JVI.01326-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S. 2003. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171:4304–4310 [DOI] [PubMed] [Google Scholar]
- 40.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. 2005. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25:2002–2009. 10.1523/JNEUROSCI.4474-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panda D, Dinh PX, Beura LK, Pattnaik AK. 2010. Induction of interferon and interferon signaling pathways by replication of defective interfering particle RNA in cells constitutively expressing vesicular stomatitis virus replication proteins. J. Virol. 84:4826–4831. 10.1128/JVI.02701-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisenacher K, Krug A. 2012. Regulation of RLR-mediated innate immune signaling—it is all about keeping the balance. Eur. J. Cell Biol. 91:36–47. 10.1016/j.ejcb.2011.01.011 [DOI] [PubMed] [Google Scholar]
- 43.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 44.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dragan AI, Hargreaves VV, Makeyeva EN, Privalov PL. 2007. Mechanisms of activation of interferon regulator factor 3: the role of C-terminal domain phosphorylation in IRF-3 dimerization and DNA binding. Nucleic Acids Res. 35:3525–3534. 10.1093/nar/gkm142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669. 10.1093/emboj/17.22.6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, Liu Y, Cao X. 2013. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152:467–478. 10.1016/j.cell.2013.01.011 [DOI] [PubMed] [Google Scholar]
- 48.Bibeau-Poirier A, Servant MJ. 2008. Roles of ubiquitination in pattern-recognition receptors and type I interferon receptor signaling. Cytokine 43:359–367. 10.1016/j.cyto.2008.07.012 [DOI] [PubMed] [Google Scholar]
- 49.Tan J, Qiao W, Wang J, Xu F, Li Y, Zhou J, Chen Q, Geng Y. 2008. IFP35 is involved in the antiviral function of interferon by association with the viral tas transactivator of bovine foamy virus. J. Virol. 82:4275–4283. 10.1128/JVI.02249-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabile A, Rhodes-Feuillette A, Jaoui FZ, Tobaly-Tapiero J, Giron ML, Lasneret J, Peries J, Canivet M. 1996. In vitro studies on interferon-inducing capacity and sensitivity to IFN of human foamy virus. Res. Virol. 147:29–37. 10.1016/0923-2516(96)80237-8 [DOI] [PubMed] [Google Scholar]
- 51.Rua R, Lepelley A, Gessain A, Schwartz O. 2012. Innate sensing of foamy viruses by human hematopoietic cells. J. Virol. 86:909–918. 10.1128/JVI.06235-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parvatiyar K, Barber GN, Harhaj EW. 2010. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 285:14999–15009. 10.1074/jbc.M110.109819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. 2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9:930–936. 10.1038/embor.2008.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Metzger MB, Hristova VA, Weissman AM. 2012. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 125:531–537. 10.1242/jcs.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maharaj NP, Wies E, Stoll A, Gack MU. 2012. Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J. Virol. 86:1358–1371. 10.1128/JVI.06543-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Yang B, Hu Y, Zheng Y, Zhou H, Wang Y, Ma Y, Mao K, Yang L, Lin G, Ji Y, Wu X, Sun B. 2013. Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J. Immunol. 191:3393–3399. 10.4049/jimmunol.1300740 [DOI] [PubMed] [Google Scholar]
- 57.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. 1990. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61:49–59. 10.1016/0092-8674(90)90214-Y [DOI] [PubMed] [Google Scholar]
- 58.Pagliuca A, Bartoli PC, Saccone S, Della Valle G, Lania L. 1995. Molecular cloning of ID4, a novel dominant negative helix-loop-helix human gene on chromosome 6p21.3-p22. Genomics 27:200–203. 10.1006/geno.1995.1026 [DOI] [PubMed] [Google Scholar]