

ABSTRACT
The integration of retroviruses into the host genome following nonrandom genome-wide patterns may lead to the deregulation of gene expression and oncogene activation near the integration sites. Slow-transforming retroviruses have been widely used to perform genetic screens for the identification of genes involved in cancer. To investigate the involvement of avian leukosis virus subgroup J (ALV-J) integration in myeloid leukosis (ML) in chickens, we utilized an ALV-J insertional identification platform based on hybrid capture target enrichment and next-generation sequencing (NGS). Using high-definition mapping of the viral integration sites in the chicken genome, 241 unique insertion sites were obtained from six different ALV-J-induced ML samples. On the basis of previous statistical definitions, MYC, TERT, and ZIC1 genes were identified as common insertion sites (CIS) of provirus integration in tumor cells; these three genes have previously been shown to be involved in the malignant transformation of different human cell types. Compared to control samples, the expression levels of all three CIS genes were significantly upregulated in chicken ML samples. Furthermore, they were frequently, but not in all field ML cases, deregulated at the mRNA level as a result of ALV-J infection. Our findings contribute to the understanding of the relationship between multipathotypes associated with ALV-J infection and the molecular background of tumorigenesis.
IMPORTANCE ALV-Js have been successfully eradicated from chicken breeding flocks in the poultry industries of developed countries, and the control and eradication of ALV-J in China are now progressing steadily. To further study the pathogenesis of ALV-J infections, it will be necessary to elucidate the in vivo viral integration and tumorigenesis mechanism. In this study, 241 unique insertion sites were obtained from six different ALV-J-induced ML samples. In addition, MYC, TERT, and ZIC1 genes were identified as the CIS of ALV-J in tumor cells, which might be a putative “driver” for the activation of the oncogene. In addition, the CIS genes showed deregulated expression compared to nontumor samples. These results have potentially important implications for the mechanism of viral carcinogenesis.
INTRODUCTION
Avian leukosis virus subgroup J (ALV-J) belongs to the Alpharetrovirus genus of the Retroviridae family. It was first isolated in commercial broiler breeder chickens with myeloid leukosis (ML) in the United Kingdom in 1991 (1) and has since spread to many other countries such as Japan, the United States, and Israel (2–4). As the spread of the disease is associated with diverse pathotypes and could result in enormous economic losses, ALV-J-induced disease in layer-type chickens has become one of the most important problems facing the global poultry industry. In addition to the fact that the higher incidence of lymphoid leukosis (LL) or erythroblastosis (EB) in susceptible chickens is induced by the ALV subgroups A (ALV-A) and B (ALV-B) (5), ALV-J was also found to be the main cause of ML and hemangioma after a long latent period, as well as a wide variety of other tumors at a lower incidence (6–9). Further studies have found that virus- and/or cell lineage-specific determinants are also important for tumor development, but the oncogenicity of ALV-J remains unclear.
Knowledge of the chromatin conformation of genomic regions targeted by retroviral integration is critical to understanding retroviral replication and viral pathogenesis. Early studies investigating the oncogenicity of ALV have focused on viral integration mechanisms and the activation of cellular proto-oncogenes by retroviral promoters. Proviral integrations near host genes may induce their deregulation as viral long terminal repeats (LTRs) can activate genes through enhancer or promoter insertions and create a virus-host gene fusion transcript (5, 10–12). The genomic regions that have been repeatedly hit by viral insertions in multiple independent tumors (defined as common insertion sites [CISs]) are likely to contain host genes involved in tumor development (12–14). For instance, twist, bic, and c-erbB are CIS genes identified in ALV-induced nephroblastoma, lymphoid leucosis, and erythroblastosis, respectively (5, 11, 15), and play a role in tumorigenesis. However, it has not been determined whether ALV-J-induced ML in chickens is associated with viral integration. The mapping and analysis of ALV-J integration sites in ML tumors could facilitate the identification of genomic regions or host genes that are recurrently targeted by proviral insertion and, by extension, are likely to be involved in tumor progression.
In this study, we analyzed ALV-J provirus junctions to construct a genome-wide map of integration sites in the genome of ML tumors. We also identified recurrent genomic integration sites in independent tumors. A simple, inexpensive, hybrid capture enrichment method was optimized for DNA extracted from ALV-J-positive tumor samples, and each enriched genomic library was massively sequenced in parallel on an Illumina HiSeq2000 PEI101 sequencer. We also used gene expression profiling as a global assay to analyze CIS genes directly linked to ML. CIS genes targeted by integration were significantly upregulated compared with levels in nontumor samples. Our work provides the first unbiased, genome-wide, single-base resolution ALV-J integration map for ML and has identified three ML-associated genes that are prominent in chicken ML.
MATERIALS AND METHODS
Sample selection.
Autoptical analysis was used to identify gross masses in the sternum and liver parenchyma, which were collected independently, as well as nontumoral sternum and liver from commercial broiler breeder chicken flocks in Guangdong Province, China, between November 2011 and March 2012. All chicken sampling procedures were approved by the Animal Care and Use Committee of Guangdong Province, China. Our sampling processes were assisted by local authorities and veterinarians. All animal research was conducted under the guidance of the SCAU's Institutional Animal Care and Use Committee. Diagnosis of tissue samples was based on characteristic microscopic lesions and molecular analyses. Microscopically, the tumor cells were observed as relatively uniform large myeloid cells, and lymphoid cell hyperplasia was observed in the liver. The results of PCR tests on the genomic DNA of tissues and viral isolation assays, as described in a previous study, were ALV-J positive (16). Six ALV-J-positive ML livers were arbitrarily selected for further proviral integration study (samples 001L to 006L). Considering the cell uniformity, we selected the corresponding infected ML sternums and uninfected normal sternum samples for microarray expression profiling.
Preparation of sample DNA libraries and capture probes.
Genomic DNA was extracted from frozen tumor tissues using an EZNA SQ tissue DNA kit (Omega Bio-Tek, Norcross, GA, USA). Sample genomic DNA libraries were constructed according to the Illumina PE Sample Preparation Protocol (17). Capture probes were generated using a MyGenostics Gencap Custom Enrichment Kit (MyGenostics, Baltimore, MD, USA) and were based on a series of overlapping PCR products tiled across the ALV-J genome (GenBank accession number Z46390).
Targeted capture and sequencing.
Each library was hybridized with target enrichment capture probes according to the MyGenostics GenCap Enrichment standard protocol (http://www.mygenostics.com/mygenostics-gencap-whole-exome-enrichment-protocol.pdf). The hybrid products were then purified and sequenced using an Illumina HiSeq2000 PEI101 sequencer according to the manufacturer's instructions (Illumina, San Diego, CA, USA).
Mapping and analysis of ALV-J integration sites.
Crude sequence reads were first aligned to a reference ALV-J genome (accession number Z46390) using quality-weighted alignments within the Burrows-Wheeler Aligner and Short Oligonucleotide Analysis software package (18, 19). Further analysis to identify viral integration sites was performed using the BLAT tool of the University of California, Santa Cruz (UCSC), Chicken Genome Browser Gateway (http://genome.ucsc.edu/cgi-bin/hgGateway) against the November 2011 version of the chicken genome sequence. Integration site sequences were considered authentic only if they contained the ALV-J LTR terminal sequence up to the 5′-CA-3′ integration dinucleotide and 20 bp or more of chicken genomic sequence. These sequences also had to match the genome with >95% identity using default parameters in the BLAT ranking (galGal4 assembly of the chicken genome). Sequences were discarded as mapping to multiple sites when they had more than one match on the chicken genome. Genomic features were annotated when their genomic coordinates overlapped for ≥1 nucleotide with a ±50-kb interval around each integration site. To validate the integrated ALV-J sequence, primer pairs were designed to span both the chicken and viral gene sequences at the insertion sites. PCR validation was performed on both tumors and normal liver tissues using a Veriti 96-Well Thermal Cycler (Life Technologies, Guangzhou, China). For each PCR product, we used Applied Biosystems 3730 capillary DNA analyzers to perform dye terminator Sanger sequencing (Applied Biosystems, Foster City, CA, USA).
Identification of the CIS gene was performed by bioinformatic analyses of the UCSC Chicken Genome Browser Gateway. For this analysis, a CIS was defined as a genomic region of <100 kb targeted by at least four different integrations from independent tumors, in accordance with the statistical definition of CIS developed in previous studies (20, 21). The closest gene to the CIS genomic region was considered a CIS gene.
Isolation of total RNA and microarray expression profiling.
Total RNA from four ALV-J-positive ML sternums and three uninfected nontumor sternum samples was extracted using TRIzol according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The Chicken 4-by-44K Gene Expression Array, version 2, Agilent Array platform, an oligonucleotide-based array containing 43,803 Gallus gallus probe sets, was used for gene expression analysis. Sample preparation and microarray hybridization were performed using the manufacturer's standard protocols. We performed a single microarray for each sample as this is the widely accepted standard when Agilent commercial microarrays that contain internal quality controls are used (22). Quantile normalization and subsequent data processing were performed using the GeneSpring GX, version 11.5.1, software package (Agilent Technologies, Santa Clara, CA, USA). Fold change expression values refer to the mean ± standard deviation (SD) for each group. Differential gene expression in tumors relative to their expression in normal tissues was analyzed by a pairwise two-tailed t test. We performed clustering analysis with unsupervised hierarchical methods using a Euclidean distance metric in Multiple Experiment Viewer (MEV), version 4.8, software (23). Gene set enrichment analysis (GSEA [http://www.broadinstitute.org/gsea/index.jsp]) was used to analyze the microarray data (24) to evaluate sets of gene differences between four ML masses and three nontumor sternum samples.
To validate the microarray data, the expression of three CIS genes was determined by quantitative real-time PCR (qRT-PCR) analysis of three independent biological replicates. We prepared the cDNA of ML and nontumor sternum samples using random decamer primers and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Beijing, China). Primers used for qRT-PCR were designed using primer3 software (http://primer3.wi.mit.edu/). Primer pairs (see Table S1 in the supplemental material) were selected based on specificity as determined by dissociation curves. qRT-PCR was performed using a 7500 Fast Real-Time PCR system (Applied Biosystems) with SYBR green PCR Master Mix (Applied Biosystems) according to the manufacturer's guidelines. For assay validation, purified products were sequenced to verify correct target amplification. We calculated the relative expression level of each gene using the formula 2−ΔΔCT, where CT is threshold cycle, normalized to the chicken housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH; GenBank no. NM_204305), and represented it as fold change relative to the mean of nontumor samples. Standard deviations were calculated using the relative expression ratios of three replicates for each gene measured. Statistical analyses were performed using GraphPad Prism, version 5, software (GraphPad Software, San Diego, CA, USA). A P value of <0.05 was considered significant.
Western blotting.
To validate the differences in protein levels between normal and tumor tissue samples, Western blotting was performed on three representative ALV-J-positive ML sternums and three uninfected nontumor sternum samples. Total cellular proteins were extracted from all samples using radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) supplemented with protease inhibitor cocktail (GenStar, Beijing, China) (25). Total protein (20 mg) was subjected to SDS-PAGE analysis and transferred onto nitrocellulose membranes (Roche, Dassel, Germany) using an iBlot dry blotting system (Invitrogen). Membranes were blocked with 5% skimmed milk for 1 h at 37°C and then incubated overnight at 4°C with specific mouse anti-c-Myc antibody (Novus Biologicals, Littleton, CO, USA), rabbit anti-ZIC1 antibody (Epitomics, Burlingame, CA, USA), and rabbit anti-GADPH antibody (Santa Cruz Biotechnology, Dallas, TX, USA). Membranes were washed three times in phosphate-buffered saline with 0.1% Tween 20, pH 7.4 (PBST), and then incubated at 37°C for 1 h with IRDye 700DX-conjugated anti-rabbit IgG or IRDye 800-conjugated anti-mouse IgG (1:8,000 in PBST; Rockland Immunochemicals, Gilbertsville, PA, USA) as a secondary antibody. Membranes were washed a further three times in PBST; then the signal was visualized and analyzed with an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA).
Gene expression in chicken livers.
Fifty individual chicken samples (25 ALV-free normal livers and 25 ALV-J-infected ML-affected livers) were collected and used for gene expression analysis. Total RNA isolation, cDNA preparation, and gene expression assays for each CIS gene (MYC, TERT, and ZIC1) were performed as previously described. We calculated the relative expression level of each gene using the 2−ΔΔCT method as described above.
Microarray data accession number.
Data sets for the microarray analysis of the whole transcriptome of four ALV-J-positive ML sternums and three uninfected nontumor sternum samples were deposited in the Gene Expression Omnibus (GEO) database under accession number GSE48674.
RESULTS
High-definition mapping of ALV-J proviral integrations in ML cases.
We performed the targeted capture and sequencing of tumor genomes for six ALV-J-positive ML livers (designated 001L to 006L). DNA was extracted, and virus-genome junctions were enriched by MyGenostics GenCap Target Enrichment technology. Enriched target sequences were massively sequenced in parallel on an Illumina HiSeq2000 sequencer, yielding 51.79 Mb of raw sequence reads from all ML samples. Preliminary statistical analysis of the sequencing results showed that up to 20% of the total sequence mapped to the ALV-J genome (see Table S2 in the supplemental material).
As previously described, we relied on off-target coverage flanking the capture regions to identify viral integration sites (26, 27). Raw sequence reads containing a complete retroviral terminal sequence were processed through an automated bioinformatic pipeline that eliminated short and disqualified sequences from each tumor and were mapped on the UCSC galGal4 chicken genome. This resulted in more than 2,000 putative viral junction fragments being isolated, sequenced, and mapped from six individual liver tumors. Following elimination of the duplicate sequences from each tumor, 241 unique retroviral integration sites were identified (see Table S3 in the supplemental material). Local features at each integration site were also determined. The 241 unique ALV-J insertion sites were randomly distributed across the whole chicken genome, with a few common integrations (Fig. 1). To confirm the validity of the viral insertion sites, we randomly selected 23 integration sites in the six affected genes (ZIC1, EGFR, HGF, TERT, MYC, and NDUFS6) for PCR analysis. Sanger sequencing of the PCR products successfully validated 73.9% of these integration sites and demonstrated that viral insertion sites were identical to those identified using data from next-generation sequencing (NGS) (see Table S4).
FIG 1.

ALV-J integration sites in the chicken genome. The chicken chromosomes are shown numbered at both sides of the linear chromosomes. Note that due to the incomplete status of the draft chicken genome sequence, gene-dense regions are not represented. Each integration site is shown as a lollipop above the linear chromosomes. For each chromosome, the color of the lollipop and the dashes on the bar indicate integration within the gene TSS-proximal region, inside genes (intragenic), and outside genes (intergenic). The annotation of ALV-J integration sites can be found in Fig. S1 in the supplemental material. The software used to draw the ideogram was an R implementation for extending the Grammar of Graphics for Genomic Data (50).
Common insertion site genes.
Proviral tagging analysis has previously been used as a powerful way to uncover genes involved in oncogenesis, where tumors were induced by proviral integrations and the genomic loci of these proviruses were identified (28, 29). We hypothesized that putative ML causal genes would be repeatedly targeted by ALV-J proviral integrations at a higher frequency than expected in a random distribution. Comparing insertions retrieved from ML livers induced by ALV-J infection, we observed few common genes affected by this virus. A summary of the 13 genes targeted more than once is shown in Table 1.
TABLE 1.
Summary of genes represented more than once in a library of 241 insertion sites from ML samples
| Gene symbol | Gene name or description | No. of hits | Reported function relevant to cancera |
|---|---|---|---|
| MYC | V-myc myelocytomatosis viral oncogene homolog | 7 | Cell cycle, apoptosis, and cellular transformation |
| TERT | Telomerase reverse transcriptase | 4 | Cellular senescence |
| ZIC1 | Zic family member 1 | 5 | Proliferation, invasion, and survival |
| NDUFS6 | NADH dehydrogenase Fe-S protein 6 | 3 | Oxidative phosphorylation |
| PARK2 | Parkinson protein 2, E3 ubiquitin protein ligase | 2 | Tumor suppressor |
| PXDN | Peroxidasin homolog | 2 | Tumor cell adhesion |
| LOC100858325 | Protein Shroom1-like | 2 | NA |
| LOC100858243 | Exostosin-1-like | 2 | NA |
| LOC100858205 | Exostosin-1-like | 2 | NA |
| HGF | Hepatocyte growth factor | 2 | Cancer cell migration and invasion |
| FAM49A | Family with sequence similarity 49, member A | 2 | NA |
| EGFR | Epidermal growth factor receptor | 2 | Cell proliferation, survival, migration, and differentiation |
| DCLK1 | Doublecortin-like kinase 1 | 2 | Candidate CSC marker, radioresistance, and tumor aggressiveness |
CSC, cancer stem cell; NA, not available.
Based on previously reported statistical definitions (20, 21), at least four different integrations from independent tumors are necessary to define a CIS. Therefore, although several of the genes listed in Table 1 with two or three integrations are validated human cancer genes, they were not analyzed further in this study as fewer than four integrations could have occurred by chance. We therefore identified three putative CIS genes: MYC (targeted by seven integrations), TERT (four integrations), and ZIC1 (five integrations). The genomic locations of the tumor-specific ALV-J integration sites in these three genes are shown in Fig. 2 and Table S3 in the supplemental material. In addition, none of the CISs found in ML tumors were targeted by ALV-J integrations retrieved from tumor-free infected livers (n = 10) (data not shown). However, as so many insertions were reported, our study suggests that there are a large number of “passenger” integration events, indicating that the ALV-J multiplicity of infection (MOI) in ML may be high.
FIG 2.

Mapping of ALV-J common insertion sites. The ALV-J insertion sites in the recurrently affected genes, MYC, TERT, and ZIC1, were mapped to the UCSC galGal4 chicken genome. Each blue arrow represents the location of an ALV integration site identified from one clinical sample in this study. The forward and reverse orientations of proviruses in CISs are shown by the up and down blue arrows. Chr, chromosome. Boxes and EX represent exons, and the open arrows show the orientation of the genes.
Gene expression profiling.
Integrations targeting MYC, TERT, and ZIC1 were repeatedly found in independent liver ML samples. For whole-transcriptome studies, we performed microarray analysis to examine the whole transcriptome of four ALV-J-positive ML sternums and three uninfected nontumor sternum samples (data deposited in GEO database under accession number GSE48674). Over 3,000 genes and expressed sequence tags (ESTs) were shown to be expressed at significantly different levels between the two groups (P < 0.05; estimated false-discovery rate, 0.007). Hierarchical unsupervised clustering identified five main clusters (Fig. 3a), of which clusters 1, 3, and 4 consisted of genes or ESTs that were mainly underexpressed in ML samples compared with nontumor tissues. Among these were genes for molecule metabolic processes (PTGS2), the extracellular matrix (ECM) and ECM-receptor status (CD36 and CD44), cell adhesion (ACTN2 and FN1), and glycolysis/gluconeogenesis (ACSS1 and ALDH1A3).
FIG 3.

Transcriptome deregulations in ALV-J-induced ML tumors. (a) Heat map and dendrogram showing hierarchical unsupervised clustering analysis of ALV-J-induced ML tumors and nontumor samples. Expression levels in the heat maps are color coded from blue (low) to red (high). (b) Expression profile of ALV-J-induced ML samples (T, in black) compared to three nontumor samples (N, in white) by GSEA. Heat map representations of the most upregulated (top) and downregulated (bottom) genes of oxidative phosphorylation between the two groups (from blue, low expression, to red, high expression) are shown. GSEA statistics: NES, normalized enrichment score; FDR q value, false discovery rate; FWER P value, family-wise error rate. The enrichment plot at bottom shows the overrepresentation at the top and bottom of the ranked gene set. (c) Expression profile of ALV-J-induced ML samples (T, in black) compared to three nontumor samples (N, in white) by GSEA. Heat map representations of the most upregulated (top) and downregulated (bottom) genes of the ErbB signaling pathway in tumors versus expression in nontumor samples are shown. The enrichment plot is as described for panel b (see also Table S5 and Note S1 in the supplemental material).
Cluster 2 genes were highly upregulated in most ML samples and were either directly or indirectly linked to the actin cytoskeleton in cancer cell migration/invasion (BRAF and PAK1) or the epidermal growth factor receptor (erbB2 and erbB3). Cluster 5 genes were also upregulated in ML samples. These included HGF, MYC oncogene, and CLDN15. GSEA was used to compare the expression profiles of ML samples with nontumor tissues, and we observed the common downregulation of genes involved in oxidative phosphorylation, ECM-receptor interaction, focal adhesion, and apoptosis (Fig. 3b; see also Fig. S2a to c in the supplemental material). We also observed the upregulation of genes involved in cancer, cell cycle, and growth (see Table S5). Several erythroblastic leukemia viral oncogene homolog (ErbB) signaling pathway genes and Janus kinase/signal transducers and activators of transcription (JAK-STAT) signaling pathway genes were upregulated exclusively in ALV-J-induced ML tumors (Fig. 3c; see also Fig. S2d in the supplemental material).
The newly identified cancer genes are relevant to the chicken ML phenotype.
Regardless of the distribution of ALV-J insertion sites, all three CIS genes showed a significant upregulation of expression in tumor samples compared to control samples (Fig. 4). qRT-PCR analysis of representative samples validated the gene expression data obtained by microarray analysis (Fig. 4b to d). Comparative analysis of gene transcription levels indicated that RNA levels of MYC, TERT, and ZIC1 were significantly higher in ML sternum samples than in uninfected nontumor sternums (P = 0.0037, P = 0.0008, and P = 0.0191, respectively, for qRT-PCR; P < 0.0001, P = 0.0006, and P < 0.0001, respectively, for microarray analysis; unpaired t test). The significant recurrence of integration in the three ALV-J-affected genes and resultant expression level changes indicate that they may play important roles in ML. Furthermore, Western blot analysis revealed that MYC proto-oncogene and ZIC1 protein levels were upregulated only in ML tumors induced by ALV-J integration and not in normal samples (Fig. 4e). We did not determine the protein level of the TERT protein because of the absence of a specific anti-chicken-TERT antibody.
FIG 4.

ALV-J integrations at CISs upregulate the targeted genes. (a) Heat map showing expression levels of recurrent ALV-J integrated genes in ML samples and nontumor samples. Magenta, low expression; cyan, high expression. (b to d) Expression fold changes for MYC (b), TERT (c), and ZIC1 (d) for nontumor samples (non-tum) and ML samples from the qRT-PCR and Agilent microarray collections. In the box plots, the middle line represents the median (50th percentile), with the bottom and top of the box representing the 25th and 75th percentiles of the data, respectively. The ends of the whiskers represent the lowest and highest data within the 1.5 interquartile range. The interquartile range was defined as the distance between the lower and upper quartiles of the data. Their associated P values were calculated by an unpaired t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (e) Western blot analysis detected high expression of the MYC and ZIC1 proteins only in ML samples, not in the normal samples. Sample identification numbers are indicated above the panel as follows: T, tumor samples; N, nontumor samples. The results are representative of three independent experiments.
Upregulation of MYC, TERT, and ZIC1 expression has previously been observed in human cancers (30–32), suggesting that their activation is required for tumor initiation and/or progression. To survey these CIS genes in ALV-J-induced tumors, we performed qRT-PCR on ALV-J-positive ML livers and uninfected nontumor livers from a tissue collection available at our institution. MYC was found to be upregulated in 60 to 70% of all ML samples and was significantly upregulated in ML samples with respect to nontumor livers (P < 0.05, unpaired t test) (Fig. 5a). In the limited collection, TERT and ZIC1 expression was also significantly higher in ML samples than in nontumor livers (P < 0.001 and P < 0.001, respectively, unpaired t test) (Fig. 5b and c).
FIG 5.
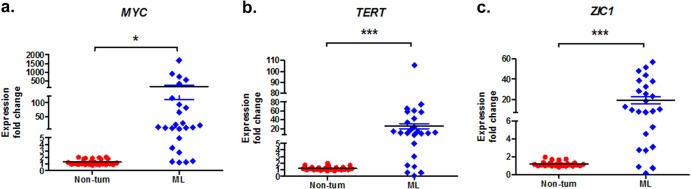
Expression analysis of newly identified CIS genes. (a to c) Expression fold changes for MYC, TERT, and ZIC1 for nontumor samples (non-tum) and ML samples from the collection. qRT-PCR was performed to detect the expression level of CIS genes. Black lines, mean; colored whiskers, SD. P values were determined by unpaired t test as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Since retroviral integration is not, in principle, site specific (33), the common sites of repeated viral integration in independent tumors could represent the tumor selection process in which cells that have had their gene(s) targeted by a provirus give rise to a tumor (13). Mapping integration sites in tumors provides powerful molecular tags to discover the potential genotoxicity of retrovirus integration in a specific genome region or disease context. By focusing on these insertions, target enrichment methods in which genomic regions are selectively captured from a DNA sample before sequencing can more effectively achieve a higher sensitivity with much less sequence and will be a valuable strategy for the detailed interrogation of retrovirus integration (34, 35).
To further understand the full spectrum of ALV-J integration distribution in avian ML tumors and to define this role in tumor pathogenesis, we used an ALV-J-specific targeted sequencing strategy involving hybrid capture coupled with Illumina HiSeq 2000 sequencing to produce a catalog of insertion sites from six ML samples. Several high-throughput techniques for the identification of retroviral integration sites have been established since 2007 (36). Here, we report on a simpler and less expensive method than the existing ones, which mapped over 20% of the total sequence back to the ALV-J genome. However, this level of efficiency is far from optimal, being slightly less than that of more expensive methods (37). Interestingly, the length of the target capture region was shown to be inversely proportional to the capture efficiency (38), so we plan to replicate our study with the 3′ terminal LTR sequence of ALV-J-specific targeted enrichment, which is shorter than the region examined in the present study, to improve our capture efficiency.
Ultimately, we retrieved more than 2,000 ALV-J insertions from six tumors, from which 241 unique viral integration sites were identified and mapped on the draft chicken genome sequence. Thus, the percentage of duplicates (88%) is unexpectedly high. Considering the sources and characteristics of the samples, this may result from tumor cell clonality or biological selection of viral integrations. As ALV-J is a group of chronic RNA tumor viruses that typically cause mono- or oligoclonal tumors with a long latency of 3 to 12 months, tumorigenesis caused by nondefective ALV-J is not induced by the presence of a virally modified proto-oncogene in the virus genome but rather from mutations caused by insertion of the proviral retrovirus into the host genome (29). The malignant ML cell therefore arises when several of its specific regulatory pathways have been distorted by ALV integration with a long latency. Since these pathways consist of cascades of functionally connected genes, the provirus has to integrate into one particular gene to deregulate the entire pathway and lead to malignant transformation. This integration is more likely to result from the selective expansion of tumor cells carrying insertions at this locus than chance alone. Moreover, most tumor cells derive from a single infected cell and may carry a large number of insertions of varying clonality (39). In our ongoing study, we hope to identify a more efficient sample-processing method without artificial bias to further explore these viral integration events while reflecting the relative proportions of insertions from the starting DNA.
CIS analysis is a classical way to discover genes involved in oncogenesis. The fundamental assumption made in this analysis is that finding proviral integrations in close proximity more frequently in multiple independent tumors than would be predicted randomly provides evidence that the genes near the integrations are involved in tumor formation (21). Tumorigenesis induced by ALV insertional mutagenesis in the gene promoter has also been reported in past decades (40). Taking into account the integration preferences of the retroviruses, the evidence suggests both a natural tendency to generate CISs and to integrate into genes that are potentially relevant to tumor. We therefore used a much more stringent criterion for our analysis (20, 21) by taking a CIS of four or more ALV-J integrations into tumors to denote statistical significance. Although we found that MYC, TERT, and ZIC1 genes were CIS genes in independent chicken ML samples, which might be a putative “driver” for oncogene activation, why and how these regions were targeted remain unknown. Furthermore, all three genes showed elevated expression in ML samples relative to nontumor samples. As the transcriptional deregulation of other host genes is likely to result from the secondary effects of overexpression of CIS genes such as MYC, these data suggest that the mutation represented by CIS gene integration may have important roles in tumor phenotype, ML development, and transcriptome alteration.
The functions of the three CIS genes in ML development are not well known. However, MYC and TERT are common oncogenes that are mutated in retrovirally induced, usually hematopoietic, tumors (31, 41). MYC, the gene with the maximum number of ALV-J integrations in our study, is one of the most highly amplified oncogenes in many different cancers. It also plays a pivotal role in cell proliferation, growth, apoptosis, differentiation, and stem cell self-renewal (30, 42). MYC expression is highly regulated, so its deregulated expression is sufficient to drive oncogenesis in some transgenic mouse tissues (43). In particular, the observation of fusion transcript ALV-J Gag-Myc in ALV-J-transformed bone marrow cells demonstrated effective retroviral integration into the regulatory region of MYC, leading to the loss of the first exon and the activation of this oncogene (44). In our study, the ALV-J provirus mostly integrated into the region proximal to the MYC transcription start site (TSS) in the forward transcriptional orientation, where many transcriptional regulatory motifs are located. Moreover, the ErbB signaling pathway was specifically deregulated in ML samples. These results suggest that the aberrantly elevated expression of MYC in chicken ML samples is caused by the insertional mutation of ALV-J, which in turn induces myelocytomatosis transformation.
TERT encodes the reverse transcriptase catalytic subunit of the enzyme telomerase, which adds telomeric repeats to the ends of chromosomes. Genome-wide association studies have previously implicated the TERT-CLPTM1L locus in susceptibility to a variety of human cancers, suggesting that TERT is a “pan-cancer susceptibility locus” (45). In addition, Yang et al. showed that telomerase is frequently activated by ALV-A integration in chicken rapid-onset B cell lymphomas, which indicated that retroviral upregulation of cellular TERT by insertional activation is important to initiate or enhance tumor progression (12). Consistent with known reports (31), our present study identified repeated ALV-J integrations into TERT in a region 0.2 to 10 kb upstream of the TERT transcriptional promoter region and mainly in the opposite transcriptional orientation to TERT. This indicates that TERT is an ALV-J CIS in ML. Moreover, human TERT is thought to be expressed in stem cells and cancer cells (31), so the significantly higher TERT mRNA level observed in ML tissues harboring viral integrations suggests that ALV-J proviral integration in TERT may have a significant impact on tumorigenesis.
Another major finding in our study is that ZIC1 was identified as a novel ALV-J CIS gene in ML samples. Indeed, the retroviral integration appeared to contribute to abnormal ZIC1 expression. ZIC1 encodes a transcription factor that is an important member of the ZIC family of C2H2-type zinc finger proteins and is specifically expressed in cerebellar granule cells and their precursors, where it is involved in neurogenesis developmental processes. ZIC1 overexpression in chickens has been shown to block neural tube differentiation (46), while its elevated expression in humans has been reported in endometrial cancer and desmoid tumors (32, 47). These findings suggest that ZIC1 overexpression may be associated with ML formation in chicken. It is important to note, however, that its overexpression also results in the inactivation of Shh, phosphatidylinositol 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) signaling pathways, as well as the regulation of multiple downstream targets that are essential for the development and progression of gastric cancer, as reported by Zhong et al. (48). Therefore, the oncogenic role of ZIC1 in cancer remains obscure.
A high level of CIS gene mRNA was also found in some field ALV-J-induced ML samples. This might result from insertional activation of CIS genes or other gene(s) that directly or indirectly control(s) CIS gene expression. It is important to note that although proto-oncogene MYC plays a critical role in tumor development in birds and mammals (30), its activation alone is insufficient for full malignancy to develop and that cooperation with other proto-oncogenes is required to induce lymphomas in both murine and avian systems (5, 49). In the context of the present study, this suggests that TERT, ZIC1, or other genes are likely to have a cooperative interaction with MYC in oncogenesis.
ALV-Js have been successfully eradicated from chicken breeding flocks in the poultry industries of developed countries, and the control and eradication of ALV-J in China are now progressing steadily. To further study the pathogenesis of ALV-J infections, it will be necessary to elucidate the in vivo viral integration and tumorigenesis mechanism. Unlike previous studies that reported the distribution or targeting bias of ALV integration in the genomes of different cell lines, we focused on such integrations in chicken tumor samples and the targeting of regions that resulted in the overexpression of CIS genes. We hypothesize that the recurrent integration of ALV-J into specific regions of the host genome acts as an early cancer-driving event as CIS genes showed deregulated expression compared to nontumor samples. However, our findings found no direct evidence to confirm the functional effects of CIS gene expression levels on ML development. Therefore, to validate the oncogenic potential of all identified genes in vivo and to uncover their molecular mechanisms behind ALV-J infection, further extensive experiments are currently in progress.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Modern Agriculture Talents Support Program (2012; number 160), a Key Project from the Agricultural Ministry (CARS-42-G09), the Key Program of Science and Technology Development of Guangdong Province (2012A0201000001), the Natural Science Foundation of Guangdong Province (S2011010001946), the NSFC-Guangdong Union Foundation (U0831002), and the Public Industry Research Program of the Ministry of Agriculture, China (201203055).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 26 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02995-13.
REFERENCES
- 1.Payne LN, Brown SR, Bumstead N, Howes K, Frazier JA, Thouless ME. 1991. A novel subgroup of exogenous avian leukosis virus in chickens. J. Gen. Virol. 72:801–807. 10.1099/0022-1317-72-4-801 [DOI] [PubMed] [Google Scholar]
- 2.Payne LN, Gillespie AM, Howes K. 1992. Myeloid leukaemogenicity and transmission of the HPRS-103 strain of avian leukosis virus. Leukemia 6:1167–1176 [PubMed] [Google Scholar]
- 3.Fadly AM, Smith EJ. 1999. Isolation and some characteristics of a subgroup J-like avian leukosis virus associated with myeloid leukosis in meat-type chickens in the United States. Avian Dis. 43:391–400. 10.2307/1592636 [DOI] [PubMed] [Google Scholar]
- 4.Nakamura K, Ogiso M, Tsukamoto K, Hamazaki N, Hihara H, Yuasa N. 2000. Lesions of bone and bone marrow in myeloid leukosis occurring naturally in adult broiler breeders. Avian Dis. 44:215–221. 10.2307/1592529 [DOI] [PubMed] [Google Scholar]
- 5.Tam W, Hughes SH, Hayward WS, Besmer P. 2002. Avian bic, a gene isolated from a common retroviral site in avian leukosis virus-induced lymphomas that encodes a noncoding RNA, cooperates with c-myc in lymphomagenesis and erythroleukemogenesis. J. Virol. 76:4275–4286. 10.1128/JVI.76.9.4275-4286.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Payne LN, Gillespie AM, Howes K. 1991. Induction of myeloid leukosis and other tumours with the HPRS-103 strain of ALV. Vet. Rec. 129:447–448. 10.1136/vr.129.20.447 [DOI] [PubMed] [Google Scholar]
- 7.Cheng Z, Liu J, Cui Z, Zhang L. 2010. Tumors associated with avian leukosis virus subgroup J in layer hens during 2007 to 2009 in China. J. Vet. Med. Sci. 72:1027–1033. 10.1292/jvms.09-0564 [DOI] [PubMed] [Google Scholar]
- 8.Zhang HN, Lai HZ, Qi Y, Zhang XT, Ning ZY, Luo KJ, Xin CA, Cao WS, Liao M. 2011. An ALV-J isolate is responsible for spontaneous haemangiomas in layer chickens in China. Avian Pathol. 40:261–267. 10.1080/03079457.2011.560142 [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Yun B, Qin L, Pan W, Qu Y, Liu Z, Wang Y, Qi X, Gao H, Wang X. 2012. Molecular epidemiology of avian leukosis virus subgroup J in layer flocks in China. J. Clin. Microbiol. 50:953–960. 10.1128/JCM.06179-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayward WS, Neel BG, Astrin SM. 1981. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 290:475–480. 10.1038/290475a0 [DOI] [PubMed] [Google Scholar]
- 11.Goodwin RG, Rottman FM, Callaghan T, Kung HJ, Maroney PA, Nilsen TW. 1986. c-erbB activation in avian leukosis virus-induced erythroblastosis: multiple epidermal growth factor receptor mRNAs are generated by alternative RNA processing. Mol. Cell. Biol. 6:3128–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F, Xian RR, Li Y, Polony TS, Beemon KL. 2007. Telomerase reverse transcriptase expression elevated by avian leukosis virus integration in B cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 104:18952–18957. 10.1073/pnas.0709173104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pajer P, Pecenka V, Karafiat V, Kralova J, Horejsi Z, Dvorak M. 2003. The twist gene is a common target of retroviral integration and transcriptional deregulation in experimental nephroblastoma. Oncogene 22:665–673. 10.1038/sj.onc.1206105 [DOI] [PubMed] [Google Scholar]
- 14.Pajer P, Pecenka V, Kralova J, Karafiat V, Prukova D, Zemanova Z, Kodet R, Dvorak M. 2006. Identification of potential human oncogenes by mapping the common viral integration sites in avian nephroblastoma. Cancer Res. 66:78–86. 10.1158/0008-5472.CAN-05-1728 [DOI] [PubMed] [Google Scholar]
- 15.Raines MA, Lewis WG, Crittenden LB, Kung HJ. 1985. c-erbB activation in avian leukosis virus-induced erythroblastosis: clustered integration sites and the arrangement of provirus in the c-erbB alleles. Proc. Natl. Acad. Sci. U. S. A. 82:2287–2291. 10.1073/pnas.82.8.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Liu X, Liu H, Xu C, Liao Y, Wu X, Cao W, Liao M. 2013. Isolation, identification, and phylogenetic analysis of two avian leukosis virus subgroup J. strains associated with hemangioma and myeloid leukosis. Vet. Microbiol. 166:356–364. 10.1016/j.vetmic.2013.06.007 [DOI] [PubMed] [Google Scholar]
- 17.Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010:pdb.prot5448. 10.1101/pdb.prot5448 [DOI] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967. 10.1093/bioinformatics/btp336 [DOI] [PubMed] [Google Scholar]
- 20.Abel U, Deichmann A, Bartholomae C, Schwarzwaelder K, Glimm H, Howe S, Thrasher A, Garrigue A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Fischer A, Jaeger D, von Kalle C, Schmidt M. 2007. Real-time definition of non-randomness in the distribution of genomic events. PLoS One 2:e570. 10.1371/journal.pone.0000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu X, Luke BT, Burgess SM. 2006. Redefining the common insertion site. Virology 344:292–295. 10.1016/j.virol.2005.08.047 [DOI] [PubMed] [Google Scholar]
- 22.Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, Bao W, Fang H, Kawasaki ES, Hager J, Tikhonova IR, Walker SJ, Zhang L, Hurban P, de Longueville F, Fuscoe JC, Tong W, Shi L, Wolfinger RD. 2006. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat. Biotechnol. 24:1140–1150. 10.1038/nbt1242 [DOI] [PubMed] [Google Scholar]
- 23.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. 2006. TM4 microarray software suite. Methods Enzymol. 411:134–193. 10.1016/S0076-6879(06)11009-5 [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng SZ, Cao WS, Liao M. 2011. The PI3K/Akt pathway is involved in early infection of some exogenous avian leukosis viruses. J. Gen. Virol. 92:1688–1697. 10.1099/vir.0.030866-0 [DOI] [PubMed] [Google Scholar]
- 26.Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2:E234. 10.1371/journal.pbio.0020234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barr SD, Leipzig J, Shinn P, Ecker JR, Bushman FD. 2005. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 79:12035–12044. 10.1128/JVI.79.18.12035-12044.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kool J, Berns A. 2009. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Cancer 9:389–399. 10.1038/nrc2647 [DOI] [PubMed] [Google Scholar]
- 29.Uren AG, Kool J, Berns A, van Lohuizen M. 2005. Retroviral insertional mutagenesis: past, present and future. Oncogene 24:7656–7672. 10.1038/sj.onc.1209043 [DOI] [PubMed] [Google Scholar]
- 30.Dang CV. 2012. MYC on the path to cancer. Cell 149:22–35. 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armanios M, Greider CW. 2005. Telomerase and cancer stem cells. Cold Spring Harb. Symp. Quant. Biol. 70:205–208. 10.1101/sqb.2005.70.030 [DOI] [PubMed] [Google Scholar]
- 32.Brill E, Gobble R, Angeles C, Lagos-Quintana M, Crago A, Laxa B, Decarolis P, Zhang L, Antonescu C, Socci ND, Taylor BS, Sander C, Koff A, Singer S. 2010. ZIC1 overexpression is oncogenic in liposarcoma. Cancer Res. 70:6891–6901. 10.1158/0008-5472.CAN-10-0745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coffin JM, Hughes SH, Varmus HE. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 34.Leproust E. 2012. Target enrichment strategies for next generation sequencing. MLO Med. Lab. Obs. 44:26–27 [PubMed] [Google Scholar]
- 35.Mertes F, Elsharawy A, Sauer S, van Helvoort JM, van der Zaag PJ, Franke A, Nilsson M, Lehrach H, Brookes AJ. 2011. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genomics 10:374–386. 10.1093/bfgp/elr033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. 2007. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 17:1186–1194. 10.1101/gr.6286907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoppman-Chaney N, Peterson LM, Klee EW, Middha S, Courteau LK, Ferber MJ. 2010. Evaluation of oligonucleotide sequence capture arrays and comparison of next-generation sequencing platforms for use in molecular diagnostics. Clin. Chem. 56:1297–1306. 10.1373/clinchem.2010.145441 [DOI] [PubMed] [Google Scholar]
- 38.Hodges E, Rooks M, Xuan Z, Bhattacharjee A, Benjamin Gordon D, Brizuela L, Richard McCombie W, Hannon GJ. 2009. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 4:960–974. 10.1038/nprot.2009.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neel BG, Hayward WS, Robinson HL, Fang J, Astrin SM. 1981. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell 23:323–334. 10.1016/0092-8674(81)90128-8 [DOI] [PubMed] [Google Scholar]
- 40.Shih CK, Linial M, Goodenow MM, Hayward WS. 1984. Nucleotide sequence 5′ of the chicken c-myc coding region: localization of a noncoding exon that is absent from myc transcripts in most avian leukosis virus-induced lymphomas. Proc. Natl. Acad. Sci. U. S. A. 81:4697–4701. 10.1073/pnas.81.15.4697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudley JP, Mertz JA, Rajan L, Lozano M, Broussard DR. 2002. What retroviruses teach us about the involvement of c-Myc in leukemias and lymphomas. Leukemia 16:1086–1098. 10.1038/sj.leu.2402451 [DOI] [PubMed] [Google Scholar]
- 42.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 43.Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, Jenkins NA, Copeland NG. 2002. New genes involved in cancer identified by retroviral tagging. Nat. Genet. 32:166–174. 10.1038/ng949 [DOI] [PubMed] [Google Scholar]
- 44.Chesters PM, Howes K, McKay JC, Payne LN, Venugopal K. 2001. Acutely transforming avian leukosis virus subgroup J strain 966: defective genome encodes a 72-kilodalton Gag-Myc fusion protein. J. Virol. 75:4219–4225. 10.1128/JVI.75.9.4219-4225.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beesley J, Pickett H, Johnatty S, Chen X, Li JJ, Rider D, Stutz M, Lambrecht D, Chang-Claude J, Dork T. 2012. Functional polymorphisms in the TERT promoter are associated with risk of serious ovarian and breast cancer. Hered. Cancer Clin. Pract. 10:A86. 10.1186/1897-4287-10-S2-A86 [DOI] [Google Scholar]
- 46.Ebert PJ, Timmer JR, Nakada Y, Helms AW, Parab PB, Liu Y, Hunsaker TL, Johnson JE. 2003. Zic1 represses Math1 expression via interactions with the Math1 enhancer and modulation of Math1 autoregulation. Development 130:1949–1959. 10.1242/dev.00419 [DOI] [PubMed] [Google Scholar]
- 47.Wong YF, Cheung TH, Lo KW, Yim SF, Siu NS, Chan SC, Ho TW, Wong KW, Yu MY, Wang VW, Li C, Gardner GJ, Bonome T, Johnson WB, Smith DI, Chung TK, Birrer MJ. 2007. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene 26:1971–1982. 10.1038/sj.onc.1209986 [DOI] [PubMed] [Google Scholar]
- 48.Zhong J, Chen S, Xue M, Du Q, Cai J, Jin H, Si J, Wang L. 2012. ZIC1 modulates cell-cycle distributions and cell migration through regulation of sonic hedgehog, PI(3)K and MAPK signaling pathways in gastric cancer. BMC Cancer 12:290. 10.1186/1471-2407-12-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams JM, Cory S. 1992. Oncogene co-operation in leukaemogenesis. Cancer Surv. 15:119–141 [PubMed] [Google Scholar]
- 50.Yin T, Cook D, Lawrence M. 2012. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 13:R77. 10.1186/gb-2012-13-8-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.