

Abstract
Natural killer cells belong to the family of innate lymphoid cells comprising the frontline defense against infected and transformed cells. Development and activation of natural killer cells is highly dependent on interleukin-15 signaling. However, very little is known about the transcription program driving this process. The transcription factor Runx3 is highly expressed in natural killer cells, but its function in these cells is largely unknown. We show that loss of Runx3 impaired interleukin-15-dependent accumulation of mature natural killer cells in vivo and under culture conditions and pregnant Runx3−/− mice completely lack the unique population of interleukin-15-dependent uterine natural killer cells. Combined chromatin immunoprecipitation sequencing and differential gene expression analysis of wild-type versus Runx3-deficient in vivo activated splenic natural killer cells revealed that Runx3 cooperates with ETS and T-box transcription factors to drive the interleukin-15-mediated transcription program during activation of these cells. Runx3 functions as a nuclear regulator during interleukin-15-dependent activation of natural killer cells by regulating the expression of genes involved in proliferation, maturation, and migration. Similar studies with additional transcription factors will allow the construction of a more detailed transcriptional network that controls natural killer cell development and function.
INTRODUCTION
Natural killer cells (NKC) belong to the family of innate lymphoid cells, originating from a common innate lymphoid progenitor (1). NKC are prearmed and constitute a key frontline defense against infected and transformed cells. Their functional responses are regulated by the combined action of a wide variety of germ line-encoded activating and inhibitory receptors. Killing of their target cells is mediated predominantly by release of perforin and granzyme-containing granules. In addition, NKC protect the host by producing inflammatory cytokines, including gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF), that influence the adaptive immune system (2, 3). Interestingly, at early stages of pregnancy, specialized uterine NKC (uNKC) play important constructive roles in endometrial remodeling, angiogenesis, and placentation (4).
The developmental stages of NKC have been best characterized in bone marrow (BM), their main production site in adult mice. Common lymphoid progenitors (CLP) in the BM give rise to pre-NK precursors (pre-NKP) (5) that yield NKP, characterized by expression of interleukin 2 (IL-2)/IL-15 receptor β chain but still lacking expression of other NKC lineage-specific markers. Subsequent developmental stages, including immature (iNKC) and mature (mNKC), are associated with acquisition of signature cell surface receptors, cytokine receptors, and specific integrins (2, 6). NKC development is highly dependent on IL-15 signaling. Mice lacking IL-15, its receptors, or downstream signaling components have a profound reduction in the number of peripheral NKC and a block in their differentiation (7–9).
Information about the molecular mechanisms underlying NKC differentiation and activation is emerging through identification of transcription factors (TFs) essential for their development (E4BP4, Id2, and Ets1) or maturation and function (Blimp1, T-bet, and Eomes) (10–13). However, very little is known about the transcription program mediated by these and other NKC TFs during cell development and function. Runx3 is highly expressed in NKC. It belongs to a small TF family of three members that are key regulators of lineage-specific gene expression in different cell types, including hematopoietic cells (14, 15). Each RUNX gene is transcriptionally regulated by two distinct promoters, designated P1 and P2 (14). RUNX TFs transcriptionally activate or repress their target genes by cooperation with other coactivators and/or corepressors and by recruiting acetyltransferases, histone deacetylases, and the polycomb repressing complex PRC1 (16–20).
Of the three RUNX TFs, Runx1 and Runx3 play important roles in hematopoiesis (15, 21–23). Previous observations have suggested that they play a role in NKC development. Mice transplanted with fetal liver cells derived from essential RUNX cofactor CBFβ-hypomorphic mice lack iNKC (24). Since CBFβ is essential for the function of both Runx1 and Runx3, the question of which Runx TF contributes to this severe NK phenotype remained open. Transgenic mice expressing a dominant negative form of RUNX exhibit decreased expressions of NK maturation markers (25). Of the two RUNX TFs (Runx1 and Runx3), Runx3 expression is higher in NKC (24, 25). Runx3 is also highly expressed in CD8+ T cells, and its loss impairs their function (22, 23). Both NKC and CD8+ T cells are cytotoxic lymphocytes that use similar effector machineries (26). However, the function of Runx3 in NKC is still largely unknown.
Here we have demonstrated that Runx3 is expressed from the NKP stage onwards and regulates the IL-15-responsive transcription program in these cells. In the absence of Runx3, IL-15-dependent accumulation of mature NKC is impaired. Interestingly, the unique uNKC subset that exhibits extensive IL-15-dependent proliferation at early gestation stages is completely missing at implantation sites of Runx3-deficient mice. Using combined chromatin immunoprecipitation-sequencing (ChIP-seq) and gene expression analysis of in vivo IL-15/IL-15Rα (IL-15/Rα)-treated splenic NKC, we identified a subset of Runx3-regulated genes that control the phenotypic outcome during the in vivo NKC response to IL-15, thus providing insights into the molecular mechanisms underlying Runx3 function in NKC.
MATERIALS AND METHODS
Mice.
Generation of Runx3−/−, Runx3P1-AFP/+, and Runx3P2-GFP/+ mice was described previously (27, 28). Rag−/− γc−/− females were obtained from Taconic farms (stock number 004111-MM). All experiments involving mice were conducted with adult (6- to 12-week-old) mice and were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Weizmann Institute of Science (permit number 4690812-2). All efforts were made to minimize mouse suffering.
FACS analysis.
For fluorescence-activated cell sorter (FACS) analysis, cells were reacted with the following fluorescence-labeled antibodies from e-Bioscience (San Diego, CA) targeting the following: NKp46 (29A1.4), CD122 (TMb1), CD3 (145-2CII), CD49b (DX5), CD11b (MI/70), CD27 (LG.7F9), KLRG1 (2F1), CD43 (R2/60), CD11c (N418), B220 (RA3-6B2), Ly49E/F (CM4), CD226 (10E5), c-Kit (2B8), CD81 (EAT2), CD107 (1D4B), annexin V (kit), fixable viability dye eFluor 780, and IFN-γ (XMG1.2). Intracellular IFN-γ was measured following activation of splenocytes for 5 h with IL-12 (10 ng/ml; R&D Systems) and IL-18 (20 ng/ml; MBL International, Woburn, MA). For NKC IFN-γ determination, cells were treated with brefeldin A (B-7651; Sigma, Israel) for the last 4 h, labeled with cell surface antibodies, fixed, permeabilized, and stained. Degranulation was analyzed under similar conditions in the presence of anti-CD107a-phycoerythrin. To record cells active in DNA synthesis, 2 mg BrdU was injected into IL-15/IL-15Rα-treated mice. Three hours later, mice were sacrificed and BrdU incorporation into NKC was determined using a BrdU Flow kit (BD Biosciences, San Jose, CA) and LSRII flow cytometer (BD Biosciences). Data were analyzed by using the software program FlowJo version 9.0 (TreeStar, Ashland, OR).
NKC culture and proliferation assay.
NKC from spleens of WT and Runx3−/− mice were enriched by negative selection using an NK cell isolation kit (R&D Systems) and cultured for 7 days at 0.5 × 106/ml in RPMI medium containing 10% fetal calf serum (FCS), 1% nonessential amino acids, 50 μM β-mercaptoethanol, and recombinant human IL-2 (1,000 U/ml; Biological Industries, Beit Haemek, Israel) or recombinant mouse IL-15 (1,000 U/ml; Peprotec, Israel). At the end of the 7-day culture period, all cells were NKp46+. To monitor cell proliferation, freshly isolated NKC were labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Life Technology) and cultured in either IL-2 or IL-15 as described above. Cell division-dependent CFSE dilution was determined at day 6 by FACS analysis.
In vivo NKC activation by IL-2 and IL-15/Rα.
Mouse IL-15 (0.5 μg) and recombinant mouse IL-15Rα (3 μg; R&D Systems) were mixed, incubated for 30 min at 37°C, diluted in 200 μl phosphate-buffered saline (PBS), and injected intraperitoneally into wild-type (WT) and Runx3−/− mice. Unless otherwise stated, two injections were given 48 h apart, and mice were sacrificed 48 h after the second injection. IL-2 (5 × 104 U) was injected intraperitoneally daily for 4 days.
IHC and in vitro cytotoxicity assay.
Immunohistochemistry (IHC) was conducted essentially as described previously (29). Briefly, paraffin sections were incubated with rabbit anti-Runx3 (1:1,000) and/or DBA (1:1,000) (L6533 lectin from Dolichos biflorus [horse gram], biotin conjugated; Sigma), or with goat anti-green fluorescent protein (anti-GFP) (1:200) (Abcam, Cambridge, United Kingdom). Periodic acid-Schiff (PAS) staining was conducted as described elsewhere (30).
A cytotoxicity assay was conducted with WT and Runx3−/− splenocytes as described by Gazit et al. (31).
Adoptive transfer of fetal liver cells.
E14.5 fetal liver cells of WT or Runx3−/− C57BL/6 embryos were injected intravenously (i.v.) into Rag−/− γc−/− females (each recipient was injected with cells derived from one embryo). Implantation sites were analyzed at gestation day 10.5 (gd10.5), ∼2 months posttransfer.
ChIP-seq and gene expression analysis.
ChIP was performed essentially as described previously (20). Briefly, cross-linked chromatin from approximately 40 × 106 NKC (duplicate, each from three mice) was prepared and fragmented to an average size of ∼200 bp by 40 cycles of sonication (30 s of sonication and 30 s of rest each) in 15-ml tubes using the Bioruptor UCD-200 sonicator (Diagenode, Liege, Belgium). For immunoprecipitation of chromatin, either our in-house rabbit anti-Runx3 antibody or rabbit preimmune serum was used. DNA was purified using QIAquick spin columns (Qiagen), the two samples were combined, and sequencing was performed using an Illumina (San Diego, CA) Hiseq2000 instrument according to the manufacturer's instructions. Runx3-bound regions were identified by analysis of the uniquely aligned reads of anti-Runx3 ChIP (60, 770, 073) versus preimmune serum (54, 424, 471) using the software program MACS1.4 (32). Peak distribution and percentage of Runx3 peaks, including RUNX binding sites (RBS), were determined using the program CisGenome. Enrichment for promoter peak regions was calculated using CEAS (Cistrom) (33). The GREAT algorithm with default parameters was used to determine the genes corresponding to Runx3-bound peaks. De novo discovery of TF binding sites was conducted using the program Homer with a motif length of 8 and CpG normalization.
For gene expression analysis, RNA was prepared from either IL-15-cultured or sorted spleen CD27+ CD11b−, CD27+ CD11b+, and CD27− CD11b+ NKC subsets (triplicates) using the micro-RNeasy kit (Qiagen). Purified RNA was reverse transcribed, amplified, and labeled using the Affymetrix GeneChip whole-transcript sense target labeling kit (Santa Clara, CA). Labeled cDNA was hybridized to either Affymetrix mouse 430.2 microarrays (cultured NK) or Affymetrix mouse gene 1.0ST microarrays (sorted spleen NK subsets) according to the manufacturer's instructions. Microarrays were scanned using the GeneChip scanner 3000 7G, and statistical analysis of data was performed using the Partek Genomics Suite 6.5 (Partek Inc., St. Louis, MO) software. CEL files (containing raw expression measurements) were imported to Partek GS. The data were preprocessed and normalized using the RMA (robust multichip average) algorithm (34) with GC correction and GC-RMA for in vivo and cultured NK cells, respectively. To identify differentially expressed genes, fold changes were calculated. Gene lists were created by filtering the genes based on the following: knockout/wild type (KO/WT) ratio ≥ 1.5; P < 0.05; and signal above the background level in at least one microarray.
qRT-PCR.
RNA from iNKC and mNKC (two biological repeats) was reverse transcribed with SuperScript III reverse transcriptase (Invitrogen, Life Technologies). Quantitative PCRs (qPCRs) were carried out using the miScript SYBR green PCR kit (Qiagen) at a melting temperature (Tm) of 55°C using β actin as a calibrator in a LightCycler 480 instrument (Roche). The fold change between Runx3−/− and WT expression was calculated using the Excel-based REST software program. PCR primers are listed in Table S1 in the supplemental material.
Statistics.
Statistical analysis was conducted using a paired two-tailed Student t test, and P values of <0.05 were considered significant. All data are presented as means ± standard errors (SE).
Microarry data accession number.
All microarray and ChIP-seq data are available in the GEO public database under accession number GSE52625.
RESULTS
Expression pattern of Runx3 during NKC development.
Western blot analysis revealed that Runx3 is highly expressed in mouse and human spleen NKC (Fig. 1A), as was also observed by Ohno et al. (25). To determine the developmental stage at which Runx3 expression is acquired, we used two Runx3 knock-in (KI) mouse strains, P1-AFP (Runx3P1-AFP/+) and P2-EGFP (Runx3P2-EGFP/+), harboring altered GFP (AFP) or enhanced green fluorescent protein (EGFP) inserted at the P1 or P2 ATG codon, respectively (27). Analysis of compound P1/P2-GFP mice (double heterozygote Runx3P1-AFP/+; P2-EGFP/+ mice) showed that ∼60% of BM NKP (Lin− NK1.1− CD122+ NKG2D+) express GFP (Fig. 1B), indicating that Runx3 expression starts at the NKP stage. In subsequent stages (iNK and mNK) marked by NK1.1 expression, the majority of BM NKC express Runx3 as evidenced by GFP expression in 94% of the Lin− NK1.1+ cells (Fig. 1C). Further analysis of Runx3P1-AFP/+ or Runx3P2-EGFP/+ mice revealed that P2 is the main active promoter in BM, spleen, and blood NKC (Fig. 1D; see also Fig. S1A in the supplemental material).
FIG 1.

Runx3 is expressed in NKC from the NKP stage, and its loss affects their maturation. (A) Western blot analysis of FACS-sorted mouse spleen NKC (NK1.1+ CD3−), IL-2-cultured mouse spleen NKC, and IL-2-cultured human blood NKC reveals expression of Runx3. (B) NKP cells express Runx3. BM of compound Runx3P1-AFP/+/P2-EGFP/+ mice was analyzed for GFP expression in Lin− (Lin = Ter119, B220, Gr1, CD3, CD8, and CD11b) NK1.1− NKG2D+ CD122+ NKPs. (C) Most NK1.1+ NKC express Runx3. Lin− NK1.1+ cells of compound Runx3P1-AFP/+/P2-EGFP/+ mice were analyzed for GFP expression. (D) Analysis of Runx3P1-AFP/+ and Runx3P2-EGFP/+ expression in BM. CD122+ CD3− BM lymphocytes were analyzed for coexpression of NKp46 and either P1- or P2-derived GFP expression. (E) Percentages of NKp46+ NKC in BM and spleen of WT and Runx3−/− mice. (F) Bar graphs showing the frequency of WT and Runx3−/− NKC subsets out of total NKp46+ spleen cells under resting conditions. Mean values are shown for the four maturation stages of NKC (n = 5). Significance, WT versus Runx3−/− NKC: ∗, P < 0.05. CD11b, CD11b+ CD27−; DP, CD11b+ CD27+; CD27, CD11b− CD27+; DN, CD11b− CD27−.
Runx3 participates in NKC maturation.
To evaluate the function of Runx3 during NKC development, we characterized cells from WT and Runx3−/− mice. Under resting conditions, loss of Runx3 caused a slight reduction in the number of BM and spleen NKC, which did not reach statistical significance (Fig. 1E). However, its absence significantly affected the BM and splenic NKC maturation state, determined by expression of CD11b and CD27 (Fig. 1F; see Fig. S2A, B, and C in the supplemental material). The CD11b− CD27− and CD11b− CD27+ profiles mark iNKC, while the CD11b+ CD27+ and CD11b+ CD27− profiles mark mNKC (35). Loss of Runx3 resulted in a 1.6-fold-increased frequency of splenic iNKC (Fig. 1F; see also Fig. S2B). On the other hand, the frequency of the CD11b+ CD27+ subpopulation of splenic mNKC decreased 2-fold in the absence of Runx3 (Fig. 1F; see also Fig. S2B). Similar results were obtained when KLRG1, which marks the most mature NKC (36), was used in combination with CD11b (see Fig. S2D).
Loss of Runx3 had no impact on production of IFN-γ (see Fig. S1B in the supplemental material), and neither did it affect the in vitro cytotoxicity of NKC toward tumor target cells (see Fig. S1C). Finally, FACS analysis indicated that expression of the activating receptor NKp46 (Ncr1), previously suggested to be regulated by Runx3 (37), was not changed in Runx3-deficient splenic NKC as was the expression of several other characteristic markers, including NK1.1, DX5, B220, and CD43 (data not shown).
Taken together, the results indicate that while loss of Runx3 significantly attenuated NKC maturation, it did not impair their ability to produce IFN-γ or kill target cells in vitro.
Runx3 is required for proliferation of cultured splenic NKC.
IL-15 plays a pivotal role in NKC development, homeostasis, and activation (9, 38, 39). The maturation defect and slight reduction in cell number in Runx3−/− mice under resting conditions led us to address whether Runx3 was involved in IL-15-induced signaling. IL-15 and IL-2 bind to the same β and γ receptor subunits, and under culture conditions, these two cytokines promote NKC activation (40). When grown in the presence of IL-2 or IL-15, cultures of Runx3−/− cells contained 3 and 2.5 times fewer NKC, respectively, than WT cultures (Fig. 2A). The lower number of Runx3−/− NKC resulted from impaired proliferation, as evidenced by the CFSE cell division assay (Fig. 2B), and not from increased apoptosis (Fig. 2C). These results imply a Runx3-dependent cell-intrinsic role in IL-2- and IL-15-dependent proliferation of NKC.
FIG 2.
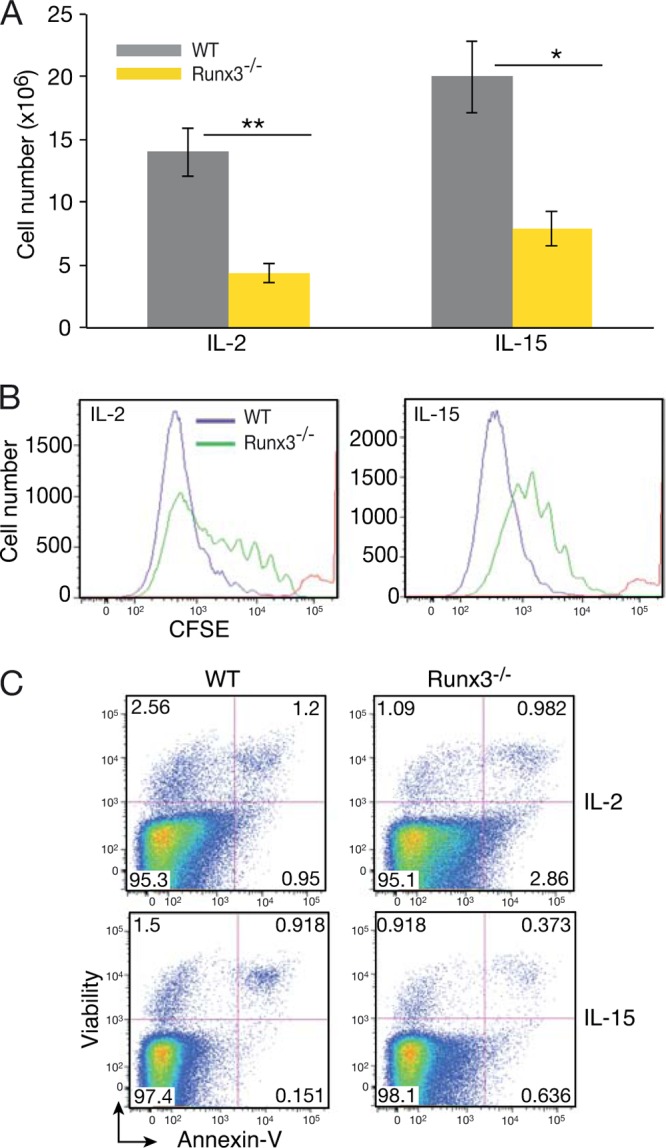
Impaired IL-2- and IL-15-induced proliferation of cultured primary Runx3−/− NKC. (A) Numbers of WT and Runx3−/− NKC (DX5+ or NKp46+) accumulating following 7 days of culture in the presence of IL-2 (n = 22) or IL-15 (n = 8). Significance: ∗∗, P < 0.01; ∗, P < 0.05. (B) CFSE dilution assay of WT and Runx3−/− NKC on day 6 of culture in the presence of IL-2 or IL-15. The red line indicates CFSE intensity before culturing, immediately after cell labeling. (C) Analysis of apoptotic (annexin V-positive) cells in WT and Runx3−/− NKC at day 6 of culture with IL-2 or IL-15.
Runx3 promotes IL-2/IL-15-dependent accumulation of mature NKC in vivo.
The profound proliferation defect of cultured Runx3−/− NKC led us to examine their in vivo response to IL-2 or IL-15. It was previously shown that IL-15/Rα complexes mimic the in vivo trans presentation of IL-15 together with its receptor by antigen-presenting cells and elicit a more efficient response than IL-15 alone (41). Administration of IL-15/Rα to WT or Runx3−/− mice evoked a pronounced increase in the number of BM and splenic NKC (Fig. 3A; see also Fig. S2E in the supplemental material). Of note, the number of WT splenic NKC that accumulated in response to IL-15/Rα accounts for ∼40% of the total number of spleen cells (see Fig. S2F). Importantly, the increase in Runx3−/− splenic NKC was considerably lower than that of the WT (i.e., 20-fold in the WT versus 10-fold for Runx3−/−) (Fig. 3A; see also Fig. S2E). Accordingly, during the active proliferation phase a significantly lower proportion of Runx3−/− BM and spleen NKC incorporated 5-bromodeoxyuridine (BrdU) than was the case for WT NKC (Fig. 3B). Injection of IL-2 elicited a significantly lower (3-fold) increase in the number of WT splenic NKC compared to IL-15/Rα and an even lower increase in Runx3−/− NKC (Fig. 3A; see also Fig. S2E). Differences between WT and Runx3−/− mice in NKC numbers were also observed in other peripheral organs, including lungs, liver (data not shown), and the peritoneal cavity (see Fig. S3A) but not in BM (Fig. 3A; see also Fig. S2E).
FIG 3.

Impaired accumulation of mature Runx3−/− splenic NKC following IL-15/Rα or IL-2 activation in vivo. (A) Percentages of NKp46+ NKC in WT and Runx3−/− BM and spleen under resting conditions (T0) (n = 8 and n = 12, respectively) and following injection of IL-15/Rα (n = 13 and n = 16, respectively) or IL-2 (n = 5). Significance: ∗∗, P < 0.001. (B) BrdU incorporation in BM (upper panels) and spleen (lower panels) NKC (NKp46+ cells) 24 h after the second IL-15/Rα injection. (C) Bar graphs showing the frequency of WT and Runx3−/− NKC subsets in spleen following IL-15/Rα activation. Mean values are shown for the four populations (n = 5). Significance, WT versus Runx3−/− NKC: ∗∗, P < 0.001; ∗, P < 0.005. (D) Impaired maturation of Runx3−/− NKC under resting and activated conditions. Percentages of iNK (CD27− CD11b− and CD27+ CD11b−) cells in WT and Runx3−/− spleen under resting conditions (T0) and following administration of IL-15/Rα. Significance: WT versus Runx3−/− at T0 and IL-15, P < 0.01. (E) IFN-γ production by WT and Runx3−/− spleen NKC following IL-15 activation in vivo.
Analysis of WT splenic NKC from IL-15/Rα-treated mice has shown enhanced maturation (Fig. 3C) compared to that of cells in the resting state (Fig. 1F). This IL-15/Rα-mediated enhanced maturation was accompanied by an ∼4-fold reduction in iNKC (CD27−CD11b− and CD27+ CD11b−), from 30.9% ± 4.5% to 8.5% ± 0.22% (Fig. 3D). In contrast, Runx3−/− iNKC were reduced by only 1.4-fold (54.5% ± 3.1% to 36.9% ± 3.1%) (Fig. 3D). Similar results were obtained using the differentiation markers CD11b/KLRG1 (see Fig. S2D in the supplemental material). Lack of Runx3 also attenuated BM and peritoneal NKC maturation in IL-15/Rα-treated mice (see Fig. S2C and S3B). Thus, in vivo activation of NKC by IL-15/Rα exacerbated the Runx3−/− NKC phenotype. Taken together, the outcome of these in vivo experiments indicates that Runx3 plays an important role in IL-15-induced accumulation of mature NKC.
We next analyzed IFN-γ production following IL-12/18-mediated activation to evaluate the impact of IL-15 activation on NKC function. More than 70% of IL-15/Rα-activated WT or Runx3−/− NKC produced IFN-γ (Fig. 3E), with the amount per cell being somewhat higher in Runx3−/− NKC. Moreover, the proportions of IL-15/Rα-activated splenic NKC expressing CD107 were similarly high (∼85%) in WT and Runx3−/− cells (see Fig. S2G in the supplemental material). Thus, despite the profound effect of Runx3 deficiency on accumulation of mature NKC under both resting and activated conditions, the production of IFN-γ was not affected. The cytotoxic potential of Runx3−/− NKC was similar to that of the WT as reflected by CD107 expression and by Prf1 and GzmB mRNA levels (see Fig. S2G and H). However, we cannot exclude the possibility that Runx3 deficiency also affects the cytotoxic mediator protein level.
Implantation sites of Runx3−/− pregnant mice lack uNKC.
Massive IL-15-dependent uNKC proliferation occurs in the decidua at early stages of pregnancy. Embryo implantation induces transformation of endometrial stromal cells into decidual cells, and as early as the fifth gestation day, uNKC are detectable in the decidua's mesometrial side. These cells rapidly proliferate, and by gd10 to gd12, they reach their maximal numbers, comprising ∼70% of all decidual leukocytes (4). Runx3 is highly expressed in uNKC as evidenced by costaining with anti-Runx3 antibody and DBA lectin (Fig. 4A), which marks both early and terminally differentiated uNKC (42).
FIG 4.

Runx3−/− pregnant mice lack uNKC. (A) DBA and anti-Runx3 immunostaining show Runx3 expression in uNKC. (B) WT and Runx3−/− implantation site sections stained with PAS (a to d, E11; panels c and d represent enlarged boxed regions in panels a and b, respectively; arrowheads in panel c mark uNKC) or DBA (e and f, E6.5; g to j, E11; panels i and j represent enlarged regions in panels g and h, respectively). (C) IHC of Runx3 (upper panels) and DBA (lower panels) on sections of E10.5 implantation sites of pregnant Rag−/− γc−/− mice to which WT (left panels) or Runx3−/− (right panels) fetal liver cells were transferred ∼2 months earlier. Bars, 1 mm (Ba and b), 50 μm (Bc, d, i, and j), 250 μm (Be to h), and 100 μm (C).
Because Runx3 is involved in IL-15-dependent NKC proliferation, we checked whether loss of Runx3 affects the immense accumulation of uNKC. At gd11, WT DBA-stained implantation sites showed the expected vast accumulation of uNKC. In sharp contrast, no uNKC were found in Runx3−/− implantation sites (Fig. 4B, g to j). To determine whether Runx3−/− uNKC might have existed earlier during pregnancy but failed to proliferate, we analyzed deciduae of gd6.5 pregnant mice. In WT deciduae, uNKC were clearly detected at gd6.5, whereas Runx3−/− deciduae completely lacked uNKC even at this early stage (Fig. 4B, e and f). To rule out the possibility that lack of Runx3 caused a loss of DBA-reacting material, thereby precluding detection of uNKC, decidua sections were stained with periodic acid-Schiff (PAS) reagent, which marks mature NKC. PAS-positive cells were clearly detected in WT but not in Runx3−/− decidua (Fig. 4B, a to d), confirming the finding that Runx3-deficient mice lack uNKC.
Dendritic cells (DC) play an essential role in embryo implantation (43) and might be driving the recruitment of NKC to implantation sites (44). Our previous observation that the function of Runx3−/− DC was impaired (45, 46) raised the possibility that Runx3−/− DC fail to promote recruitment of uNKC to embryo implantation sites. Analysis of uNKC at implantation sites of pregnant Runx3fl/fl/CD11c-Cre mice lacking Runx3 expression specifically in DC revealed normal numbers of uNKC (see Fig. S4 in the supplemental material), indicating that lack of Runx3 expression in DC was not the underlying cause for the complete absence of uNKC in Runx3−/− pregnant mice.
Histopathological analysis revealed that nonpregnant Runx3−/− uteri were atrophic, as was also reported by Sakuma et al. (47). An atrophic process in the uterus could explain the absence of uNKC due to impaired production of progesterone-induced IL-15, which is crucial for uNKC development (48). However, injection of progesterone (0.2 mg/day) into Runx3−/− mice between gd2.5 and gd5.5 followed by decidua analysis at gd7.5 to gd9.5 revealed no accumulation of uNKC (see Table S2 in the supplemental material). Together these data suggest that as in the above-noted proliferation/maturation defect of Runx3−/− NKC, loss of intrinsic NKC Runx3 function also caused the complete absence of uNKC.
We next performed cell transfer experiments to further validate the uNKC-autonomous function of Runx3. Because Runx3−/− mice are not viable on a C57BL/6 background beyond embryonic stages (49), we transferred WT or Runx3−/− embryonic day 14.5 (E14.5) fetal liver cells into Rag−/− γc−/− mice which lack NKC and analyzed implantation sites of pregnant recipients. While implantation sites of pregnant recipients of WT fetal liver cells contained DBA+ uNKC, recipients of Runx3−/− fetal liver cells completely lacked these cells (Fig. 4C). Taken together, these results demonstrated that Runx3 plays an important cell-autonomous function in uNKC development.
Runx3 P1/P2 promoter switch in NKCs.
Interestingly, among the various NKC populations, uNKC are the only one in which in vivo Runx3 expression is regulated by the P1 promoter (Fig. 5A). However, pregnant homozygote Runx3P1-AFP/P1-AFP mice, which lack P1 activity, contain a normal level of uNKC due to Runx3 expression by P2 (Fig. 5B). Thus, when P1 activity is lost, P2 is activated and compensates for lack of P1. A P1/P2 promoter switch also occurs in cultured splenic NKC. FACS analysis of freshly isolated or day 1 IL-2/IL-15-cultured splenic NKC from heterozygote Runx3P1-AFP/+ or Runx3P2-EGFP/+ mice revealed that Runx3 expression was mediated predominantly by P2 (Fig. 5C). However, during subsequent days in culture, a promoter usage switch occurred, reflected in a marked reduction in P2 usage and a sharp increase in P1 usage (Fig. 5C). Consequently, day 7 WT NKC cultures expressed high levels of Runx3 P1 RNA and protein isoform and low levels of P2 products (Fig. 5D and E). Interestingly, this marked switch to P1 usage did not abrogate Runx3P1-AFP/P1-AFP NKC capacity to respond normally to IL-2-induced proliferation (Fig. 5F), by switching to usage of P2 (Fig. 5D and E). The results from in vivo uNKC and cultured splenic NKC demonstrate that although Runx3 P1 is the major active promoter in these cells, when it is inactivated, P2 is not silenced and thereby compensates for the loss of P1-derived Runx3.
FIG 5.

Runx3 promoter usage in NKC. (A) E11 implantation site sections of Runx3P1-AFP/+ or Runx3P2-EGFP/+ pregnant mice stained with anti-GFP. (B) E11 implantation sites of Runx3P1-AFP/P1-AFP mice analyzed by DBA IHC (upper panel) or Runx3 (lower panel). Bars (A and B), 50 μm. (C) FACS analysis of GFP expression in IL-2-cultured NKC of Runx3P1-AFP/+ and Runx3P2-EGFP/+ mice. (D) RT-PCR analysis of Runx3 common and P1- and P2-specific transcripts in RNA prepared from WT and Runx3P1-AFP/P1-AFP NKC on day 7 of culture with IL-2. (E) Western blot documenting Runx3 expression in IL-2-cultured WT and Runx3P1-AFP/P1-AFP NKC. The 46-kDa Runx3 P1 isoform is clearly detected in the WT lane, while in the Runx3P1-AFP/P1-AFP lane, the two typical P2 Runx3 45- and 43-kDa isoforms (76) are detected. (F) Analysis of WT and Runx3P1-AFP/P1-AFP NKC numbers following 7 days in culture with IL-2.
Runx3-mediated transcriptional program in developing NKC.
The data described above suggested that Runx3 functions in NKC as a nuclear mediator of IL-15/IL-2 signaling. We next sought to identify Runx3-responsive genes that participate in this Runx3-mediated regulation. First, we analyzed the overall alterations in gene expression resulting from loss of Runx3, using in vivo IL-15-activated splenic NKC. Three subpopulations of WT and Runx3−/− activated NKC were analyzed: iNKC (CD27+ CD11b−), mNKC (CD27+ CD11b+), and mNKC (CD27− CD11b+). Analysis revealed pronounced changes in gene expression, underscoring the central role of Runx3 in NKC activation. Specifically, in a combined count for the 3 subpopulations, a total of 891 genes were Runx3 responsive, exhibiting differential expression in the absence of Runx3 (KO/WT ≥ 1.5; P < 0.05). Of these 891 genes, 193 were differentially expressed uniquely in CD27+ CD11b− populations, 59 in CD27+ CD11b+ populations, and 273 in CD27− CD11b+ populations, and 366 genes were common in at least two populations (Fig. 6A).
FIG 6.

Integrated analysis of Runx3 ChIP-seq and gene expression data of in vivo IL-15-activated NKC. (A) Intersection of differentially expressed genes (Runx3−/−/WT ≥ 1.5-fold; P < 0.05) in the three spleen NKC subpopulations of WT and Runx3−/− mice (CD27, CD27+ CD11b−; DP, CD27+ CD11b+; CD11b, CD11b+ CD27−). (B) Pie chart showing the genomic distribution of Runx3 peaks in in vivo IL-15/Rα-activated NKC. (C) Analysis of Runx3 peaks in NKC in vivo. Runx3 peak numbers, corresponding genes, and percentages of peaks with RUNX motif (RCCRCA) in resting NK cells (68) and in in vivo IL-15-activated NKC. (D) De novo-discovered enriched TF motifs in Runx3 peaks in in vivo IL-15/Rα-activated NKC. (E) In vivo IL-15-activated Runx3-regulated genes: intersection between the list of genes containing Runx3 peaks with a RUNX motif and the list of differentially expressed genes (Runx3−/−/WT ≥ 1.5-fold; P < 0.05) in the three spleen NKC populations.
To selectively map the genomic occupancy of Runx3, we performed ChIP-seq analysis using in vivo IL-15/Rα-activated splenic NKC. Data analysis revealed 19,647 Runx3-bound regions (ChIP-seq peaks), corresponding to 12,255 genes (Fig. 6C). Significantly, 68% of these peaks were already present in resting NKC, and most (82%) of the genes occupied by Runx3 in activated NKC were already bound by Runx3 at the resting state (Fig. 6C). Location analysis of the Runx3-occupied regions relative to the nearest transcription start sites (TSSs) of the annotated genes showed that 44% of the peaks were localized at promoter regions (Fig. 6B), defined as ±1 kb from the TSS. This finding represents a marked enrichment, considering that promoters constitute only ∼2% of the genome (10-fold enrichment of peak regions residing in promoters; P = 1.3e−322). The remaining ∼56% of the peaks are remote from the TSS, with the majority located at a distance above 5 kb from the nearest TSS (Fig. 6B), indicating that Runx3 regulates a substantial number of its targets through long-range enhancer-promoter interactions. To further characterize the Runx3-bound regions, we analyzed the abundance of RBS. We found that ∼90% of the peaks contained an RBS bearing the RCCRCA (R = A/G) consensus motif (Fig. 6C). De novo motif finding using the software program Homer (50) identified overrepresented motifs and indicated potential Runx3-collaborating TFs. Besides enrichment for RBS, the other three top-scoring enriched motifs were of ETS, T-box, and AP-1 TFs (Fig. 6D). Members of these three families of TFs were previously shown to collaborate with RUNX TFs (20, 23) and to serve essential functions in NKC (51–53). ChIP-seq of T-bet was performed in Th1 cells (54) in which Runx3 expression is induced in a T-bet-dependent manner (55). Comparison revealed that 7,027 out of 19,647 (36%) of the Runx3-bound regions in NKC overlap T-bet peaks in Th1 cells.
Analysis of the Runx3-bound regions using the program GREAT (56), which predicts biological functions from occupancy landscapes, revealed enrichment for ontology terms implicating T cell activation, IL-2-mediated signaling events, and NKC-mediated cytotoxicity (see Table S3 in the supplemental material). To identify Runx3-regulated genes, we integrated the ChIP-Seq and gene expression data sets. The group of 891 Runx3-responsive genes was intersected with the group of genes corresponding to RUNX motif-containing Runx3 peaks. This analysis revealed a significant overlap (P < 2.2e−16 in both Pearson chi-square and Fisher exact tests) and singled out 655 genes (i.e., ∼73% of Runx3-responsive genes), which we defined as direct-Runx3-regulated genes in IL-15-activated NKC (Fig. 6E; see also Table S4 in the supplemental material). Of these genes, the expression of 356 and 299 genes was down- or upregulated, respectively, in response to loss of Runx3. Analysis of these Runx3-regulated genes by using the “Ingenuity Pathway” gene annotation tool highlighted overrepresented biological functions, including cell growth and proliferation, cell death, and cell movement (see Table S5). Significantly, the majority (94%) of Runx3-regulated genes contained the ETS motif in the Runx3-bound regions (Genomatix Genome analyzer) (http://www.genomatix.de/solutions/genomatix-genome-analyzer.html), and half of them included a common T-bet peak in Th1 cells (see Fig. S5 in the supplemental material). These observations strongly suggest that Runx3 cooperates with ETS and T-box TFs to regulate the transcription program of NKC development and activation.
Runx3-regulated genes are involved in proliferation, migration, and NKC-specific functions.
Some selected NK cell-associated Runx3 direct target genes are shown in Fig. 7A. Differential expression of a Runx3-regulated gene subset was confirmed by qRT-PCR or by FACS analysis comparing WT and Runx3−/− NKC (Fig. 7B and C). Among these genes, Prdm1 (Blimp1), Kit, and Tnfrsf9 are of special interest, since their expression is induced by IL-15 and they are known to be involved in NKC proliferation (57–59). Expression of Prdm1 was upregulated in Runx3−/− iNKC (CD27+ CD11b−) (Fig. 7A and B), and it could contribute to the proliferation defect. The finding that loss of Prdm1 promotes NKC proliferation supports this possibility (58). Kit is expressed mainly in iNKC, and its increase in response to IL-2/IL-15 evokes efficient iNKC proliferation (57). It could thus participate in the Runx3−/− proliferation defect, since loss of Runx3 resulted in a significant decrease of Kit expression in iNKC (Fig. 7A and C). Reduced expression of Tnfrsf9 in Runx3−/− NKC (Fig. 7A and B) is also consistent with the impaired proliferation phenotype, since Tnfrsf9 is known to promote proliferation of IL-2/IL-15-activated NKC (59). Of potential relevance to the proliferation defect is also the serine-threonine-tyrosine kinase encoded by Styk1, which shares homology with the fibroblast and platelet-derived growth factor receptors (60). Styk1 is highly expressed in NKC (61), and its expression was markedly reduced in Runx3−/− NKC (Fig. 7A and B), consistent with its involvement in their proliferation defect. Interestingly, these 4 genes (Prdm1, Kit, Tnfrsf9, and Styk1) were differentially expressed in IL-15-activated Runx3-deficient primary cultured NKC that exhibited impaired proliferation (see Table S6 in the supplemental material).
FIG 7.

Runx3-regulated genes in NKC. (A) Heat map showing Runx3−/−/WT fold expression (log 2 ratio) for a subset of Runx3-regulated genes in in vivo IL-15-activated iNKC (CD27, CD27+ CD11b−) and mNKC (CD11b, CD11b+ CD27−). (B) qRT-PCR analysis of Runx3-regulated genes in in vivo IL-15-activated spleen iNK (CD27+ CD11b−) and mNK (CD11b+ CD27−) cell populations. (C) FACS analysis of Runx3-regulated genes: Kit expression in iNKC (CD27+ CD11b−) 24 h after the second IL-15/Rα injection and CD226, CD81, Cxcr3, and Itgαx in spleen NKC 48 h after the second IL-15/Rα injection.
The subset of Runx3-regulated genes includes receptor and integrin coding genes that play important roles in NKC function. For example, the four members of nectin binding receptors (Crtam, CD96, CD226, and Tigit) that regulate effector function of cytotoxic lymphocytes (62, 63) are among Runx3-regulated genes (Fig. 7A and B). In the absence of Runx3, expression of Crtam and Cd96 was downregulated while that of Cd226 and Tigit was upregulated (Fig. 7A, B, and C). Additional membrane-associated proteins that were scored as Runx3 targets are tetraspanins, shown to regulate NKC cytokine production, proliferation, and migration (64). Loss of Runx3 caused increased expression of Cd81 and Tspan32 (Fig. 7A to C). Finally, absence of Runx3 also affected the expression of chemokine receptors (Cxcr3, Cxcr4, and Cx3cr1) and integrins (Itgαx and Itgβ7) (Fig. 7A to C), which are important for NKC migration and adhesion. Of these three chemokine receptors, Cxcr3 is a known target of T-bet (65). Interestingly, in both NK and Th1 cells, Cxcr3 is cobound by Runx3 and T-bet (see Fig. S5 and S6 in the supplemental material).
DISCUSSION
In the present study, we investigated the in vivo function of Runx3 in adult mouse NKC. We found that under resting conditions, Runx3 is largely dispensable for NKC development and function, although it is required for NKC maturation. Importantly, however, we demonstrated that in response to in vivo activation of NKC by IL-15, Runx3 serves as a nuclear mediator of IL-15 signaling. Accordingly, in the absence of Runx3, IL-15-induced accumulation of mature NKC is impaired and uNKC are completely missing at implantation sites of pregnant mice.
The observation that Runx3−/− NKC failed to proliferate properly under culture conditions led us to conclude that Runx3 cell-autonomous function in NKC is essential for IL-2/IL-15-induced proliferation. Conditional KO mice lacking expression of Runx3 only in NKC could certainly support the conclusion of Runx3 cell-autonomous function in NKC. However, our attempts to use Runx3fl/fl/Ncr1-Cre mice resulted in inefficient excision of Runx3 loxed alleles. Due to the proliferation disadvantage of Runx3−/− cells, the proliferation of Runx+/+ cells precluded our ability to obtain and analyze the NKC-specific Runx3−/− phenotype. Transfer experiments would not solve this problem, since it involves the entire hematopoietic system where additional cell types lacking Runx3 exist, including CD8+ T cells and DC. We did use transfer experiments to show requirement of hematopoietic cell-autonomous function of Runx3 for development of uNKC in mice with a WT uterus.
Starting at the NKP stage and throughout their development, NKC express the IL-2/IL-15 receptor β (IL-2/IL-15Rβ), compatible with the essential role of IL-15 during in vivo NKC development (66). IL-15 signaling is also important for NKC activation. Although controversial (26), published data suggest that during viral infection, increased type I IFN in DC leads to production of IL-15. DC trans present IL-15 in complex with IL-15Rα to NKC and thereby promote their activation and proliferation (67). In this regard, it is of interest to note that Runx3 was recently identified as one of the sustained effector genes induced in murine cytomegalovirus (MCMV)-infected Ly49H+ NKC during clonal expansion (61). The induction of the T-bet TF at early stages postinfection (61) fits nicely with the indications that Runx3 and T-box TFs collaborate in regulating target genes during IL-15 activation. Our analysis indicates that Runx3 deficiency does not cause a maturation block but likely affects the in vivo accumulation of NKC. This conclusion is supported by the fact that following IL-15 activation, the differences in NKC number and maturation state were more pronounced than under steady-state conditions. These observations lead us to speculate that fetal development of Runx3−/− NKC is grossly normal. Accordingly, although Runx3 is expressed throughout development of NKC, its absence leads to a mild phenotype possibly due to compensation by other RUNX family members. This possibility is supported by the observation that under resting conditions, expression of Runx1 is upregulated 2.4-fold in Runx3−/− NKC while Runx2 is prominently expressed in both WT and Runx3−/− NKC (68).
Not much is known about the transcriptional program regulating NKC response to IL-15/Rα. It was previously reported that the TFs Blimp1 and Ets1 negatively regulate NKC proliferation in response to IL-15 (53, 58), while the ETS TF-family member Pu.1 positively regulates the proliferative response to IL-2 (51). More recently, members of the E2F TF family known to control the cell cycle were implicated in NKC IL-15 signaling (67). Only a few Runx3 target genes were identified using NK cell lines (37, 69), and no information is available about the Runx3-mediated transcriptional program regulating the NKC response to IL-15. We have used in vivo IL-15/Rα-treated splenic NKC to identify Runx3-regulated genes. These genes were differentially expressed in WT versus Runx3−/− IL-15/Rα-treated NKC (i.e., Runx3 responsive) and occupied by Runx3 in ChIP-seq analysis. This subset of Runx3-regulated genes transcriptionally controls the phenotypic outcome of NKC response to IL-15. Of particular relevance to these findings is the observation that in response to IL-15/Rα, Runx3 positively regulates the expression of Kit, Tnfrsf9 and Styk1, which are likely involved in IL-15/Rα signaling, including the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways (57), thereby promoting NKC proliferation. Loss of Runx3 resulted in reduced expression of these genes, contributing to the proliferative defect of IL-15/Rα-activated Runx3−/− NKC. The tetraspanin gene Tspan32 and Blimp1/Prdm1 are negatively regulated by Runx3 in NKC. Significantly, Tarrant et al. (70) have reported that Tspan32 acts as a negative regulator of lymphocyte cell proliferation. Thus, increased expression of Tspan32 and that of Blimp1/Prdm1 could contribute to the Runx3−/− NKC proliferation defect. Prdm1, Tnfrsf9, Styk1, and most of the other genes shown in Fig. 7A as Runx3 direct target genes include at least one Runx3 peak overlapping a T-bet peak observed in Th1 cells, further strengthening the indication of collaborative activity of these TFs.
uNKC are involved in decidual formation and vascular remodeling at early stages of pregnancy (4). uNKC deficiencies were previously found in IL-15−/− mice that have residual amounts of improperly developed NKC (71) and in IL-11−/− or Hoxa-10−/− mice bearing developmentally impaired deciduae (72, 73). However, T-bet−/− mice that have an overall reduction in NKC number do contain normal numbers of uNKC (71). Thus, although the general NKC phenotype of Runx3−/− mice is significantly milder than that of IL-15−/− mice, both mouse strains completely lack uNKC, underscoring the important role of Runx3 in IL-15-mediated signaling in NKC development.
The origin of uNKC is unclear. They are either derived by in situ proliferation of tissue-resident NKC precursors or recruited from the circulation (71). Transplantable uNKC progenitors have been identified in primary and secondary lymphoid tissues but not in uterine segments (71). This observation is consistent with the scenario that the few uNKC detected at early pregnancy implantation sites were recruited from the circulation, lymph nodes, or BM and proliferated massively in the decidua, generating the large number of uNKC seen at midgestation. Our inability to detect even a trace amount of uNKC in Runx3−/− decidua at early pregnancy (i.e., gestation day 6.5) may suggest that in addition to impaired proliferation, Runx3−/− uNKC progenitors suffer from attenuated migration. Compatible with this hypothesis, loss of Runx3 affected splenic NKC expression of the chemokine receptors Cxcr3 and Cxcr4, implicated in IL-15-dependent NKC migration to the decidua (74). Of potential relevance is the possibility that the human KIR2DL4 receptor plays a role in uNKC function (75) and the finding that IL-2/IL-15-induced expression of KIR2DL4 was regulated by RUNX3 in cooperation with the ETS1 TF (69). While the mouse ortholog of KIR2DL4 is not known, it is tempting to speculate that it might contribute to the lack of uNKC in Runx3−/− mice.
In conclusion, our analysis revealed that Runx3 plays an important role as a nuclear mediator during IL-15 activation of NK cells. By cooperation with ETS and T-box TFs, Runx3 orchestrates the transcription regulation of genes involved in proliferation, maturation, migration, and function of NK cells. The comprehensive study of Runx3 function at the cellular and molecular levels presented here is an obligatory first step toward elucidating the largely unknown transcription program operating during NKC development and activation. Similar studies with additional TFs will allow the construction of a more detailed transcriptional network that regulates NKC development and function.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the help of Rafi Saka and Ofira Higfa in animal husbandry, Kalanit Raanan from the Department of Veterinary Resources with histological preparations, Daniela Amann-Zalcennstein and Shirley Horn-Saban for help in DNA sequencing and gene expression analysis, and A. Sharp and E. Ariel for help with FACS analysis. We thank E. Tzehoval and L. Eisenbach for help with the in vitro cytotoxicity assay.
This study was supported by individual Israel Science Foundation grants to Y.G. and D.L.
Footnotes
Published ahead of print 13 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01202-13.
REFERENCES
- 1.Spits H, Di Santo JP. 2011. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 12:21–27. 10.1038/ni.1962 [DOI] [PubMed] [Google Scholar]
- 2.Huntington ND, Vosshenrich CA, Di Santo JP. 2007. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat. Rev. Immunol. 7:703–714. 10.1038/nri2154 [DOI] [PubMed] [Google Scholar]
- 3.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. 2008. Functions of natural killer cells. Nat. Immunol. 9:503–510. 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 4.Bilinski MJ, Thorne JG, Oh MJ, Leonard S, Murrant C, Tayade C, Croy BA. 2008. Uterine NK cells in murine pregnancy. Reprod. Biomed. 16:218–226. 10.1016/S1472-6483(10)60577-9 [DOI] [PubMed] [Google Scholar]
- 5.Fathman JW, Bhattacharya D, Inlay MA, Seita J, Karsunky H, Weissman IL. 2011. Identification of the earliest natural killer cell-committed progenitor in murine bone marrow. Blood 118:5439–5447. 10.1182/blood-2011-04-348912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yokoyama WM, Kim S, French AR. 2004. The dynamic life of natural killer cells. Annu. Rev. Immunol. 22:405–429. 10.1146/annurev.immunol.22.012703.104711 [DOI] [PubMed] [Google Scholar]
- 7.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. 1995. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc. Natl. Acad. Sci. U. S. A. 92:377–381. 10.1073/pnas.92.2.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki H, Duncan GS, Takimoto H, Mak TW. 1997. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor beta chain. J. Exp. Med. 185:499–505. 10.1084/jem.185.3.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vosshenrich CA, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, Di Santo JP. 2005. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J. Immunol. 174:1213–1221 http://www.jimmunol.org/content/174/3/1213 [DOI] [PubMed] [Google Scholar]
- 10.Martin-Fontecha A, Lord GM, Brady HJ. 2011. Transcriptional control of natural killer cell differentiation and function. Cell. Mol. Life Sci. 68:3495–3503. 10.1007/s00018-011-0800-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boos MD, Ramirez K, Kee BL. 2008. Extrinsic and intrinsic regulation of early natural killer cell development. Immunol. Res. 40:193–207. 10.1007/s12026-007-8006-9 [DOI] [PubMed] [Google Scholar]
- 12.Hesslein DG, Lanier LL. 2011. Transcriptional control of natural killer cell development and function. Adv. Immunol. 109:45–85. 10.1016/B978-0-12-387664-5.00002-9 [DOI] [PubMed] [Google Scholar]
- 13.Ramirez K, Kee BL. 2010. Transcriptional regulation of natural killer development. Curr. Opin. Immunol. 22:193–198. 10.1016/j.coi.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levanon D, Groner Y. 2004. Structure and regulated expression of mammalian RUNX genes. Oncogene 23:4211–4219. 10.1038/sj.onc.1207670 [DOI] [PubMed] [Google Scholar]
- 15.de Bruijn MF, Speck NA. 2004. Core-binding factors in hematopoiesis and immune function. Oncogene 23:4238–4248. 10.1038/sj.onc.1207763 [DOI] [PubMed] [Google Scholar]
- 16.Lutterbach B, Hiebert SW. 2000. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene 245:223–235. 10.1016/S0378-1119(00)00014-7 [DOI] [PubMed] [Google Scholar]
- 17.Levanon D, Goldstein RE, Bernstein Y, Tang H, Goldenberg D, Stifani S, Paroush Z, Groner Y. 1998. Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proc. Natl. Acad. Sci. U. S. A. 95:11590–11595. 10.1073/pnas.95.20.11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarmus M, Woolf E, Bernstein Y, Fainaru O, Negreanu V, Levanon D, Groner Y. 2006. Groucho/transducin-like Enhancer-of-split (TLE)-dependent and -independent transcriptional regulation by Runx3. Proc. Natl. Acad. Sci. U. S. A. 103:7384–7389. 10.1073/pnas.0602470103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida H, Kitabayashi I. 2008. Chromatin regulation by AML1 complex. Int. J. Hematol. 87:19–24. 10.1007/s12185-007-0004-0 [DOI] [PubMed] [Google Scholar]
- 20.Pencovich N, Jaschek R, Tanay A, Groner Y. 2011. Dynamic combinatorial interactions of RUNX1 and cooperating partners regulates megakaryocytic differentiation in cell line models. Blood 117:e1–e14. 10.1182/blood-2010-07-295113 [DOI] [PubMed] [Google Scholar]
- 21.Collins A, Littman DR, Taniuchi I. 2009. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat. Rev. Immunol. 9:106–115. 10.1038/nri2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, Rao A. 2009. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 206:51–59. 10.1084/jem.20081242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djuretic IM, Cruz-Guilloty F, Rao A. 2009. Regulation of gene expression in peripheral T cells by Runx transcription factors. Adv. Immunol. 104:1–23. 10.1016/S0065-2776(08)04001-7 [DOI] [PubMed] [Google Scholar]
- 24.Guo Y, Maillard I, Chakraborti S, Rothenberg EV, Speck NA. 2008. Core binding factors are necessary for natural killer cell development and cooperate with Notch signaling during T-cell specification. Blood 112:480–492. 10.1182/blood-2007-10-120261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohno S, Sato T, Kohu K, Takeda K, Okumura K, Satake M, Habu S. 2008. Runx proteins are involved in regulation of CD122, Ly49 family and IFN-gamma expression during NK cell differentiation. Int. Immunol. 20:71–79. 10.1093/intimm/dxm120 [DOI] [PubMed] [Google Scholar]
- 26.Sun JC, Lanier LL. 2011. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat. Rev. Immunol. 11:645–657. 10.1038/nri3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levanon D, Bernstein Y, Negreanu V, Bone KR, Pozner A, Eilam R, Lotem J, Brenner O, Groner Y. 2011. Absence of Runx3 expression in normal gastrointestinal epithelium calls into question its tumour suppressor function. EMBO Mol. Med. 3:593–604. 10.1002/emmm.201100168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, Kremer E, Otto F, Brenner O, Lev-Tov A, Groner Y. 2002. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 21:3454–3463. 10.1093/emboj/cdf370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levanon D, Brenner O, Negreanu V, Bettoun D, Woolf E, Eilam R, Lotem J, Gat U, Otto F, Speck N, Groner Y. 2001. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates nonredundant functions during mouse embryogenesis. Mech. Dev. 109:413–417. 10.1016/S0925-4773(01)00537-8 [DOI] [PubMed] [Google Scholar]
- 30.Zhang JH, Yamada AT, Croy BA. 2009. DBA-lectin reactivity defines natural killer cells that have homed to mouse decidua. Placenta 30:968–973. 10.1016/j.placenta.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 31.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O. 2006. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7:517–523. 10.1038/ni1322 [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, Pape UJ, Poidinger M, Chen Y, Yeung K, Brown M, Turpaz Y, Liu XS. 2011. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12:R83. 10.1186/gb-2011-12-8-r83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa Y, Smyth MJ. 2006. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 176:1517–1524 http://www.jimmunol.org/content/176/3/1517 [DOI] [PubMed] [Google Scholar]
- 36.Huntington ND, Tabarias H, Fairfax K, Brady J, Hayakawa Y, Degli-Esposti MA, Smyth MJ, Tarlinton DM, Nutt SL. 2007. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J. Immunol. 178:4764–4770 http://www.jimmunol.org/content/178/8/4764 [DOI] [PubMed] [Google Scholar]
- 37.Lai CB, Mager DL. 2012. Role of runt-related transcription factor 3 (RUNX3) in transcription regulation of natural cytotoxicity receptor 1 (NCR1/NKp46), an activating natural killer (NK) cell receptor. J. Biol. Chem. 287:7324–7334. 10.1074/jbc.M111.306936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castillo EF, Stonier SW, Frasca L, Schluns KS. 2009. Dendritic cells support the in vivo development and maintenance of NK cells via IL-15 trans-presentation. J. Immunol. 183:4948–4956. 10.4049/jimmunol.0900719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elpek KG, Rubinstein MP, Bellemare-Pelletier A, Goldrath AW, Turley SJ. 2010. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc. Natl. Acad. Sci. U. S. A. 107:21647–21652. 10.1073/pnas.1012128107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma A, Koka R, Burkett P. 2006. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu. Rev. Immunol. 24:657–679. 10.1146/annurev.immunol.24.021605.090727 [DOI] [PubMed] [Google Scholar]
- 41.Dubois S, Mariner J, Waldmann TA, Tagaya Y. 2002. IL-15Ralpha recycles and presents IL-15 in trans to neighboring cells. Immunity 17:537–547. 10.1016/S1074-7613(02)00429-6 [DOI] [PubMed] [Google Scholar]
- 42.Paffaro VA, Jr, Bizinotto MC, Joazeiro PP, Yamada AT. 2003. Subset classification of mouse uterine natural killer cells by DBA lectin reactivity. Placenta 24:479–488. 10.1053/plac.2002.0919 [DOI] [PubMed] [Google Scholar]
- 43.Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V, Mor G, Keshet E, Dekel N, Neeman M, Jung S. 2008. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J. Clin. Invest. 118:3954–3965. 10.1172/JCI36682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karsten CM, Behrends J, Wagner AK, Fuchs F, Figge J, Schmudde I, Hellberg L, Kruse A. 2009. DC within the pregnant mouse uterus influence growth and functional properties of uterine NK cells. Eur. J. Immunol. 39:2203–2214. 10.1002/eji.200838844 [DOI] [PubMed] [Google Scholar]
- 45.Fainaru O, Woolf E, Lotem J, Yarmus M, Brenner O, Goldenberg D, Negreanu V, Bernstein Y, Levanon D, Jung S, Groner Y. 2004. Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J. 23:969–979. 10.1038/sj.emboj.7600085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fainaru O, Shseyov D, Hantisteanu S, Groner Y. 2005. Accelerated chemokine receptor 7-mediated dendritic cell migration in Runx3 knockout mice and the spontaneous development of asthma-like disease. Proc. Natl. Acad. Sci. U. S. A. 102:10598–10603. 10.1073/pnas.0504787102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakuma A, Fukamachi H, Ito K, Ito Y, Takeuchi S, Takahashi S. 2008. Loss of Runx3 affects ovulation and estrogen-induced endometrial cell proliferation in female mice. Mol. Reprod. Dev. 75:1653–1661. 10.1002/mrd.20904 [DOI] [PubMed] [Google Scholar]
- 48.Okada H, Nakajima T, Sanezumi M, Ikuta A, Yasuda K, Kanzaki H. 2000. Progesterone enhances interleukin-15 production in human endometrial stromal cells in vitro. J. Clin. Endocrinol. Metab. 85:4765–4770. 10.1210/jcem.85.12.7023 [DOI] [PubMed] [Google Scholar]
- 49.Brenner O, Levanon D, Negreanu V, Golubkov O, Fainaru O, Woolf E, Groner Y. 2004. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proc. Natl. Acad. Sci. U. S. A. 101:16016–16021. 10.1073/pnas.0407180101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38:576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colucci F, Samson SI, DeKoter RP, Lantz O, Singh H, Di Santo JP. 2001. Differential requirement for the transcription factor PU.1 in the generation of natural killer cells versus B and T cells. Blood 97:2625–2632. 10.1182/blood.V97.9.2625 [DOI] [PubMed] [Google Scholar]
- 52.Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL. 2012. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36:55–67. 10.1016/j.immuni.2011.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramirez K, Chandler KJ, Spaulding C, Zandi S, Sigvardsson M, Graves BJ, Kee BL. 2012. Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity 36:921–932. 10.1016/j.immuni.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, Sun HW, Vahedi G, Hakim O, Handon R, Schwartzberg PL, Hager GL, O'Shea JJ. 2011. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity 35:919–931. 10.1016/j.immuni.2011.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. 2007. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 8:145–153. 10.1038/ni1424 [DOI] [PubMed] [Google Scholar]
- 56.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28:495–501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benson DM, Jr, Yu J, Becknell B, Wei M, Freud AG, Ferketich AK, Trotta R, Perrotti D, Briesewitz R, Caligiuri MA. 2009. Stem cell factor and interleukin-2/15 combine to enhance MAPK-mediated proliferation of human natural killer cells. Blood 113:2706–2714. 10.1182/blood-2008-05-159285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kallies A, Carotta S, Huntington ND, Bernard NJ, Tarlinton DM, Smyth MJ, Nutt SL. 2011. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood 117:1869–1879. 10.1182/blood-2010-08-303123 [DOI] [PubMed] [Google Scholar]
- 59.Wilcox RA, Tamada K, Strome SE, Chen L. 2002. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J. Immunol. 169:4230–4236 http://www.jimmunol.org/content/169/8/4230 [DOI] [PubMed] [Google Scholar]
- 60.Liu L, Yu XZ, Li TS, Song LX, Chen PL, Suo TL, Li YH, Wang SD, Chen Y, Ren YM, Zhang SP, Chang ZJ, Fu XY. 2004. A novel protein tyrosine kinase NOK that shares homology with platelet-derived growth factor/fibroblast growth factor receptors induces tumorigenesis and metastasis in nude mice. Cancer Res. 64:3491–3499. 10.1158/0008-5472.CAN-03-2106 [DOI] [PubMed] [Google Scholar]
- 61.Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, Best JA, Goldrath AW, Lanier LL. 2012. Molecular definition of the identity and activation of natural killer cells. Nat. Immunol. 13:1000–1009. 10.1038/ni.2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanietsky N, Mandelboim O. 2010. Paired NK cell receptors controlling NK cytotoxicity. FEBS Lett. 584:4895–4900. 10.1016/j.febslet.2010.08.047 [DOI] [PubMed] [Google Scholar]
- 63.Chan CJ, Andrews DM, Smyth MJ. 2012. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr. Opin. Immunol. 24:246–251. 10.1016/j.coi.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 64.Jones EL, Demaria MC, Wright MD. 2011. Tetraspanins in cellular immunity. Biochem. Soc. Trans. 39:506–511. 10.1042/BST0390506 [DOI] [PubMed] [Google Scholar]
- 65.Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, Weinmann AS. 2006. T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J. Biol. Chem. 281:11992–12000. 10.1074/jbc.M513613200 [DOI] [PubMed] [Google Scholar]
- 66.Castillo EF, Schluns KS. 2012. Regulating the immune system via IL-15 transpresentation. Cytokine 59:479–490. 10.1016/j.cyto.2012.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, Crozat K, Bessou G, Zucchini N, Robbins SH, Vivier E, Kalinke U, Ferrier P, Dalod M. 2012. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe 12:571–584. 10.1016/j.chom.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 68.Lotem J, Levanon D, Negreanu V, Leshkowitz D, Friedlander G, Groner Y. 2013. Runx3-mediated transcriptional program in cytotoxic lymphocytes. PLoS One 8:e80467. 10.1371/journal.pone.0080467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Presnell SR, Zhang L, Chlebowy CN, Al-Attar A, Lutz CT. 2012. Differential transcription factor use by the KIR2DL4 promoter under constitutive and IL-2/15-treated conditions. J. Immunol. 188:4394–4404. 10.4049/jimmunol.1103352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tarrant JM, Groom J, Metcalf D, Li R, Borobokas B, Wright MD, Tarlinton D, Robb L. 2002. The absence of Tssc6, a member of the tetraspanin superfamily, does not affect lymphoid development but enhances in vitro T-cell proliferative responses. Mol. Cell. Biol. 22:5006–5018. 10.1128/MCB.22.14.5006-5018.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Croy BA, van den Heuvel MJ, Borzychowski AM, Tayade C. 2006. Uterine natural killer cells: a specialized differentiation regulated by ovarian hormones. Immunol. Rev. 214:161–185. 10.1111/j.1600-065X.2006.00447.x [DOI] [PubMed] [Google Scholar]
- 72.Ain R, Trinh ML, Soares MJ. 2004. Interleukin-11 signaling is required for the differentiation of natural killer cells at the maternal-fetal interface. Dev. Dyn. 231:700–708. 10.1002/dvdy.20183 [DOI] [PubMed] [Google Scholar]
- 73.Rahman MA, Li M, Li P, Wang H, Dey SK, Das SK. 2006. Hoxa-10 deficiency alters region-specific gene expression and perturbs differentiation of natural killer cells during decidualization. Dev. Biol. 290:105–117. 10.1016/j.ydbio.2005.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carlino C, Stabile H, Morrone S, Bulla R, Soriani A, Agostinis C, Bossi F, Mocci C, Sarazani F, Tedesco F, Santoni A, Gismondi A. 2008. Recruitment of circulating NK cells through decidual tissues: a possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood 111:3108–3115. 10.1182/blood-2007-08-105965 [DOI] [PubMed] [Google Scholar]
- 75.Rajagopalan S, Long EO. 2012. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. U. S. A. 109:20596–20601. 10.1073/pnas.1208248109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bone KR, Gruper Y, Goldenberg D, Levanon D, Groner Y. 2010. Translation regulation of Runx3. Blood Cells Mol. Dis. 45:112–116. 10.1016/j.bcmd.2010.04.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.