

ABSTRACT
HIV-1 infection is characterized by the rapid generation of genetic diversity that facilitates viral escape from immune selection and antiretroviral therapy. Despite recombination's crucial role in viral diversity and evolution, little is known about the genomic factors that influence recombination between highly similar genomes. In this study, we use a minimally modified full-length HIV-1 genome and high-throughput sequence analysis to study recombination in gag and pol in T cells. We find that recombination is favored at a number of recombination hot spots, where recombination occurs six times more frequently than at corresponding cold spots. Interestingly, these hot spots occur near important features of the HIV-1 genome but do not occur at sites immediately around protease inhibitor or reverse transcriptase inhibitor drug resistance mutations. We show that the recombination hot and cold spots are consistent across five blood donors and are independent of coreceptor-mediated entry. Finally, we check common experimental confounders and find that these are not driving the location of recombination hot spots. This is the first study to identify the location of recombination hot spots between two similar viral genomes with great statistical power and under conditions that closely reflect natural recombination events among HIV-1 quasispecies.
IMPORTANCE The ability of HIV-1 to evade the immune system and antiretroviral therapy depends on genetic diversity within the viral quasispecies. Retroviral recombination is an important mechanism that helps to generate and maintain this genetic diversity, but little is known about how recombination rates vary within the HIV-1 genome. We measured recombination rates in gag and pol and identified recombination hot and cold spots, demonstrating that recombination is not random but depends on the underlying gene sequence. The strength and location of these recombination hot and cold spots can be used to improve models of viral dynamics and evolution, which will be useful for the design of robust antiretroviral therapies.
INTRODUCTION
The high level of genetic diversity is one of the main contributors to immune system and drug treatment failure during HIV-1 infection. This diversity is generated primarily by the error-prone reverse transcriptase during DNA synthesis, a process that results in approximately one mutation every three replication cycles (1–4). Moreover, each HIV-1 virion contains two copies of the RNA genome, allowing the reverse transcriptase to switch between the two copackaged RNA genomes. This process of recombination also influences HIV-1's sequence diversity by generating a progeny that is a genetic mix of the two parental strains (5). Recombination occurs much more frequently than mutation and is a powerful force that influences the evolution of the HIV-1 genome (for a review, see reference 4). Investigations into locations of inter/intrasubtype recombination indicate that sequence identity is sufficient to explain most breakpoint locations (6–9). This is unsurprising, as sequence similarity between genomic partners is a strict requirement for efficient recombination (7, 10–12). Given that the vast majority of HIV-1 infections are not the result of coinfections with multiple divergent viral strains but are initiated from a single virion, a model system that measures recombination between genetically similar genomes rather than inter/intrasubtypes will better approximate the quasispecies in vivo (13–15). However, little is known about recombination likely to be found within the viral quasispecies of an infected individual, because it is difficult to detect recombination between genetically similar genomes. Understanding recombination is a critical piece in the puzzle of HIV-1's evolutionary history and may help with the development of future treatments or with vaccine design.
Measuring recombination involves analyzing the progeny of heterozygous virions (virions containing two genetically different genomes) to determine where recombination breakpoints exist and at what frequency they are generated. Studies to date have measured recombination rates in a number of elegant ways. The use of retroviral reporter systems, where correctly positioned recombination will recreate a functional foreign gene insert conferring antibiotic resistance or fluorescence (16–18), allows for the rapid screening of recombinants but does not allow the measurement of recombination on the natural HIV-1 sequence. A more direct method of detecting recombination is through the sequencing of reverse transcription products derived from an authentic HIV-1 replication cycle. Importantly, recombination can be observed only when it leads to the generation of chimeric molecules. That is, template switching between identical genomes, or an even number of template switches between two genetic loci, will lead to no genetic changes and will go unobserved. Thus, to detect recombination on the native HIV-1 genome, genetically different strains must be utilized. Previous studies have leveraged sequence differences between highly divergent but naturally occurring subtypes to measure intra- or intersubtype recombination (19–22). However, as the overall sequence similarity between RNA templates is a major driving force governing recombination (6, 7, 10, 12), and the majority of infected individuals harbor viral populations that are known to be genetically similar (14, 23), measurements of recombination between genetically divergent strains will reflect only the special case of inter/intrasubtype recombination but will not reflect recombination among the genetically similar HIV-1 genomes found in most viral quasispecies.
To address these issues, we developed a minimally codon-modified HIV-1 genome and showed that this could be used to directly measure recombination under conditions where sequence similarity between RNA templates remains high (24). Using Sanger sequencing of single-round reverse transcription products in the absence of selection, we showed that recombination does not occur randomly. This is in agreement with studies showing that recombination rates depend on a complex set of factors, such as the availability of nucleotide (nt) substrates (25–27), the RNA template itself (7, 12, 28), overall sequence similarity (6, 7, 10, 12), and local sequence context of recombining sequences (28–30). Using both in vitro assays and single-cycle HIV-1 vectors, recombination hot spots have been identified in the untranslated regions (UTRs) (30–32), in gag (29, 33), and in env (28, 34). However, only limited information on recombination is available within other regions of the HIV-1 genome (33). We and others have attempted to use direct sequencing to locate recombination hot spots within the HIV-1 genome (24, 33, 35), but the large amount of sequencing data required made it impossible to draw firm conclusions with strong statistical support.
In this study, we made use of next-generation sequencing to perform a comprehensive analysis of HIV-1 recombination using the marker method, with two marker configurations in gag and pol that allow recombination to be measured over 13 and 26 regions, respectively. This configuration is uniquely high resolution, with regions (separated by adjacent marker points) ranging from 21 to 159 nucleotides in length. Additionally, the system has broad coverage within gag and pol. We develop a statistical approach for comparing recombination rates and find that the recombination is not constant along the genome but varies with nucleotide position. This variation is statistically significant, with some regions showing a 6-fold difference in recombination rate. We identify 7 hot spots and 3 cold spots in gag and 5 hot spots and 7 cold spots in pol. Hot spots appear in gag at the beginning of the matrix, the matrix-capsid junction, and the capsid-p2 junction and in pol at the protease-p51 junction. We found no hot spots around regions that have been implicated with protease inhibitor and reverse transcriptase inhibitor drug resistance mutations. We also analyze recombination rates using a virus with a completely different set of engineered marker points and find that differences in recombination rate are not simply due to our silent marker manipulation of the viral sequence. Our results show that the viral gene region is a strong independent predictor of recombination rate.
MATERIALS AND METHODS
Molecular clones.
pDRNLMKlow (GenBank accession no. KC771033) and pDRNLMKhigh (GenBank accession no. KC771034) are minimally modified plasmids based on the prototypic HIV-1 strain pDRNL43. pDRNL43 is itself a derivative of pNL43, which originates from Ron Desrosiers (New England Primate Research Center) and is modified to remove 1.5 kb of cellular DNA flanking the HIV-1 genome in the pNL43 construct (36). The modified plasmids are altered in gag to include 17 and 15 marker points and in pol to include 16 and 34 marker points for pDRNLMKlow and pDRNLMKhigh, respectively. Marker points consist of, where possible, two single base pair changes in adjacent codons. This strategy allows us to distinguish easily between mutations introduced during the experimental procedure and real recombination. Furthermore, these marker points do not change any viral protein sequence or known RNA sequence elements, such a splice sites, and were rationally designed to minimize structural changes to the HIV-1 genome. Sequences were synthesized commercially (GenScript) and cloned into the ApaI and SpeI (gag) and XbaI and NotI (pol) sites of pDRNL43. pDRNLMKlow and pDRNLMKhigh were converted from the X4 tropic phenotype to the R5 phenotype, to generate pDRNLAD8MKlow and pDRNLAD8MKhigh by exchanging the Env gene from the pDRNLAD8 using the EcoRI and BamHI restriction sites. These modifications were well tolerated, as the protein processing profile and the abilities to establish infection via reverse transcription were not affected, enabling us to accurately quantify the recombination processes during primary cell infection.
Recombination assay.
We produced pools of homozygous virus (virus containing identical genomes) by transfecting wild-type (WT) and marker virus plasmids separately and produced heterozygous virus (virus containing two different genomes) by cotransfection of the wild-type and marker plasmids. Viral particles from clarified transfection supernatants were further purified by sequential filtration through 0.8-μm and 0.45-μm sterile syringe filters (Sartorius). Purified virus was then concentrated by ultracentrifugation through a 20% sucrose cushion using an L-90 ultracentrifuge (Beckman Coulter) at 100,000 × g for 1 h at 4°C. Pellets were resuspended in medium, and the virus was quantified by enzyme-linked immunosorbent assay (ELISA) (Vironostika). Concentrated virus stocks were supplemented with 2 mM MgCl2 and treated with 90 units/ml of Benzonase (Sigma) for 15 min at 37°C before infection to remove any contaminating plasmid DNA. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of HIV-1-seronegative blood donors (supplied by the Red Cross Blood Bank Service, Melbourne, Australia) by density gradient centrifugation over Ficoll-Plaque Plus (Amersham Biosciences). The identities of the blood donors from Red Cross are anonymous. Peripheral blood lymphocytes (PBLs) were purified from PBMCs and stimulated in medium (2 × 106 cells/ml) supplemented with 10 μg/ml phytohemagglutinin (PHA) (Murex Diagnostics) and 10 units/ml human interleukin-2 (IL-2) (Roche Applied Science) for 2 days in Teflon-coated jars. After 2 days, PBLs were resuspended in fresh medium containing 10 units/ml human IL-2 (Roche Applied Science) and incubated for a further 2 days before infection. Stimulated PBLs were infected with equal amounts of either homozygous or heterozygous virus, as determined by an HIV-1 antigen (p24 CA) micro-ELISA. Heat-inactivated (2 h at 56°C) control infections were carried out to confirm efficient removal of plasmid DNA for each sample. Six hours postinfection, 10 μg/ml T-20 (NIH AIDS Reagent Program) was added to the cells to prevent second-round replication. At 24 h post-PBL infection, cells were lysed and full-length reverse transcriptase products were quantified. Reverse transcription products were amplified using 10 sets of primers, generating 10 overlapping PCR amplicons (see “Primers” below). The following 2-step cycling conditions were chosen to minimize PCR-induced recombination, as previously described (37): initial copy number, 2,500; denaturation, 98°C for 30 s, followed by 72°C for 2 min for 29 cycles. PCR products for sequencing were created by pooling at least 4 independent PCRs per condition. Unique 6-nucleotide identifiers (barcodes) were attached using a modified parallel tagged sequencing protocol to allow multiplexing on the same sequencing run (38). Emulsion PCR and sequencing were performed at the Institute for Immunology and Infectious Diseases (IIID), Perth, Australia, according to standard GS FLX titanium procedures. In order to avoid resampling, we generated our sequencing libraries in such a way as to ensure that it contained PCR products generated from over 10,000,000 original DNA molecules per plate of 454 sequencing run, whereas a single 454 sequencing run has a sequencing capacity of ∼1 million reads. We note that in any event, resampling per se would not lead to an increase in recombination rates.
Primers.
Overlapping PCR amplicons for sequencing were generated using 10 sets of primers: G1(2945)Fw (GAGATGGGTGCGAGAGCGTC) and G1(3314)Rv (TGTGTCAGCTGCTGCTTGCTG), G2(3236)Fw (ACCAAGGAAGCCTTAGATAAGATAGAGGAAGAG) and G2(3679)Rv (TGAAGGGTACTAGTAGTTCCTGCTATGTCACTTC), G3(3584)Fw (GATAGATTGCATCCAGTGCATGCAG) and G3(3955)Rv (GCTTTTAAAATAGTCTTACAATCTGGGTTCGC), G4(3793)Fw (TCTGGACATAAGACAAGGACCAAAGG) and G4(4195)Rv (ACATTTCCAACAGCCCTTTTTCCTAG), P1(4433)Fw (GCGACCCCTCGTCACAATAAAGATAG) and P1(4884)Rv (GAGTATTGTATGGATTTTCAGGCCCAAT), P2(4695)Fw (CACTTTAAATTTTCCCATTAGTCCTATTGAGACTG) and P2(5110)Rv (ACTAGGTATGGTAAATGCAGTATACTTCCTGAAG), P3(4951)Fw (AAGAGAACTCAAGATTTCTGGGAAGTTCA) and P3(5325)Rv (CTCAGTTCCTCTATTTTTGTTCTATGCTGC), P4(5233)Fw (CCAGACATAGTCATCTATCAATACATGGATGA) and P4(5618)Rv (CCAGTTCTAGCTCTGCTTCTTCTGTTAGTG), P5(5503)Fw (TGGGCAAGTCAGATTTATGCAGG) and P5(5934)Rv (GTGGCTTGCCAATACTCTGTCCAC), P6(5774)Fw (GAATGAAGGGTGCCCACACTAATG) and P6(6166)Rv (GCAAAGCTAGATGAATTGCTTGTAACTCAG).
Data processing.
In order to align, process, and categorize the very large volume of sequencing data (>1 million sequences) that result from next-generation sequencing, we used EMBOSS needle (39) and custom software written in BioRuby (39). After alignment to the genome, each sequence read was processed to identify regions that cover two markers points. Each region was then classified as recombination observed (if marker endpoints switched between marker type and wild-type virus) or recombination not observed (if marker endpoints were identical). It is important to note that our marker points were designed so that all marker points contained at least two mutations in usually adjacent codons. Consequently, it is very unlikely that mutations introduced by the experimental setup, infection process, or sequencing will artificially signal recombination. This is confirmed by the low rates of recombination in our controls. However, several marker points did exhibit poor sequence quality and alignment (regions PH1, PH2, PH3, PH4, PH5, PL1, PL2, and PL3, likely due to the presence of indels (either naturally or introduced by the marker point). As 454 sequencing has known issues with homopolymer sequences (40), and the sequence quality around these markers is vital for our analysis, the marker points showing poor sequence alignment (shown in black in Fig. 3) are excluded from the analysis. These excluded markers and bordering regions represented a small fraction (∼10%) of the precleaned data.
FIG 3.

Schematic of marker configurations and how to compare between them. (A) In this study, recombination is measured between wild-type virus and a marker system with silent codon modification markers that do not affect any viral proteins or packaging (marker configuration MKhigh). To test that these codon modifications do not influence our recombination rate measurements, a second marker system virus is created where the codon modifications occur at different nucleotide positions (marker configuration MKlow). (B) To compare between marker configurations, MKhigh is used to predict what would be measured as the recombination rate if MKlow was used. This prediction can then be directly compared to the experimental results for MKlow. For each interval in MKlow, the MKhigh prediction is calculated by averaging the overlapping MKhigh interval's recombination rate and weighting this average by the proportion of overlap.
Recombination rates.
Recombination rates and confidence intervals were calculated in the statistical package R (41) using the linear model function (lm) on the optimal recombination rate (r) over all genome regions. For each interval, the recombination rate is calculated as r = [−ln(1 − 2a)]/2L, where L is the nucleotide length of the genome region and a is the proportion of heterozygous sequences that contain a recombination for that region. This equation compensates for the probability of multiple (and therefore unobserved) recombination events between marker points (24). The number of heterozygous sequences is expected to be 50%; however, this is directly estimated from the homozygous sequence frequency of each virus type using the method described by Schlub et al. (24). The calculated recombination rate will represent an average recombination rate for each interval, as the precise nucleotide position of the recombination event cannot be determined within the interval where parental sequences are identical.
Comparing recombination rates.
We use two distinct marker configurations, where codon modifications occur on different nucleotides, to test if the choice of marker nucleotide position influences recombination rate fluctuations. To compare the results from the two configurations, we use marker system 1 to predict the recombination rate in marker configuration 2 and correlate this prediction with the experimental data for marker configuration 2. For each region in marker configuration 2, the prediction is calculated as the weighted average of recombination rates in overlapping regions from marker configuration 1, where the weighting is the proportion of overlap (see Fig. 3B).
Correlations between data sets are performed in the statistical package R (41), using the cor.test function. Correlations are Pearson correlations unless otherwise stated. When correlating between marker configurations 1 and 2, adjacent regions in the marker configuration 1 prediction of marker configuration 2 will not be independent if a region from configuration 1 overlaps with two regions in configuration 2. To check whether this influences the correlation results presented, we define the dependence between two predictions that share an overlapping marker configuration 1 region to be the minimum weighting (percentage of overlap) for those overlapping regions. Predictions with a dependence value over 10% are systematically removed to keep the maximal amount of data. The correlation coefficients and corresponding P values resulting from this removal do not change substantially from those presented in the figures, and no significance levels or conclusions would be changed. Additionally, using the nonparametric Spearman rank correlation instead of the Pearson correlation does not change the significance of correlation coefficient nor any of the conclusions.
Controls for experimentally associated recombination.
Our primary focus is on the viral recombination induced during reverse transcription of the HIV-1 genome in vitro. However, recombination can also be experimentally induced at different stages of the procedure, such as during transfection of cell with plasmid, during PCR amplification, or during sequencing (37, 42–44). To ensure that the recombination rates presented are representative of the recombination rates experienced during a single cycle of HIV-1 replication, we comprehensively measured potential sources of artificial recombination.
To measure any background recombination that might arise as a result of plasmid transfection and PCR amplification, we performed a number of controls. First, RNA was extracted from heterozygous virus using phenol chloroform-based TRI reagent (Sigma-Aldrich) according to the manufacturer's recommendations and reverse transcribed into cDNA using SuperScriptIII (SSIII) (Invitrogen Life Technologies) and gene-specific primer GAG4(4195)R (5′ ACATTTCCAACAGCCCTTTTTCCTAG 3′). This measured the transfection recombination rate to be approximately 5 × 10−6 recombination events per nucleotide per round of infection (REPN), which corresponds to 0.25% of the total recombination rate reported in this study. To control for potential recombination during in vitro cell-free reverse transcription, we also performed the same reverse transcription and processing on a mix of homozygous WT virus and homozygous MK virus (mixed in equal quantities [based on p24 values] prior to RNA extraction and reverse transcribed in parallel with RNA extracted from heterozygous virus). We measure this rate to be 3 × 10−6 REPN (representing over half of the recombination occurring during our transfection control). Given that the recombination induced by SSIII is not present in our regular assay, this indicates that recombination occurring during transfection is even lower than our measured 5 × 10−6 REPN rate. Reverse transcription was performed in the presence and absence of SSIII, the latter condition providing a control for any plasmid contamination carried over from transfection. Real-time PCR was used to estimate viral cDNA copy number against a standard curve based on plasmid pDRNL(AD8) using primers GAG1(2945)F (5′ GAGATGGGTGCGAGAGCGTC 3′) and GAG1 (3314)R (5′ TGTGTCAGCTGCTGCTTGCTG 3′). Again, template viral cDNA was amplified using optimized PCR conditions outlined above in “Recombination assay.”
To assess background recombination introduced by PCR, we amplified a 1:1 mixture of WT and MK plasmid and sequenced the resulting DNA (PCR control plasmid). As a more stringent PCR control, we infected cells with an equal mixture of homozygous wild-type and homozygous marker virus and subsequently PCR amplified and sequenced the resultant cDNA (PCR control cDNA). As each infection is the product of a homozygous virion, any intravirion recombination will be effectively “silent” (since both strands are identical). Thus, any recombination observed between WT and MK virus must have occurred due to chimera formation during PCR amplification (or less likely due to recombination occurring between virions in the infected cell). We calculate the average cumulative background rate of PCR-induced recombination to be 2.9 × 10−4 REPN, well below that of the recombination rate in the experimental sample. Three regions (GH1, PH23, and PH25) did exhibit a higher risk of recombination in some (but not all) controls. As a precaution, these were removed from all data analysis (see Fig. 3). After removal, the average induced recombination rate was 2.2 × 10−4 REPN.
Generalized linear models.
Generalized linear models (GLMs) were performed in the statistical package R (41), using the glm function with a binomial error distribution. For each region, the relationship between the estimated parameter (recombination rate) and experimental data (number of observed recombinations) depends on region nucleotide length and the proportion of heterozygous sequences (see the equation given above). To compensate for these factors and ensure the binomial error distribution, a custom link function identical to the equation given above was used. The factors viral phenotype, blood sample donor, and interval region were tested with a process of forward addition. Statistical significance of the covariates was tested using a chi-square test during an analysis of deviance.
Nucleotide sequence accession numbers.
The sequences determined in this study were deposited under accession numbers KC771033 and KC771034
RESULTS
Experimental system.
We developed a system that can measure recombination between highly similar genomes by rationally designing codon modifications into the full-length HIV-1 genome. This system contains no foreign gene inserts that could alter the folding of the RNA genome, and we avoided RNA sequences that were known to fold into functional RNA structures, such as splice or frameshifting sites. We further minimized structural changes to the RNA genome by using only silent adenine-to-guanine or cytosine-to-thymine (uracil) substitutions. That is, while all genetic changes have the potential to alter RNA structure, adenine and guanine both form Watson-Crick base pairs with the RNA base uracil. Similarly, cytosine and thymine (uracil) both form Watson-Crick base pairs with the guanine. We reasoned that these substitutions are likely to have the least impact on global RNA structure, as they do not disrupt preexisting base pairing. Finally, wherever possible, substitutions were made only if they occurred naturally in the HIV sequence compendium (45). These codon modifications do not change the ability to establish infection and the synthesis of viral cDNA via reverse transcription. These modifications create 39 genome regions ranging from 21 nt to 159 nt in length, over which recombination can be studied. We produced pools of homozygous virus (virus containing identical genomes) by transfecting wild-type and marker virus plasmids separately and produced a mixture of homozygous and heterozygous virus (virus containing two different genomes) by cotransfection of the wild-type and marker plasmids. We performed a single-round infection in peripheral blood mononuclear cells (PBMCs) with pools of heterozygous and homozygous virions, after which recombination can be detected with high-throughput sequencing of cDNA. The recombination rate between marker points was calculated with equations that (i) estimate the ratio of heterozygous to homozygous infections, (ii) compensate for the nucleotide length over which recombination is measured, and (iii) compensate for the probability of multiple (unobserved) recombination events between marker points (24) (see Materials and Methods).
Recombination rate fluctuates within gag and pol.
We first measured the recombination rate across our two regions of interest in gag and pol. We sequenced approximately 86,000 genome regions pooled from 5 donors and measured an average recombination rate of 2.0 × 10−3 recombination events per nucleotide per round of infection (REPN), corresponding to approximately 19 or 20 recombination events per genome (95% confidence interval of 1.8 × 10−3 to 2.2 × 10−3 REPN). When we segregated our data into the two regions, gag and pol, we found weak evidence for a different recombination rate, with an average recombination rate of 2.3 × 10−3 and 1.8 × 10−3 REPN, respectively (P = 0.07, t test on interval recombination rates). An advantage of our high-resolution marker system is the ability to investigate if recombination levels change with nucleotide position. Interestingly, we found a large level of fluctuation in recombination rate in different segments of the genome, where individual genome region rates vary from 0.51 × 10−3 REPN to 3.4 × 10−3 REPN, a greater-than-6-fold difference (Fig. 1). This indicates that the recombination rate is not constant along the HIV-1 genome and that recombination hot and cold spots may exist.
FIG 1.
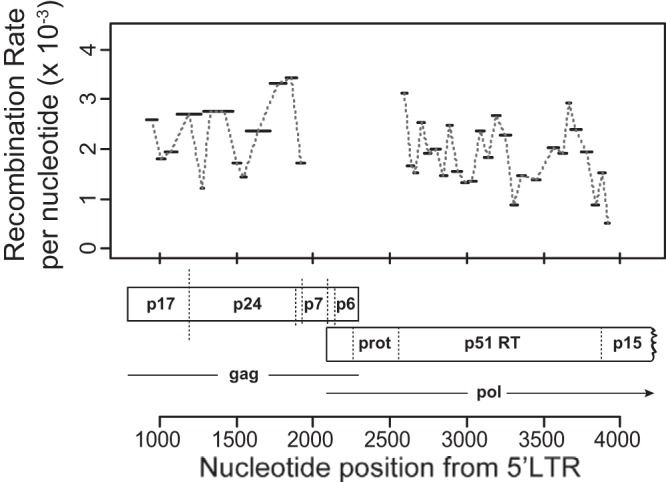
Recombination rate variation in gag and pol. Recombination rates were measured in 39 genome regions ranging from 21 nt to 159 nt in length (denoted by horizontal bars) in gag and pol. The average number of recombination events per nucleotide per round of infection (REPN) is shown on the y axis, with nucleotide position relative to the beginning of the NL43 5′ long terminal repeat (LTR) shown on the x axis.
To investigate this further, we sought to determine if the locations of putative recombination hot spots were consistent across two viral phenotypes that enter different subpopulations of T-lymphocytes via distinct coreceptors (CCR5 and CXCX4) and between unrelated blood donors. We found a significant and high correlation for the recombination rates in identical intervals when we compared between the R5 and X4 viral phenotype (r = 0.69, P < 0.0001; Fig. 2A and B) and between blood donors (Fig. 2C, Table 1). This provides strong evidence that the locations of putative recombination hot spots are similar between these groups and also constant across multiple independent infection experiments, indicating a systematic change in recombination rate along the genome.
FIG 2.

Recombination rate hot spots are consistent between viral phenotypes and PBMC blood donors. (A, C) Recombination rates are compared between two viral phenotypes, R5 and X4, and between 5 blood donors, with the average number of recombination events per nucleotide per round of infection (REPN) shown on the y axis and nucleotide position relative to the beginning of the NL43 5′ LTR shown on the x axis. (B) Correlation between the recombination rates of two viruses differing in viral phenotype, with REPN shown on both axes.
TABLE 1.
Between-patient correlations for R5a
| Donor | Donor 2 |
Donor 3 |
Donor 4 |
Donor 5 |
||||
|---|---|---|---|---|---|---|---|---|
| r | P | r | P | r | P | r | P | |
| 1 | 0.58 | 0.003 | 0.71 | <0.001 | 0.58 | <0.001 | 0.66 | <0.001 |
| 2 | 0.44 | 0.04 | 0.58 | 0.003 | 0.64 | <0.001 | ||
| 3 | 0.54 | 0.001 | 0.61 | <0.001 | ||||
| 4 | 0.63 | <0.001 | ||||||
To investigate whether recombination hot and cold spot locations are similar across different donors, the recombination rates for each interval and donor were calculated (see Fig. 2C). The pairwise correlations on the interval-specific recombination rate across donors were all positive and significant, indicating that recombination hot and cold spot locations are consistent across donors.
The recombination rates presented above theoretically include the cumulative effect of experimentally induced recombination during DNA transfection and subsequent PCR (46). To demonstrate that these experimentally induced rates are not the source of recombination hot spots, we independently measured the experimentally induced recombination rates (see Materials and Methods). We addressed whether transfection-induced recombination could influence our recombination rates by directly measuring recombination rates on RNA extracted from heterozygous virions produced from cells cotransfected with WT and MK plasmids. We used SuperScript III (RNaseH−, recombination defective) to reverse transcribe RNA before subjecting it to PCR and sequencing using the same conditions as the experimental samples. This experiment measured the accumulation of recombination due to transfection, in vitro SuperScript III reverse transcription, and PCR. This rate was calculated to be 5 × 10−6 REPN. For completeness, we also included two controls to dissect the contribution of PCR-induced recombination and a further control to measure the contribution of SuperScript III recombination (see Materials and Methods). Although we did see some variation in the level of experimental recombination between experimental replicates, under all cases, we found that overall recombination rates were too low to introduce significant bias, in agreement with our previous results (24). We also measured the rate of recombination for each interval and found that the infrequent experimental recombination was not localized to hot spots but evenly spread over gag and pol regions (data not shown). Three regions (GH1, PH23, and PH25) did exhibit a higher risk of recombination in some (but not all) controls. As a precaution, these were removed from the analysis for this paper (see Materials and Methods). To further check that these low levels of recombination are not driving the recombination hot spots, we correlate the recombination rate between intervals in our experimental and biological sample. We found that the recombination rates following infection do not significantly correlate with the experimentally induced recombination rate (PCR cDNA recombination rate, r = 0.02, P = 0.93; transfection recombination rate, r = 0.03, P = 0.93) (data not shown). Therefore, the rates presented in this study are not biased by the experimental method and provide an accurate view of HIV-1 recombination hot spots within the genome regions defined by our marker points.
Recombination rate hot spots are not a product of experimental marker design.
The HIV-1 genome used in this study includes a number of introduced silent codon modifications to act as markers for recombination. These modifications were designed so that they did not alter any viral proteins or known RNA elements. However, as nucleotide sequence can influence recombination frequencies (47), we sought to investigate whether the choice of codon modifications was driving the variation in recombination rate observed in Fig. 1. To test this, we created an additional viral phenotype, MKlow, with more broadly spaced marker points at different nucleotide positions within gag and pol (original phenotype, MKhigh) (Fig. 3A, schematic of two marker systems). As with MKhigh, these modifications do not change the viral protein sequence or the in vitro infectivity of the virus (data not shown). If the putative recombination hot spots measured in MKhigh (Fig. 1) are purely driven by sequence disruption due to codon modification, then the location of the hot spots in marker system MKlow will be different (as markers are at different nucleotide positions). Conversely, if the hot spot locations for MKhigh and MKlow are similar, then this provides evidence that the variations in recombination rates are intrinsic to the viral genome and not a product of our codon modification.
The regions that measure the recombination rate in the two marker systems do not perfectly align (due to different marker codon nucleotide position; Fig. 3), which makes it difficult to directly compare recombination rates at different sites between the two marker systems. To overcome this, the recombination rates from marker system MKhigh were interpolated to predict the recombination rate using the new (more broadly spaced) marker system MKlow (Fig. 3B, schematic of interpolation between marker systems; Materials and Methods). In this way, the recombination rates expected from the experimental rates in MKhigh and the overlap between MKhigh and MKlow can be compared with the experimentally observed rates for MKlow using a correlation analysis. Although this interpolation from high resolution to low does reduce the information available in the high resolution and does increase variability, making a correlation harder to detect, it is necessary to directly compare the resolutions. We found that the recombination rate between marker sets is significantly correlated (r = 0.42, P = 0.03 for R5; r = 0.72, P < 0.001 for X4; Fig. 4A to D), indicating that in general genomic regions with a high/low recombination rate in MKhigh also have a high/low recombination rate in MKlow. Therefore, recombination hot spot locations are consistent between the marker systems, and these hot spots are not driven by the experimental codon modification.
FIG 4.

Recombination hot spots are not a product of marker design. To check if recombination rate hot spots are driven by the choice of silent codon modifications, we measured the recombination rate in two different marker configurations, MKhigh and MKlow, for CCR5(R5)-tropic viruses (A) and CXCR4(X4)-tropic virus (C) and performed viral replicates of identical viruses (E). (A, C, D) Recombination rates, with the average number of recombination events per nucleotide per round of infection (REPN) shown on the y axis and nucleotide position relative to the beginning of the NL43 5′ LTR shown on the x axis. (B, D) Correlations between the recombination rates of MKhigh and MKlow viruses, with REPN shown on both axes. (F) Correlation between the recombination rates of MKhigh replicate infections with REPN shown on both axes. Correlations are Pearson product moment correlations.
Finally, recombination rate variation may be influenced by other experimental factors and sampling error (together called “random variation” for simplicity). To estimate how much random variation exists for this study, we correlate two identical experiments with identical marker systems (both MKhigh). If there were zero random variation, these two results should be identical and correlate perfectly. Therefore, any deviation here provides a measure for the random variation in this study (Fig. 4E and F). We found a high rate of correlation between experimental replicates (Fig. 4F, r = 0.78, P < 0.001), further highlighting that putative recombination hot spots are intrinsic to the HIV-1 genome and not a product of other experimental factors.
Identifying the recombination hot and cold spots.
We have shown that our procedure reliably estimates local recombination rate changes in gag and pol and that these changes are consistent across viral phenotypes, blood donors, and codon marker systems. Thus, the identified changes of recombination rate across the viral sequences are intrinsic to the HIV-1 genome. However, to accurately determine hot or cold spots with recombination rates significantly different from the average, a number of additional factors need to be considered. These include the estimated number of sequences sampled for each interval, the variance introduced by unrelated blood donors, and the variance introduced by the target cell (controlled by the two viral phenotypes, CCR5 and CXCR4). Generalized linear models (GLMs) provide an analytic framework for investigating the relationship between recombination rate and genomic position while accounting for the factors listed above. Generalized linear models generalize a multiple regression analysis, allowing for the binomial distribution of our sequence recombination data and the adjustment for interval nucleotide length when calculating recombination rates.
We used a process of forward addition to test and build the final GLM and to identify which covariates are significantly associated with recombination rate (Table 2). We find that recombination rate is significantly associated with viral phenotype (X4/R5, P < 0.001) and blood sample donor (P < 0.001) (chi-square test on analysis of deviance). We also find that the interval along the genome over which recombination is measured is also significantly associated with recombination rate (P < 0.001). The final model, which includes viral phenotype, blood sample donor, and interval, provides very strong evidence that the recombination rate is not constant over gag and pol and that this result is consistent over viral phenotype and blood sample donor. This final GLM estimates the recombination rate parameter for each interval. By calculating the standard error for these parameter estimates, the intervals with a recombination rate significantly different from the average rate, that is, recombination hot/cold spots, can be identified.
TABLE 2.
Generalized linear models fitteda
| Model no. | Description | Residual deviance | df (no. of parameters) | P value (when compared to model no.) |
|---|---|---|---|---|
| 1 | One avg recombination rate | 1,883 | 274 (1) | |
| 2 | Rate depends on virus | 1,813 | 273 (2) | <0.001 (1) |
| 3 | Rate depends on donor | 1,470 | 270 (5) | <0.001 (1) |
| 4 | Rate depends on virus and donor | 1,424 | 269 (6) | <0.001 (1, 2, or 3) |
| 5 | Rate depends on virus, donor, and interval | 696 | 231 (44) | <0.001 (4) |
Generalized linear models (GLMs) are a good analytic framework for investigating the effects of nucleotide position on recombination rate after accounting for the confounding effects of virus phenotype and blood donor. To build up the appropriate complexity for this analysis, a base model (model 1) with one average recombination rate fitted to all of the data pooled together was created. We next fitted more complex models with a recombination rate for each virus (model 2), a recombination rate for each donor (model 3), and a recombination rate that depends on both donor and phenotype (model 4). These models increase the complexity of the analysis, which is reflected in the increase in number of parameters and decrease in the degrees of freedom (df column). However, this increased complexity is statistically justified, as the reduction in deviance (a measure of error in the model) is sufficiently large. This indicates that viral phenotype and donor are confounding effects and should be included in the final model. In the final model, recombination rates depend on phenotype, donor, and genome interval (model 5). This model's increase in complexity is also justified by the reduction in deviance. The final model shows that genome position is an independent predictor for recombination rate, that the hot and cold spots we observe in our data are statistically significant, and that the location of recombination hot and cold spots are consistent across viral phenotypes and donors.
Over the 39 regions, we found 12 statistically significant recombination hot spots and 10 statistically significant recombination cold spots (Fig. 5, Table 3). Interestingly, these hot and cold spots are unequally distributed in gag and pol, with gag containing seven of the 12 hot spots yet only three of the 10 cold spots. In gag, hot spots appear to cluster around gene junctions, at the beginning of the matrix, the matrix-capsid junction, and the capsid-p2 junction (Fig. 5B). In pol, we find one hot spot at the protease-p51 junction (Fig. 5B) but find no hot spots in genome regions containing mutations that have been implicated in drug resistance. Therefore, recombination is less likely to influence the generation of multidrug-resistant HIV-1 within these regions compared to regions of the HIV-1 genome containing recombination hot spots for the generation of recombinant HIV-1.
FIG 5.

Ninety-five-percent confidence intervals for the recombination rate in each region for the R5 phenotype. We fit a generalized linear model to the data set to calculate the statistical significance of recombination hot and cold spots, after accounting for confounding factors such as viral phenotype and donor. The model estimates the standard error in recombination rate for each genome region, from which a 95% confidence interval is obtained. Those intervals that do not overlap the average rate are bolded. (A) Recombination rate per nucleotide for each genome segment in R5 averaged over all donors. Horizontal bars represent the length of the genome region. Ninety-five-percent confidence intervals are Bonferroni corrected for multiple comparisons. (B) Statistically significant hot and cold spots corresponding to genome location.
TABLE 3.
Locations of hot spots and cold spotsa
| Interval | RR difference to mean (× 10−3) | P value | Nt position start (from 5′ LTR) | Nt position end (from 5′ LTR) | Interval length (nt) | Amino acid 5′ interval | Amino acid 3′ interval |
|---|---|---|---|---|---|---|---|
| GH2 | 0.38 | <0.001 | 912 | 984 | 72 | E42 | Q65 |
| GH3 | −0.09 | 984 | 1032 | 48 | P66 | T81 | |
| GH4 | −0.10 | 1032 | 1113 | 81 | I82 | Q108 | |
| GH5 | 0.51 | <0.001 | 1113 | 1266 | 153 | N109 | V159 |
| GH6 | −0.74 | <0.001 | 1266 | 1287 | 21 | E160 | P166 |
| GH7 | 0.49 | <0.001 | 1287 | 1374 | 87 | E167 | Q195 |
| GH8 | 0.55 | <0.001 | 1374 | 1476 | 102 | A196 | R229 |
| GH9 | −0.31 | 1476 | 1524 | 48 | E230 | E245 | |
| GH10 | −0.56 | <0.001 | 1524 | 1560 | 36 | Q246 | P257 |
| GH11 | 0.32 | <0.05 | 1560 | 1719 | 159 | V258 | S310 |
| GH12 | 0.95 | <0.001 | 1719 | 1821 | 102 | Q311 | E344 |
| GH13 | 1.11 | <0.001 | 1821 | 1896 | 75 | E345 | Q369 |
| GH14 | −0.31 | <0.05 | 1896 | 1947 | 51 | V370 | Q386 |
| PH6 | 0.78 | <0.05 | 2573 | 2615 | 42 | V8 | K22 |
| PH7 | −0.22 | 2615 | 2651 | 36 | Q22 | L34 | |
| PH8 | −0.55 | 2651 | 2681 | 30 | V34 | E44 | |
| PH9 | 0.55 | 2681 | 2726 | 45 | G44 | P59 | |
| PH10 | −0.17 | 2726 | 2771 | 45 | V59 | L74 | |
| PH11 | 0.11 | 2771 | 2825 | 54 | V74 | L92 | |
| PH12 | −0.63 | <0.001 | 2825 | 2870 | 45 | G92 | T107 |
| PH13 | 0.45 | 2870 | 2909 | 39 | V107 | L120 | |
| PH14 | −0.02 | 2909 | 2966 | 57 | D120 | T139 | |
| PH15 | −0.54 | <0.05 | 2966 | 3011 | 45 | P139 | K154 |
| PH16 | −0.46 | 3011 | 3065 | 54 | G154 | R172 | |
| PH17 | 0.32 | 3065 | 3116 | 51 | K172 | V189 | |
| PH18 | −0.10 | 3116 | 3167 | 51 | G189 | R206 | |
| PH19 | 0.57 | <0.01 | 3167 | 3218 | 51 | Q206 | K223 |
| PH20 | 0.43 | <0.05 | 3218 | 3290 | 72 | E223 | P247 |
| PH21 | −0.93 | <0.001 | 3290 | 3326 | 36 | E247 | K259 |
| PH22 | −0.46 | <0.001 | 3326 | 3383 | 57 | L259 | Q278 |
| PH24 | −0.49 | <0.01 | 3425 | 3479 | 54 | V292 | L310 |
| PH26 | −0.13 | 3530 | 3599 | 69 | A327 | K350 | |
| PH27 | 0.11 | 3599 | 3650 | 51 | T350 | Q367 | |
| PH28 | 0.68 | <0.01 | 3650 | 3680 | 30 | L367 | T377 |
| PH29 | 0.41 | <0.05 | 3680 | 3746 | 66 | E377 | E399 |
| PH30 | 0.46 | 3746 | 3815 | 69 | A399 | L422 | |
| PH31 | −0.72 | <0.01 | 3815 | 3860 | 45 | V422 | A437 |
| PH32 | −0.35 | 3860 | 3905 | 45 | E437 | L452 | |
| PH33 | −1.29 | <0.001 | 3905 | 3930 | 25 | G453 | D460 |
Using the final GLM (Table 2, model 5), we predicted the recombination rate for each interval after adjusting for the effects of viral phenotype and donor variability (Fig. 5). From the estimate of standard error for each interval, we determined which regions are significantly different to the average recombination rate across gag and pol regions. Intervals without P values were not significant at the 0.05 level.
DISCUSSION
The high replication rate of HIV-1 and high rates of mutation and recombination lead to remarkable adaptability of the virus in the face of intense evolutionary pressure. Recombination is thought to make natural selection more efficient by breaking linkages between mutations (48–50). That is, recombination helps to maintain genetic diversity by breaking linkages between advantageous and deleterious mutations while also facilitating the removal of deleterious mutations by bringing them together in the same genome. Importantly, recombination can also pair advantageous mutations, which can facilitate the acquisition of multidrug resistance leading to treatment failure (48–54). Recombination may also be an important mechanism by which the virus eventually escapes immune control (55–58). However, recombination also has the potential to inhibit adaptation and evolution depending on epistasis and genetic drift (51). Consequently, an improved understanding of recombination is important for understanding the evolutionary history of HIV-1 and may help to guide the design of robust antiretroviral therapies.
There have been many studies showing that even in the absence of selection, recombination does not occur randomly on the HIV-1 genome, highlighting the presence of additional factors governing the recombination process (11, 19, 28–35, 59). However, many of these studies do not measure recombination rate in their natural genome context, or they measure recombination between highly divergent genomes that may not be most representative of the situation in vivo, where we expect recombination between closely related members of the viral quasispecies. Here, we present a system that allows the study of recombination between highly similar genomes that mimic the HIV-1 quasispecies within an HIV-1-infected patient. We delineate the process of retroviral recombination through infection of primary T lymphocytes with a minimally codon-modified full-length virus. An advantage of this method is that we can target specific areas of the genome while controlling the length of interval and hence the accuracy of our study. We have previously used a similar system to analyze recombination rates in a small region of gag (37). In this case, we were unable to draw conclusions about the location of recombination hot spots, primarily because this requires analysis of large numbers of sequences (19, 35, 37). In this study, we applied next-generation sequencing to systematically measure high-resolution recombination rates in gag and pol. These two genome regions were chosen because of their importance in the generation of drug-resistant virus and immune escape mutations (60).
We have optimized this system and shown that it is not biased by confounding factors related to experimentally induced recombination and for the occurrence of multiple template switches over intervals of various lengths (24, 37). Using two independent sets of marker modification, we show that putative recombination hot spots are not due to modifications introduced by our marker system. Indeed, there is a high correlation of recombination hot spots between our two systems. Notably, regardless of viral phenotype and blood donor, we demonstrate greater-than-6-fold recombination rate changes across gag and pol. These changes are consistent regardless of viral phenotype (r = 0.68, P < 0.001) and blood donor (r = 0.44 to 0.71, P = <0.001 to 0.04). We identify 12 genome regions with significantly higher rates of recombination and 10 genome regions with significantly lower rates of recombination.
It is instructive to compare our recombination hot spots between closely related genomes with those identified in natural HIV-1 sequences. Surprisingly, the gag hot/cold spots identified in our study match closely with those identified by analyzing patient sequences (6, 9, 61). This is surprising, because regions of sequence similarity are presumed to drive intersubtype recombination, and one would not expect to see the impact of local recombination hot spots after so many confounding factors, such as selection for functional proteins, drug resistance, or selection from the immune system (9, 62). One of the most comprehensive studies, by Simon-Loriere and colleagues, analyzed sequences retrieved from the Los Alamos National Laboratory HIV sequence database (http://www.hiv.lanl.gov) and provides evidence of recombination (9). Their study identified two hot spots and one cold spot in the capsid of gag, corresponding to our regions GH5 to GH8, GH12 and GH13, and GH9 and GH10. These regions also corresponded to hot and cold spot clusters in our analysis. The hot region spanning GH5 to GH8 does include a subregion with a strong and significant cold spot (GH6; −0.73 × 10−3; P < 0.0001) that is not present in the Simon-Loriere study. However, this subregion may have been missed in their data set, as the segment GH6 is only 21 bp in length, and they averaged their recombination breakpoints using a sliding window of 200 nt. It is interesting to note that these two hot regions span the matrix-capsid and capsid-P2 junctions of Gag. Indeed, it has been proposed that the distribution of RNA structures along the HIV-1 genome has evolved to facilitate gene swapping in a way that maximizes genetic diversity while minimizing the chance that the resulting progeny is impaired (9, 61). Our study does not directly address this issue, as our marker points were designed to minimize structural changes to the genome. However, our data showing the position of hot and cold spots in the genome will help to inform future mechanistic studies into the factors that influence recombination.
Within pol, some of our hot spots do not match those found by analyzing patient sequence databases. In our data set, we observe a hot spot near the beginning of p51 (PH6; 0.74 × 10−5; P < 0.05) that is followed by a region of intermediate recombination ending with a strong recombination cold spot at PH12 (−0.66 × 10−5; P < 0.0001). In the Simon-Loriere study, they identify a broad hot spot beginning at region PH6 and peaking at PH11. Thus, where their study finds one of their strongest hot spots, we find a region of intermediate recombination ending with one of our coldest spots at Ph12. As this region contains important resistance mutations, such as the thymidine analogue mutations (60), the detection of hot spots for recombination in the in vivo data could be evidence for selection. Similarly, we identify a cold spot (PH31; −0.75 × 10−5; P < 0.001) that falls close to the p51-RNase H junction, which was labeled as a hot spot for recombination in the Simon-Loriere study. On the other hand, we identify hot spot PH19 (0.54 × 10−5; <0.05), which falls within an unstructured peptide loop of the reverse transcriptase enzyme (63). Interestingly, this hot region, PH19 to PH21, corresponds exactly to some of the most highly structured RNA in the HIV-1 genome, as measured by selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) chemistry (63). Indeed, RNA structures are proposed to favor recombination by causing reverse transcriptase to pause on the template (12, 27, 64–66), and mechanistic studies demonstrate that the presence of RNA structure is often a feature of recombination hot spots (34, 67). It has been previously reported that HIV-1 gene junctions are both enriched in RNA structure and thus more recombinogenic than other regions of the HIV-1 genome (61, 63). We anecdotally note that our recombination hot spots do seem to be enriched at gene junctions, with the exception of the RNase H junction. This suggests that local fluctuations in recombination rates could drive the evolution of the RNA genome on a global scale. Further investigation of these genomic locations is warranted, as the molecular mechanisms that cause recombination hot and cold spots may shed further light on the higher-level organization of the HIV-1 genome.
As recombination is thought to facilitate viral evolution by intermixing immune escape and drug resistance mutations within HIV-1 gag and pol, knowledge of how recombination rates vary within these particular (68) regions of the viral genome is of importance for designing antiviral strategies. From a therapeutic viewpoint, the shuffling of resistance mutations within gag and pol could impact the generation of multidrug-resistant viruses (48–50). In general, the further apart genomic regions are, the less likely they will be linked together, and the easier it will be to shuffle mutations between these regions. For genomic regions that are close together, it should be easier to generate an RT double mutation where the resistance mutations are separated by a recombination hot spot. Our data suggest that the major reverse transcriptase drug resistance mutations lie in a relatively stable region of the genome, theoretically reducing the risk that they will be brought together by recombination. It is important to note, however, that an important prerequisite for recombination is the copackaging of genetically distinct genomes into viral particles via efficient coinfection of cells. Early studies suggested that these conditions were likely to be fulfilled in vivo, with between 75 and 80% of infected spleen cells harboring at least two or more proviruses, with most of these cells harboring genetically distinct proviruses (69). More recent studies on both CD4+ T cells and infected spleen cells contradict this view and show that the majority of cells are only singly infected (68, 70). Nevertheless, there is ample evidence that at least some recombination does occur in vivo and that it is functionally relevant to immune escape and the generation of multidrug-resistant HIV-1 (48–52, 54–58, 68). Furthermore, it is possible that the location of recombination hot spots may be more important under scenarios of low coinfection than under scenarios where the conditions for recombination are rampant. It will be important to test this assertion by including the possibility of recombination hot spots in models of HIV-1 dynamics.
All together, our data provide unique insights into HIV-1 recombination occurring between highly similar genomes likely to be found in the majority of infected individuals. Our results demonstrate that recombination does not occur randomly, and we identify recombination hot spots and cold spots in gag and pol. Importantly, our recombination hot/cold spots match closely with those found by analysis of patient sequence databases, indicating that, for gag and pol, the recombinogenic properties of the RNA genome itself, rather than sequence similarity, is likely to be the main driver of recombinant genomes circulating in the human population. Further studies into this area may ultimately prove crucial in developing robust antiviral strategies against HIV-1.
ACKNOWLEDGMENT
We thank Matteo Negroni for critical reading of the manuscript.
Footnotes
Published ahead of print 26 December 2013
REFERENCES
- 1.Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. 2010. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84:9864–9878. 10.1128/JVI.00915-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mansky LM. 1996. Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retroviruses 12:307–314. 10.1089/aid.1996.12.307 [DOI] [PubMed] [Google Scholar]
- 4.Smyth RP, Davenport MP, Mak J. 2012. The origin of genetic diversity in HIV-1. Virus Res. 169:415–429. 10.1016/j.virusres.2012.06.015 [DOI] [PubMed] [Google Scholar]
- 5.Hu WS, Temin HM. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U. S. A. 87:1556–1560. 10.1073/pnas.87.4.1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Archer J, Pinney JW, Fan J, Simon-Loriere E, Arts EJ, Negroni M, Robertson DL. 2008. Identifying the important HIV-1 recombination breakpoints. PLoS Comput. Biol. 4:e1000178. 10.1371/journal.pcbi.1000178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baird HA, Galetto R, Gao Y, Simon-Loriere E, Abreha M, Archer J, Fan J, Robertson DL, Arts EJ, Negroni M. 2006. Sequence determinants of breakpoint location during HIV-1 intersubtype recombination. Nucleic Acids Res. 34:5203–5216. 10.1093/nar/gkl669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baird HA, Gao Y, Galetto R, Lalonde M, Anthony RM, Giacomoni V, Abreha M, Destefano JJ, Negroni M, Arts EJ. 2006. Influence of sequence identity and unique breakpoints on the frequency of intersubtype HIV-1 recombination. Retrovirology 3:91. 10.1186/1742-4690-3-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon-Loriere E, Galetto R, Hamoudi M, Archer J, Lefeuvre P, Martin DP, Robertson DL, Negroni M. 2009. Molecular mechanisms of recombination restriction in the envelope gene of the human immunodeficiency virus. PLoS Pathog. 5:e1000418. 10.1371/journal.ppat.1000418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An W, Telesnitsky A. 2002. Effects of various sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 76:7897–7902. 10.1128/JVI.76.15.7897-7902.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magiorkinis G, Paraskevis D, Vandamme AM, Magiorkinis E, Sypsa V, Hatzakis A. 2003. In vivo characteristics of human immunodeficiency virus type 1 intersubtype recombination: determination of hot spots and correlation with sequence similarity. J. Gen. Virol. 84:2715–2722. 10.1099/vir.0.19180-0 [DOI] [PubMed] [Google Scholar]
- 12.Song M, Balakrishnan M, Chen Y, Roques BP, Bambara RA. 2006. Stimulation of HIV-1 minus strand strong stop DNA transfer by genomic sequences 3′ of the primer binding site. J. Biol. Chem. 281:24227–24235. 10.1074/jbc.M603097200 [DOI] [PubMed] [Google Scholar]
- 13.Novitsky V, Wang R, Margolin L, Baca J, Rossenkhan R, Moyo S, van Widenfelt E, Essex M. 2011. Transmission of single and multiple viral variants in primary HIV-1 subtype C infection. PLoS One 6:e16714. 10.1371/journal.pone.0016714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557. 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J. Virol. 83:3556–3567. 10.1128/JVI.02132-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Powell D, Hu WS. 2006. High frequency of genetic recombination is a common feature of primate lentivirus replication. J. Virol. 80:9651–9658. 10.1128/JVI.00936-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Rhodes TD, Hu WS. 2005. Comparison of the genetic recombination rates of human immunodeficiency virus type 1 in macrophages and T cells. J. Virol. 79:9337–9340. 10.1128/JVI.79.14.9337-9340.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 101:4204–4209. 10.1073/pnas.0306764101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang J, Jetzt AE, Sun G, Yu H, Klarmann G, Ron Y, Preston BD, Dougherty JP. 2002. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76:11273–11282. 10.1128/JVI.76.22.11273-11282.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jetzt AE, Yu H, Klarmann GJ, Ron Y, Preston BD, Dougherty JP. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 74:1234–1240. 10.1128/JVI.74.3.1234-1240.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chin MPS, Rhodes TD, Chen JB, Fu W, Hu WS. 2005. Identification of a major restriction in HIV-1 intersubtype recombination. Proc. Natl. Acad. Sci. U. S. A. 102:9002–9007. 10.1073/pnas.0502522102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iglesias-Sanchez MJ, Lopez-Galindez C. 2002. Analysis, quantification, and evolutionary consequences of HIV-1 in vitro recombination. Virology 304:392–402. 10.1006/viro.2002.1657 [DOI] [PubMed] [Google Scholar]
- 23.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289. 10.1084/jem.20090378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlub TE, Smyth RP, Grimm AJ, Mak J, Davenport MP. 2010. Accurately measuring recombination between closely related HIV-1 genomes. PLoS Comput. Biol. 6:e1000766. 10.1371/journal.pcbi.1000766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeiffer JK, Topping RS, Shin NH, Telesnitsky A. 1999. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J. Virol. 73:8441–8447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Operario DJ, Balakrishnan M, Bambara RA, Kim B. 2006. Reduced dNTP interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J. Biol. Chem. 281:32113–32121. 10.1074/jbc.M604665200 [DOI] [PubMed] [Google Scholar]
- 27.Svarovskaia ES, Delviks KA, Hwang CK, Pathak VK. 2000. Structural determinants of murine leukemia virus reverse transcriptase that affect the frequency of template switching. J. Virol. 74:7171–7178. 10.1128/JVI.74.15.7171-7178.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galetto R, Moumen A, Giacomoni V, Veron M, Charneau P, Negroni M. 2004. The structure of HIV-1 genomic RNA in the gp120 gene determines a recombination hot spot in vivo. J. Biol. Chem. 279:36625–36632. 10.1074/jbc.M405476200 [DOI] [PubMed] [Google Scholar]
- 29.Shen W, Gao L, Balakrishnan M, Bambara RA. 2009. A recombination hot spot in HIV-1 contains guanosine runs that can form a G-quartet structure and promote strand transfer in vitro. J. Biol. Chem. 284:33883–33893. 10.1074/jbc.M109.055368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moumen A, Polomack L, Roques B, Buc H, Negroni M. 2001. The HIV-1 repeated sequence R as a robust hot-spot for copy-choice recombination. Nucleic Acids Res. 29:3814–3821. 10.1093/nar/29.18.3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen ES, Jeeninga RE, Damgaard CK, Berkhout B, Kjems J. 2003. Dimerization and template switching in the 5′ untranslated region between various subtypes of human immunodeficiency virus type 1. J. Virol. 77:3020–3030. 10.1128/JVI.77.5.3020-3030.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikkelsen JG, Rasmussen SV, Pedersen FS. 2004. Complementarity-directed RNA dimer-linkage promotes retroviral recombination in vivo. Nucleic Acids Res. 32:102–114. 10.1093/nar/gkh159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dykes C, Balakrishnan M, Planelles V, Zhu Y, Bambara RA, Demeter LM. 2004. Identification of a preferred region for recombination and mutation in HIV-1 gag. Virology 326:262–279. 10.1016/j.virol.2004.02.033 [DOI] [PubMed] [Google Scholar]
- 34.Galetto R, Giacomoni V, Veron M, Negroni M. 2006. Dissection of a circumscribed recombination hot spot in HIV-1 after a single infectious cycle. J. Biol. Chem. 281:2711–2720. 10.1074/jbc.M505457200 [DOI] [PubMed] [Google Scholar]
- 35.Chin MP, Lee SK, Chen J, Nikolaitchik OA, Powell DA, Fivash MJ, Jr, Hu WS. 2008. Long-range recombination gradient between HIV-1 subtypes B and C variants caused by sequence differences in the dimerization initiation signal region. J. Mol. Biol. 377:1324–1333. 10.1016/j.jmb.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibbs JS, Regier DA, Desrosiers RC. 1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retrovir. 10:343–350. 10.1089/aid.1994.10.343 [DOI] [PubMed] [Google Scholar]
- 37.Smyth RP, Schlub TE, Grimm A, Venturi V, Chopra A, Mallal S, Davenport MP, Mak J. 2010. Reducing chimera formation during PCR amplification to ensure accurate genotyping. Gene 469:45–51. 10.1016/j.gene.2010.08.009 [DOI] [PubMed] [Google Scholar]
- 38.Meyer M, Stenzel U, Myles S, Prufer K, Hofreiter M. 2007. Targeted high-throughput sequencing of tagged nucleic acid samples. Nucleic Acids Res. 35:e97. 10.1093/nar/gkm566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T. 2010. BioRuby: bioinformatics software for the Ruby programming language. Bioinformatics 26:2617–2619. 10.1093/bioinformatics/btq475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The R Development Core Team. 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 42.Thompson JR, Marcelino LA, Polz MF. 2002. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR.' Nucleic Acids Res. 30:2083–2088. 10.1093/nar/30.9.2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyerhans A, Vartanian JP, Wain-Hobson S. 1990. DNA recombination during PCR. Nucleic Acids Res. 18:1687–1691. 10.1093/nar/18.7.1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson RA, Eliason SL. 1986. Recombination of homologous DNA fragments transfected into mammalian cells occurs predominantly by terminal pairing. Mol. Cell. Biol. 6:3246–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuiken C, Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B. 2010. HIV Sequence Compendium 2010, LA-UR 10–03684 Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM [Google Scholar]
- 46.Di Giallonardo F, Zagordi O, Duport Y, Leemann C, Joos B, Kunzli-Gontarczyk M, Bruggmann R, Beerenwinkel N, Gunthard HF, Metzner KJ. 2013. Next-generation sequencing of HIV-1 RNA genomes: determination of error rates and minimizing artificial recombination. PLoS One 8:e74249. 10.1371/journal.pone.0074249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Powell RLR, Lezeau L, Kinge T, Nyambi PN. 2010. Longitudinal quasispecies analysis of viral variants in HIV type 1 dually infected individuals highlights the importance of sequence identity in viral recombination. AIDS Res. Hum. Retrovir. 26:253–264. 10.1089/aid.2009.0174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charpentier C, Nora T, Tenaillon O, Clavel F, Hance AJ. 2006. Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients. J. Virol. 80:2472–2482. 10.1128/JVI.80.5.2472-2482.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown RJ, Peters PJ, Caron C, Gonzalez-Perez MP, Stones L, Ankghuambom C, Pondei K, McClure CP, Alemnji G, Taylor S, Sharp PM, Clapham PR, Ball JK. 2011. Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J. Virol. 85:6024–6037. 10.1128/JVI.00131-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wain-Hobson S, Renoux-Elbe C, Vartanian JP, Meyerhans A. 2003. Network analysis of human and simian immunodeficiency virus sequence sets reveals massive recombination resulting in shorter pathways. J. Gen. Virol. 84:885–895. 10.1099/vir.0.18894-0 [DOI] [PubMed] [Google Scholar]
- 51.Bretscher MT, Althaus CL, Muller V, Bonhoeffer S. 2004. Recombination in HIV and the evolution of drug resistance: for better or for worse? Bioessays 26:180–188. 10.1002/bies.10386 [DOI] [PubMed] [Google Scholar]
- 52.Vijay NN, Vasantika Ajmani R, Perelson AS, Dixit NM. 2008. Recombination increases human immunodeficiency virus fitness, but not necessarily diversity. J. Gen. Virol. 89:1467–1477. 10.1099/vir.0.83668-0 [DOI] [PubMed] [Google Scholar]
- 53.Kellam P, Larder BA. 1995. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J. Virol. 69:669–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mostowy R, Kouyos RD, Fouchet D, Bonhoeffer S. 2011. The role of recombination for the coevolutionary dynamics of HIV and the immune response. PLoS One 6:e16052. 10.1371/journal.pone.0016052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Streeck H, Li B, Poon AF, Schneidewind A, Gladden AD, Power KA, Daskalakis D, Bazner S, Zuniga R, Brander C, Rosenberg ES, Frost SD, Altfeld M, Allen TM. 2008. Immune-driven recombination and loss of control after HIV superinfection. J. Exp. Med. 205:1789–1796. 10.1084/jem.20080281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu SL, Mittler JE, Nickle DC, Mulvania TM, Shriner D, Rodrigo AG, Kosloff B, He X, Corey L, Mullins JI. 2002. Selection for human immunodeficiency virus type 1 recombinants in a patient with rapid progression to AIDS. J. Virol. 76:10674–10684. 10.1128/JVI.76.21.10674-10684.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishimura Y, Shingai M, Lee WR, Sadjadpour R, Donau OK, Willey R, Brenchley JM, Iyengar R, Buckler-White A, Igarashi T, Martin MA. 2011. Recombination-mediated changes in coreceptor usage confer an augmented pathogenic phenotype in a nonhuman primate model of HIV-1-induced AIDS. J. Virol. 85:10617–10626. 10.1128/JVI.05010-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi B, Kitchen C, Weiser B, Mayers D, Foley B, Kemal K, Anastos K, Suchard M, Parker M, Brunner C, Burger H. 2010. Evolution and recombination of genes encoding HIV-1 drug resistance and tropism during antiretroviral therapy. Virology 404:5–20. 10.1016/j.virol.2010.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wooley DP, Bircher LA, Smith RA. 1998. Retroviral recombination is nonrandom and sequence dependent. Virology 243:229–234. 10.1006/viro.1998.9052 [DOI] [PubMed] [Google Scholar]
- 60.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. 2011. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 19:156–164 [PMC free article] [PubMed] [Google Scholar]
- 61.Simon-Loriere E, Martin DP, Weeks KM, Negroni M. 2010. RNA structures facilitate recombination-mediated gene swapping in HIV-1. J. Virol. 84:12675–12682. 10.1128/JVI.01302-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galli A, Kearney M, Nikolaitchik OA, Yu S, Chin MP, Maldarelli F, Coffin JM, Pathak VK, Hu WS. 2010. Patterns of human immunodeficiency virus type 1 recombination ex vivo provide evidence for coadaptation of distant sites, resulting in purifying selection for intersubtype recombinants during replication. J. Virol. 84:7651–7661. 10.1128/JVI.00276-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716. 10.1038/nature08237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klarmann GJ, Schauber CA, Preston BD. 1993. Template-directed pausing of DNA synthesis by HIV-1 reverse transcriptase during polymerization of HIV-1 sequences in vitro. J. Biol. Chem. 268:9793–9802 [PubMed] [Google Scholar]
- 65.Roda RH, Balakrishnan M, Kim JK, Roques BP, Fay PJ, Bambara RA. 2002. Strand transfer occurs in retroviruses by a pause-initiated two-step mechanism. J. Biol. Chem. 277:46900–46911. 10.1074/jbc.M208638200 [DOI] [PubMed] [Google Scholar]
- 66.Gao L, Balakrishnan M, Roques BP, Bambara RA. 2007. Insights into the multiple roles of pausing in HIV-1 reverse transcriptase-promoted strand transfers. J. Biol. Chem. 282:6222–6231. 10.1074/jbc.M610056200 [DOI] [PubMed] [Google Scholar]
- 67.Hanson MN, Balakrishnan M, Roques BP, Bambara RA. 2005. Effects of donor and acceptor RNA structures on the mechanism of strand transfer by HIV-1 reverse transcriptase. J. Mol. Biol. 353:772–787. 10.1016/j.jmb.2005.08.065 [DOI] [PubMed] [Google Scholar]
- 68.Josefsson L, Palmer S, Faria NR, Lemey P, Casazza J, Ambrozak D, Kearney M, Shao W, Kottilil S, Sneller M, Mellors J, Coffin JM, Maldarelli F. 2013. Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 9:e1003432. 10.1371/journal.ppat.1003432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. 2002. Recombination: multiply infected spleen cells in HIV patients. Nature 418:144. 10.1038/418144a [DOI] [PubMed] [Google Scholar]
- 70.Josefsson L, King MS, Makitalo B, Brannstrom J, Shao W, Maldarelli F, Kearney MF, Hu WS, Chen J, Gaines H, Mellors JW, Albert J, Coffin JM, Palmer SE. 2011. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. U. S. A. 108:11199–11204. 10.1073/pnas.1107729108 [DOI] [PMC free article] [PubMed] [Google Scholar]