

ABSTRACT
Herpes simplex virus 1 (HSV-1) encodes Us3 protein kinase, which is critical for viral pathogenicity in both mouse peripheral sites (e.g., eyes and vaginas) and in the central nervous systems (CNS) of mice after intracranial and peripheral inoculations, respectively. Whereas some Us3 substrates involved in Us3 pathogenicity in peripheral sites have been reported, those involved in Us3 pathogenicity in the CNS remain to be identified. We recently reported that Us3 phosphorylated HSV-1 dUTPase (vdUTPase) at serine 187 (Ser-187) in infected cells, and this phosphorylation promoted viral replication by regulating optimal enzymatic activity of vdUTPase. In the present study, we show that the replacement of vdUTPase Ser-187 by alanine (S187A) significantly reduced viral replication and virulence in the CNS of mice following intracranial inoculation and that the phosphomimetic substitution at vdUTPase Ser-187 in part restored the wild-type viral replication and virulence. Interestingly, the S187A mutation in vdUTPase had no effect on viral replication and pathogenic effects in the eyes and vaginas of mice after ocular and vaginal inoculation, respectively. Similarly, the enzyme-dead mutation in vdUTPase significantly reduced viral replication and virulence in the CNS of mice after intracranial inoculation, whereas the mutation had no effect on viral replication and pathogenic effects in the eyes and vaginas of mice after ocular and vaginal inoculation, respectively. These observations suggested that vdUTPase was one of the Us3 substrates responsible for Us3 pathogenicity in the CNS and that the CNS-specific virulence of HSV-1 involved strict regulation of vdUTPase activity by Us3 phosphorylation.
IMPORTANCE Herpes simplex virus 1 (HSV-1) encodes a viral protein kinase Us3 which is critical for pathogenicity both in peripheral sites and in the central nervous systems (CNS) of mice following peripheral and intracranial inoculations, respectively. Whereas some Us3 substrates involved in Us3 pathogenicity in peripheral sites have been reported, those involved in Us3 pathogenicity in the CNS remain to be identified. Here, we report that Us3 phosphorylation of viral dUTPase (vdUTPase) at serine 187 (Ser-187), which has been shown to promote the vdUTPase activity, appears to be critical for viral virulence in the CNS but not for pathogenic effects in peripheral sites. Since HSV proteins critical for viral virulence in the CNS are, in almost all cases, also involved in viral pathogenicity at peripheral sites, this phosphorylation event is a unique report of a specific mechanism involved in HSV-1 virulence in the CNS.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) encodes at least two protein kinases, Us3 and UL13 (1–6). HSV-1 Us3 is a serine/threonine protein kinase with an amino acid sequence that is conserved in the subfamily Alphaherpesvirinae (1, 2). The Us3 protein and its enzymatic activity have been suggested to play a critical role in viral replication and pathogenicity, based on studies showing that recombinant Us3-null mutant viruses and recombinant viruses encoding catalytically inactive Us3 (Us3 kinase-dead mutant viruses) have impaired growth properties in cell cultures and reduced pathogenicity and replication in mouse models (7–11).
In experimental animal models of HSV-1 infection, viral pathogenicity in peripheral sites (e.g., eyes and vaginas) and in central nervous systems (CNS) was studied by peripheral and intracranial inoculations, respectively. These are semi-independent indicators of viral virulence since viral proteins or their domains that are important for viral pathogenicity in peripheral sites are often not required for pathogenicity in the CNS in mouse models of HSV-1 infection (12–15). HSV-1 Us3 has been reported to be critical for viral pathogenicity in both peripheral sites and in the CNS in mice, based on studies showing that Us3-null and Us3 kinase-dead mutant viruses had significantly reduced viral replication and pathogenicity in the eyes and brains of mice following ocular and intracranial inoculation, respectively (7, 10, 11).
HSV-1 Us3 has been considered to be a multifunctional protein regulating various aspects of cellular and viral functions (5). Supporting to this hypothesis, more than 15 putative viral and cellular substrates of Us3 have been reported (16–27). However, among them, only a limited substrates (18–28), including gB, UL31, Us3 itself, UL47, viral dUTPase (vdUTPase), and tuberous sclerosis complex 2, have been shown both to be physiological Us3 substrates in infected cells and directly linked with Us3 functions in infected cells (11, 15, 24–28). Among these functional Us3 substrates, gB, Us3 itself, and UL47 have been shown to be involved in pathogenicity in peripheral sites, based on data showing that amino acid substitutions in the Us3 phosphorylation sites of these substrates significantly impaired viral replication and pathogenic effects in the eyes of mice after ocular inoculation (11, 15, 25). In contrast, these mutations had no effect on viral pathogenicity in mice following intracranial inoculation (11, 15, 25). Thus, although mechanisms of Us3 pathogenicity in peripheral sites have been gradually elucidated, the mechanisms of Us3 pathogenicity in the CNS remain unknown at present.
We recently reported that Us3 phosphorylated vdUTPase at serine 187 (Ser-187) in infected cells (17). dUTPases are known to catalyze hydrolysis of dUTP to dUMP and pyrophosphate and play a role in accurate replication of DNA genomes by preventing the misincorporation of dUTP into replicating DNA (29–32). HSV-1 dUTPase (vdUTPase) is encoded by the UL50 gene and is conserved throughout the Herpesviridae family (33, 34). Us3 phosphorylation of vdUTPase at Ser-187 appeared to regulate optimal activity of vdUTPase in infected cells, based on the observations that an alanine replacement of vdUTPase Ser-187 (S187A), as well as a Us3 kinase-dead mutation, significantly reduced vdUTPase activity in infected cells, whereas a phosphomimetic substitution at vdUTPase Ser-187 restored the wild-type vdUTPase activity (17). Furthermore, replication of a recombinant virus carrying the S187A or enzyme-dead mutation in vdUTPase was significantly impaired in a human neuroblastoma SK-N-SH cells at a multiplicity of infection (MOI) of 5, but not at an MOI of 0.01, whereas replication in human carcinoma HEp-2 and simian kidney epithelial Vero cells was not affected (17). These observations suggested that Us3 phosphorylation of vdUTPase at Ser-187 promoted viral replication in a manner dependent on cell types and MOIs by regulating optimal enzymatic activity of vdUTPase (17). Notably, it has been reported that a recombinant HSV-1 carrying a null mutation in vdUTPase was less virulent than wild-type virus, replicated less well in the CNS and reactivated less efficiently in a mouse model of HSV-1 infection (35), suggesting that vdUTPase was critical for viral replication and pathogenicity in vivo. Taking this together with our recent report described above (17), it is reasonable to hypothesize that Us3 phosphorylation of vdUTPase at Ser-187 is involved in viral replication and pathogenicity in vivo, especially in the CNS. In the present study, to test this hypothesis, we examined the effects of this phosphorylation on viral replication and pathogenicity in vivo and presented the data showing that the phosphorylation was required for efficient viral replication and virulence in the CNS of mice following intracranial inoculation. Surprisingly, we have also shown that the phosphorylation was not required for pathogenicity at peripheral sites following inoculation at these sites, i.e., the eyes and vaginas of mice. Thus, Us3 phosphorylation of vdUTPase at Ser-187 appeared to be involved in the CNS-specific viral replication and virulence.
MATERIALS AND METHODS
Cells and viruses.
Vero and rabbit skin cells were described previously (36, 37), as was HSV-1 wild-type strain HSV-1(F) (38). A number of the recombinant viruses were described previously (17, 24): recombinant virus YK511, encoding an enzymatically inactive Us3 mutant in which lysine at Us3 residue 220 was replaced with methionine (Us3K220M); recombinant virus YK513, in which the Us3K220M mutation in YK511 was repaired (Us3K220M-repair); recombinant virus YK750, in which the vdUTPase gene was disrupted by insertion of a foreign gene cassette containing an I-SceI site, a kanamycin resistance gene, and 37 bp of the vdUTPase sequence encoding codons 181 to 193 in which Ser-187 was replaced with alanine (ΔvdUTPase); recombinant virus YK751, encoding vdUTPase with an alanine substituted for Ser-187, that was constructed by excision of a part of the foreign gene cassette, including the I-SceI site and the kanamycin resistance gene in YK750 vdUTPase gene (vdUTPaseS187A); recombinant virus YK752, in which the vdUTPaseS187A mutation in YK751 was repaired (vdUTPaseΔ/SA-repair); recombinant virus YK753 with the vdUTPaseS187D mutation (vdUTPaseS187D); recombinant virus YK754, in which the vdUTPaseS187D mutation in YK753 was repaired (vdUTPaseS187D-repair); recombinant virus YK759, encoding an enzymatically inactive vdUTPase mutant in which aspartic acid at vdUTPase residue 97 was replaced with alanine (vdUTPaseD97A); and recombinant virus YK760, in which the vdUTPaseD97A mutation in YK759 was repaired (vdUTPaseD97A-repair). We note that YK752 (vdUTPaseΔ/SA-repair) can be the repaired virus for both YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A), based on the sequential construction strategy of the recombinant viruses as described above and previously (17).
Mutagenesis of viral genomes and generation of recombinant HSV-1.
Recombinant viruses YK755 encoding ICP22 with alanines substituted for Ser-5, Ser-22, Thr-162, and Ser-167 (ICP22S5A/S22A/T162A/S167A) (Fig. 1), and YK757 encoding UL51 with alanine substituted for threonine at residue Thr-190 (UL51T190A) (Fig. 1) were generated by the two-step Red-mediated mutagenesis procedure using Escherichia coli GS1783 containing pYEbac102 as described previously (24, 39), except using the primers listed in Table 1. Recombinant viruses YK756 and YK758 in which the ICP22S5A/S22A/T162A/S167A and UL51T190A mutations in YK755 and YK757, respectively (Fig. 1), were repaired were generated as described previously (24, 39), except using the primers listed in Table 1 and pBS-ICP22-Kan (40).
FIG 1.
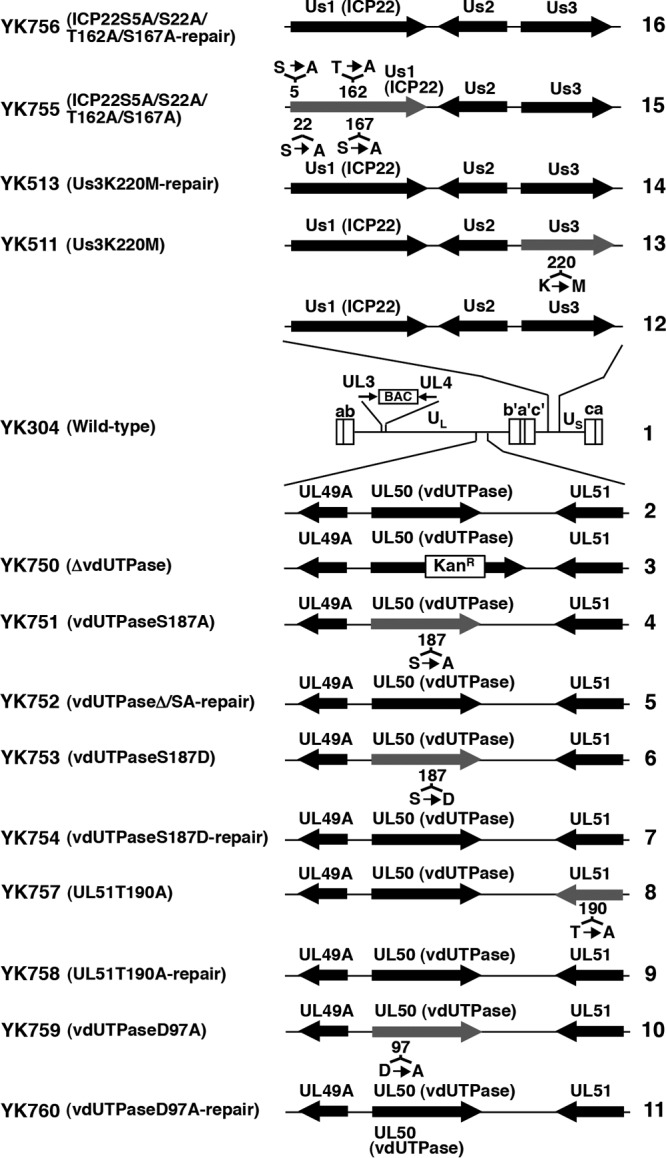
Schematic diagrams of the genome structure of wild-type and recombinant viruses used in the present study. Line 1, Wild-type HSV-1(YK304) genome carrying a bacmid (BAC) in the intergenic region between UL3 and UL4. Line 2, domain carrying the UL49A, UL50 (vdUTPase) and UL51 open reading frames. Lines 3 to 7, 10, and 11, recombinant viruses with a mutation in the UL50 (vdUTPase) gene. Lines 8 and 9, recombinant viruses with a mutation in the UL51 gene. Lines 13 and 14, recombinant viruses with a mutation in the Us3 gene. Lines 15 and 16, recombinant viruses with mutations in the Us1 (ICP22) gene.
TABLE 1.
Primer sequences for the construction of recombinant viruses
| Mutation | Orientationa | Sequence (5′–3′) |
|---|---|---|
| ICP22S5A/S22A | F | CACTGTGGTCCTCCGGGACGTTTTCTGGATGGCCGACATTGCCCCAGGCGCTTTTGCGCCTTGTGTAAAAGCGCGGCGTCCCGCTCTCCGAGCCAGGATGACGACGATAAGTAGGG |
| R | GGCGGGAAGGGCGCTTGCGCTTGCGCGTGCCCAGGGGCGGGGCTCGGAGAGCGGGACGCCGCGCTTTTACACAAGGCGCAAAAGCGCCTGGGGCCAACCAATTAACCAATTCTGATTAG | |
| ICP22T162A/S167A | F | GCGGGTCCGGTCTACCCGGGAAACGCAGCCCCGGGCCCCCGCCCCGTCGGCCCCAGCCCCAAATGCAATGAGGATGACGACGATAAGTAGGG |
| R | TCTGGGCCTGGCGCACCGAGCGCCGTAGCATTGCATTTGGGGCTGGGGCCGACGGGGCGGGGGCCCGGGGCAACCAATTAACCAATTCTGATTAG | |
| ΔICP22 | F | GCGGGGGGAAGCCACTGTGGTCCTCCGGGACGTTTTCTGGGTCCGGTCGCCCCGACCCCCAGGATGACGACGATAAGTAGGG |
| R | TTTTATTTGGGGACATACAAGGGGGTCGGGGCGACCGGAC-CCAGAAAACGTCCCGGAGGACAACCAATTAACCAATTCTGATTAG | |
| ΔICP22-repair | F | GCGGGGGGAAGCCACTGTGGTCCTCCGGGACGTTTTCTGGATGGCCGACATTTCCCCAGGCGC |
| R | CTTTTATTTGGGGACATACAAGGGGGTCGGGGCGACCGGACTCACGGCCGGAGAAACGTGT | |
| UL51T190A | F | GCTTGGGGTGACCGAGGCGCCCTCCTTGGGGCACCCCCACGCACCGCCCCCGGAGGTTACAGGATGACGACGATAAGTAGGG |
| R | CGTTTCGGGCGGCAGGCGCCAGCGTAACCTCCGGGGGCGGTGCGTGGGGGTGCCCCAAGGCAACCAATTAACCAATTCTGATTAG | |
| UL51T190A-repair | F | GCTTGGGGTGACCGAGGCGCCCTCCTTGGGGCACCCCCACACGCCGCCCCCGGAGGTTACAGGATGACGACGATAAGTAGGG |
| R | CGTTTCGGGCGGCAGGCGCCAGCGTAACCTCCGGGGGCGGCGTGTGGGGGTGCCCCAAGGCAACCAATTAACCAATTCTGATTAG |
F, forward; R, reverse.
Animal studies.
Female ICR mice were purchased from Charles River. For intracranial infection, 3-week-old mice were infected intracranially with 102 PFU of each of the indicated viruses as described previously (36). Mice were monitored daily, and mortality from 1 to 14 days postinfection was attributed to the inoculated virus. Virus titers in the brains of mice were determined as described previously (41). For ocular infection, 5-week-old mice were infected with 2 × 106 PFU/eye of each of the indicated viruses as described previously (11). The scoring scale for the severity of herpes stromal keratitis (HSK) was described previously (11). Virus titers in the tear films of mice were determined as described previously (11). For intravaginal infection, 4-week-old mice were each injected subcutaneously in the neck ruff with 1.67 mg of medroxyprogesterone (Depo-M; Vesco) in 100 μl at 7 days prior to viral infection. The treated mice were then infected intravaginally with 5 × 106 PFU of each of the indicated viruses (42). Mice were monitored daily until 14 days postinfection for survival and the severity of vaginal disease using a scoring system of 0 (no sign of disease), 1 (slight genital erythema and edema), 2 (moderate genital inflammation), 3 (purulent genital lesions), 4 (hind-limb paralysis), and 5 (death) as described previously (42). Virus titers in the vaginal secretions of mice were determined as described previously (42). All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval19-26).
Statistical analysis.
Differences in virus titers, HSK scores, and vaginal lesion scores were statistically analyzed by using the two-tailed Student t test. Differences in mortality of infected mice were statistically analyzed by using a log-rank test.
RESULTS
Effect of phosphorylation of vdUTPase Ser-187 on viral replication and virulence in the CNS of mice.
To investigate the relevance of phosphorylation of vdUTPase Ser-187 in viral replication and virulence in the CNS of mice, 3-week-old female ICR mice were inoculated intracranially with 102 PFU of YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), or YK511 (Us3K220M) or the repaired viruses YK752 (vdUTPaseΔ/SA-repair) and YK513 (Us3K220M-repair). The survival of the infected mice was monitored for 14 days postinfection, and virus titers in infected mouse brains were assayed at 3 days postinfection. As shown in Fig. 2A and B, all or most of the mice infected with YK750 (ΔvdUTPase), YK511 (Us3K220M), or YK751 (vdUTPaseS187A) survived, but the repaired viruses YK752 (vdUTPaseΔ/SA-repair) and YK513 (Us3K220M-repair) killed 68 to 75% of infected mice. These results are in agreement with previous reports (10, 11, 35) that the null mutation in vdUTPase and the kinase-dead mutation in Us3 significantly reduced mortality in mice after intracranial inoculation. Similarly, the vdUTPase S187A mutation also significantly reduced mortality in mice (Fig. 2A and C), indicating that vdUTPase Ser-187 was required for efficient virulence in the CNS of mice.
FIG 2.
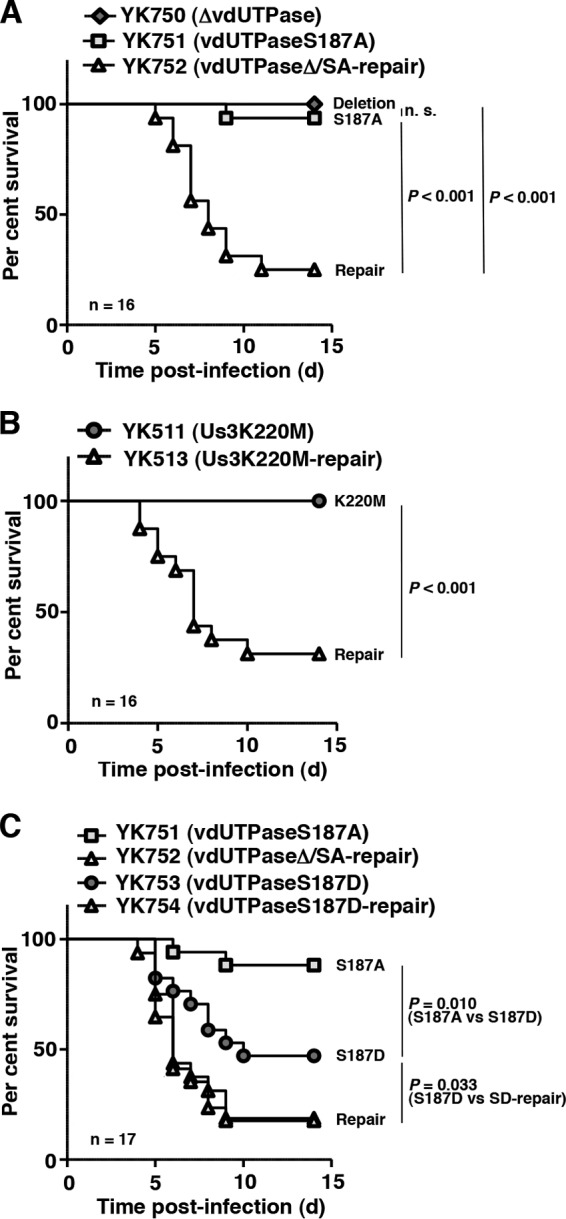
Effect of mutations in vdUTPase or the kinase-dead mutation in Us3 on viral virulence in the CNS of mice following intracranial inoculation. (A to C) Sixteen (A and B) or seventeen (C) 3-week-old female ICR mice were infected intracranially with YK750 (ΔvdUTPase) (A), YK751 (vdUTPaseS187A) (A and C), YK752 (vdUTPaseΔ/SA-repair) (A and C), YK511 (Us3K220M) (B), YK513 (Us3K220M-repair) (B), YK753 (vdUTPaseS187D) (C), or YK754 (vdUTPaseS187D-repair) (C) and monitored for 14 days. P represents the statistical significance value according to the log-rank test. n.s., not significant.
To further investigate the linkage between phosphorylation of vdUTPase Ser-187 and virulence in the CNS of mice, we also studied the virulence of YK753 (vdUTPaseS187D) carrying a phosphomimetic mutation in vdUTPase Ser-187 and its repaired virus YK754 (vdUTPaseS187D-repair) and of YK751 (vdUTPaseS187A) and its repaired virus YK752 (vdUTPaseΔ/SA-repair) in the CNS of mice. As shown in Fig. 2C, after intracranial inoculation, the survival of mice infected with YK753 (vdUTPaseS187D) was significantly less than that of mice infected with YK751 (vdUTPaseS187A), and the survival of mice infected with YK752 (vdUTPaseΔ/SA-repair) or YK754 (vdUTPaseS187D-repair) was even less than that of mice infected with YK753 (vdUTPaseS187D). This suggested that a negatively charged amino acid at vdUTPase Ser-187, due to either the phosphorylation of Ser-187 or an S187D substitution, was required for efficient virulence in the CNS of mice. Furthermore, in agreement with the effects of null, S187A, and S187D mutations in vdUTPase, and the kinase-dead mutation in Us3 on the survival of mice following intracranial inoculation, the null and S187A mutations in vdUTPase, and the kinase-dead mutation in Us3 significantly reduced viral replication in the brains of mice (Fig. 3A and B), whereas the S187D mutation in vdUTPase had little effect (Fig. 3C). Taken together, these results suggested that Us3 phosphorylation of vdUTPase Ser-187 was required for efficient viral virulence and replication in the CNS of mice following intracranial inoculation.
FIG 3.
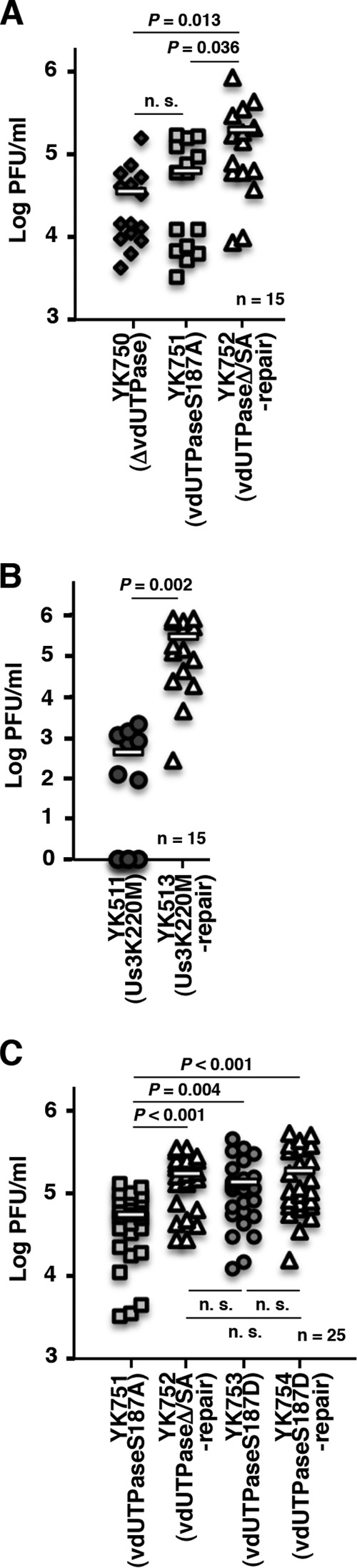
Effect of mutations in vdUTPase or the kinase-dead mutation in Us3 on viral replication in the CNS of mice following intracranial inoculation. (A to C) Fifteen (A and B) or twenty-five (C) 3-week-old female mice were infected intracranially with YK750 (ΔvdUTPase) (A), YK751 (vdUTPaseS187A) (A and C), YK752 (vdUTPaseΔ/SA-repair) (A and C), YK511 (Us3K220M) (B), YK513 (Us3K220M-repair) (B), YK753 (vdUTPaseS187D) (C), or YK754 (vdUTPaseS187D-repair) (C). At 3 days postinfection, the brains of infected mice were harvested, and virus titers were assayed. Each data point is the virus titer in the brain of one mouse. The horizontal bars indicate the mean for each group. P represents the statistical significance value according to the two-tailed Student t test. n.s., not significant.
Effect of phosphorylation of vdUTPase Ser-187 on viral replication and pathogenicity at peripheral sites in mice.
We previously reported that HSV-1 Us3 kinase activity is critical for development of herpes stroma keratitis (HSK) and viral replication in the eyes of mice after ocular inoculation (11). Us3 kinase activity is also critical for development of pathogenicity and viral replication in the vaginas of mice after vaginal inoculation, based on the observation that the Us3 kinase-dead mutation significantly reduced the pathogenicity scores of vaginal lesions and the virus titers in infected vaginas (Fig. 4A and 5A).
FIG 4.
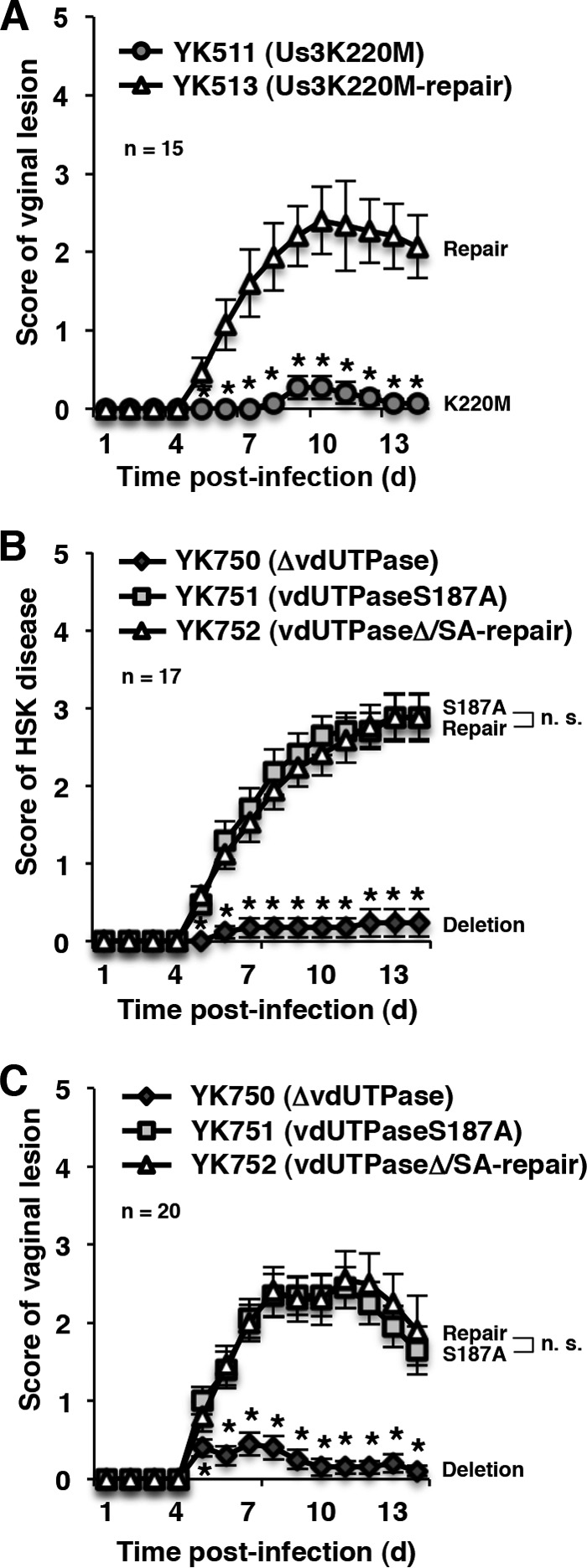
Effect of mutations in vdUTPase or the kinase-dead mutation in Us3 on viral pathogenicity in peripheral sites of mice following peripheral inoculation. (A) Fifteen 5-week-old female ICR mice pretreated with Depo-M were infected vaginally with YK511 (Us3K220M) or YK513 (Us3K220M-repair) and scored for vaginal disease every day for 14 days. Each data point is the mean ± the standard error of the scores. (B) Seventeen 5-week-old female ICR mice were ocularly infected with YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), or YK752 (vdUTPaseΔ/SA-repair) and scored for HSK every day for 14 days. Each data point is the mean ± the standard error of the scores. (C) Twenty 5-week-old female ICR mice pretreated with Depo-M were infected vaginally with YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), or YK752 (vdUTPaseΔ/SA-repair). The experiments were performed as described for panel A. Each data point is the mean ± the standard error of the scores. Asterisks represent the statistical significance value (*, P < 0.05) according to the two-tailed Student t test. n.s., not significant.
FIG 5.
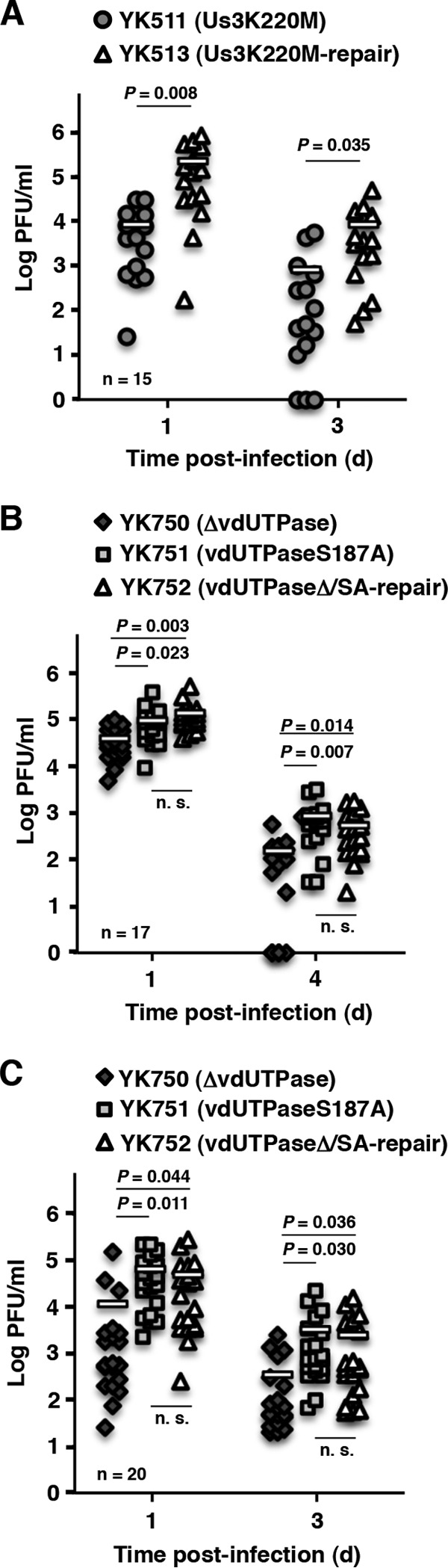
Effect of mutations in vdUTPase or the kinase-dead mutation in Us3 on viral replication in peripheral sites of mice following peripheral inoculation. (A) The vaginal secretions of infected mice at 1 and 3 days postinfection in the experiment described in Fig. 4A were harvested, and virus titers were assayed. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean for each group. (B) The tear films of infected mice at 1 and 4 days postinfection in the experiment described in Fig. 4B were harvested, and virus titers were assayed. Each data point is the virus titer in the tear film of one mouse. The horizontal bars indicate the mean for each group. (C) The vaginal secretions of infected mice at 1 and 3 days postinfection in the experiment described in Fig. 4C were harvested, and virus titers were assayed. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean for each group. P represents the statistical significance value according to the two-tailed Student t test. n.s., not significant.
To examine whether the phosphorylation of vdUTPase Ser-187 was also involved in HSV-1 pathogenicity at peripheral sites, we carried out two series of experiments. In the first series of experiments, 5-week-old female ICR mice were infected ocularly with 2 × 106 PFU/eye of YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), or YK752 (vdUTPaseΔ/SA-repair). The infected mice were observed daily for development of HSK for 14 days postinfection, and virus titers in tear films were determined at 1 and 4 days postinfection. In agreement with the reduction in virulence of YK750 (ΔvdUTPase) in mice following intracranial inoculation, mice ocularly infected with YK750 (ΔvdUTPase) exhibited significantly reduced severity of HSK compared to mice infected with the repaired virus YK752 (vdUTPaseΔ/SA-repair) (Fig. 4B), and virus titers in the tear films of mice infected with YK750 (ΔvdUTPase) were significantly lower than those of mice infected with YK752 (vdUTPaseΔ/SA-repair) (Fig. 5B). In contrast, although the pathogenicity of YK751 (vdUTPaseS187A) was significantly attenuated in mice following intracranial inoculation (Fig. 2A and C), mice infected with YK751 (vdUTPaseS187A) exhibited a severity of HSK almost identical to mice infected with YK752 (vdUTPaseΔ/SA-repair) (Fig. 4B), and the virus titers in the tear films of mice infected with YK751 (vdUTPaseS187A) were similar to those of mice infected with YK752 (vdUTPaseΔ/SA-repair) (Fig. 5B).
In the second series of experiments, 5-week-old female ICR mice pretreated with Depo-M were infected vaginally with 5 × 106 PFU of YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), or YK752 (vdUTPaseΔ/S187A-repair). Disease progression was observed daily for 14 days postinfection, and the virus titers in vaginal secretions were determined at 1 and 3 days postinfection. In agreement with the results in the mouse HSK model, the null mutation in vdUTPase significantly reduced the development of vaginal disease and viral replication in mouse vaginas, whereas the vdUTPase S187A mutation had no significant effect on vaginal pathogenicity or viral replication (Fig. 4C and 5C).
Taken together, these results indicated that phosphorylation of vdUTPase Ser-187 was not required for HSV-1 pathogenicity and replication in the eyes and vaginas of mice following ocular and vaginal inoculation, respectively, although the vdUTPase protein itself was critical for efficient viral pathogenicity and replication in peripheral sites of mice.
Effect of phosphorylation of other viral proteins on viral pathogenesis and replication in mice.
To confirm the specificity of the role of phosphorylation of vdUTPase Ser-187 in viral pathogenesis and replication in mice, we constructed recombinant viruses YK755 (ICP22S5A/S22A/T162A/S167A) and YK757 (UL51T190A), in which the phosphorylation sites in ICP22 and UL51 identified by phosphoproteomic analyses in previous studies (17, 43) were replaced with alanines, and their repaired viruses YK756 (ICP22S5A/S22A/T162A/S167A-repair) and YK758 (UL51T190A-repair) (Fig. 1), and used them to infect cultured cells and mice. Alanine substitutions for the phosphorylation sites in ICP22 and UL51 had no effect on viral growth in Vero cells at multiplicities of infection (MOIs) of 5 and 0.01, or on the survival of mice after intracranial inoculation (Fig. 6). These mutations also had no effect on viral replication and pathogenicity in the eyes of mice following ocular inoculation (Fig. 7A to F). These results indicated that the phosphorylation of ICP22 Ser-5, Ser-22, Thr-162, and Ser-167, and of UL51 Thr-190 was not required for viral replication and pathogenesis in mice after intracranial and ocular inoculation.
FIG 6.
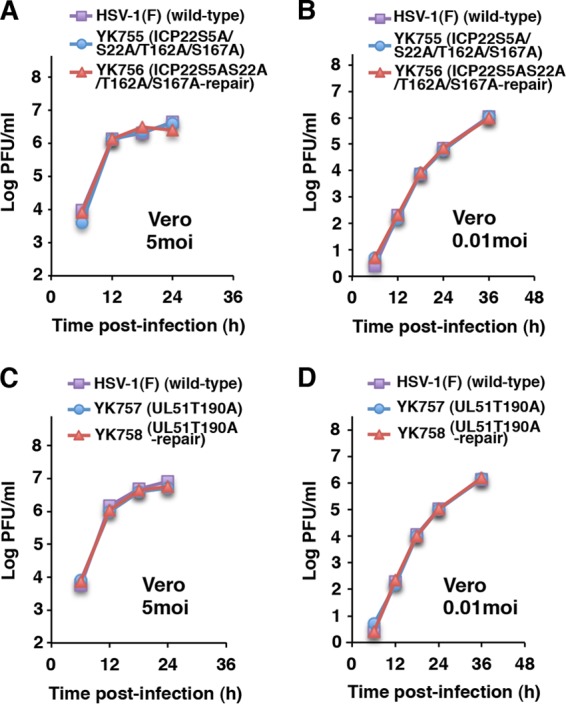
Effect of phosphorylation of other viral proteins on viral replication in Vero cells. (A to D) Vero cells were infected at an MOI of 5 (left panels) or 0.01 (right panels) with wild-type HSV-1(F), YK755 (ICP22S5A/S22A/T162A/S167A), and YK756 (ICP22S5A/S22A/T162A/S167A-repair) (A and B) or wild-type HSV-1(F), YK757 (UL51T190A), and YK758 (UL51T190A-repair) (C and D). The total virus from the cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells. The data are representative of three independent experiments.
FIG 7.
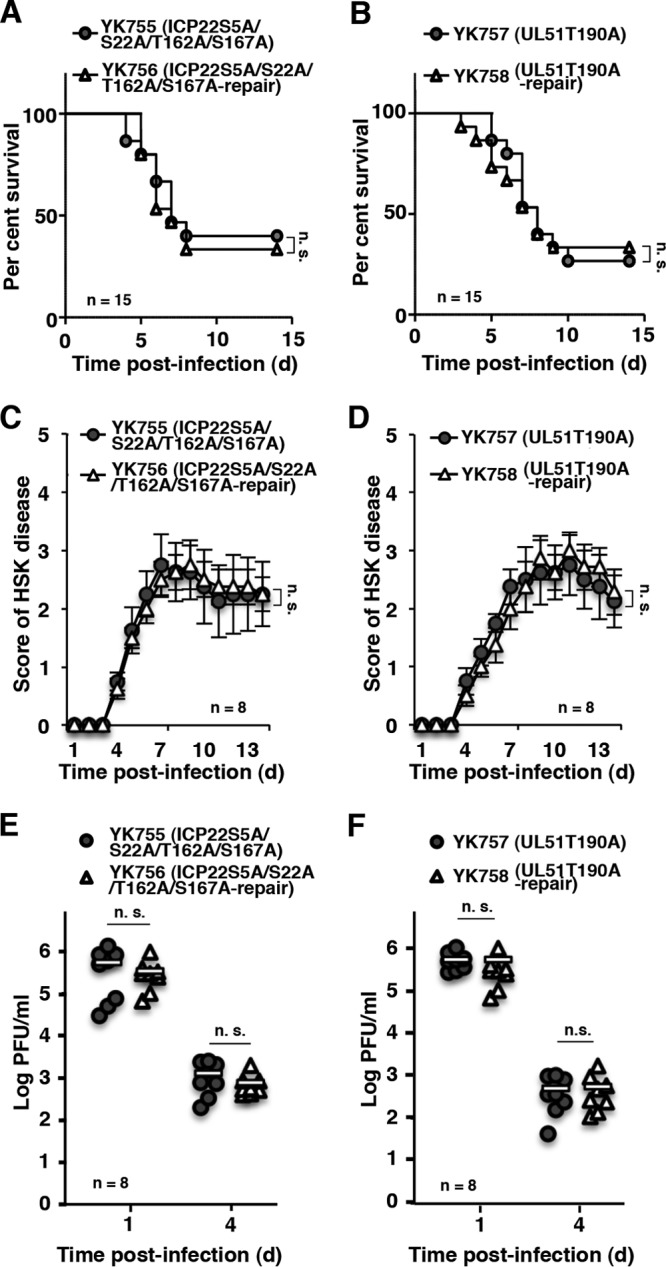
Effect of phosphorylation of other viral proteins on viral pathogenicity and replication in mice. (A and B) Fifteen 3-week-old female ICR mice were infected intracranially with YK755 (ICP22S5A/S22A/T162A/S167A) and YK756 (ICP22S5A/S22A/T162A/S167A-repair) (A) or YK757 (UL51T190A) and YK758 (UL51T190A-repair) (B), and the survival of infected mice was monitored for 14 days postinfection. (C and D) Eight 5-week-old female ICR mice were ocularly infected and scored for severity of HSK every day for 14 days postinfection with YK755 (ICP22S5A/S22A/T162A/S167A) and YK756 (ICP22S5A/S22A/T162A/S167A-repair) (C) or with YK757 (UL51T190A) and YK758 (UL51T190A-repair) (D). Each data point is the mean ± the standard error of the observations. (E and F) The tear films of infected mice at 1 and 4 days postinfection in the experiments described in panels C and D, respectively, were harvested, and virus titers were assayed. Each data point is the virus titer in the tear film of one mouse. The horizontal bars indicate the mean for each group. The statistical significance values were analyzed according to the log-rank test (A and B) or the two-tailed Student t test (C to F). n.s., not significant.
Effect of vdUTPase enzymatic activity on viral replication and pathogenesis in mice.
Our previous and present observations that Us3 phosphorylation of vdUTPase Ser-187 was required for optimal vdUTPase enzymatic activity in infected cells (17) and for viral virulence in the mouse CNS (Fig. 2A and C) led us to hypothesize that vdUTPase enzymatic activity regulated viral virulence, in particular in the CNS of mice. To investigate this hypothesis, we examined the replication and pathogenicity of YK759 (vdUTPaseD97A) encoding an enzymatically inactive vdUTPase mutant in which vdUTPase Asp-97 was substituted with alanine and its repaired virus YK760 (vdUTPaseD97A-repair) in mice following intracranial, ocular, and vaginal inoculations. In agreement with the observations with YK751 (vdUTPaseS187A) and its repaired virus YK752 (vdUTPaseΔ/S187A-repair), following intracranial inoculation, there was a significant increase in mortality and progeny virus titers in the CNS of mice inoculated with YK760 (vdUTPaseD97A-repair) compared to mice inoculated with YK759 (vdUTPaseD97A) (Fig. 8A and 9A). In contrast, the level of HSK, vaginal disease and viral replication were similar in the eyes and vaginas of mice inoculated with either YK759 (vdUTPaseD97A) or YK760 (vdUTPaseD97A-repair) (Fig. 8B and C and Fig. 9B and C). These results supported our hypothesis that vdUTPase enzymatic activity specifically regulated viral virulence and replication in the CNS of mice following intracranial inoculation but was not required for viral pathogenicity and replication in the eyes and vaginas of mice following ocular and vaginal inoculation. Furthermore, these results suggested that a currently unidentified vdUTPase activity, unrelated to its enzymatic activity, was critical for viral pathogenicity and replication in peripheral sites of mice following peripheral inoculation.
FIG 8.
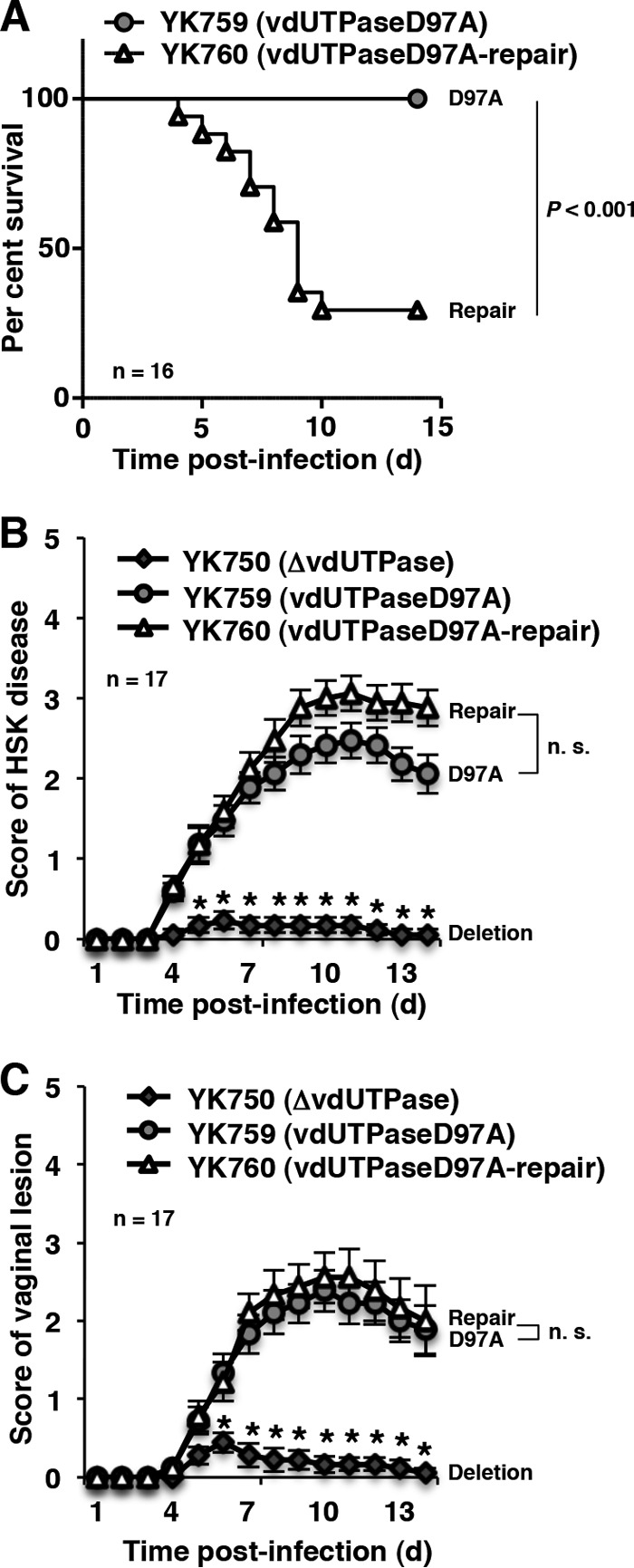
Effect of the enzymatically inactive mutation in vdUTPase on viral pathogenicity in mice. (A) Sixteen 3-week-old female ICR mice were infected intracranially with YK759 (vdUTPaseD97A) and YK760 (vdUTPaseD97A-repair) and monitored for 14 days. (B) Seventeen 5-week-old female ICR mice were ocularly infected with YK750 (ΔvdUTPase), YK759 (vdUTPaseD97A), and YK760 (vdUTPaseD97A-repair) and scored for HSK as described for Fig. 4B. Each data point is the mean ± the standard error of the scores. (C) Seventeen 5-week-old female ICR mice pretreated with Depo-M were infected with YK750 (ΔvdUTPase), YK759 (vdUTPaseD97A), and YK760 (vdUTPaseD97A-repair), and the score of vaginal disease in mice was analyzed as described for Fig. 4C. Each data point is the mean ± the standard error of the scores. P represents the statistical significance value according to the log-rank test (A) or the two-tailed Student t test (B and C). n.s., not significant.
FIG 9.
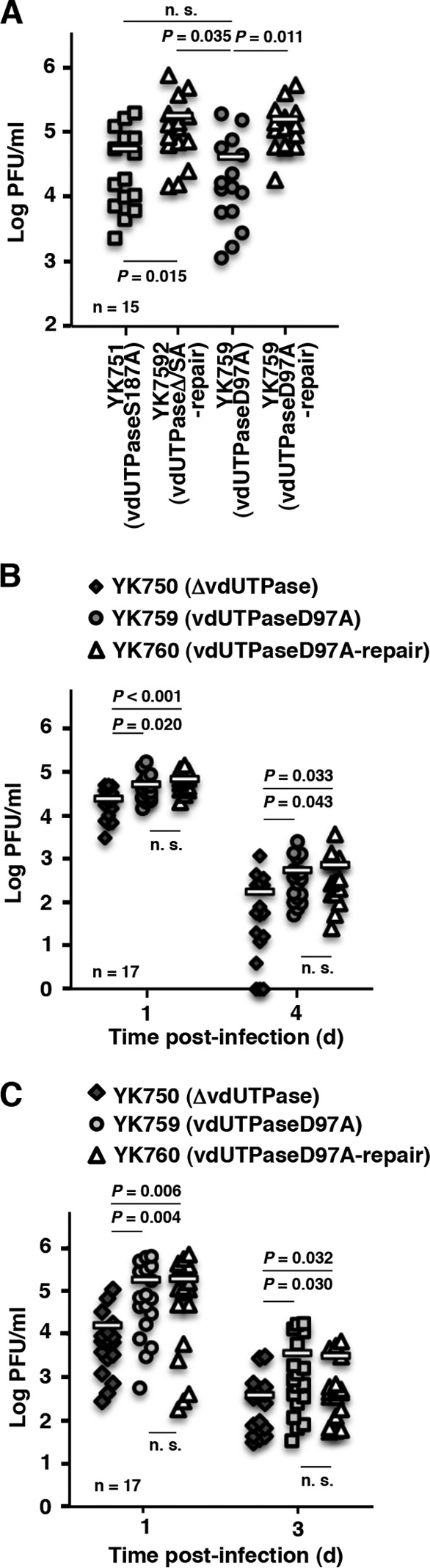
Effect of the enzymatically inactive mutation in vdUTPase on viral replication in mice. (A) Fifteen 3-week-old female mice were infected intracranially with YK751 (vdUTPaseS187A) YK752 (vdUTPaseΔ/SA-repair), YK759 (vdUTPaseD97A), or YK760 (vdUTPaseD97A-repair). At 3 days postinfection, the brains of infected mice were harvested, and virus titers were assayed. Each data point is the virus titer in the brain of one mouse. The horizontal bars indicate the mean for each group. (B) The tear films of infected mice at 1 and 4 days postinfection in the experiment described in Fig. 8B were harvested, and virus titers were assayed. Each data point is the virus titer in the tear film of one mouse. The horizontal bars indicate the mean for each group. (C) The vaginal secretions of infected mice at 1 and 3 days postinfection in the experiment described in Fig. 8C were harvested, and virus titers were assayed. Each data point is the virus titer in the vaginal secretions of one mouse. The horizontal bars indicate the mean for each group. P represents the statistical significance value according to the two-tailed Student t test. n.s., not significant.
DISCUSSION
We previously reported that vdUTPase Ser-187 is a physiological Us3 phosphorylation site in infected cells and that this phosphorylation promoted HSV-1 replication in a manner dependent on cell types and MOIs by regulating the optimal enzymatic activity of vdUTPase (17). However, the significance of Us3 phosphorylation of vdUTPase Ser-187 in viral replication and pathogenicity in vivo was uncertain. In the present study, we have shown that the vdUTPase S187A mutation significantly reduced mortality and viral replication in the brains of mice following intracranial inoculation and that the phosphomimetic substitution at vdUTPase Ser-187 in part restored the wild-type mortality and viral replication. In addition, the Us3 kinase-dead and vdUTPase enzyme-inactive mutations showed the phenotypes similar to the vdUTPase S187A mutation in mice following intracranial inoculation. Collectively, these observations suggested that the regulation of vdUTPase activity by Us3 phosphorylation of vdUTPase Ser-187 was critical for viral replication and virulence in the CNS of mice following intracranial inoculation. This is the first identification of a Us3 substrate involved in Us3 pathogenicity in the CNS of mice following intracranial inoculation. The more striking feature in the present study was that the phosphorylation, as well as the enzymatic activity, of vdUTPase played no role in viral replication and pathogenicity in the eyes and vaginas of mice following ocular and vaginal inoculation, respectively. To our knowledge, HSV proteins or their domains critical for viral replication and virulence in the CNS of mice following intracranial inoculation are, in almost all cases, also involved in pathogenicity in peripheral sites, such as eyes and vaginas, following peripheral (ocular and vaginal) inoculation (7, 10, 11, 15, 27, 41, 44–59). Therefore, this phosphorylation reaction, which promoted the vdUTPase activity, is a unique indication of a specific mechanism involved in HSV-1 replication and virulence in the CNS of mice following intracranial inoculation. We should note that HSV thymidine kinases (TKs) might behave similarly in the phosphorylation and the enzymatic activity of vdUTPase. However, roles of the TKs in viral replication at the periphery have been controversial, although the importance of the TKs in viral replication and virulence in the CNS was well established (48). Thus, several groups reported that the null mutations in the TKs had no obvious effect on viral replication in eyes of mice or guinea pigs following ocular inoculation (46, 60, 61), whereas other groups demonstrated that these mutations significantly impaired viral replication in eyes, ears, or vaginas of mice following ocular, ear, or vaginal inoculation, respectively (49, 62–64).
Our observations raised the interesting question of why this phosphorylation was needed for infection of the CNS of mice but not for infection of peripheral sites. Many viruses, including poxviruses, adenoviruses, D-type retroviruses, and African swine fever virus (ASFV), encode dUTPases in addition to herpesviruses (29, 65–67). In agreement with our recent report that HSV-1 dUTPase activity was required for efficient viral replication in a manner dependent on cell types and MOIs, replication of recombinant ASFV and D-type retroviruses (e.g., equine infectious anemia virus, feline immunodeficiency virus, and caprine arthritis encephalitis virus) carrying mutations in each of the viral dUTPases was severely affected in specific cell types, especially in nondividing cells, whereas replication in actively dividing cells was only minimally decreased (68–72). Based on these observations, it has long been thought, but not proven, that viruses encode a vdUTPase to compensate for low cellular dUTPase activity if that is the situation in their host cells, e.g., in resting and differentiated cells, such as neurons and macrophages, where cellular dUTPase activity has been suggested to be low (70, 73). Therefore, one possible explanation for the unique mechanism mediated by Us3 phosphorylation of vdUTPase for the CNS-specific viral replication and virulence may be that the vdUTPase enzymatic activity was upregulated by Us3 phosphorylation of vdUTPase Ser-187 to compensate for the low cellular endogenous dUTPase activity in the CNS of mice for efficient viral replication and virulence. Further experiments to directly prove this hypothesis, e.g., data that overexpression of cellular dUTPase compensates for the reduction in replication and/or pathogenesis of viruses carrying mutations in vdUTPase in cell cultures and/or in vivo, will be needed to clarify this subject and are in progress in this laboratory.
Us3 phosphorylation of vdUTPase Ser-187 was not essential for vdUTPase activity but appeared to optimize vdUTPase activity in infected cells, based on our recent report that the S187A mutation in vdUTPase only reduced vdUTPase enzymatic activity in infected cells to 68% of that in wild-type virus-infected cells (17). In contrast, the data presented here indicated that this phosphorylation was critical for efficient viral replication and virulence in the CNS of mice following intracranial inoculation. These results suggested that vdUTPase enzymatic activity in infected cells was tightly regulated by Us3 phosphorylation of vdUTPase Ser-187, and this strict regulation was critical for efficient viral replication and virulence in the CNS of mice. dUTPase hydrolysis of dUTP to dUMP and pyrophosphate may have a dual effect on HSV-1 replication in the CNS: the prevention of the misincorporation of dUTP into replicating viral genomes and the promotion of dTTP synthesis (29, 74). In the CNS, composed of mostly nondividing cells, the deoxynucleoside triphosphate (dNTP) pools are limited since cellular genome replication does not take place, and therefore vdUTPase activity might unbalance the dNTP pools. It is well established that appropriately balanced dNTP pools are critical for the maintenance of cell homeostasis (75). It has been reported that an imbalance in dNTP pools can result in cell death, dysfunction of mitochondria, mutation, recombination, enhanced sensitivity to mutagens, and chromosome breakage, exchange, or loss. Thus, we suggest that the Us3 phosphorylation site in vdUTPase may have evolved to tightly regulate vdUTPase enzymatic activity in its host cells, such as the CNS.
We have shown that the vdUTPase null mutation reduced viral replication and pathogenicity in mice, both in the CNS following intracranial inoculation and in peripheral sites following inoculation at these sites. In contrast, S187A and D97A mutations in vdUTPase, which reduced or abolished vdUTPase enzymatic activity, both reduced viral replication and pathogenesis in the CNS of mice following intracranial inoculation, but not in the eyes and vaginas of mice following inoculation in these sites. These results indicated that a currently unidentified activity of the vdUTPase protein, which is unrelated to its enzymatic activity, was critical for viral replication and pathogenesis at peripheral sites of mice. Two recent reports are in agreement with this conclusion. It has been reported that murine gammaherpesvirus-68 (MHV-68) and Kaposi's sarcoma-associated herpesvirus (KSHV) vdUTPases inhibited type I interferon signaling by inducing degradation of the type I interferon receptor protein (76) and cell surface expression of NKp44L, a ligand for natural killer cell activating receptor NKp44 (77), respectively, independent of their enzymatic activity (76, 77). In addition, Epstein-Barr virus (EBV) vdUTPase has been reported to function as a pathogen-associated molecular pattern, to be recognized by Toll-like receptor 2, and to induce expression of several pro- and anti-inflammatory cytokines when unstimulated resting human peripheral blood mononuclear cells and human monocyte-derived macrophages were cultured with purified EBV vdUTPase proteins (78, 79). The biological significance of the immune modulations mediated by KSHV and EBV vdUTPases in the context of viral infection remains to be determined, and this KSHV vdUTPase function has been shown to not be conserved in homologs in other herpesviruses, including HSV-1 and EBV (77). However, it is of interest to investigate whether an HSV-1 vdUTPase activity unrelated to its enzymatic activity is involved in the immune modulations observed with MHV-68 and EBV vdUTPases. Furthermore, HSV-1 vdUTPase is a major virion component (17, 80), indicating that it has a role in the viral replication cycle, perhaps as a structural protein in virion maturation. Further studies to clarify the activity of vdUTPase unrelated to its enzymatic activity are needed and are in progress in our laboratories.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for cientific Research from the Japan Society for the Promotion of Science, a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases and the Global COE Program “Center of Education and Research for the Advanced Genome-Based Medicine: For Personalized Medicine and the Control of Worldwide Infectious Diseases,” a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology of Japan, and grants from the Takeda Science Foundation, the Naito Foundation, and the Tokyo Biochemical Research Foundation.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.McGeoch DJ, Davison AJ. 1986. Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res. 14:1765–1777. 10.1093/nar/14.4.1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frame MC, Purves FC, McGeoch DJ, Marsden HS, Leader DP. 1987. Identification of the herpes simplex virus protein kinase as the product of viral gene Us3. J. Gen. Virol. 68(Pt 10):2699–2704 [DOI] [PubMed] [Google Scholar]
- 3.Chee MS, Lawrence GL, Barrell BG. 1989. Alpha-, beta- and gammaherpesviruses encode a putative phosphotransferase. J. Gen. Virol. 70(Pt 5):1151–1160 [DOI] [PubMed] [Google Scholar]
- 4.Smith RF, Smith TF. 1989. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 63:450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deruelle MJ, Favoreel HW. 2011. Keep it in the subfamily: the conserved alphaherpesvirus Us3 protein kinase. J. Gen. Virol. 92:18–30. 10.1099/vir.0.025593-0 [DOI] [PubMed] [Google Scholar]
- 6.Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J. Virol. 85:1158–1173. 10.1128/JVI.01369-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. 10.1016/0042-6822(88)90417-5 [DOI] [PubMed] [Google Scholar]
- 8.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 Ul31, Ul34, and Us3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952. 10.1128/JVI.76.17.8939-8952.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: Ul34 protein modification and the Us3-Ul34 catalytic relationship. J. Virol. 78:399–412. 10.1128/JVI.78.1.399-412.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 83:11624–11634. 10.1128/JVI.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783. 10.1128/JVI.00103-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber PC, Levine M, Glorioso JC. 1987. Rapid identification of nonessential genes of herpes simplex virus type 1 by Tn5 mutagenesis. Science 236:576–579. 10.1126/science.3033824 [DOI] [PubMed] [Google Scholar]
- 13.Balan P, Davis-Poynter N, Bell S, Atkinson H, Browne H, Minson T. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins Gg, Ge, Gi, or the putative Gj. J. Gen. Virol. 75(Pt 6):1245–1258 [DOI] [PubMed] [Google Scholar]
- 14.Izumi KM, Stevens JG. 1990. Molecular and biological characterization of a herpes simplex virus type 1 (HSV-1) neuroinvasiveness gene. J. Exp. Med. 172:487–496. 10.1084/jem.172.2.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162. 10.1128/JVI.01447-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1 serine/threonine kinase Us3 hyperphosphorylates Irf3 and inhibits beta interferon production. J. Virol. 87:12814–12827. 10.1128/JVI.02355-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J. Virol. 88:655–666. 10.1128/JVI.02710-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Patenode C, Roizman B. 2011. Us3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 108:14632–14636. 10.1073/pnas.1111942108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the Us3 gene mediates posttranslational modification of the phosphoprotein encoded by the Ul34 gene J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein α22 mediated by the Ul13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. U. S. A. 90:6701–6705. 10.1073/pnas.90.14.6701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munger J, Roizman B. 2001. The Us3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of bad and prevents bad-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 98:10410–10415. 10.1073/pnas.181344498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mou F, Forest T, Baines JD. 2007. Us3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470. 10.1128/JVI.00380-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, Roller RJ. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both Ul34 and Us3. J. Virol. 81:10792–10803. 10.1128/JVI.00196-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein Ul47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613. 10.1128/JVI.00845-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250–261. 10.1128/JVI.01451-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24:2627–2639. 10.1101/gad.1978310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191. 10.1128/JVI.00090-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vertessy BG, Toth J. 2009. Keeping uracil out of DNA: physiological role, structure, and catalytic mechanism of dUTPases. Acc. Chem. Res. 42:97–106. 10.1021/ar800114w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sedwick WD, Brown OE, Glickman BW. 1986. Deoxyuridine misincorporation causes site-specific mutational lesions in the lacI gene of Escherichia coli. Mutat. Res. 162:7–20. 10.1016/0027-5107(86)90066-7 [DOI] [PubMed] [Google Scholar]
- 31.Kunz BA, Kohalmi SE. 1991. Modulation of mutagenesis by deoxyribonucleotide levels. Annu. Rev. Genet. 25:339–359. 10.1146/annurev.ge.25.120191.002011 [DOI] [PubMed] [Google Scholar]
- 32.Bessman MJ, Lehman IR, Adler J, Zimmerman SB, Simms ES, Kornberg A. 1958. Enzymatic synthesis of deoxyribonucleic acid. iii. the incorporation of pyrimidine and purine analogues into deoxyribonucleic acids. Proc. Natl. Acad. Sci. U. S. A. 44:633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGeehan JE, Depledge NW, McGeoch DJ. 2001. Evolution of the dUTPase gene of mammalian and avian herpesviruses. Curr. Protein Peptide Sci. 2:325–333 [DOI] [PubMed] [Google Scholar]
- 34.Preston VG, Fisher FB. 1984. Identification of the herpes simplex virus type 1 gene encoding the dUTPase. Virology 138:58–68. 10.1016/0042-6822(84)90147-8 [DOI] [PubMed] [Google Scholar]
- 35.Pyles RB, Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 66:6706–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 39.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 40.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of P53 in herpes simplex virus 1 replication. J. Virol. 87:9323–9332. 10.1128/JVI.01581-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J. Virol. 85:5003–5015. 10.1128/JVI.02314-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison LA, Da Costa XJ, Knipe DM. 1998. Influence of mucosal and parenteral immunization with a replication-defective mutant of HSV-2 on immune responses and protection from genital challenge. Virology 243:178–187. 10.1006/viro.1998.9047 [DOI] [PubMed] [Google Scholar]
- 43.Bell C, Desjardins M, Thibault P, Radtke K. 2013. Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 12:1820–1829. 10.1021/pr301157j [DOI] [PubMed] [Google Scholar]
- 44.Idowu AD, Fraser-Smith EB, Poffenberger KL, Herman RC. 1992. Deletion of the herpes simplex virus type 1 ribonucleotide reductase gene alters virulence and latency in vivo. Antivir. Res. 17:145–156. 10.1016/0166-3542(92)90048-A [DOI] [PubMed] [Google Scholar]
- 45.Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, Coen DM. 1989. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology 173:276–283. 10.1016/0042-6822(89)90244-4 [DOI] [PubMed] [Google Scholar]
- 46.Gordon YJ, Gilden DM, Becker Y. 1983. HSV-1 thymidine kinase promotes virulence and latency in the mouse. Invest. Ophthalmol. Vis. Sci. 24:599–602 [PubMed] [Google Scholar]
- 47.Coen DM. 1994. Acyclovir-resistant, pathogenic herpesviruses. Trends Microbiol. 2:481–485. 10.1016/0966-842X(94)90652-1 [DOI] [PubMed] [Google Scholar]
- 48.Field HJ, Wildy P. 1978. The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J. Hyg. (Lond.) 81:267–277. 10.1017/S0022172400025109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDermott MR, Smiley JR, Leslie P, Brais J, Rudzroga HE, Bienenstock J. 1984. Immunity in the female genital tract after intravaginal vaccination of mice with an attenuated strain of herpes simplex virus type 2. J. Virol. 51:747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandt CR, Kintner RL, Pumfery AM, Visalli RJ, Grau DR. 1991. The herpes simplex virus ribonucleotide reductase is required for ocular virulence. J. Gen. Virol. 72(Pt 9):2043–2049 [DOI] [PubMed] [Google Scholar]
- 51.Ramachandran S, Davoli KA, Yee MB, Hendricks RL, Kinchington PR. 2010. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J. Virol. 84:8811–8820. 10.1128/JVI.00496-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poffenberger KL, Idowu AD, Fraser-Smith EB, Raichlen PE, Herman RC. 1994. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch. Virol. 139:111–119. 10.1007/BF01309458 [DOI] [PubMed] [Google Scholar]
- 53.Perng GC, Thompson RL, Sawtell NM, Taylor WE, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1995. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol. 69:3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science 250:1262–1266. 10.1126/science.2173860 [DOI] [PubMed] [Google Scholar]
- 55.MacLean CA, Robertson LM, Jamieson FE. 1993. Characterization of the Ul10 gene product of herpes simplex virus type 1 and investigation of its role in vivo. J. Gen. Virol. 74(Pt 6):975–983 [DOI] [PubMed] [Google Scholar]
- 56.Smith TJ, Morrison LA, Leib DA. 2002. Pathogenesis of herpes simplex virus type 2 virion host shutoff (Vhs) mutants. J. Virol. 76:2054–2061. 10.1128/jvi.76.5.2054-2061.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka M, Kato A, Satoh Y, Ide T, Sagou K, Kimura K, Hasegawa H, Kawaguchi Y. 2012. Herpes simplex virus 1 Vp22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J. Virol. 86:5264–5277. 10.1128/JVI.06913-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duffy C, Lavail JH, Tauscher AN, Wills EG, Blaho JA, Baines JD. 2006. Characterization of a Ul49-null mutant: Vp22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664–8675. 10.1128/JVI.00498-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pyles RB, Thompson RL. 1994. Evidence that the herpes simplex virus type 1 uracil DNA glycosylase is required for efficient viral replication and latency in the murine nervous system. J. Virol. 68:4963–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobson JG, Ruffner KL, Kosz-Vnenchak M, Hwang CB, Wobbe KK, Knipe DM, Coen DM. 1993. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia. J. Virol. 67:6903–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tenser RB, Miller RL, Rapp F. 1979. Trigeminal ganglion infection by thymidine kinase-negative mutants of herpes simplex virus. Science 205:915–917. 10.1126/science.224454 [DOI] [PubMed] [Google Scholar]
- 62.Efstathiou S, Kemp S, Darby G, Minson AC. 1989. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J. Gen. Virol. 70(Pt 4):869–879 [DOI] [PubMed] [Google Scholar]
- 63.Tenser RB, Ressel S, Dunstan ME. 1981. Herpes simplex virus thymidine kinase expression in trigeminal ganglion infection: correlation of enzyme activity with ganglion virus titer and evidence of in vivo complementation. Virology 112:328–341. 10.1016/0042-6822(81)90638-3 [DOI] [PubMed] [Google Scholar]
- 64.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc. Natl. Acad. Sci. U. S. A. 86:4736–4740. 10.1073/pnas.86.12.4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elder JH, Lerner DL, Hasselkus-Light CS, Fontenot DJ, Hunter E, Luciw PA, Montelaro RC, Phillips TR. 1992. Distinct subsets of retroviruses encode dUTPase. J. Virol. 66:1791–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baldo AM, McClure MA. 1999. Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J. Virol. 73:7710–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McClure MA. 2001. Evolution of the dut gene: horizontal transfer between host and pathogen in all three domains of life. Curr. Protein Pept Sci. 2:313–324. 10.2174/1389203013381062 [DOI] [PubMed] [Google Scholar]
- 68.Turelli P, Petursson G, Guiguen F, Mornex JF, Vigne R, Querat G. 1996. Replication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J. Virol. 70:1213–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Threadgill DS, Steagall WK, Flaherty MT, Fuller FJ, Perry ST, Rushlow KE, Le Grice SF, Payne SL. 1993. Characterization of equine infectious anemia virus dUTPase: growth properties of a dUTPase-deficient mutant. J. Virol. 67:2592–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Payne SL, Elder JH. 2001. The role of retroviral dUTPases in replication and virulence. Curr. Protein Pept. Sci. 2:381–388. 10.2174/1389203013381008 [DOI] [PubMed] [Google Scholar]
- 71.Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH. 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U. S. A. 92:7480–7484. 10.1073/pnas.92.16.7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oliveros M, Garcia-Escudero R, Alejo A, Vinuela E, Salas ML, Salas J. 1999. African swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J. Virol. 73:8934–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 2501–2601 In Knipe DM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott/The Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 74.Shlomai J, Kornberg A. 1978. Deoxyuridine triphosphatase of Escherichia coli: purification, properties, and use as a reagent to reduce uracil incorporation into DNA. J. Biol. Chem. 253:3305–3312 [PubMed] [Google Scholar]
- 75.Kunz BA, Kohalmi SE, Kunkel TA, Mathews CK, McIntosh EM, Reidy JA. 1994. International commission for protection against environmental mutagens and carcinogens: deoxyribonucleoside triphosphate levels—a critical factor in the maintenance of genetic stability. Mutat. Res. 318:1–64 [DOI] [PubMed] [Google Scholar]
- 76.Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, Sun R. 2011. The anti-interferon activity of conserved viral dUTPase ORF54 Is essential for an effective MHV-68 infection. PLoS Pathog. 7:e1002292. 10.1371/journal.ppat.1002292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Madrid AS, Ganem D. 2012. Kaposi's sarcoma-associated herpesvirus Orf54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J. Virol. 86:8693–8704. 10.1128/JVI.00252-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glaser R, Litsky ML, Padgett DA, Baiocchi RA, Yang EV, Chen M, Yeh PE, Green-Church KB, Caligiuri MA, Williams MV. 2006. EBV-encoded dUTPase induces immune dysregulation: implications for the pathophysiology of EBV-associated disease. Virology 346:205–218. 10.1016/j.virol.2005.10.034 [DOI] [PubMed] [Google Scholar]
- 79.Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. 2009. The EBV-encoded dUTPase activates NF-κB through the Tlr2 and Myd88-dependent signaling pathway. J. Immunol. 182:851–859 [DOI] [PubMed] [Google Scholar]
- 80.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]