

ABSTRACT
Anti-HIV-1 envelope glycoprotein (Env) antibodies without broadly neutralizing activity correlated with protection in the RV144 clinical trial, stimulating interest in other protective mechanisms involving antibodies, such as antibody-dependent cell-mediated cytotoxicity (ADCC). Env epitopes targeted by many antibodies effective at mediating ADCC are poorly exposed on the unliganded Env trimer. Here we investigated the mechanism of exposure of ADCC epitopes on Env and showed that binding of Env and CD4 within the same HIV-1-infected cell effectively exposes these epitopes. Env capacity to transit to the CD4-bound conformation is required for ADCC epitope exposure. Importantly, cell surface CD4 downregulation by Nef and Vpu accessory proteins and Vpu-mediated BST-2 antagonism modulate exposure of ADCC-mediating epitopes and reduce the susceptibility of infected cells to this effector function in vitro. Significantly, Env conformational changes induced by cell surface CD4 are conserved among Env from HIV-1 and HIV-2/SIVmac lineages. Altogether, our observations describe a highly conserved mechanism required to expose ADCC epitopes that might help explain the evolutionary advantage of downregulation of cell surface CD4 by the HIV-1 Vpu and Nef proteins.
IMPORTANCE HIV-1 envelope epitopes targeted by many antibodies effective at mediating antibody-dependent cell-mediated cytotoxicity (ADCC) are poorly exposed on the unliganded envelope trimer. Here we investigated the mechanism of exposure of these epitopes and found that envelope interaction with the HIV-1 CD4 receptor is required to expose some of these epitopes. Moreover, our results suggest that HIV-1 CD4 downregulation might help avoid the killing of HIV-1-infected cells by this immune mechanism.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) entry, mediated by the trimeric viral envelope glycoproteins (Env) complex, is a critical step of the viral infectious cycle. Env trimer is the only virus-specific antigen present at the surface of viral particles; as such, it is highly exposed and thus elicits neutralizing and nonneutralizing antibodies. The mature HIV-1 Env trimer is derived from proteolytic cleavage of a trimeric gp160 precursor (1, 2) and is composed of the exterior gp120 and transmembrane gp41 subunits. The gp120 is retained on the trimer via noncovalent interactions with the gp41 ectodomain (3–5). The gp120 glycoprotein is responsible for interactions with the initial receptor, CD4 (6, 7). CD4 binding triggers conformational changes in gp120 that promote its interaction with one of the chemokine receptors, CCR5 or CXCR4 (8–15). CD4 binding also induces conformational changes within the HIV-1 Env trimer that result in the exposure of a helical heptad repeat (HR1) segment of the gp41 ectodomain (16–19). Eventually, the conformational transition of the gp41 ectodomain into a six-helix bundle composed of the HR1 and HR2 heptad repeat regions results in the fusion of the viral and target cell membranes (20–22). As the major viral determinant recognized by anti-HIV-1 antibodies, the HIV-1 Env trimer represents a likely candidate for a vaccine immunogen. Interestingly, there is increasing evidence suggesting a role of Fc-mediated effector function in controlling or preventing HIV-1 transmission. Studies in macaques infected with simian immunodeficiency virus (SIV) show an inverse correlation between Fc-mediated effector functions and viral loads or rate of disease progression (23–25). Of note, this inverse correlation was also observed in several studies on HIV-1-infected individuals (26–30, 64). It has been recently suggested that antibody-dependent cellular cytotoxicity (ADCC) responses apply significant immune pressure on HIV-1 (31), highlighting the potential impact of ADCC on viral progression. Recently, a potential correlation between high levels of ADCC-mediating antibodies and HIV-1 acquisition in the RV144 trial was suggested for a subset of individuals with low plasma IgA anti-Env antibody levels (32, 33). Indeed, Env-specific plasma IgA/IgG ratios were shown to be higher in infected than in uninfected RV144 vaccine recipients (34). Accordingly, efficient ADCC-inducing monoclonal antibodies (MAbs) were isolated from a subset of RV144 subjects (33). Therefore, ADCC-mediating antibodies may contribute to the partial protection observed in the RV144 trial, stimulating renewed interest in the mechanism of recognition of these antibodies (33, 35). Here we investigated whether interaction of CD4 and Env modulates some of the ADCC-mediating antibody recognition.
MATERIALS AND METHODS
Cells.
The 293T human embryonic kidney and HOS cell lines (obtained from ATCC and the NIH AIDS Research and Reference Reagent Program, respectively) were grown at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 5% fetal bovine serum (FBS) (Sigma) and 100 μg/ml of penicillin-streptomycin (Wysent). CEM.NKR cells (obtained from David Evans) (25) were grown at 37°C and 5% CO2 in RPMI 1640 medium (Gibco) containing 10% fetal bovine serum (R10).
Ficoll density gradient-isolated and cryopreserved human peripheral blood mononuclear cells (PBMCs) from healthy donors (HIV and hepatitis C virus [HCV] seronegative), who gave written informed consent under research protocols approved by the CRCHUM, were thawed and kept at 37°C and 5% CO2 in RPMI 1640 containing 10% FBS and 100 μg/ml penicillin-streptomycin for at least 16 h before subsequent experiments. CD4+ T cells were isolated using a negative-isolation kit (EasySep human CD4+ T cell enrichment kit; Stemcell) according to manufacturer's specifications and were then activated for 48 h with 5 μg/ml phytohemagglutinin (PHA) (Sigma) and 100 U/ml interleukin-2 (IL-2) (NIH AIDS Reagent). Activated CD4+ T cells were maintained in culture in the presence of 100 U/ml IL-2 for another 48 h before infection.
Plasmids and site-directed mutagenesis.
Mutations were introduced individually into the previously described pSVIIIenv vector expressing HIV-1YU2 or pcDNA3.1 expressing the codon-optimized HIV-1JRFL envelope glycoproteins (5, 36) or into a previously reported pcDNA3.1 human CD4 expressor (37). Site-directed mutagenesis was performed using the QuikChange II XL site-directed mutagenesis protocol (Stratagene). When indicated, a stop codon was introduced to replace the codon for Gly 711, truncating the cytoplasmic tail (ΔCT) and enhancing cell surface expression of selected HIV-1YU2 and HIV-1JRFL envelope glycoproteins. The HIV-1JRFL Env-eYFP expressor was generated by inserting the codon-optimized sequence of HIV-1JRFL Env (residues 1 to 711) at the N terminus of enhanced yellow fluorescent protein (eYFP) (from BD Clontech). The CD4-eCFP fusion protein was previously described (38). Transmitted/founder (T/F) envelope expressors from clades C (C1086) (39–41) and D (190049) were previously described (42), HIV-2 7312A (43, 44) and SIV (45) envelope expressors were previously described. The ADA Env was introduced into the pNL4.3 infectious molecular clone (46) through insertion of the SalI-BamHI fragment from NLHXADA (47). Subsequently, the BamHI-GFP-IRES.Nef-XhoI portion of the HxBru.ADA.GFP.IRES.Nef− construct (48) was transferred to the NL4.3 ADA intermediate to generate NL4.3.ADA.GFP.IRES.Nef. The isogenic NL4.3.ADA.GFP.IRES.Nef− Vpu− variant was generated by overlapping PCR using BamHI and SalI cloning sites and NL4.3.ADA.GFP.IRES.Nef and NL4.3Udel (49) as templates, respectively. An XhoI frameshift was made to the Nef sequence of NL4.3.ADA.GFP.IRES.Nef to generate NL4.3.ADA.GFP.IRES.Nef− and of NL4.3.ADA.GFP.IRES.Nef Vpu− to make NL4.3.ADA.GFP.IRES Nef− Vpu−.
Antibodies and ligands.
Anti-HIV-1 gp120 monoclonal antibodies directed against the inner domain (A32 and C11) were previously described (50, 51). Antibodies directed against the outer domain (2G12 and PGT121), CD4-induced epitopes (17b, 412d, and 48d), the CD4-binding site (VRC01, VRC03, and b12), and quaternary-dependent epitopes (PG9 and PG16) were obtained from Peter Kwong (VRC, NIAID), Dennis Burton (Scripps), and IAVI. The monoclonal antibody against a CD4-induced epitope on HIV-2 Env (1.4H) was previously reported (52). The anti-cluster A Abs (L9-i1, L9-i2, N5-i5, N12-i3, and N26-i1) were previously reported (35). The A32 Fab and the A32-blockable antibodies isolated from RV144 vaccinees (CH29, CH38, CH40, CH51, CH52, CH54, CH55, CH57, CH77, CH80, CH81, CH89, CH91, CH92, and CH94) were recently described (33). CD4-Ig is a fusion protein in which the N-terminal two domains of CD4 are linked to the Fc component of immunoglobulin G (53). The monoclonal antibody anti-CD4 OKT4 (BioLegend) binds to the D3 domain of CD4 and was used to measure cell surface levels of CD4. The secondary goat anti-mouse and anti-human antibodies coupled to Alexa Fluor 594 and 647 (Invitrogen), respectively, were used in flow cytometry experiments.
Viral production and infection of T cells.
Vesicular stomatitis virus G (VSVG)-pseudotyped NL4.3 green fluorescent protein (GFP) ADA-based viruses were produced by calcium phosphate transfection of 293T cells with our panel of pNL4.3-GFP-ADA-based HIV-1 proviral vectors and VSVG-encoding vector. Two days after transfection, cell supernatants were harvested and concentrated by ultracentrifugation for 1 h at 29,000 rpm on a 20% sucrose cushion. Pellets were harvested in fresh DMEM, and aliquots were stored at −80°C until use. These viruses were then used to infect approximately 20% of CEM.NKR and primary CD4+ T cells by spin infection at 300 × g for 1 to 2 h in 96-well plates at 7°C.
Cell surface staining and ADCC measurement by flow cytometry.
For cell surface staining, CEM.NKR or primary CD4+ T cells were incubated for 30 min at room temperature (RT) 48 h postinfection with 1 μg/ml OKT4, 2G12, A32, CH54, CH94, and anti-cluster A-recognizing Abs in phosphate-buffered saline (PBS). Cells were then washed once with PBS and stained with 1 μg/ml goat anti-mouse (AF-594) and anti-human (AF-647) secondary antibodies for 20 min in PBS. After one more PBS washing, cells were fixed in a 2% PBS-formaldehyde solution. For evaluation of A32-mediated antibody-dependent cellular cytotoxicity (ADCC), CEM.NKR infected cells were stained with viability (AquaVivid; Invitrogen) and cellular (eFluor670; eBiosciences) markers for 20 min and then washed twice in R10 (Gibco). Target cells were then mixed with PBMC effectors cells at an effector/target (E/T) ratio of 10:1 in 96-well V-bottom plates (Corning) and preincubated for 5 min at RT before addition of 5 μg/ml of the monoclonal A32 antibody. After 15 min of incubation at RT, cocultures were centrifuged for 1 min at 300 × g and incubated at 37°C for 6 h before being fixed in a 2% PBS-formaldehyde solution. Samples were analyzed on an LSRII cytometer (BD Biosciences), and data analysis was performed using FlowJo vX.0.6 (Tree Star). The percentage of cytotoxicity was calculated with the following formula: (percentage of GFP-positive cells in T plus E) − (percentage of GFP-positive cells in T plus E plus A32)/(percentage of GFP-positive cells in T). Figure S3 in the supplemental material shows the gating strategy and formula allowing the calculation of A32-mediated cytotoxicity.
Cell-based ELISA.
Detection of trimeric Env on the surface of HOS cells was performed by cell-based enzyme-linked immunosorbent assay (ELISA), as described previously (54). Briefly, HOS cells were seeded in 96-well plates (2 × 104 cells per well) and transfected the next day with 150 ng of envelope expressors or proviruses together with 1.7, 3.5, or 7 ng per well of a pcDN3.1 vector expressing human wild-type or mutant CD4 molecules. When the pSVIII envelope expressor was used, it was cotransfected with 0.01 μg of a Tat-expressing plasmid per well, using the standard polyethylenimine (PEI) (Polyscience Inc., PA, USA) transfection method. Two days later, cells were washed twice with blocking buffer (10 mg/ml nonfat dry milk, 1.8 mM CaCl2, 1 mM MgCl2, 25 mM Tris [pH 7.5], and 140 mM NaCl) and then incubated for 1 h at RT with 20 nM CD4-Ig or anti-HIV-1, HIV-2/SIV Env monoclonal antibodies. All ligands were diluted in blocking buffer. A horseradish peroxidase (HRP)-conjugated antibody specific for the Fc region of human IgG (Pierce) was then incubated with the samples for 45 min at RT. For all conditions, cells were washed 5 times with blocking buffer and 5 times with washing buffer. HRP enzyme activity was determined after the addition of 30 μl per well of a 1:1 mix of Western Lightning oxidizing and luminol reagents (Perkin-Elmer Life Sciences). Light emission was measured with an LB 941 TriStar luminometer (Berthold Technologies).
Immunoprecipitation of envelope glycoproteins.
For pulse-labeling experiments, 3 × 105 293T cells were transfected by the calcium phosphate method with codon-optimized vectors expressing the HIV-1YU2 envelope glycoprotein variants (5, 54). One day after transfection, cells were metabolically labeled for 16 h with 100 μCi/ml [35S]methionine-cysteine ([35S] protein labeling mix; Perkin-Elmer) in Dulbecco's modified Eagle's medium lacking methionine and cysteine and supplemented with 5% dialyzed fetal bovine serum. Precipitation of radiolabeled HIV-1YU2 envelope glycoproteins from medium was performed with various amounts of A32 for 1 h at 37°C in the presence of 50 μl of 10% protein A-Sepharose (American BioSciences).
Statistical analyses.
Statistics were analyzed using GraphPad Prism version 6.01. Unpaired t tests were used to test the significance of differences between means.
RESULTS
Exposure of Env ADCC-mediating epitopes by coexpressed CD4.
It was recently reported that recipients of the ALVAC-HIV/AIDSVAX B/E vaccine in the RV144 trial induced ADCC responses mediated by antibodies that were competed by the A32 Fab fragment (33); these results indicate that these ADCC-mediating antibodies may recognize an Env epitope similar to or overlapping the A32 epitope. The A32 antibody recognizes a discontinuous epitope on the surface of the inner domain of the gp120 exterior Env (65). Interestingly, we observed that the interaction of A32, C11 (an antibody that recognizes the gp120 β-sandwich and N and C termini) (50, 51), RV144 MAbs (33), and the recently described anti-cluster A class of Abs (35) with Env is greatly increased upon coexpression of the CD4 receptor in a dose-dependent manner (see Materials and Methods) (Fig. 1A to D). We interpreted this observation as an indication that this increased antibody binding was dependent on the ability of Env and CD4 to interact, because changes in the HIV-1 gp120 CD4-binding site (D368A) or in the gp120-binding site (F43H) of CD4 that are known to decrease gp120-CD4 interaction (55, 56) decreased the exposure of epitopes recognized by those antibodies (Fig. 1A to D). For some RV144 antibodies (CH29 and CH38), expression of the CD4 F43H variant did not decrease Env recognition as much as for A32. This could be due to the fact that this CD4 variant, although it decreases Env interaction, it does not completely abrogate CD4/Env interaction (55), and therefore a suboptimal Env-CD4 interaction might be sufficient to expose epitopes recognized by these antibodies. Expression levels of Env at the cell surface were not affected by the coexpression of the wild-type or mutated (F43H) CD4 receptor (see Fig. S1 in the supplemental material), indicating that the observed differences in binding were not a result of variable levels of surface Env expression.
FIG 1.

Interaction of HIV-1 Env with coexpressed CD4 induces conformational changes that expose inner domain as well as complex CD4-induced epitopes. (A to D) Interaction of coexpressed CD4 with HIV-1YU2ΔCT Env enhances recognition by A32 (A), C11 (B), ADCC-mediating antibodies isolated from RV144 trial recipients (C) (33), and cluster A-recognizing antibodies (D) (35). Epitope exposure requires CD4-gp120 interaction as shown by decreased recognition of these epitopes with either a mutant of CD4 (F43H) with decreased capacities to interact with gp120 or a CD4-binding site Env variant (D368A). (E and F) Coexpression of Env and CD4 also induces conformational changes in Env that enhance CD4i epitopes (17b and 412D) (E) and decrease recognition by quaternary-dependent Abs such as PG9 and PG16 (F). Data shown are representative of those obtained in at least 3 independent experiments performed in triplicate (mean ± standard deviation [SD]). Env signals were normalized to that obtained with the gp120 outer domain-recognizing antibody 2G12 (A, B, C, and D), and this ratio was normalized to the absence of coexpressed CD4 for panels E and F. −, absence of CD4. The increasing blue bar indicates a stepwise increase in the amount of CD4 expressor being transfected. wt, wild type.
These conformational changes were not limited to the exposure of inner domain epitopes but also resulted in the exposure of complex CD4i (17b and 412d) epitopes comprising elements of the outer domain and bridging sheet of gp120 Env (Fig. 1E). The coexpression of CD4 apparently promoted the opening of the Env trimer, as indicated by decreased recognition by quaternary-dependent Abs such as PG9 and PG16 (57) (Fig. 1F). These conformational changes were dramatically decreased by introducing the F43H-binding site mutation in CD4, indicating that proper Env-CD4 interaction is required for these changes to occur. Meaningfully, cell surface CD4 competed for ligands that recognize the CD4-binding site such as CD4-Ig, b12, VRC03, and VRC01 (Fig. 2), suggesting that CD4 recognizes its binding site in a manner similar to the recognition of Env in the context of viral particles.
FIG 2.
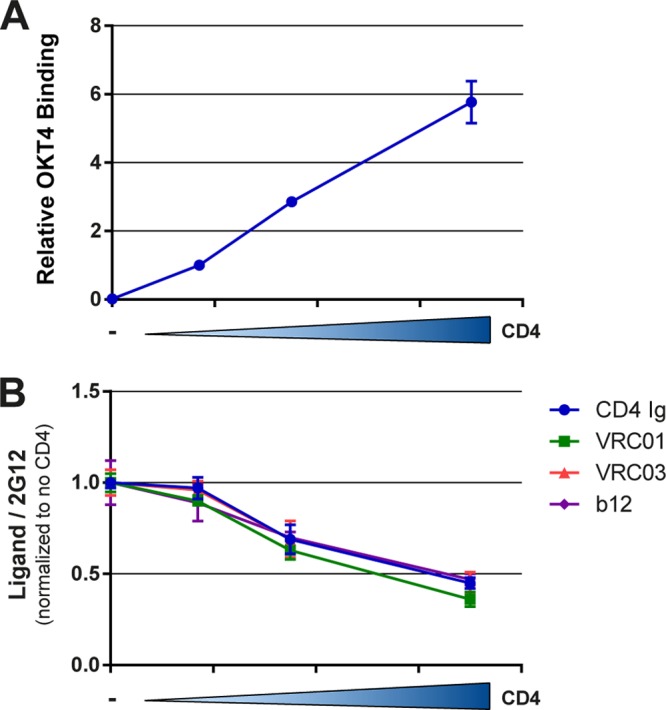
Coexpressed CD4 competes for ligands that recognize the Env CD4-binding site. Cells expressing HIV-1YU2ΔCT Env together with increasing concentrations of human CD4 were stained by the anti-CD4 OKT4 antibody (A) or the CD4-binding site ligands CD4-Ig, b12, VRC03, and VRC01 (B), using the cell-based ELISA described in Materials and Methods. Data are representative of those obtained in at least three independent experiments performed in triplicate (mean ± SD). Signals were normalized to that obtained with the gp120 outer domain-recognizing antibody 2G12 in the absence of coexpressed CD4.
The transition of Env to the CD4-bound conformation is required for efficient interaction with ADCC-mediating antibodies.
Consistent with the necessity that Env interacts with cell surface CD4 to expose CD4i epitopes, these conformational changes required the ability of Env to reach the CD4-bound conformation. Indeed, a mutation (H66A) in layer 1 of the gp120, which is known to impede the transition to the CD4-bound conformation (5, 54), decreased the CD4-induced exposure of A32 and C11 epitopes. Conversely, an S375W substitution, which fills the Phe43 cavity and predisposes to a CD4-bound conformation (58), enhanced exposure of these epitopes and was sufficient to revert the phenotype of the layer 1 variant (Fig. 3). Thus, the ability of Env to transition from the unbound to the CD4-bound conformation is a prerequisite to expose inner domain ADCC epitopes.
FIG 3.
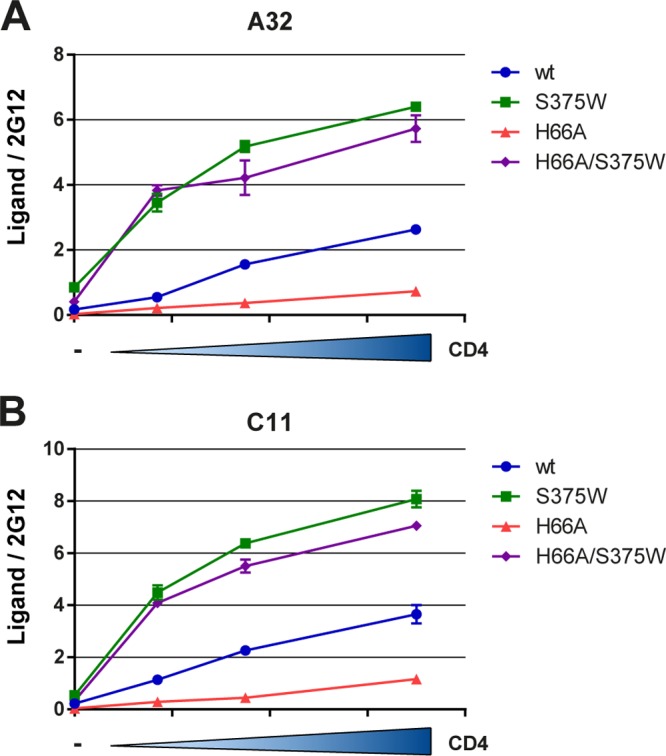
Conformational changes induced by coexpressed CD4 require Env to transit to the CD4-bound conformation. The HIV-1YU2ΔCT layer 1 (H66A) Env variant with a decreased propensity to sample the CD4-bound conformation (5, 54) exhibits decreased exposure of A32 (A) and C11 (B) epitopes upon coexpression of CD4. However, a gp120 change that fills the Phe43 cavity and favors a conformation closer to the CD4-bound conformation (58) enhances the CD4-induced exposure of these epitopes and is sufficient to revert the phenotype of the layer 1 variant. Data shown are representative of those obtained in at least 3 independent experiments performed in triplicate (mean ± SD). Signals were normalized to that obtained with the gp120 outer domain-recognizing antibody 2G12.
Of note, Env conformational changes induced by coexpressed CD4 were observed in Envs from laboratory-adapted, primary, and transmitted/founder (T/F) HIV-1 isolates as well as Envs from the HIV-2/SIV lineage (Fig. 4), indicating that this mechanism is highly conserved among primate immunodeficiency virus Envs. In addition, and consistent with previous reports indicating that CD4 could interact with Env within the endoplasmic reticulum (ER) (59, 60), we observed that Env-CD4 interaction occurs within the same cell, as evaluated by fluorescence resonance energy transfer (FRET) analysis (see Fig. S2 in the supplemental material). Moreover, we obtained evidence suggesting that CD4 coexpression results in the formation of Env-CD4 complexes at the cell surface, since these complexes are recognized by the anti-cluster A N26-i1 antibody (Fig. 1D), which selectively binds gp120-CD4 complexes (see Fig. S4 in the supplemental material).
FIG 4.

Envelope conformational changes induced by coexpressed CD4 are conserved among HIV-1 and HIV-2/SIVmac Env. Laboratory-adapted HIV-1HxBc2 (A), primary HIV-1JRFL (B), HIV-1ADA (C), transmitted/founder HIV-1 clades C (C1086) (D) and D (190049) (E), and HIV-27312 (F) and SIVmac239 (G) envelope glycoprotein expressors were transfected into HOS cells together with increasing concentrations of a human CD4 expressor. At 48 hours posttransfection, Env conformation was assessed by cell-based ELISA with antibodies A32 and C11 recognizing the HIV-1 gp120 inner domain, as described in Materials and Methods. For HIV-2 and SIVmac239 envelopes, the recently described CD4i (1.4H) antibody was used (52). Data are representative of those obtained in at least three independent experiments performed in triplicate (mean ± SD). Signals were normalized to that obtained with the gp120 outer domain-recognizing antibody 2G12 or PGT121 (for C1086) for HIV-1 Env. For HIV-2 and SIVmac239 Envs, signals were normalized to that obtained with serum from SIV-infected macaques.
HIV-1 accessory proteins Nef and Vpu prevent the exposure of epitopes recognized by anti-Env ADCC-mediating antibodies at the surface of infected cells.
HIV-1 accessory proteins Nef and Vpu are known to decrease cell surface levels of CD4 (68, 69). We therefore asked whether these proteins could indirectly affect Env conformation by modulating CD4 levels at the cell surface. We first evaluated whether Env conformational changes induced by CD4 were observed when the envelope glycoproteins were expressed by replication-competent proviruses in our cell-based ELISA. As expected, Env conformational changes depended on proper Env-CD4 interactions, since the D368A Env mutant expressed in the proviral context was unable to be recognized by A32 or C11 despite high levels of cell surface CD4 (Fig. 5). Notably, proviruses lacking the ability to downregulate CD4, due to deletions of their viral accessory proteins Nef and Vpu, presented Envs with higher exposure of the A32 and C11 epitopes (Fig. 5). We then asked whether Env conformational changes at the surface of T cells could be modulated by cell surface levels of CD4. We therefore infected a T cell line (CEM-NKR) (25) with viruses lacking nef, vpu, or nef and vpu. While deletion of vpu alone modestly affected cell surface CD4 levels, nef deletion had a more pronounced effect (Fig. 6A). Nef and Vpu are known to act together, through different cellular mechanisms, to decrease cell surface levels of CD4 (68, 69). Accordingly, deletion of both genes impaired the ability of HIV-1 to downregulate CD4 to extents that were not achieved by deleting nef or vpu alone. HIV-1-mediated CD4 downregulation was completely abrogated only when, in addition to deleting nef and vpu, the ability of Env to interact with CD4 was decreased by the D368A mutation. This is in agreement with the notion that Env-CD4 interaction plays a role in CD4 downregulation (60). Deletion of the vpu gene also resulted in enhanced levels of Env at the cell surface as measured by the outer domain-specific 2G12 antibody (Fig. 6B). In addition to its role in CD4 degradation, Vpu also antagonizes a restriction factor, Tetherin/BST-2, which normally inhibits retroviral release (66, 70). Therefore, viruses produced in the absence of Vpu remain trapped at the cell surface by Tetherin/BST-2 and likely account for the observed enhancement of 2G12 staining.
FIG 5.
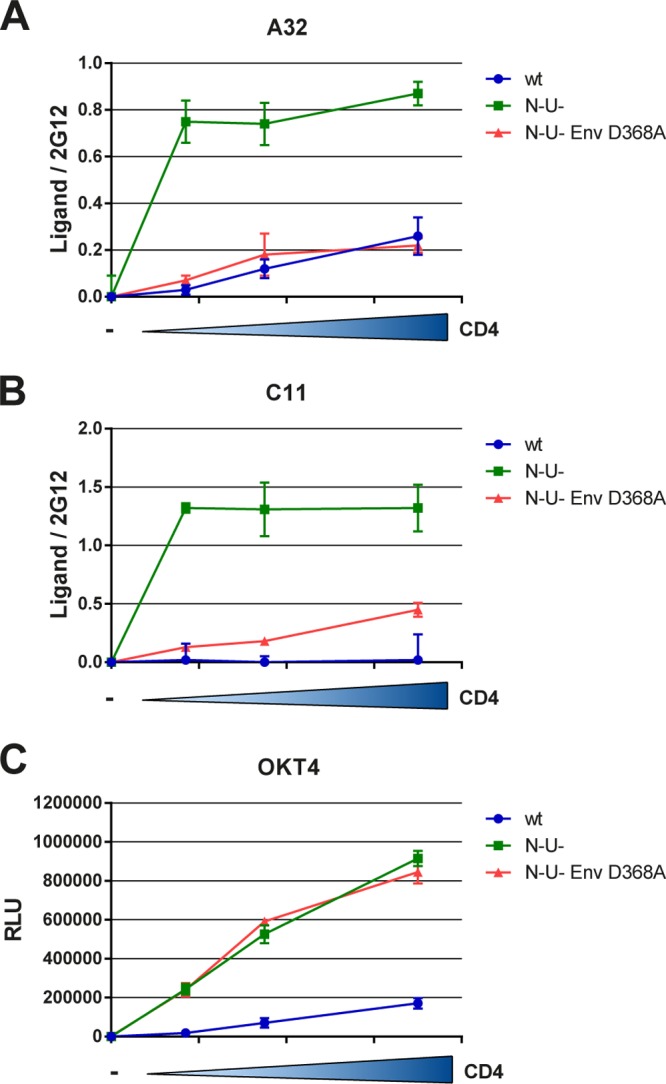
Env conformational changes induced by coexpressed CD4 are conserved when Env is expressed within replication-competent proviruses and are indirectly modulated by Nef and Vpu proteins. pNL4.3 GFP ADA and pNL4.3 GFP ADA Vpu− Nef− were transfected into permissive, BST2-free HOS cells (66) together with increasing concentrations of a human CD4 expressor. At 48 hours posttransfection, Env conformation was assessed by cell-based ELISA with A32 (A) and C11 (B) Abs, as described in Materials and Methods. Importantly, Env conformational changes require CD4-gp120 interaction, as shown by decreased recognition by these Abs of an Env variant with a change (D368A) in the CD4-binding site. (C) Anti-CD4 OKT4 antibody was used to monitor levels of CD4. Data are representative of those obtained in at least three independent experiments performed in triplicate ± SD. Signals were normalized to that obtained with the gp120 outer domain-recognizing antibody 2G12 (A and B).
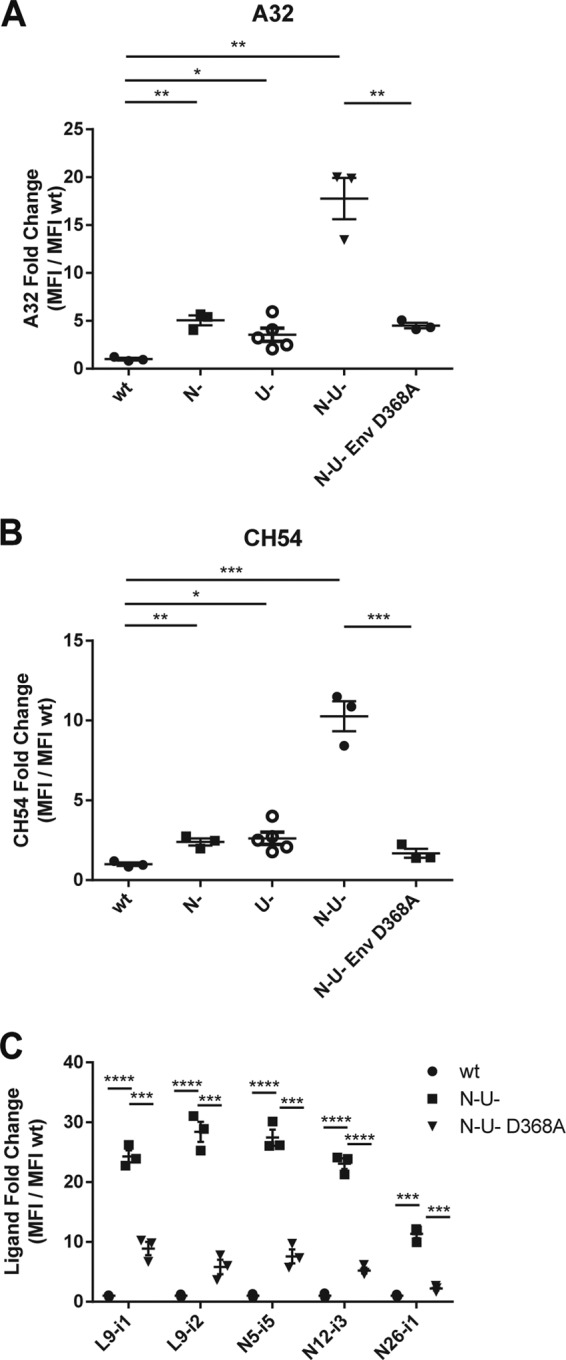
FIG 6.

Env conformational changes induced by surface CD4 increase susceptibility of HIV-infected cells to antibody-dependent cellular cytotoxicity. (A to D) CEM.NKR cells infected with VSV-G pseudotyped NL4.3 GFP ADA either wild-type (wt), Nef− (N−), Vpu− (U−), or Nef− Vpu− (N− U−) encoding wt or D368A Env were stained against surface CD4 (OKT4 Ab) (A) or Env 2G12 (B), A32 (C), and CH54 (D) at 48 h postinfection and analyzed by flow cytometry as described in Materials and Methods. Signals were normalized to the mean signal of the wt virus (fold change) for Env epitopes and to the mock control for surface CD4. (E) Alternatively, the susceptibility to A32-mediated lysis by PBMC effector cells of those cells was analyzed by flow cytometry as described in Materials and Methods. (F) Preincubating with the Fab fragment of A32 prevented A32-mediated lysis. (G) Susceptibility to A32-, CH54-, CH94-, L9-i1-, L9-i2-, N5-i5-, N12-i3-, and N26-i1-mediated lysis by PBMC effector cells of wt versus N-U-infected CEM.NKR cells was analyzed as described for panel E. Data shown are the results of at least three independent infections ± standard error of the mean (SEM). ADCC was measured using PBMCs from three different healthy donors using the gating and formula presented in Fig. S3 in the supplemental material. Statistical significance was tested using an unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
We hypothesized that an adequate level of CD4 and Env at the cell surface must be achieved and, in addition, Env must be able to engage CD4 efficiently in order to detect Env conformational changes with inner domain-recognizing Abs such as CH54 and A32. Indeed, deletion of nef and vpu alone resulted in a significant but only modest increase in A32 and CH54 staining (Fig. 6C and D). However, when both genes were deleted in combination, both CH54 and A32 staining were significantly enhanced. Moreover, this enhancement depended on the ability of Env to engage CD4, because CH54 or A32 staining of cells infected with a nef− vpu− virus encoding the D368A Env variant was decreased (Fig. 6C and D) despite having similar levels of Env at the cell surface as measured by 2G12 (Fig. 6B). Importantly, infection of activated primary CD4+ T cells from three different healthy donors with our panel of viruses resulted in a similar pattern of A32, CH54, and anti-cluster A antibody cell surface staining (Fig. 7), supporting the hypothesis that efficient interaction of Env ADCC-mediating Abs requires a threshold of CD4 and Env at the surface of infected cells as well as their ability to interact together.
FIG 7.
Nef and Vpu prevent the exposure of A32-like epitopes at the surface of infected primary CD4+ T cells. PHA-activated CD4+ T cells isolated from PBMCs of three healthy donors were infected with VSV-G-pseudotyped NL4.3 GFP ADA, either wild type (wt), Nef− (N−), Vpu− (U−), or Nef− Vpu− (N− U−), encoding wt or D368A Env and were stained with A32 (A), CH54 (B), and anti-cluster A (C) Abs at 48 h postinfection and analyzed by flow cytometry as described in Materials and Methods. Signals were normalized to the mean signal of the wt virus (fold change). Data shown are the results of at least three independent infections ± SEM. Statistical significance was tested using an unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
We then asked whether the observed enhancement of A32 staining at the surface of HIV-1-infected cells correlated with enhanced susceptibility to ADCC mediated by PBMCs from healthy individuals. While deletion of nef or vpu only modestly enhanced A32 recognition at the surface of HIV-1-infected cells (Fig. 6C), it resulted in a significant increase in ADCC (Fig. 6E) suggesting that a low threshold of ADCC-mediating Abs recognition at the surface of HIV-1-infected cells is sufficient to sensitize cells to ADCC. Interestingly, the ability of PBMCs to mediate ADCC was at its maximum only when they were in contact with cells infected with the nef− vpu− virus (Fig. 6E). This was also observed for RV144 CH54, CH94, and the anti-cluster A class of antibodies, indicating that this result is not limited to A32 (Fig. 6G). Remarkably, the interaction between Env and CD4 was critical for A32-mediated ADCC because cells infected with the nef− vpu− virus but coding for Env D368A were significantly less susceptible to be killed by the same PBMCs. Of note, the observed A32-mediating killing was specific to A32, since it was completely blocked by preincubation of the target cells with an A32 Fab fragment (Fig. 6F).
The A32 epitope is potentially accessible in the CD4-bound Env trimer.
Finally, we asked how A32 could recognize Env at the surface of infected cells. A recent report (35) supported our previous observations indicating that A32 interaction with HIV-1 gp120 was modulated by the inner domain layers 1 and 2 (5). Here we expanded this analysis by analyzing the contribution of the recently described layer 3 (5, 54, 61). Interestingly, two highly conserved residues located at the interface between layers 2 and 3 (T248, W479) also modulated A32 interaction with gp120. Notably, these residues were shown to be critical for the transition of Env to the CD4-bound conformation (54). This is in agreement with the notion that the epitope recognized by A32, and perhaps by other inner domain-directed Abs such as RV144 Abs, are occluded in the unliganded trimer and must be exposed by cell surface CD4 in order to be recognized efficiently (Fig. 8).
FIG 8.
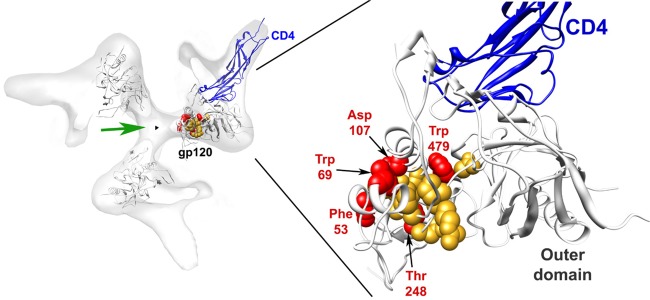
The A32 epitope is potentially accessible in the CD4-bound Env trimer. The cryoelectron tomographic map of the CD4-bound and 17b Fab-bound HIV-1 Env trimer (EMDB 5020) (67) is viewed from the perspective of the target cell. The density associated with the 17b antibody Fab has been removed for clarity. The Env trimer axis is designated with a black triangle. Three CD4-bound gp120 cores (PDB 3DNO) were fit to the density map, and the CD4-bound gp120 with complete N and C termini (PDB 3JWO) (61) was aligned with one subunit. On this subunit, CD4 domains 1 and 2 are shown (blue ribbon). The gp120 residues implicated by mutagenesis in binding the A32 antibody in this study and a previous study (5) are depicted in CPK mode (red, significant effects on A32 binding; orange, moderate effects on binding). The hypothesized angle of approach of A32-like antibodies that mediate ADCC is represented by the green arrow.
DISCUSSION
ADCC utilizes the IgG Fc-gamma receptor (FcγR, FCγRIIIa, or CD16a) expressed on natural killer (NK) cells but also in other immune cell types (e.g., macrophages and neutrophils) as a means of bringing these cells into contact with antibody-coated cells (62). The Fab portion of the antibody binds the antigen at the surface of the target cell, and the Fc portion binds FcγR on the effector cells. Because FcγR is able to form a complex with the ITAM-containing CD3ξ and/or FCεRI, the interaction of FcγR with cell-bound IgG Abs can result in the activation of these cells to mediate cytolysis by releasing perforin and granzymes (63). Interestingly, there is increasing evidence supporting a role of Fc-mediated effector function in controlling or preventing HIV-1 transmission. Studies in macaques infected with simian immunodeficiency virus (SIV) show an inverse correlation between Fc-mediated effector functions and viral loads or decreased disease progression (23–25). Of note, this inverse correlation was also observed in several studies with HIV-1-infected individuals (26–30, 64). Importantly, it has been recently suggested that antibody-dependent cellular cytotoxicity (ADCC) responses apply significant immune pressure on HIV-1 (31), highlighting the potential impact of ADCC on viral progression. Recently, a potential correlation between high levels of ADCC-mediating Abs and HIV-1 acquisition in the RV144 trial was identified for a subset of individuals presenting low plasma IgA anti-Env antibody levels (32, 33). Accordingly, high plasma levels of IgA Abs specific for the C1 region of Env were shown to block the binding and effector function of known ADCC-mediating Abs (34). Moreover, efficient ADCC-inducing MAbs were isolated from a subset of RV144 subjects, suggesting that at least partial protection in the RV144 trial might be due to ADCC-mediating Abs (33), thus driving renewed interest in the mechanism of recognition of these Abs (33, 35). However, additional work is needed to understand how these ADCC-mediating antibodies work. RV144 ADCC-mediating monoclonal Abs have been described to preferentially use the VH1 gene family, and their effector function is largely blocked by competition with the A32 Fab fragment (33), which recognizes a discontinuous epitope on the surface of the inner domain of gp120 (65), thus suggesting that they all recognize a common/overlapping epitope. Accordingly, a recently reported group of MAbs targeting Env epitopes exposed by CD4 binding (anti-cluster A Abs) and able to mediate ADCC were shown to recognize an epitope blocked by A32 Fab (35) and by the specific gp120 inner domain C11 MAb (50, 51). Detailed structural information on the epitope recognized by A32 is still missing; however, we recently mapped the A32 epitope to the inner domain layers 1 and 2 and identified residues W69 and D107 as key players in this interaction (5). We have now extended these results to some layer 3 residues (T248 andW479) involved in the transition to the CD4-bound conformation (54), which is consistent with our findings that the epitope recognized by this antibody is not well exposed in the unbound trimeric Env, since its interaction depends on the exposure of the inner domain layers, which are exposed only upon CD4 interaction (Fig. 8).
We observed that interaction of A32, C11, and RV144 MAbs with Env was greatly increased upon coexpression of the CD4 receptor in a dose-dependent manner. Importantly, this was dependent on the ability of Env and CD4 to engage efficiently, since changes in CD4 or Env that altered this interaction decreased exposure of these epitopes (Fig. 1). Of note, cell surface CD4 competed for ligands that recognize the CD4-binding site, suggesting that cell surface CD4 recognizes its binding site in a manner similar to the recognition of Env in the context of viral particles (Fig. 2). Strikingly, Env conformational changes induced by cell surface CD4 were observed among several primate immunodeficiency virus Envs, suggesting that this mechanism is highly conserved (Fig. 4).
Viruses lacking the ability to downregulate CD4 or that presented high levels of Env at the surface of infected cells, due to deletions of their viral accessory proteins Nef and Vpu, presented Envs with higher exposure of Env ADCC-mediating epitopes (Fig. 5, 6 and 7) and resulted in a significant increase of elimination of these cells by ADCC (Fig. 6). Maximal sensitization of HIV-1-infected cells to elimination by ADCC required both high levels of cell surface CD4 and enhanced expression of Env at the surface of infected cells. Finally, the ability of Env and CD4 to efficiently engage was also required for efficient elimination of HIV-1-infected cells by ADCC.
Altogether, these observations support the notion that efficient viral release and cell surface CD4 downregulation result in the removal of Env-CD4 complexes targeted by ADCC-mediating antibodies. Thus, interaction of Env and CD4 is important for exposure of ADCC-mediating epitopes, suggesting that one consequence of Vpu and Nef activity might be downregulation of potential ADCC epitopes from the surface of HIV-1-infected cells. Therefore, enhancing Env levels at the surface of infected cells and targeting the ability of Vpu and Nef to downregulate CD4 could potentially render HIV-1-infected cells susceptible to ADCC and thus have therapeutic utility. Additional efforts are required to address the potential role of ADCC in decreasing viral progression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dennis Burton, Pascal Poignard, and IAVI for their generous gifts of PGT121, PG9, and PG16 MAbs, Peter Kwong for VRC01, and David Evans for the CEM-NKR cell line. We thank Éric Cohen for providing the CCR5-tropic NL4.3.ADA.IRES.GFP WT and NL4.3.ADA.IRES.GFP ΔNefΔU, generated by Jonathan Richard and Rajesh Murali, as well as Rafick Sekaly for the pECD4-CFP plasmid. We thank David Baltimore for sending the pNL4.3 Nef IRES GFP provirus. We also thank Éric Cohen and Tram N. Q. Pham for helpful discussions on this study and for sharing their fluorescence-activated cell sorting (FACS)-based ADCC assay. We thank Dominique Gauchat, in charge of the CRCHUM cytometry core facility, for technical support and Jean-Luc Petit for laboratory coordination.
This work was supported by Canada Foundation for Innovation Program Leader grant 29866, by CIHR operating grant 257792, by FRQS Establishment of Young Scientist grant 24639, by a Grand Challenges Explorations grant from the Bill and Melinda Gates Foundation (OPP1042670) to A.F., by the Center for HIV/AIDS Vaccine Immunology and Immunogen Design (U19 AI100645), by the National Institutes of Health (AI67854), by the International AIDS Vaccine Initiative, and by the late William F. McCarty-Cooper. A.F. is the recipient of FRSQ Chercheur Boursier Junior 1 fellowship 24639. M.V. was supported by CIHR Doctoral Research Award 291485.
We have no conflicts of interest to report.
Footnotes
Published ahead of print 18 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03230-13.
REFERENCES
- 1.Allan JS, Coligan JE, Barin F, McLane MF, Sodroski JG, Rosen CA, Haseltine WA, Lee TH, Essex M. 1985. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 228:1091–1094. 10.1126/science.2986290 [DOI] [PubMed] [Google Scholar]
- 2.Robey WG, Safai B, Oroszlan S, Arthur LO, Gonda MA, Gallo RC, Fischinger PJ. 1985. Characterization of envelope and core structural gene products of HTLV-III with sera from AIDS patients. Science 228:593–595. 10.1126/science.2984774 [DOI] [PubMed] [Google Scholar]
- 3.Helseth E, Olshevsky U, Furman C, Sodroski J. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J. Virol. 65:2119–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X, Mahony E, Holm GH, Kassa A, Sodroski J. 2003. Role of the gp120 inner domain beta-sandwich in the interaction between the human immunodeficiency virus envelope glycoprotein subunits. Virology 313:117–125. 10.1016/S0042-6822(03)00273-3 [DOI] [PubMed] [Google Scholar]
- 5.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37:656–667. 10.1016/j.molcel.2010.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. 10.1038/312763a0 [DOI] [PubMed] [Google Scholar]
- 7.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767–768. 10.1038/312767a0 [DOI] [PubMed] [Google Scholar]
- 8.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. 10.1126/science.272.5270.1955 [DOI] [PubMed] [Google Scholar]
- 9.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148. 10.1016/S0092-8674(00)81313-6 [DOI] [PubMed] [Google Scholar]
- 10.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666. 10.1038/381661a0 [DOI] [PubMed] [Google Scholar]
- 11.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158. 10.1016/S0092-8674(00)81314-8 [DOI] [PubMed] [Google Scholar]
- 12.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667–673. 10.1038/381667a0 [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877. 10.1126/science.272.5263.872 [DOI] [PubMed] [Google Scholar]
- 14.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183. 10.1038/384179a0 [DOI] [PubMed] [Google Scholar]
- 15.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384:184–187. 10.1038/384184a0 [DOI] [PubMed] [Google Scholar]
- 16.Furuta RA, Wild CT, Weng Y, Weiss CD. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 5:276–279. 10.1038/nsb0498-276 [DOI] [PubMed] [Google Scholar]
- 17.He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss CD. 2003. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J. Virol. 77:1666–1671. 10.1128/JVI.77.3.1666-1671.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koshiba T, Chan DC. 2003. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J. Biol. Chem. 278:7573–7579. 10.1074/jbc.M211154200 [DOI] [PubMed] [Google Scholar]
- 19.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 101:5036–5041. 10.1073/pnas.0307953101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. 10.1016/S0092-8674(00)80205-6 [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Blacklow SC, Kim PS. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 2:1075–1082. 10.1038/nsb1295-1075 [DOI] [PubMed] [Google Scholar]
- 22.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. 10.1038/387426a0 [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Asmal M, Lane S, Permar SR, Schmidt SD, Mascola JR, Letvin NL. 2011. Antibody-dependent cell-mediated cytotoxicity in simian immunodeficiency virus-infected rhesus monkeys. J. Virol. 85:6906–6912. 10.1128/JVI.00326-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banks ND, Kinsey N, Clements J, Hildreth JE. 2002. Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res. Hum. Retroviruses 18:1197–1205. 10.1089/08892220260387940 [DOI] [PubMed] [Google Scholar]
- 25.Alpert MD, Harvey JD, Lauer WA, Reeves RK, Piatak M, Jr, Carville A, Mansfield KG, Lifson JD, Li W, Desrosiers RC, Johnson RP, Evans DT. 2012. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLoS Pathog. 8:e1002890. 10.1371/journal.ppat.1002890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baum LL, Cassutt KJ, Knigge K, Khattri R, Margolick J, Rinaldo C, Kleeberger CA, Nishanian P, Henrard DR, Phair J. 1996. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J. Immunol. 157:2168–2173 [PubMed] [Google Scholar]
- 27.Forthal DN, Landucci G, Haubrich R, Keenan B, Kuppermann BD, Tilles JG, Kaplan J. 1999. Antibody-dependent cellular cytotoxicity independently predicts survival in severely immunocompromised human immunodeficiency virus-infected patients. J. Infect. Dis. 180:1338–1341. 10.1086/314988 [DOI] [PubMed] [Google Scholar]
- 28.Ljunggren K, Moschese V, Broliden PA, Giaquinto C, Quinti I, Fenyo EM, Wahren B, Rossi P, Jondal M. 1990. Antibodies mediating cellular cytotoxicity and neutralization correlate with a better clinical stage in children born to human immunodeficiency virus-infected mothers. J. Infect. Dis. 161:198–202. 10.1093/infdis/161.2.198 [DOI] [PubMed] [Google Scholar]
- 29.Chung AW, Navis M, Isitman G, Wren L, Silvers J, Amin J, Kent SJ, Stratov I. 2011. Activation of NK cells by ADCC antibodies and HIV disease progression. J. Acquir. Immune Defic. Syndr. 58:127–131. 10.1097/QAI.0b013e31822c62b9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J. 2012. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. 8:e1002739. 10.1371/journal.ppat.1002739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, Stratov I. 2011. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc. Natl. Acad. Sci. U. S. A. 108:7505–7510. 10.1073/pnas.1016048108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366:1275–1286. 10.1056/NEJMoa1113425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonsignori M, Pollara J, Moody MA, Alpert MD, Chen X, Hwang KK, Gilbert PB, Huang Y, Gurley TC, Kozink DM, Marshall DJ, Whitesides JF, Tsao CY, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Kim JH, Michael NL, Tomaras GD, Montefiori DC, Lewis GK, Devico A, Evans DT, Ferrari G, Liao HX, Haynes BF. 2012. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 86:11521–11532. 10.1128/JVI.01023-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, Poling B, Nicholson CO, Zhang R, Lu X, Parks R, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Gilbert PB, Kim JH, Michael NL, Montefiori DC, Haynes BF. 2013. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. U. S. A. 110:9019–9024. 10.1073/pnas.1301456110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, Fouts TR, Gallo RC, Devico AL, Lewis GK. 2013. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc. Natl. Acad. Sci. U. S. A. 110:E69–78. 10.1073/pnas.1217609110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J. 2012. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 19:893–899. 10.1038/nsmb.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bour S, Schubert U, Strebel K. 1995. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J. Virol. 69:1510–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moldovan MC, Sabbagh L, Breton G, Sekaly RP, Krummel MF. 2006. Triggering of T cell activation via CD4 dimers. J. Immunol. 176:5438–5445 [DOI] [PubMed] [Google Scholar]
- 39.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J. Virol. 83:3556–3567. 10.1128/JVI.02132-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraus MH, Parrish NF, Shaw KS, Decker JM, Keele BF, Salazar-Gonzalez JF, Grayson T, McPherson DT, Ping LH, Anderson JA, Swanstrom R, Williamson C, Shaw GM, Hahn BH. 2010. A rev1-vpu polymorphism unique to HIV-1 subtype A and C strains impairs envelope glycoprotein expression from rev-vpu-env cassettes and reduces virion infectivity in pseudotyping assays. Virology 397:346–357. 10.1016/j.virol.2009.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medjahed H, Pacheco B, Desormeaux A, Sodroski J, Finzi A. 2013. The HIV-1 gp120 major variable regions modulate cold-inactivation. J. Virol. 87:4103–4111. 10.1128/JVI.03124-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baalwa J, Wang S, Parrish NF, Decker JM, Keele BF, Learn GH, Yue L, Ruzagira E, Ssemwanga D, Kamali A, Amornkul PN, Price MA, Kappes JC, Karita E, Kaleebu P, Sanders E, Gilmour J, Allen S, Hunter E, Montefiori DC, Haynes BF, Cormier E, Hahn BH, Shaw GM. 2013. Molecular identification, cloning and characterization of transmitted/founder HIV-1 subtype A, D and A/D infectious molecular clones. Virology 436:33–48. 10.1016/j.virol.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao F, Yue L, Robertson DL, Hill SC, Hui H, Biggar RJ, Neequaye AE, Whelan TM, Ho DD, Shaw GM, et al. 1994. Genetic diversity of human immunodeficiency virus type 2: evidence for distinct sequence subtypes with differences in virus biology. J. Virol. 68:7433–7447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med. 201:1407–1419. 10.1084/jem.20042510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finzi A, Pacheco B, Xiang SH, Pancera M, Herschhorn A, Wang L, Zeng X, Desormeaux A, Kwong PD, Sodroski J. 2012. Lineage-specific differences between human and Simian immunodeficiency virus regulation of gp120 trimer association and CD4 binding. J. Virol. 86:8974–8986. 10.1128/JVI.01076-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westervelt P, Gendelman HE, Ratner L. 1991. Identification of a determinant within the human immunodeficiency virus 1 surface envelope glycoprotein critical for productive infection of primary monocytes. Proc. Natl. Acad. Sci. U. S. A. 88:3097–3101. 10.1073/pnas.88.8.3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. 2010. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115:1354–1363. 10.1182/blood-2009-08-237370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schubert U, Bour S, Willey RL, Strebel K. 1999. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J. Virol. 73:887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson JE, Holton D, Elliott S, Ho DD. 1992. Distinct antigenic sites on HIV gp120 identified by a panel of human monoclonal antibodies. J. Cell. Biochem. Suppl. 16E:Q449 [Google Scholar]
- 51.Moore JP, Willey RL, Lewis GK, Robinson J, Sodroski J. 1994. Immunological evidence for interactions between the first, second, and fifth conserved domains of the gp120 surface glycoprotein of human immunodeficiency virus type 1. J. Virol. 68:6836–6847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong R, Li H, Georgiev I, Changela A, Bibollet-Ruche F, Decker JM, Rowland-Jones SL, Jaye A, Guan Y, Lewis GK, Langedijk JP, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2012. Epitope mapping of broadly neutralizing HIV-2 human monoclonal antibodies. J. Virol. 86:12115–12128. 10.1128/JVI.01632-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chowdhury IH, Koyanagi Y, Takamatsu K, Yoshida O, Kobayashi S, Yamamoto N. 1991. Evaluation of anti-human immunodeficiency virus effect of recombinant CD4-immunoglobulin in vitro: a good candidate for AIDS treatment. Med. Microbiol. Immunol. 180:183–192 [DOI] [PubMed] [Google Scholar]
- 54.Desormeaux A, Coutu M, Medjahed H, Pacheco B, Herschhorn A, Gu C, Xiang SH, Mao Y, Sodroski J, Finzi A. 2013. The highly conserved layer-3 component of the HIV-1 gp120 inner domain is critical for CD4-required conformational transitions. J. Virol. 87:2549–2562. 10.1128/JVI.03104-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brand D, Srinivasan K, Sodroski J. 1995. Determinants of human immunodeficiency virus type 1 entry in the CDR2 loop of the CD4 glycoprotein. J. Virol. 69:166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. 10.1126/science.1178746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R, Wang L, Hendrickson WA, Doyle ML, Sodroski J. 2002. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 76:9888–9899. 10.1128/JVI.76.19.9888-9899.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin RA, Nayak DP. 1996. Mutational analysis of HIV-1 gp160-mediated receptor interference: intracellular complex formation. Virology 220:461–472. 10.1006/viro.1996.0333 [DOI] [PubMed] [Google Scholar]
- 60.Hoxie JA, Alpers JD, Rackowski JL, Huebner K, Haggarty BS, Cedarbaum AJ, Reed JC. 1986. Alterations in T4 (CD4) protein and mRNA synthesis in cells infected with HIV. Science 234:1123–1127. 10.1126/science.3095925 [DOI] [PubMed] [Google Scholar]
- 61.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166–1171. 10.1073/pnas.0911004107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peltz GA, Grundy HO, Lebo RV, Yssel H, Barsh GS, Moore KW. 1989. Human Fc gamma RIII: cloning, expression, and identification of the chromosomal locus of two Fc receptors for IgG. Proc. Natl. Acad. Sci. U. S. A. 86:1013–1017. 10.1073/pnas.86.3.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanier LL. 2008. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9:495–502. 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lambotte O, Pollara J, Boufassa F, Moog C, Venet A, Haynes BF, Delfraissy JF, Saez-Cirion A, Ferrari G. 2013. High antibody-dependent cellular cytotoxicity responses are correlated with strong CD8 T cell viral suppressive activity but not with B57 status in HIV-1 elite controllers. PLoS One 8:e74855. 10.1371/journal.pone.0074855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wyatt R, Moore J, Accola M, Desjardin E, Robinson J, Sodroski J. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 69:5723–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. 10.1038/nature06553 [DOI] [PubMed] [Google Scholar]
- 67.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113. 10.1038/nature07159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz O, Dautry-Varsat A, Goud B, Maréchal V, Subtil A, Heard JM, Danos O. 1995. Human immunodeficiency virus type 1 Nef induces accumulation of CD4 in early endosomes. J. Virol. 69:528–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 66:7193–7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. 10.1016/j.chom.2008.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.