

Abstract
Broken-symmetry density functional theory (BS-DFT) calculations are assessed for redox energetics [Cu(SCH3)2]1–/0, [Cu(NCS)2]1–/0, [FeCl4]1–/0, and [Fe(SCH3)4]1–/0 against vertical detachment energies (VDE) from valence photoelectron spectroscopy (PES), as a prelude to studies of metalloprotein analogs. The M06 and B3LYP hybrid functionals give VDE that agree with the PES VDE for the Fe complexes, but both underestimate it by ∼400 meV for the Cu complexes; other hybrid functionals give VDEs that are an increasing function of the amount of Hartree–Fock (HF) exchange and so cannot show good agreement for both Cu and Fe complexes. Range-separated (RS) functionals appear to give a better distribution of HF exchange since the negative HOMO energy is approximately equal to the VDEs but also give VDEs dependent on the amount of HF exchange, sometimes leading to ground states with incorrect electron configurations; the LRC-ωPBEh functional reduced to 10% HF exchange at short-range give somewhat better values for both, although still ∼150 meV too low for the Cu complexes and ∼50 meV too high for the Fe complexes. Overall, the results indicate that while HF exchange compensates for self-interaction error in DFT calculations of both Cu and Fe complexes, too much may lead to more sensitivity to nondynamical correlation in the spin-polarized Fe complexes.
I. Introduction
Redox properties are essential functional characteristics of electron transfer proteins, and most electron transfer proteins are metalloproteins. However, while experimentally synthesized analogs of the iron–sulfur proteins have played a critical role in understanding their redox properties, the synthesis of redox site analogs for the copper proteins has been difficult due to the flexible coordination of copper. So far, spectroscopically similar analogs have been synthesized,1−5 but they exhibit weak structural similarity to even the simplest copper protein redox sites. Thus, computational chemistry provides a route to understanding these proteins.6
However, determining reduction potentials of redox-active metalloproteins by computational chemistry methods is challenging. For instance, quantum mechanical calculations of first-row transition metals found in these proteins must include electron correlation. Since advances in density functional theory (DFT)7 include functionals with effective electron correlation, DFT is usually the method of choice because of computational efficiency. However, unlike conventional ab initio molecular orbital theory,8 DFT suffers from the lack of a clear pathway for improvement. Currently, DFT calculations with hybrid (i.e., including some Hartree–Fock (HF) exchange) or hybrid meta (i.e., in addition, including the spin kinetic energy densities) generalized gradient approximation (GGA) density functionals appear more accurate for treating transition-metal systems than local density approximations9 and even some post-HF methods.10,11 Moreover, efforts have been made to develop range-separated (RS) functionals in which the HF exchange is treated differently at different ranges for further improving upon global hybrids.9,12−14 However, benchmarking of density functionals and basis sets by comparisons with experimental measurements of structural and energetic properties of relevant compounds remain essential, with a goal of a balance of accuracy and computational efficiency. In particular, valence photoelectron spectroscopy (PES) of well-defined gaseous metal complexes has become important for testing the electronic structure and energetics, independent of environmental perturbations such as solvent, crystal field, or surrounding protein,15−17 since the calculation can be performed under the same conditions without any approximations for the environment.
Our previous benchmark studies for analogs of Fe–S proteins18 used gaseous PES experimental data,15,16,19,20 the ligand–metal bond covalency from X-ray absorption spectroscopy (XAS),21,22 and structures from X-ray crystallography.23 Good structures can be obtained using broken-symmetry (BS) DFT with the B3LYP functional7 and a double-ζ basis set with polarization functions such as 6-31G**. In addition, good single-point energies of these structures can be obtained when diffuse functions are added to the sulfurs, even comparable to coupled cluster (CC) methods.8 The calculated vertical (VDE) and adiabatic (ADE) detachment energies agree well with the gaseous PES data of [1Fe], [2Fe–2S], and [4Fe–4S] protein analogs, while the calculated percentage of ligand character of the Fe 3d orbitals correlate well with ligand K-edge XAS measurements. Furthermore, calculations at this level also give reliable inner-sphere reduction free energies for reduction potential calculations of iron–sulfur proteins, including redox couples not accessible in the PES experiments.24
Since spectroscopically and structurally similar analogs of copper proteins have not yet been synthesized, understanding the structural, electronic, and redox properties of simple copper complexes is especially important for benchmarking calculations of copper redox site analogs. Furthermore, calculations with reliable methods may lead to better design strategies for synthesizing new analogs. In the current article, Cu–S and Cu–N bonding interactions are investigated in the simple Cu complexes, [CuX2]n (X = SCH3, NCS; n = −1 and 0), as a prelude to investigations of the type I copper site found in the blue copper proteins. The type-I copper site consists of a distorted tetrahedral structure with four ligands: two histidines, a cysteine, and a weak axial fourth ligand, which is usually a methionine. Since our long-term goal is to find functionals and basis sets that work well for the transition metals found in metalloprotein redox sites, which include mixed metal sites, the Fe complexes, [FeX4]n (X = Cl, SCH3; n = −1 and 0), which were used in our previous studies for redox sites of Fe–S proteins, are also studied. We focus on contrasting the ability of the methods to handle the closed shell, low-spin Cu complexes (S = 1/2) with the open shell, high-spin Fe complexes (S = 5/2 and 2) rather than a statistical survey of many different transition-metal complexes. While a general criteria is good agreement of geometries with the X-ray crystallographic structures, our essential criteria is good agreement of energetics with PES electron detachment energies since the methods are being tested for redox calculations. Several double-ς basis sets were evaluated against two triple-ς basis sets. Also, several hybrid GGA and meta-GGA functionals and range-separated GGA and meta-GGA functionals were compared to CC methods.
II. Methods
A. Computational Methods
Spin-unrestricted DFT calculations with broken-symmetry (BS) molecular orbital (MO) wavefunctions were performed for geometries and energies. The energies were also calculated at the CCSD and CCSD(T) levels, with the inner shells up to and including the 3s and 3p orbitals frozen in the correlation calculations.8 If the energy is calculated using a different method than the geometry optimization, the notation level2/basis2//level1/basis1 is used in which level1/basis1 calculations are for the geometry optimization and level2/basis2 calculations are used for single-point energies of the level1/basis1 geometry.
Several different double- and triple-ζ basis sets were tested. The first three, DZVP2,25 def2-SVP,26 and 6-31G**,27−29 are double-ζ basis sets with polarization functions. The next two, def2-SVPD30 and 6-31++G**,27−29,31,32 contain diffuse functions, while 6-31(++)LG**28,31 has diffuse functions added only to the subscripted ligand atoms. Finally, def2-TZVPPD30 and aug-cc-pVTZ33,34 are triple-ζ basis sets with polarization and diffuse functions.
Several different hybrid and RS density functionals were also tested. The hybrid functionals tested with percentage HF exchange indicated in parentheses are the B3LYP GGA (20% HF); B3LYP*,35 a modified B3LYP GGA (15% HF); the B97 GGA (19.43% HF);36 the PBE1PBE GGA (25% HF);37 the M06 meta-GGA (27% HF);38 and the B(38HF)P86 GGA (38% HF).39 In addition, one set of RS density functionals with percentage HF exchange at short-range (SR) and at long-range (LR) and range parameter ω indicated in parentheses tested are the BNL GGA (0% SR, 100% LR, 0.33 a0–1);40,41 the CAM-B3LYP GGA (19% SR, 65% LR, 0.33 a0–1);42 and the LRC-ωPBEh GGA (20% SR, 100% LR, 0.2 a0–1).43 Finally, another set of RS density functionals with highly parametrized and reoptimized short-range DFT exchange (referred to here as highly optimized RS) are the ωB97 GGA (0% SR, 100% LR, 0.4 a0–1);44 the ωB97X GGA (15.77% SR, 100% LR, 0.3 a0–1);44 and the M11 meta-GGA (42.8% SR, 100% LR, 0.25 a0–1).45 (Note many different notations are used for RS functions; for instance, % SR = α, % LR = α+β, and ω = γ in refs (12 and 46).)
The initial wavefunctions of the oxidized open-shell species were generated either from the wavefunctions of the reduced species or of its higher spin states at a reasonable initial geometry. Since the HF and Kohn–Sham (KS) electron configurations of the ground state of open-shell Cu and Fe complexes were often different, both were used as initial electron configurations for the DFT and CC calculations to help establish that the solution was not an excited state. Stability analysis of the second derivatives of the energy with respect to occupied orbital variation in the wavefunction47 was further utilized to confirm the ground state of the complexes in the HF and DFT calculations. Stationary points in the DFT calculations that were uncertain due to a flat potential energy surface were examined further using an extra fine integration grid, with an energy convergence of 1 × 10–8 au.48 Detachment energies of complexes were approximately calculated by the difference of total electronic energies between the reduced and oxidized species using the geometry of the reduced species (VDEs) and by the negative energy of the highest occupied molecular orbital (HOMO) of the reduced species. The T1 diagnostic and the largest T2 amplitude values were determined from the CCSD(T)/def2-SVP(D)L//M06/def2-SVP calculations to evaluate multireference effects.49
All hybrid DFT calculations were performed using the NWChem program package,48 the RS DFT (except LRC-ωPBEh), CCSD, and CCSD(T) calculations utilized the Gaussian09 program package,50 and the LRC-ωPBEh calculations utilized the Q-Chem program package.51 The molecular orbital visualizations were performed using the extensible computational chemistry environment (Ecce) application software.52
B. Experimental Methods
The PES experiments were performed on a magnetic-bottle apparatus equipped with an electrospray ionization (ESI) source.53 The only modification was the shortening of the electron flight tube of the magnetic-bottle PES analyzer from 4 to 2.5 m.54 Moreover, the energy resolution of the PES spectra of both species is enhanced using our newly built cryogenically cooled ion trap operated at 20 K.55 Briefly, a 1 mM solution of Cu2O with NaSCH3 in a CH3CN/H2O mixed solvent (3:1 volume ratio) was prepared to generate the [Cu(SCH3)2]1– anion, and a 1 mM solution of Cu(CH3COO)2 with KSCN in a pure CH3CN solvent was prepared to generate the [Cu(NCS)2]1– anion. After being accumulated in the ion trap for 0.1 s, the desired anions were mass-selected via time-of-flight mass spectrometry and then decelerated before being intercepted by a probe laser beam. The laser wavelengths employed were 213 nm (5.821 eV) from a dye laser and 266 nm (4.661 eV) from a Nd:YAG laser. Photoelectron time-of-flight spectra were collected and converted to kinetic energy spectra, calibrated by the known spectra of I– and Au.54 The electron kinetic energy resolution was about 3%, i.e. 30 meV for 1 eV electrons. The VDE of each feature was measured from the maximum of each detachment band. In addition, PES VDEs of the Fe complexes were obtained from our previous studies.20,56
Crystal structures were obtained from the Cambridge Structural Database (CSD).57 No crystal structure is available for [Cu(SCH3)2]1–, so [Cu(S-t-butyl)2]1– (CSD ID: KOBVAZ)58 was used. In addition, the [Cu(NCS)2]1– (VICCIT),59 [FeCl4]1– (AHEPOT),60 and [Fe(SCH3)4]1– (JURHIN)61 were used.
III. Results and Discussion
A. Photoelectron Spectroscopy
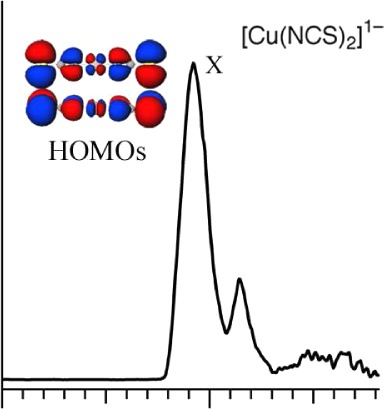
The photoelectron spectra of [Cu(SCH3)2]1– and [Cu(NCS)2]1– (Figure 1) were obtained at 20 K, which led to sharper and much better resolved peaks than at room temperature. The spectrum of [Cu(SCH3)2]1– at 266 nm (Figure 2a) reveals four prominent detachment bands, labeled as X, A, B, and C. The ground state X band is very broad, suggesting a large geometry change between the ground state of [Cu(SCH3)2]1– and its neutral, and overlaps the A feature. In contrast, the B and C features are much sharper, suggesting they are from nonbonding Cu 3d type orbitals. The spectrum of [Cu(NCS)2]1– at 157 nm (Figure 2b) shows a detachment feature labeled as X, with a short vibrational progression with frequency 1780 ± 50 cm–1. The detachment from the ground state (X band) is of relevance to redox reactions in proteins, and both the VDE and ADE from X can be determined. However, given the possible large geometric change upon oxidation of [Cu(SCH3)2]1–, we focus on the VDE for comparisons with the calculated results since crystal structures of the oxidized Cu complexes are apparently not available.
Figure 1.
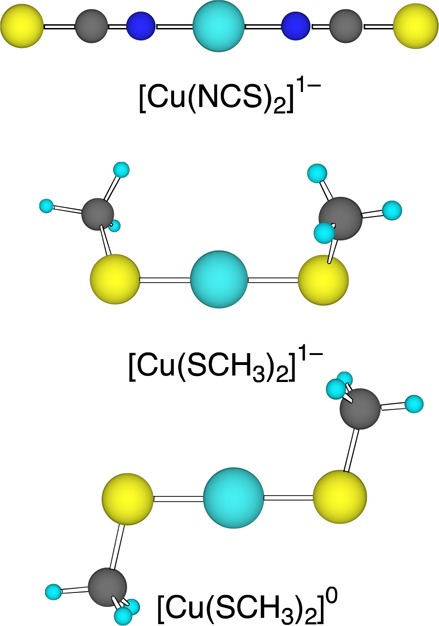
Ball-stick renderings of [Cu(NCS)2]1– and [Cu(SCH3)2]1–/0, M06/aug-cc-pVTZ geometries.
Figure 2.

Photoelectron spectra at 20 K of (a) [Cu(SCH3)2]1– at 266 nm and (b) [Cu(NCS)2]1– at 213 nm. Vibrational progressions are labeled with vertical lines.
B. Electronic Structure
KS MO interaction diagrams between the Cu1+ and the ligands for [Cu(SCH3)2]1– and [Cu(NCS)2]1– (Figure 3) were examined to consistency with the PES results. This figure is based on the M06/DZVP2 calculations but is representative of the B3LYP results as well. A full analysis of the calculated electronic structure and the PES spectrum will be presented elsewhere.
Figure 3.
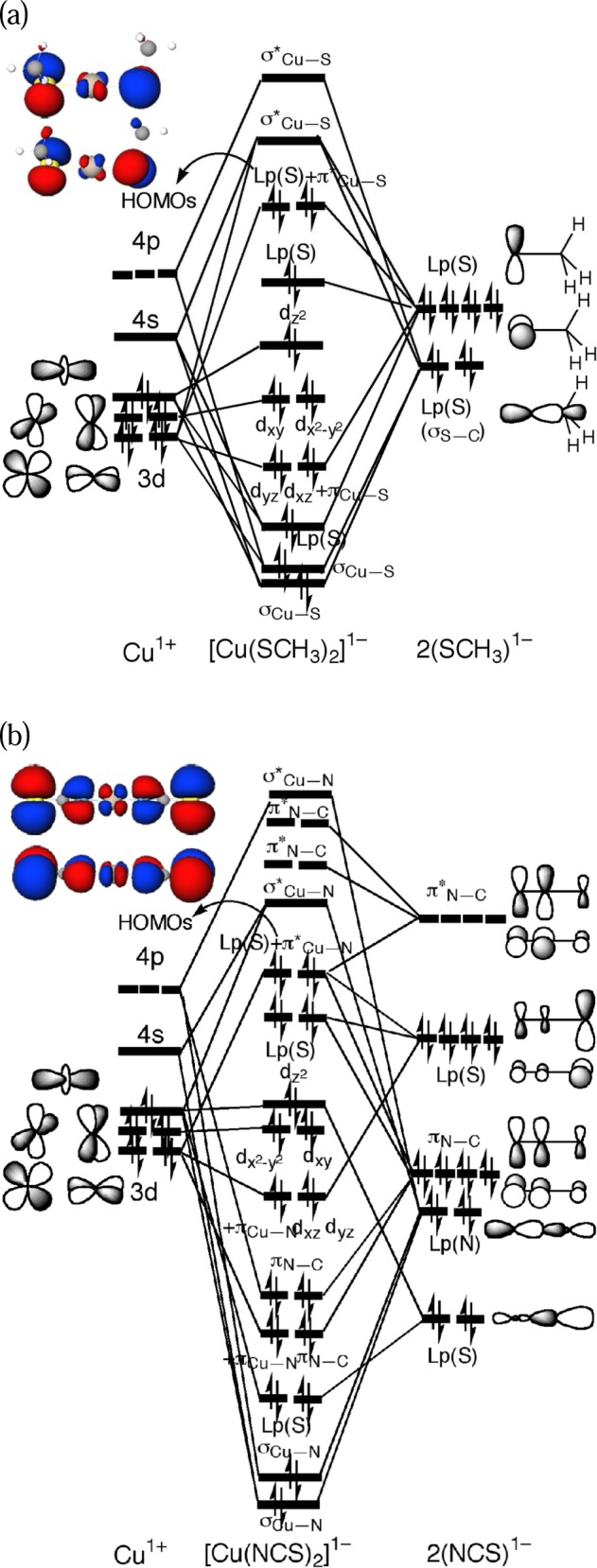
Schematic Kohn–Sham molecular orbital interaction diagrams based on M06/DZVP2 calculations (a) between Cu+ and SCH3–and the HOMOs of [Cu(SCH3)2]1– and (b) between Cu+ and NCS–and the HOMOs of [Cu(NCS)2]1–.
The two HOMOs of [Cu(SCH3)2]1– are formed by interactions between the Cu dyz and dxz orbitals and the higher-lying S lone-pairs. The two σCu–S bonds are formed by interactions of the Cu 4s, 4p, and dz2 orbitals and the lower-lying S lone-pairs with σS–C bonding character. The five occupied Cu 3d orbitals lie below the symmetric S lone-pair orbitals and above the two σCu–S orbitals and an asymmetric S lone-pair orbital with σS–C bonding character. The oxidation of [Cu(SCH3)2]1– involves the near-degenerate S lone-pair MOs, which should induce a significant structural distortion (consistent with the broad PES X band) toward a more planar structure with strong spin-polarization. Consequently, the α singly occupied MO (SOMO) and the β lowest unoccupied MO (LUMO) upon oxidation increase the Cu dxz character, while the other MOs (α HOMO-1 and β HOMO) become pure Lp(S) without any Cu 3d character. Also, since the MO (β LUMO) that is oxidized has π*Cu–S antibonding character (consistent with S K-edge XAS experiments of the redox site of the blue copper proteins62), the Cu–S bond lengths should shorten.
The two degenerate HOMOs of [Cu(NCS)2]1– are formed by interactions between the Cu dyz and dxz orbitals and the higher-lying S lone-pair orbitals with π*C–S and πN–C character, even though the metal ligates to the N; the asymmetric HOMOs are stabilized by the electron back-donation into the high-lying unoccupied π*N–C orbitals, which increases the πN–C bonding character in the HOMOs. The two σCu–N bonds are stabilized by interactions of the Cu 4s, 4p, and dz2 orbitals and the lower-lying N lone-pairs [Lp(N)] with σN–C bonding character. The five occupied Cu 3d orbitals lie below the four S lone-pair orbitals and above the four πN–C orbitals and an asymmetric S lone-pair orbital with σS–C bonding character as that in [Cu(SCH3)2]1–. The oxidation of [Cu(NCS)2]1– should differ from [Cu(SCH3)2]1– because of stabilization of the HOMOS by electron back-donation. For instance, electron detachment from the stabilized HOMO is more difficult so the VDE should be larger than that in [Cu(SCH3)2]1–, which is consistent with the PES data. In addition, the linear structure is unlikely to distort upon oxidation as does the [Cu(SCH3)2]1– structure since the degenerate HOMOs are stabilized by the back-donation. Thus, although oxidation of both [Cu(NCS)2]1– and [Cu(SCH3)2]1– involve the S lone-pair electron, the ground state X band in the PES spectra (Figure 2) of [Cu(NCS)2]1– has a very different shape from [Cu(SCH3)2]1–. Finally, the decrease in the π*Cu–N antibonding, π*C–S antibonding, and πN–C bonding character upon oxidation should lead to a decrease in the Cu–N and C–S bond lengths and an increase in the N–C bond lengths.
C. Basis Set Effects on Density Functional Theory Calculations
The basis sets were first evaluated for their performance in geometry optimization and energetics using the B3LYP and M06 hybrid functionals. First, the double-ζ sets are examined for agreement with the triple-ζ sets, and then agreement with experiment is assessed.
The complete DFT optimized geometries of [Cu(SCH3)2]1–/0 and [Cu(NCS)2]1–/0 as well as crystal structures for the reduced species are given in the Supporting Information (Tables S1 and S2). The calculations all show that [Cu(SCH3)2]1– has a gauche conformation with “C2” symmetry while [Cu(NCS)2]1– has a linear structure (Figure 1), both in agreement with the X-ray structures. All of the basis sets show similar performance for most of the geometry. However, the θS–Cu–S angle of [Cu(SCH3)2]1– is more than 20° larger in the 6-31(++)LG** and 6-31++G** (not shown) basis set compared to experiment, which causes larger deviations in the θCu–S–C angle as well, so we do not recommend adding diffuse functions into the 6-31G basis sets for geometry optimization. Also, when the same basis set is used, the M06 functional gives slightly shorter bond lengths than the B3LYP functional, while the bond angles are quite similar. Upon oxidation, similar changes are observed in the double-ζ basis sets as in the triple-ζ basis sets, although no experimental data are available for the neutral species. [Cu(SCH3)2]0 is distorted from the gauche conformation of the reduced species into a planar trans conformation with “C2h” symmetry (Figure 1), which is consistent with the broad PES X band (Figure 2a) and may give rise to a large reorganization energy upon oxidation. The decrease in the Cu–S and S–C bond lengths of [Cu(SCH3)2]0 is also consistent with the MO analysis. In addition, [Cu(NCS)2]0 remains a linear structure and its Cu–N and C–S bond lengths are slightly shorter while the N–C bond lengths are longer relative to the reduced site, consistent with the MO analysis and the vibrational progression observed for the X feature.
The metal–ligand bond lengths are perhaps the most important geometrical feature for assessing the computational methods, so they are examined for the Cu complexes and for FeCl4– and [Fe(SCH3)4]1– (Figure 4). The double-ζ basis sets are consistent with the triple-ζ basis sets and give M–L bond lengths for that are generally less than ∼0.05 Å longer than the crystal structures. However, the 6-31G** and 6-31(++)LG** basis sets give significantly shorter bond lengths for Cu(SCH3)2]1– and [Cu(NCS)2]1–, which brings the calculated results further from experiment for [Cu(NCS)2]1–/0 but closer for [Cu(SCH3)2]1–; however, the experimental M–L bond length for the latter comes from [Cu(S-t-butyl)2]1–, which may be shorter than in [Cu(SCH3)2]1– due to the better electron donating ability of the -S-t-butyl group than the -SCH3 group21 that leads to stronger interactions with the Cu. Thus, of the small double-ζ basis sets, the DZVP2 and Def2-SVP basis sets appear to give good M–L bond lengths, with results similar to the triple-ζ basis sets and the addition of diffuse functions to the latter (i.e., def2-SVPD) makes little difference. In addition, the M06 functional gives slightly shorter bond lengths than the B3LYP functional, so that the M–L bond lengths are in somewhat better agreement with experiment.
Figure 4.

Optimized M–L bond lengths for [Cu(NCS)2]1– (triangle), [Cu(SCH3)2]1– (square), FeCl4– (circle), and [Fe(SCH3)4]1– (diamond), using the B3LYP (open symbols) and M06 (solid symbols) functionals as a function of basis set plotted on a scale of the logarithm of the number of basis functions in [Cu(NCS)2]1–. From left to right, the basis sets are DZVP2, Def2-SVP, 6-31G**, 6-31(++)LG**, Def2-SVPD, Def2-TZVPPD, and aug-cc-pVTZ. The symbols are connected by dotted lines to guide the eye, and the results for the Fe complexes are shifted upward by 0.1 Å to avoid overlaps. The experimental M–L bond lengths (gray line with error indicated approximately by width of line) are also shown.
The complete DFT VDEs and ADEs of [Cu(SCH3)2]1–/0 and [Cu(NCS)2]1–/0 are given in the Supporting Information (Table S3). The VDEs for the same Cu and Fe complexes are used to examine the redox energetics (Figure 5). Of the double-ζ basis sets, the DZVP2 results are equivalent to the triple-ζ basis set with the diffuse functions results, while the def2-SVP and 6-31G** are much lower. Of the double-ζ basis sets with the diffuse functions, the 6-31(++)LG** and 6-31++G** (not shown) basis sets do not improve agreement as much for the Cu complexes as for the Fe complexes, while def2-SVPD is in good agreement with the triple-ζ basis set results, consistent with the better performance of the Karlsruhe def2 basis sets with diffuse functions in the M06 calculations of barrier heights, ionization potentials, and electron affinities.63 In addition, because the differences in geometry are small between def2-SVP and def2-SVPD, the def2-SVPD energy of the def2-SVP geometry is almost identical to that of the def2-SVPD geometry, which suggests geometry optimization with def2-SVP and a single point energy calculation with def2-SVPD for larger molecules. However, although the DZVP2 nondiffuse basis set is ∼2/3 the size of def2-SVPD diffuse basis set for these redox sites, the results are remarkably similar, presumably because the DFT optimized Gaussian DZVP2 basis sets provide high quality valence orbitals in comparison with the energies and orbitals obtained by numerical solutions of the Kohn–Sham equations.25 In particular, the smallest exponential parameters for each angular momentum are smaller than in def2-SVP, which is more diffuse than standard nondiffuse basis sets,63 indicating DZVP2 is even more diffuse. However, while the B3LYP and M06 calculations show the increase in the VDE of [Cu(NCS)2]1– over [Cu(SCH3)2]1– observed experimentally, both underestimate the experimental values considerably by ∼400 meV although M06 is generally somewhat closer than B3LYP.
Figure 5.

Calculated VDE for [Cu(NCS)2]1– (triangle), [Cu(SCH3)2]1– (square), FeCl4– (circle), and [Fe(SCH3)4]1– (diamond), using the B3LYP (open symbols) and M06 (solid symbols) functionals as a function of a basis set plotted on a scale of the logarithm of the number of basis functions in [Cu(NCS)2]1–. From left to right, the basis sets are DZVP2, Def2-SVP, 6-31G**, 6-31(++)LG**, Def2-SVPD, Def2-TZVPPD, and aug-cc-pVTZ. The symbols are connected by dotted lines to guide the eye, and the results for the Fe complexes are shifted upward by 3 eV to avoid overlaps. The experimental M–L bond lengths (gray line with error indicated approximately by width of line) are also shown.
Thus, DFT calculations using def2-SVPD agree well with those using the triple-ζ basis sets with the diffuse functions for geometry and energetics for both the Cu and Fe complexes and appear to be a good balance of speed and accuracy for larger molecules. In addition, using either DZVP2 or else calculating single point energies using def2-SVPD on def2-SVP geometries, which are very similar to def2-SVPD geometries, are options for much larger molecules or when computational speed is important. However, none of the DFT results give good agreement for energetics compared to experiment for the Cu complexes.
D. Effects of Functionals on Density Functional Theory Calculations
Given the poor energetics of B3LYP and M06 for the Cu complexes, other functionals were tested. Our strategy was first to examine trends and narrow down the functionals in BS-DFT calculations with the DZVP2 double-ζ basis set, since it compares well with larger basis sets with diffuse functions but its small size makes the calculations faster. All of the functionals give generally reasonable geometries including M–L bonds (Figure 6), so the discussion will focus on the VDE. In addition, the criterion of equality of the negative of the HOMO energy (−εHOMO) with the SCF VDE proposed by Baer and co-workers46 was examined, since they should be equal in exact KS theory.64 Then, calculations of the functionals with the best performance with the DZVP2 basis set but now using the def2-SVPD basis set are compared. The origins of differences with different functionals are discussed with the caveat that only two Cu and two Fe complexes are also examined.
Figure 6.

Optimized M–L bond lengths for [Cu(NCS)2]1– (triangle), [Cu(SCH3)2]1– (square), FeCl4– (circle), and [Fe(SCH3)4]1– (diamond), using the DZVP2 basis set for different functionals in order of increasing (short-range) HF exchange. From left to right, the hybrid functionals (open symbols) are B3LYP*, B97, B3LYP, PBE1PBE, M06, and B(38HF)P86; the RS functionals (solid symbols) are BNL, CAM-B3LYP, and LRC-ωPBEh; and the highly optimized RS functionals (gray symbols) are ωB97, ωB97X, and M11. The symbols are connected by dotted lines to guide the eye, and the results for the Fe complexes are shifted upward by 0.1 Å to avoid overlaps. The experimental M–L bond lengths (gray line with error indicated approximately by width of line) are also shown.
First, several hybrid functionals were examined with DZVP2. The VDE for these functionals generally increases with % HF exchange, despite having different exchange-correlation functionals (Figure 7). Thus, B(38HF)P86, which was optimized against experimental CuCl4 spin densities,39 agrees quite well for the Cu complexes compared to experimental VDE but is too large for the Fe complexes, and generally none of the hybrid functionals can predict VDE accurately for both the Cu and Fe complexes. In addition, −εHOMO is generally significantly lower than the SCF VDE in the hybrid functionals, although the agreement improves somewhat with the % HF exchange (Figure 7).
Figure 7.
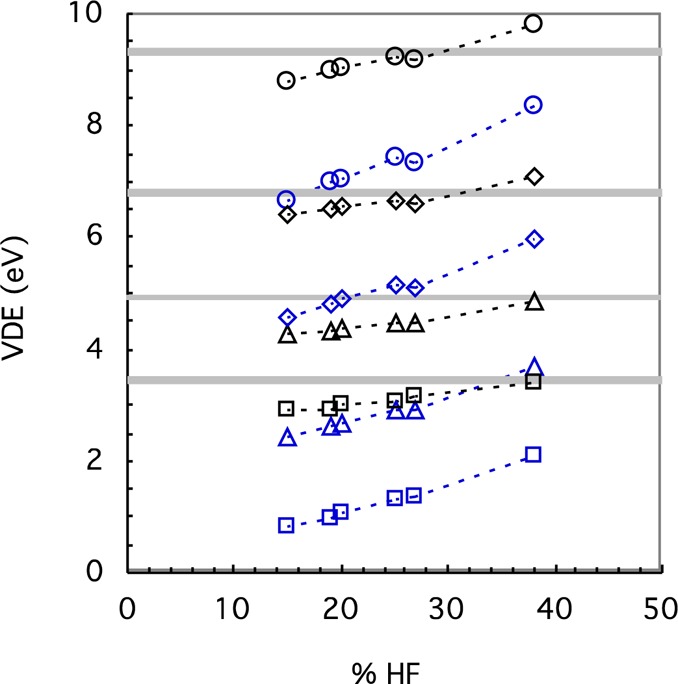
Calculated VDE from the difference in energy between the reduced form and a Franck–Condon transition (black) and from the −εHOMO (blue) for [Cu(NCS)2]1– (triangle), [Cu(SCH3)2]1– (square), FeCl4– (circle), and [Fe(SCH3)4]1– (diamond), using the DZVP2 basis set for hybrid functionals in order of increasing (short-range) HF exchange. From left to right, the functionals (open symbols) are B3LYP*, B97, B3LYP, PBE1PBE, M06, and B(38HF)P86. The symbols are connected by dotted lines to guide the eye, and the results for the Fe complexes are shifted upward by 3 eV to avoid overlaps. The experimental PES values (gray line with error indicated approximately by width of line) are also shown.
The poor prediction of VDE by the hybrid functionals may be due to (among other things) spin contamination, multireference effects, or errors in the exchange-correlation functional. Spin contamination due to the BS approach can be ruled out since the expectation values of the total spin S2 (calculated using M06/def2-SVP) of [CuL2]0 (S = 1/2), [FeL4]1– (S = 5/2), and [FeL4]0 (S = 2) with the ligands studied here are equal to 0.76, 8.77, and 6.18, respectively, which are almost the same as those of the pure states. In addition, since the HOMOs in the Cu and Fe complexes are degenerate, multireference problems are possible. Since empirically multireference character may be a problem in CCSD calculations when T1 diagnostic is greater than 0.0249 or the largest T2 amplitude of the double excitations is greater than 0.2, both conditions were checked; the conditions for both diagnostics are satisfied for the reduced states but not always for the oxidized states (Table S4). However, CCSD(T), which is often very effective in correcting for a single-reference treatment of weakly to moderately multireference problems,49 gives results that are in better agreement with experiment (Table S3), indicating a single-reference treatment is possible. DFT also appears less sensitive to multireference character than HF theory but, in a hybrid functional, is dependent on the amount of HF exchange included in the functional.49 In other words, while adding HF exchange tends to balance the self-interaction error in DFT that leads to overdelocalization, it is expected to give worse performance on static correlation65 such as the overestimated spin polarization found in systems containing transition metals.66 Since the VDE generally becomes more positive with % HF exchange for both Cu and Fe complexes in the hybrid functionals so that no functional works well for both (Figure 7); another option is to look for a better, more flexible description of exchange in the density functional.
Of the less parametrized RS functionals (BNL, CAM-B3LYP, and LRC-ωPBEh), the BNL functional (0% SR HF) appears to give a ground state with a different electron configuration from the B3LYP, M06, and CCSD(T) calculations for the vertical oxidation state of [Cu(SCH3)2]1– and too low VDE for both Cu complexes, while both CAM-B3LYP and LRC-ωPBEh with ∼20% SR HF exchange have reasonable ground states and VDE for both Cu complexes (Figure 8), which supports that some HF exchange at short-range is necessary as others have concluded.43 However, all three functionals suffer from having what appears to be the incorrect electron configuration for the ground state for at least one of the Fe complexes in the oxidized state: the ground states obtained by M06, B3LYP, and CCSD(T) are all symmetric with regard to distribution of the spins, while the ground states obtained by the RS functionals are generally asymmetric and are much like the electron configuration of the ground state obtained in a pure UHF calculation. Consequently, the VDEs are too high for the Fe complexes. The −εHOMO of the less parametrized RS functionals (BNL, CAM-B3LYP, and LRC-ωPBEh) are quite close to the VDE (Figure S1), although not so much for CAM-B3LYP since it has only 65% LR HF exchange. Of the highly optimized RS functionals (ωB97, ωB97X, and M11), the incorrect electron configuration of the oxidized ground state is obtained for most of the Cu and Fe complexes, and the VDEs are consistently too positive compared to experiment regardless of the amount of SR HF exchange (i.e., between 0 and 42.8% in Figure 8). Since they also have slightly higher −εHOMO than VDE (Figure S1), the εHOMO of the reduced state appears to have been increased too much. Thus, even for the RS functionals, no functional using default parameters (with the DZVP2 basis set) works well for both Cu and Fe complexes, although CAM-B3LYP and LRC-ωPBEh appear to work best since they at least give what appears to be the correct electron configuration of the ground state and reasonable VDE for the Cu complexes.
Figure 8.
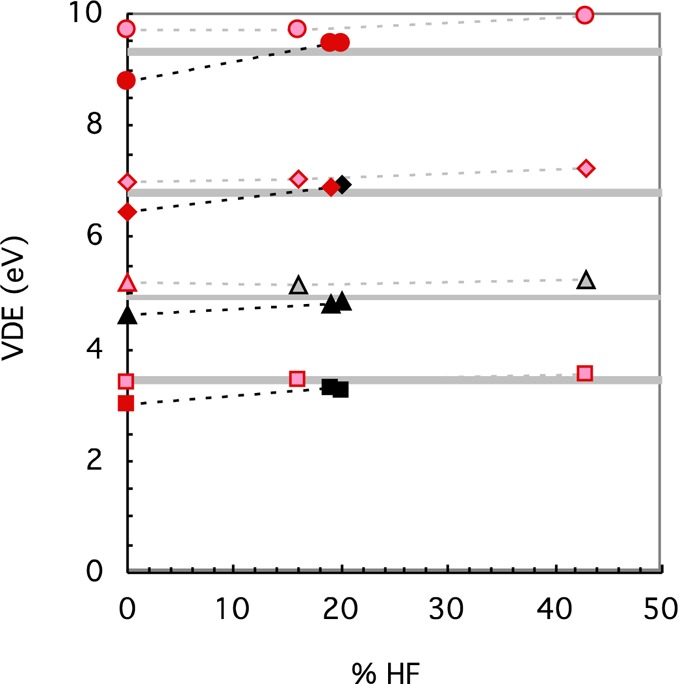
Calculated VDE from the difference in energy between the reduced form and a Franck–Condon transition for [Cu(NCS)2]1– (triangle), [Cu(SCH3)2]1– (square), FeCl4– (circle), and [Fe(SCH3)4]1– (diamond), using the DZVP2 basis set for RS functionals in order of increasing (short-range) HF exchange. From left to right, the RS functionals (solid symbols) are BNL, CAM-B3LYP, and LRC-ωPBEh, and the highly optimized RS functionals (lighter colored symbols) are ωB97, ωB97X, and M11. The functionals that give the incorrect ground state are highlighted in red. The symbols are connected by dotted lines to guide the eye, and the results for the Fe complexes are shifted upward by 3 eV to avoid overlaps. The experimental PES values (gray line with error indicated approximately by width of line) are also shown.
The M–L bond length and VDE (Tables 1 and 2, respectively) were also calculated using the def2-SVPD basis set for B3LYP, M06, CAM-B3LYP, and LRC-ωPBEh using default parameters; for comparison, appropriate values from experiment and CCSD and CCSD(T) using the def2-TZVPPD basis sets on the M06/def2-SVP geometry are also given. The “highly optimized” RS functionals were not investigated since the VDEs are too high for both the Cu and Fe complexes. Even with this larger basis set, although the RS separated functionals work well for the Cu complexes, the VDEs are too large for the Fe complexes compared to experiment and give an incorrect ground state similar to the DZVP2 results upon oxidation. However, the LRC-ωPBEh appears superior to CAM-B3LYP in the agreement of the VDE with −εHOMO, as expected since the latter has only 65% LR HF exchange.
Table 1. M–L Bond Length (Å) Using the def2-SVPD Basis Set.
| functional | [Cu(SCH3)2]1– | [Cu(NCS)2]1– | FeCl4– | [Fe(SCH3)4]1– |
|---|---|---|---|---|
| B3LYP | 2.206 | 1.828 | 2.251 | 2.326 |
| M06 | 2.187 | 1.818 | 2.226 | 2.307 |
| CAM-B3LYP (ω = 0.33) | 2.194 | 1.831 | 2.234a | 2.302 |
| CAM-B3LYP (ω = 0.20) | 2.191 | 1.820 | 2.236 | 2.306 |
| LRC-ωPBEh (47.3%ω = 0.20) | 2.194 | 1.837 | NA | NA |
| LRC-ωPBEh (20%ω = 0.20) | 2.186 | 1.828 | 2.227a | 2.293a |
| LRC-ωPBEh (20%ω = 0.10) | 2.190 | 1.825 | 2.234 | 2.301 |
| LRC-ωPBEh (10%,ω = 0.20) | 2.182 | 1.823 | 2.229 | 2.292 |
| LRC-ωPBEh (7.5%,ω = 0.20) | 2.181 | 1.822 | 2.229 | 2.291 |
| exp | 2.143(1) | 1.808(10) | 2.194(12) | 2.264(1) |
The ground state with “incorrect” electron configuration. See text.
Table 2. VDE (and −εHOMO) (eV) Using the def2-SVPD Basis Setd.
| functional | [Cu(SCH3)2]1– | [Cu(NCS)2]1– | FeCl4– | [Fe(SCH3)4]1– |
|---|---|---|---|---|
| B3LYP | 3.070 (1.259) | 4.357 (2.721) | 6.215 (4.317) | 3.740 (2.170) |
| M06 | 3.121 (1.359) | 4.524 (2.940) | 6.287 (4.435) | 3.766 (2.260) |
| CAM-B3LYP (ω = 0.33)a | 3.418 (2.787) | 4.845 (4.195) | 6.738c (6.000) | 4.152c (3.656) |
| LRC-ωPBEh (47.3%,ω = 0.2) | 3.535c (3.877) | 5.253 (5.378) | NA | NA |
| LRC-ωPBEh (20%,ω = 0.2)a | 3.360 (3.265) | 4.872 (4.708) | 6.706c (6.504) | 4.165c (4.177) |
| LRC-ωPBEh (20%,ω = 0.1) | 3.135 (2.395) | 4.517 (3.864) | 6.338 (5.524) | 3.836 (3.294) |
| LRC-ωPBEh (10%,ω = 0.2) | 3.304 (3.048) | 4.729 (4.436) | 6.364 (6.068) | 3.916 (3.883) |
| LRC-ωPBEh (7.5%,ω = 0.2) | 3.277 (2.966) | 4.694 (4.381) | 6.267 (5.959) | 3.837 (3.803) |
| CCSDb | 3.546 | 5.081 | 6.942 | 4.193 |
| CCSD(T)b | 3.433 | 4.89 | 6.519 | 3.816 |
| exp | 3.43(7) | 4.92(5) | 6.32(8) | 3.82(8) |
Default LRC parameters.
CC/def2-TZVPPD//M06/def2-SVP except [Fe(SCH3)4]1– are CC/def2-SVPD//M06/DZVP2.
The ground state with “incorrect” electron configuration upon oxidation. See text.
Best DFT values are in boldface (see text).
To investigate the source of the error in the RS functionals, the parameters altering the HF exchange were varied slightly using the def2-SVPD basis set. Again, the “highly optimized” RS functionals were not investigated because of the too high VDE cited above and because the other parameters for these functionals were optimized for a specific amount of HF exchange. In addition, CAM-B3LYP was not investigated because of the poor agreement of the VDE with the −εHOMO, and the condition of 100% LR HF exchange is becoming widely accepted. For instance, Baer and co-workers suggested that the range parameter ω is not necessarily “universal” to all molecules,67 while our results here indicate that some amount of short-range HF exchange is necessary. Decreasing ω results in less HF exchange at intermediate ranges and as ω→0, RS functionals became non-RS functionals, and RS hybrid functionals become non-RS hybrid functionals. Thus, ω was reduced in LRC-ωPBEh from the default 0.2 a0–1 to 0.1 a0–1, the correct ground state and much better agreement with experimental and CCSD(T) VDEs are obtained for the Fe complexes. However, the default ω values give better VDE for the Cu complexes as well as better agreement of the −εHOMO with the VDE for both Cu and Fe complexes. On the other hand, when the SR HF exchange was reduced to 10% and ω was kept at the default 0.2 a0–1 in LRC-ωPBEh, the VDEs for the Cu complexes are only slightly too low (0.1 to 0.2 eV) while that for the Fe complexes are slightly too high (0.07 eV) with respect to the experimental values. In addition, the agreement of the −εHOMO with VDE is somewhat better than when ω is reduced. Together, this implies that the default value of ω is close to correct, while the altering amount of SR HF in LRC-ωPBEh for transition metals may be more important, with 10% representing a possible compromise for Cu and Fe complexes.
Furthermore, a good option may be to optimize the parameters for including range-separation into the DFT exchange functional for each transition metal subject to certain additional criteria beyond the equality of −εHOMO with the SCF VDE such as those proposed by several workers.14,68 Although no attempt is made here to determine the best criteria for optimization, the optimal value of 47.3% SR HF exchange found for CuCl using the additional criteria of straight-line behavior of E(N), the energy as a function of a fractional electron number68 was examined for the Cu complexes using the LRC-ωPBEh. However, the best agreement occurs when the SR HF exchange is closer to 20 than 47.3% and ω = 0.2 a0–1 for the Cu complexes and 7.5% SR HF exchange and ω = 0.2 a0–1 are used for the Fe complexes. In addition, spin-polarization effects may be a good criteria since for the open shell, high-spin Fe complexes, spin polarization is generally important and a smaller amount of HF exchange is expected to improve performance, while for the closed shell, low-spin Cu-complexes, spin polarization tends to be less important and the correction of self-interaction error by HF exchange may be more important.
IV. Conclusions
Overall, the results here indicate that BS-DFT with RS functionals and double-ζ basis sets are able to model the electronic structure and geometry of two Cu complexes, [Cu(NCS)2]1– and [Cu(SCH3)2]1–, with metal–ligand bonds found in the blue copper protein redox sites, as well as the two Fe complexes used in previous studies for the Fe–S protein redox sites. Based on comparisons with triple-ζ basis sets with diffuse functions, the Karlsruhe double-ζ basis sets appear to be a good balance of size and accuracy for both geometry and energies as long as diffuse functions are used for energies. However, the VDE cannot be predicted reliably compared to experiment for both Cu and Fe complexes by hybrid density functionals. On the other hand, LRC-ωPBEh, a range-separated density functional, with 10% short-range and “exact” long-range HF exchange predicts VDE better than the hybrid functionals for both metals, although even better values are obtained when differing amounts of short-range HF exchange are used based on the metal type. This suggests that parameters in RS functionals chosen to balance compensating errors (i.e., HF exchange vs self-interaction error in DFT) may differ between transition metals, for instance, because of the conflicting requirements of the low-spin, closed shell Cu complexes versus the high-spin, open shell Fe complexes. Therefore, careful benchmarking of the DFT functionals for redox energetics of smaller complexes against experimental and/or CCSD(T) methods along with careful analysis of the ground states of both the oxidized and reduced forms still appears necessary for good VDE of protein redox sites containing transition metals, especially when using RS functionals. Moreover, the establishment of criteria for choosing different parameters for RS for transition metals is an important goal.
Acknowledgments
This work was supported by the National Institutes of Health under grant GM045303 (T.I.) and the National Science Foundation under grant CHE-1049717 (L.S.W.). The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of the U.S. Government. Computational resources from the William G. McGowan Foundation and Georgetown University (Matrix) are gratefully acknowledged. The calculations were performed at the EMSL, a national user facility sponsored by the U.S. DOE’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, operated for DOE by Battelle, under the grants EMSL38793 and st39962 and at Texas Advanced Computing Center (TACC) at the University of Texas at Austin under project ID TG-MCB050008N.
Supporting Information Available
Supplemental tables (S1–S4) of selected optimized geometries, calculated redox energies, and T1 and T2 values and Figure (S1) comparing VDE and −εHOMO for range-separated functionals. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Holland P. L.; Tolman W. B. J. Am. Chem. Soc. 1999, 121, 7270–7271. [Google Scholar]
- Holland P. L.; Tolman W. B. J. Am. Chem. Soc. 2000, 122, 6331–6332. [Google Scholar]
- Kitajima N.; Fujisawa K.; Morooka Y. J. Am. Chem. Soc. 1990, 112, 3210–3212. [Google Scholar]
- Kitajima N.; Fujisawa K.; Tanaka M.; Morooka Y. J. Am. Chem. Soc. 1992, 114, 9232–9233. [Google Scholar]
- Torelli S.; Belle C.; Philouze C.; Pierre J. L.; Rammal W.; Saint-Aman E. Eur. J. Inorg. Chem. 2003, 2452–2457. [Google Scholar]
- Gray H. B.; Malmstrom B. G.; Williams R. J. P. J. Biol. Inorg. Chem. 2000, 5, 551–559. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Yang W.. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, 1989. [Google Scholar]
- Levine I. N.Quantum Chemistry, 5th ed.; Prentice Hall: Upper Saddle River, NJ, 2000. [Google Scholar]
- Cramer C. J.; Truhlar D. G. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [DOI] [PubMed] [Google Scholar]
- Niu S. Q.; Hall M. B. Chem. Rev. 2000, 100, 353–405. [DOI] [PubMed] [Google Scholar]
- Niu S. Q.; Nichols J. A.; Ichiye T. J. Chem. Theory Comput. 2009, 5, 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson T. M.; Janesko B. G.; Scuseria G. E. J. Phys. Chem. A 2008, 112, 12530–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Hoyos C. A.; Janesko B. G.; Scuseria G. E. J. Phys. Chem. A 2009, 113, 11742–11749. [DOI] [PubMed] [Google Scholar]
- Refaely-Abramson S.; Sharifzadeh S.; Govind N.; Autschbach J.; Neaton J. B.; Baer R.; Kronik L. Phys. Rev. Lett. 2012, 109, 226405. [DOI] [PubMed] [Google Scholar]
- Wang X. B.; Wang L. S. J. Chem. Phys. 2000, 112, 6959–6962. [Google Scholar]
- Wang X. B.; Niu S. Q.; Yang X.; Ibrahim S. K.; Pickett C. J.; Ichiye T.; Wang L. S. J. Am. Chem. Soc. 2003, 125, 14072–14081. [DOI] [PubMed] [Google Scholar]
- Niu S. Q.; Wang X. B.; Nichols J. A.; Wang L. S.; Ichiye T. J. Phys. Chem. A 2003, 107, 2898–2907. [Google Scholar]
- Niu S. Q.; Ichiye T. Mol. Simul. 2011, 37, 572–590. [Google Scholar]
- Yang X.; Niu S. Q.; Ichiye T.; Wang L. S. J. Am. Chem. Soc. 2004, 126, 15790–15794. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang X. B.; Fu Y. J.; Wang L. S. J. Phys. Chem. A 2003, 107, 1703–1709. [Google Scholar]
- Niu S. Q.; Ichiye T. J. Phys. Chem. A 2009, 113, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S. Q.; Ichiye T. J. Am. Chem. Soc. 2009, 131, 5724–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P. V.; Holm R. H. Chem. Rev. 2004, 104, 527–559. [DOI] [PubMed] [Google Scholar]
- Perrin B. S. Jr.; Niu S.; Ichiye T. J. Comput. Chem. 2013, 34, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout N.; Salahub D. R.; Andzelm J.; Wimmer E. Can. J. Chem. 1992, 70, 560–571. [Google Scholar]
- Weigend F.; Ahlrichs R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- Hariharan P. C.; Pople J. A. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- Francl M. M.; Petro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Rassolov V.; Pople J. A.; Ratner M.; Windus T. L. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar]
- Rappoport D.; Furche F. J. Chem. Phys. 2010, 133, 134105. [DOI] [PubMed] [Google Scholar]
- Clark T.; Chandrasekhar J.; Schleyer P. V. R. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; Defrees D. J.; Pople J. A. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Dunning T. H. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar]
- Balabanov N. B.; Peterson K. A. J. Chem. Phys. 2005, 123, 64107. [DOI] [PubMed] [Google Scholar]
- Salomon O.; Reiher M.; Hess B. A. J. Chem. Phys. 2002, 117, 4729–4737. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar]
- Adamo C.; Barone V. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Chem. Phys. 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- Szilagyi R. K.; Metz M.; Solomon E. I. J. Phys. Chem. A 2002, 106, 2994–3007. [Google Scholar]
- Livshits E.; Baer R. Phys. Chem. Chem. Phys. 2007, 9, 2932–2941. [DOI] [PubMed] [Google Scholar]
- Stein T.; Kronik L.; Baer R. J. Am. Chem. Soc. 2009, 131, 2818–2820. [DOI] [PubMed] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar]
- Rohrdanz M. A.; Martins K. M.; Herbert J. M. J. Chem. Phys. 2009, 130, 054112. [DOI] [PubMed] [Google Scholar]
- Chai J. D.; Head-Gordon M. J. Chem. Phys. 2008, 128, 084106. [DOI] [PubMed] [Google Scholar]
- Peverati R.; Truhlar D. G. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar]
- Baer R.; Livshits E.; Salzner U. Annual Review of Physical Chemistry, Vol 61 2010, 61, 85–109. [DOI] [PubMed] [Google Scholar]
- Seeger R.; Pople J. A. J. Chem. Phys. 1977, 66, 3045–3050. [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma T. P.; Van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; de Jong W. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar]
- Cramer C. J.Essentials of Computational Chemistry: Theories and Models, 2nd ed.; Wiley: Chichester, West Sussex, England; Hoboken, NJ, 2004. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, 2009.
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; DiStasio R. A.; Lochan R. C.; Wang T.; Beran G. J. O.; Besley N. A.; Herbert J. M.; Lin C. Y.; Van Voorhis T.; Chien S. H.; Sodt A.; Steele R. P.; Rassolov V. A.; Maslen P. E.; Korambath P. P.; Adamson R. D.; Austin B.; Baker J.; Byrd E. F. C.; Dachsel H.; Doerksen R. J.; Dreuw A.; Dunietz B. D.; Dutoi A. D.; Furlani T. R.; Gwaltney S. R.; Heyden A.; Hirata S.; Hsu C. P.; Kedziora G.; Khalliulin R. Z.; Klunzinger P.; Lee A. M.; Lee M. S.; Liang W.; Lotan I.; Nair N.; Peters B.; Proynov E. I.; Pieniazek P. A.; Rhee Y. M.; Ritchie J.; Rosta E.; Sherrill C. D.; Simmonett A. C.; Subotnik J. E.; Woodcock H. L.; Zhang W.; Bell A. T.; Chakraborty A. K.; Chipman D. M.; Keil F. J.; Warshel A.; Hehre W. J.; Schaefer H. F.; Kong J.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [DOI] [PubMed] [Google Scholar]
- Black G. D.; Schuchardt K. L.; Gracio D. K.; Palmer B.. The Extensible Computational Chemistry Environment: A Problem Solving Environment for High Performance Theoretical Chemistry. In Computational Science - ICCS 2003 Software Version 6.4 ed.; Sloot P. M. A., Abramson D., Bogdanov A. V., Dongarra J., Eds.; Springer Verlag: Berlin, Germany, International Conference Saint Petersburg Russian Federation, Melbourne, Australia, Proceedings 2660, 2003; Vol. 10, pp 122–131. [Google Scholar]
- Wang L. S.; Ding C. F.; Wang X. B.; Barlow S. E. Rev. Sci. Instrum. 1999, 70, 1957–1966. [Google Scholar]
- Liu H. T.; Wang Y. L.; Xiong X. G.; Dau P. D.; Piazza Z. A.; Huang D. L.; Xu C. Q.; Li J.; Wang L. S. Chem. Sci. 2012, 3, 3286–3295. [Google Scholar]
- Dau P. D.; Liu H. T.; Huang D. L.; Wang L. S. J. Chem. Phys. 2012, 137, 116101. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang X. B.; Wang L. S.; Niu S. Q.; Ichiye T. J. Chem. Phys. 2003, 119, 8311–8320. [Google Scholar]
- Bruno I. J.; Cole J. C.; Edgington P. R.; Kessler M.; Macrae C. F.; McCabe P.; Pearson J.; Taylor R. Acta Crystallogr., Sect. B 2002, 58, 389–397. [DOI] [PubMed] [Google Scholar]
- Kohner-Kerten A.; Tshuva E. Y. J. Organomet. Chem. 2008, 693, 2065–2068. [Google Scholar]
- Watson W. H.; Kashyap R. P. Acta Crystallogr., Sect. C 1990, 46, 2312–2316. [Google Scholar]
- Masuda Y.; Kuratsu M.; Suzuki S.; Kozaki M.; Shiomi D.; Sato K.; Takui T.; Okada K. Polyhedron 2009, 28, 1950–1954. [Google Scholar]
- Maelia L. E.; Millar M.; Koch S. A. Inorg. Chem. 1992, 31, 4594–4600. [Google Scholar]
- Hadt R. G.; Sun N.; Marshall N. M.; Hodgson K. O.; Hedman B.; Lu Y.; Solomon E. I. J. Am. Chem. Soc. 2012, 134, 16701–16716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. J.; Xu X. F.; Truhlar D. G. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar]
- Almbladh C. O.; Vonbarth U. Phys. Rev. B 1985, 31, 3231–3244. [DOI] [PubMed] [Google Scholar]
- Cohen A. J.; Mori-Sanchez P.; Yang W. T. Science 2008, 321, 792–794. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- Kronik L.; Stein T.; Refaely-Abramson S.; Baer R. J. Chem. Theory Comput. 2012, 8, 1515–1531. [DOI] [PubMed] [Google Scholar]
- Srebro M.; Autschbach J. J. Phys. Chem. Lett. 2012, 3, 576–581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.