

Abstract
The reactive oxygen species produced continuously during oxidative metabolism are generated at very high rates in the brain. Therefore, defending against oxidative stress is an essential task within the brain. An important cellular system against oxidative stress is the thioredoxin system (TS). TS is composed of thioredoxin, thioredoxin reductase, and NADPH. This review focuses on the evidence gathered in recent investigations into the central nervous system, specifically the different brain regions in which the TS is expressed. Furthermore, we address the conditions that modulate the thioredoxin system in both, animal models and the postmortem brains of human patients associated with the most common neurodegenerative disorders, in which the thioredoxin system could play an important part.
1. Introduction
The thioredoxin system (TS) consists of an electron donor and two types of antioxidant oxidoreductase proteins: thioredoxin (Trx) and thioredoxin reductase (TrxR) and NADPH as the electron donor. Trx was first identified as a hydrogen donor for ribonucleotide reductase in Escherichia coli [1]. Trx is a small 12 kD protein that has an active conserved site, Cys-Pro-Gly-Cys, which is essential for its function as both an active oxidoreductase and an electron donor of some peroxiredoxins that are important molecules for the reduction of peroxides [2]. Trx is also a regulator of cellular functions in response to redox signals and stress, modulating various signaling pathways, transcription factors, and immunological responses [3]. Trx is an important regulator of redox balance in the cell and has been implicated as playing a role in cell survival in many conditions including cancer and neurodegenerative diseases [4]. Human cells contain 3 different thioredoxins [5]. Trx1 has been reported as cytoplasmic, Trx2 as a mitochondrial form, and a third variant highly expressed in spermatozoa. Trx1 has been located in several cell compartments such as the nucleus and the plasma membrane or as a secreted protein [6, 7]. Posttranslational modifications to cysteine on Trx1 appear critical to its localization and function in different cell types [7]. Organelles such as the mitochondrion and nucleus require Trx to preserve a local reducing environment to minimize damage from ROS leakage during mitochondrial respiration [8]. In the nucleus the activation of transcription factor requires the presence of reduced Trx [9]. Cytosolic Trx1 is important in the control of growth and apoptosis and during chronic inflammation; likewise Trx1 is also secreted as a cocytokine and for chemokine activities [5].
TrxR is a homodimer, first described in bovine tissue by Holmgren and Luthman in 1978 [10], which catalyzes the reduction of the disulfide at the Trx active site, using NADPH with one FAD cofactor per subunit and a selenocysteine active site [5, 8]. There are three distinct genes in mammals that encode three different TrxRs: the cytosolic TrxR (TrxR1), mitochondrial TrxR (TrxR2), and thioredoxin-glutaredoxin reductase (TGR o TrxR3). TrxR1 and TrxR2 are expressed in all mammalian cells and tissues, while TrxR3 is expressed in the testicles [11].
Besides Trx, TrxR can directly reduce the number of other substrates, such as peroxides (including lipid hydroperoxides), hydrogen peroxide [12, 13], and protein disulfide isomerases, which participate in the posttranslational folding and processing of cellular proteins [14]. TrxR also participates in the regeneration of some antioxidant molecules with antioxidant activity such as dehydroascorbate [15, 16], lipoic acid [17], and ubiquinone [18].
The brain is more susceptible to oxidative damage compared with other organs, due to several factors that promote the formation of reactive species: high oxygen consumption, high iron levels found in some brain regions, and high fat content of unsaturated fatty acids, accompanied by low levels and low activities of some antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (Gpx) [19]. Both Trx and TrxR are widely expressed in tissues and organs; their distribution seems to be tissue and cell specific [20], including the brain tissue in which Trx and TrxR are found.
This review discusses the expression of the TS in different brain regions and cells and the participation of the TS in neurotoxic insults and the variety of neurodegenerative disorders where oxidative stress plays a key role.
2. Protein or mRNA Expression of Trx and TrxR in the Nervous System
The identification and localization of Trx and TrxR in the different brain regions have been made possible mostly through the use of immunochemistry techniques using monoclonal and polyclonal antibodies and by in situ hybridization techniques. Differentiation between the different isoforms is not always mentioned in the reports (Table 1). However, Trx, probably Trx1, due to its cytoplasmic localization, has been detected in the human brain and that of several mammal species including the rat, gerbil, cow (a yearling calf, more precisely), and mouse [10, 21–24]. Trx and TrxR were first identified in the sciatic nerve of the rat, in which both proteins showed strong cytoplasmic immunoreactivity in the Schwann cells at the Ranvier nodes and neuronal cells [20, 21]. Studies in rats demonstrate high levels of Trx mRNA in regions with high energy demands and high activity that involves redox reactive metabolites including the substantia nigra and the subthalamic nucleus. According to the authors, this suggests that the TS participates in the maintenance of the redox homeostasis in these regions. At the same time, the C1 area of the hippocampal formation shows very small expression in contrast to CA2/CA3 and the dental gyrus of the hippocampus. These are regions in contact with peripheral blood such as the choroid plexus which expresses a significant quantity of Trx mRNA [25]. Godoy et al. (2011) reported immunoreactivity to Trx1 in the Purkinje cell layer of the rat, as well as the motor neurons of the spinal cord, ependymal cell layer, and the cells of the choroid plexus. In contrast with Trx1, TrxR1 was abundantly expressed in the glial cells of the cerebellar white matter. Trx2 (mitochondrial Trx) was detected in the axonal fibers of the cerebral cortex, striatum, cerebellar white matter, and spinal cord, while TrxR2 expression was pronounced in the cell bodies of neurons found in the Purkinje and molecular cell layers in the cerebellum [24]. Godoy also evaluated the expression of Trx1, Trx2, TrxR1, and TrxR2 in the mouse brain, assessing the presence of the protein semiquantitatively (see Table 1) [26]. The mRNA and protein localization of Trx2 and Trx1 differ in some regions such as the hippocampus, and it is proposed that posttranscriptional regulation of Trxs may occur in this region. Another important observation is that in the rat Trx1 and Trx2 expression occurs predominately in brain neurons [25, 27], while TrxR protein levels are higher in glial cells than in neurons in both rat and mice cell cultures [20, 28]. These findings suggest that the functional needs and requirements of the TS molecules are different in each type of cell [25, 27].
Table 1.
TS expression in different species and brain regions.
| Species | Protein | Findings (localization/expression) | Detection method | Reference |
|---|---|---|---|---|
| Calf | Trx | ↑ Kidney, liver, brain, thymus | Radio immunoassay, rabbit antiserum, calf liver Trx and 125I-labeled Trx | [10] |
|
| ||||
| Rat sciatic nerve | Trx | ↑ Cytoplasm of Schwann cells Nodes of Ranvier |
Immunofluorescence with specific rabbit antisera. | [21] |
| TrxR | ↓ Axoplasm of myelinated axons | |||
|
| ||||
| Gerbil brain | ADF/Trx | ↓ Ependyma, tanycytes Endotheliall cells Neuronal cell bodies |
Immunochemisty anti-human ADF antibody | [22] |
|
| ||||
| Rat brain | Trx | ↑ Paraventricular hypothalamic Nucleus Locus coeruleus Nucleus of the solitary tract |
In situ hybridization Human Trx mRNA |
[25] |
| ↓ Frontoparietal cortex Caudate/putamen Magnocellular preoptic nucleus | ||||
|
| ||||
| Human brain | ADF/Trx | White matter astrocytes Schwann cells of posterior root |
Immunochemistry | [23] |
| White matter astrocytes | In situ hybridization and semiquantitative mRNA | |||
|
| ||||
| Mouse | Trx1 |
Nucleus of granular cells in hippocampus Golgi cells of substantia nigra Purkinje cells Motor neurons of the spinal cord |
Anti-mouse Trx1 | [24] |
| Trx2 |
Golgi cells of Substantia nigra Axonal staining in cerebral cortex Axons in striatum Axonal staining in cortex Axon-bundles in striatum Golgi cells of substantia nigra |
Anti-human Trx2 | ||
| TR1 | Faint staining in hippocampus pyramidal cells | Anti-rat TrxR1 | ||
| TR2 | Golgi cells of substantia nigra Strong staining in Purkinje cells Molecullar layer in cerebellum |
Anti-rabbit TrxR2 (Santa Cruz Bio. sc-67127) |
||
|
| ||||
| Rat brain | Trx1 | ↑ Cerebellum Cortex Substantia nigra Retinal Spinal cord ↓ Striatum hippocampus |
Anti-mouse Trx1 | [26] |
| Trx2 | ↑ Striatum Substantia nigra ↓ Cerebellum Hippocampus Cortex, Retinal Spinal cord |
Anti-human Trx2 | ||
| TR1 | ↑ Cerebellum Hippocampus Striatum ↓ Cortex Substantia nigra Spinal cord Retina |
Anti-rat TrxR1 | ||
| TR2 | ↑ Cerebellum ↓ Striatum |
Anti-rabbit TrxR2 (Santa Cruz Bio. Sc-67127) |
||
↑: high protein content; ↓: low protein content. The origin of the antibodies employed is mentioned when provided in the reference cited.
Immunoreactivity of rabbit antiserum against rat liver Trx is found in the epithelial cells and secreting cells of the rat choroid plexus [20]. A human Trx homologue, adult T leukemia-derived factor (ADF/TRX), has been found to be widely expressed in the central nervous system (CNS); this includes the subcommissural organs, ependymal, tanycytes, and endothelial cells, as well as in the neuronal cell bodies of gerbils, albeit weakly [22], and the white matter astrocytes and Schwann cells in the posterior root of the human brain [23]. A truncated Trx1, thioredoxin 80 (Trx80), is present in the human brain in an aggregated form, principally in neurons [29]. The main role described for Trx80 is to activate monocytes, cause proliferation, and secrete pro- and anti-inflammatory cytokines [30].
Despite its function as an intracellular disulfide reducing protein, Trx has been found in extracellular components, secreted mainly by activated T and B lymphocytes [31]. Studies have established that different forms of stimulation can cause different cells including nerve and glial cells to secrete Trx [32]. TrxmRNA has been localized also in epithelial cells of the choroid plexus and the ependymal cells of the ventricle and the secreting cells of the choroid plexus [25, 28]. The secretion of Trx into the cerebrospinal fluid may help protect nerve cells from oxidation by environmental influences maintaining a protective microenvironment [25]. Stemme et al. (1985) [21] and LoPachin and Barber (2006) [33] showed anterograde and retrograde axoplasmic transport of Trx and TrxR in stressed rat sciatic nerve cells; this transport to synaptic terminals may be involved in thiol redox reactions related to synaptic transmission, such as membrane pore formation by the participation of specific cysteine residues that modulate regulatory proteins. These observations are indirectly supported by the evidence showing the high sensitivity of the synaptic process to modifying nucleophilic sulfhydryl groups with different electrophilic neurotoxicants. Astrocytoma cells exposed to H2O2 release Trx1 into the culture medium. The addition of this medium to neuron cultures promotes their survival in the absence of serum [34]. These observations support the view that glial cells provide neurotrophic and antioxidant support for the neurons. The levels of Trx that are secreted by cells depend on their stage of metabolic activity (Figure 1) [25].
Figure 1.
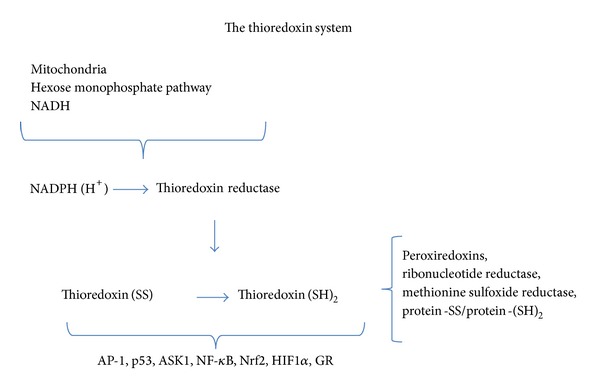
TS components are NADPH, TrxR, and Trx. NADPH is the electron donor for TrxR. Cytosolic NADPH generation principally occurs in the hexose monophosphate pathway, with mitochondrial NADPH production depending on specific dehydrogenases and the transference of electrons from NADH to NADP+ [35]. Trx acts as the reducing agent for peroxiredoxins, ribonucleotide reductase, methionine sulfoxide reductase, and disulfides in proteins including activating protein 1 (AP-1), tumour suppressor p53, apoptosis signal-regulating kinase-1 (ASK1), nuclear factor erythroid 2-related factor 2 (Nrf2), hypoxia inducible factor 1α (HIF1α), nuclear factor κB (NF-κB), and glucocorticoid receptor (GR) [36].
3. TS Modulation by Stress and Chemical Compounds
Trx expression is induced by stress, such as that produced by infectious agents, UV radiation, or O2 [37]. Furthermore, also many physicochemical agents and stimuli induce Trx gene expression, including hormones and nontoxic agents. CNS studies report a close association between the increased expression of Trx and TrxR and cell damage where oxidative stress is implicated. For example TS is upregulated in postmortem examination of AD where also oxidative stress has been documented [23, 38]. Mechanical nerve injury, such as sciatic nerve crush, induces TS components in rats [21]; as well middle cerebral artery occlusion [39, 40], hypoglossal nerve axotomy [41], and transient focal ischemia do so in both rats and gerbils [22, 42]. Exposure to several toxic chemicals that induce oxidative stress upregulates TS proteins. Enhanced immunoreactivity to Trx in the hippocampus and striatum is induced when rats are exposed to 3-nitropropionic acid, a mitochondrial complex II toxin [43]. The environmental pollutant formaldehyde has toxic effects on the CNS [44]. Trx1 expression increases in PC12 cells exposed to formaldehyde. This upregulation decreases if the exposure time increases [45]. Morphine, the most effective opioid analgesic, has pharmacological effects associated with cellular redox state [46]. SH-SY5Y cells exposed to morphine show augmented expression of Trx1, activating the opioid receptor and the phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinases (ERK) signaling pathways [47]. Trx expression is also induced without stressor components such as compounds present in dietary intake; for example, fish oil increases the activity of TrxR in rat brain [48]. While t-bhq (t-butyl hydroquinone) increases the expression and activity of TrxR1 and TrxR2 in astrocytes, it does not in neurons [49]. Also, 17-β estradiol induces Trx protein expression in SH-SYE5Y cells, while the estrogen receptor activation is ligated to the upregulation of cytoprotective genes, including Trx via a cyclic guanosine monophosphate (cGMP) mediated signaling pathway [50].
Other studies have reported a downregulation or inhibition of the TS proteins. The effect may depend on the exposure time, dose, or the nature of the compound. Mice exposed to different concentrations of arsenic for four months showed that diminished Trx1 mRNA levels were in male striatum and the female nucleus accumbens [51]. Tellurium is present in optic and electronic technology, in batteries, and as an environment contaminant [52]. Diphenyl telluride induced prominent effects in mouse brain, including decreased TrxR activity [53]. Mercury compounds are accumulated in seafood and fish and readily cross the blood brain barrier [54]. Exposure to mercury compounds reduced TrxR activity in zebra fish brains, causing neurotoxicity through oxidative stress in this target organ and other organs such as the kidney [55]. The upregulation of the TS in response to different stressors is associated with neuronal survival mechanisms, which can protect against cell or tissue damage, while the inhibition or downregulation of TS leads to increased damage and cell death.
The promoter region for the constitutive expression of Trx1 contains various transcription factor binding sites, such as transcription factor SP1, GC-rich sequence DNA-binding factor (GCF), and wild type zinc finger (WT-ZFP), while the promoter regions for the inducible expression binding sites are AP-1, activating protein 2 (AP-2), NF-κB, octamer binding transcription factor (Oct-1), polyoma enhancer activator 3 (PEA-3), myeloblastosis transcription factor (Myb), and the antioxidant-responsive element (ARE) [56]. The augmented expression of Trx1 and the activation of Nrf2 were observed in the peri-infarct regions of rats after middle artery occlusion [57] and prevented light-induced photoreceptor degeneration [58]. The presence of ARE is required for the induction of Trx1 in SH-SY5Y cells after hemin exposure and requires Nrf2 nuclear translocation downstream PI3K [59].
4. TS and Neuroprotection
Trx induction contributes to brain tolerance for and protection from toxic insults. Rats treated with selenium after receiving quinolinic acid treatment, a potent neurotoxin, show increased levels of protein and increased activity of TrxR1, ameliorating quinolinic acid damage [60]. Pretreatment with beta estradiol 3-benzoate ameliorates the injury induced by ferrous citrate in female rat brain. This protective effect is accompanied by increased Trx levels and activity [61]. The use of electroacupuncture produces clinically beneficial effects in stroke patients [62] and induces Trx expression in ischemic-reperfused rat brain [63]. Selegiline improves behavioral and cognitive functions in AD and PD. Selegiline protects SH-SY5Y cells against MPP+-induced neurotoxicity through the induction of the Trx gene via protein kinase A mediated by mitogen-activated protein kinases/extracellular signal regulated protein kinase 1/2 (MAP Erk1/2) and protooncogene protein c-Myc [64]. Microtubule associated protein-2 (MAP-2) and Tau protein are important in promoting and maintaining the neuronal cytoskeleton [65]. NOO− and H2O2 induce thiol oxidation and disulfide formation in Tau and MAP-2, thus altering the ability of proteins to improve the assembly of microtubules in vitro from purified porcine tubulin. Treatment with TrxR restores the ability of MAPs to promote microtubule assembly [66]. Trx2 prevents the neurotoxicity that results from protein misfolding, due to the protein refolding effect of Trx on scrambled (mispaired disulfide-containing) RNase A and protein disulfide isomerase activity [67, 68].
Other experimental approaches enhanced the levels of Trxs expression in vivo and in vitro. Transgenic mice that overexpress human Trxs show enhanced levels of this protein in the brain. These mice show attenuated focal cerebral ischemic damage [40], seizures, and excitotoxicity induced by kainate [69], delayed retinal neurodegeneration in Tubby mice (a mouse model for retinal degeneration and loss of visual function). Tubby mice protection ocurred via Akt survival signal pathways and by increasing both the brain-derived neurotrophic factor (BDNF) and the glial cell line-derived neurotrophic factor (GDNF). In this experimental model, Trx overexpression inhibits the ASK1/JNK pathway [70, 71]. The overexpression or administration of human recombinant Trx (rTrx) on PC12 cells attenuates MPP+ neurotoxicity [72, 73]. Overexpression of human Trx1 and Trx2 protects retinal ganglion cells against oxidative stress-induced neurodegeneration [74]. Trx2 human overexpression in SH-SY5Y neuroblastoma cells prevents apoptosis and loss of the mitochondrial membrane potential induced by tert-butyl hydroperoxide [75]. The use of human rTrx has a protective effect in which the generation of reactive oxygen species (ROS) is involved in cytotoxic mechanisms [76]. Exogenously administered human rTrx ameliorates neuronal damage after transient middle cerebral artery occlusion in mice [42], reduces oxidative/nitrative stress and neuronal apoptosis after cerebral ischemia/reperfusion injury in mice [77], and augments neurogenesis following brain ischemia/reperfusion (I/R) injury in rats [78]. Studies in vitro demonstrate that the administration of rTrx increased neuronal cell survival in murine primary cultured neurons [34].
Other studies have described the preconditioning mechanisms as neuroprotection strategies that induce TS proteins and other antioxidant proteins. In vivo studies, in rats, show that hypobaric hypoxia preconditioning enhances Trx1 and Trx2 protein expression [79, 80]. In vitro studies show that the transient serum depletion of SH-SY5Y cells produces a hormetic response increasing Trx1 levels [81], which contributes to neuronal tolerance and protection against a posterior oxidative stress exposure. This type of Trx induction belongs to adaptive group of cytoprotective responses, allowing potentially recurrent stressors the survival to potentially recurrent stressors.
Mitochondrion is considered an important source of ROS, and the antioxidants systems play a significant role in this organelle. The two major scavenging systems in this organelle are GSH and Trx2. Trx2, together with GSH, plays an important role in the detoxification of H2O2 in the mitochondria of different types of brain cells in the rat hippocampus, to a greater extent even than other enzymes such as catalase [82]. Cellular GSH concentration ranges from ~2 to 10 mM depending on cell type in different species [19, 83], while Trx (isoform not specified) baboon tissues concentrations tend to be around ≤10 μM, specifically in the brain 381 ± 110 pg/mg of protein [84]. Trx2 plays an important role in reducing other antioxidants, including peroxiredoxin 3 (Prx3), which is an antioxidant enzyme found exclusively in mitochondria [85]. Changes in Trx2 and Prx3 expression in the gerbil hippocampus after ischemic reperfusion may be associated with delayed neuronal death. The administration of Prx3 and Trx2 in ischemic brains shows substantial neuroprotective effects that reduce the oxidative stress induced by ischemia [86]. Trx2 plays an important role in the control of oxidative stress in mitochondria. Neurons with mitochondrial dysfunction (complex IV inhibition) show low levels of Trx mRNA and protein and are thus more vulnerable to H2O2. This vulnerability could be associated with the downregulation in the TS [87].
5. TS in Neuronal Development and Protection
Trx-2 and Trx-1 knockout mice present early embryonic lethality. Trx (isoform not specified) knockout mice embryos die shortly after implantation, and the concepti were resorbed prior to gastrulation, due a failed proliferation [88]. Studies in vivo and in vitro, deficient in Trx2, display increased cellular ROS, apoptosis, exencephaly, and early embryonic lethality [89, 90]. This evidence demonstrates that both Trx isoforms have essential roles in neuronal differentiation, proliferation, and survival. TS maintains a reductive environment in cells. Trx not only works as an antioxidant but also has other key biological activities, including growth control and antiapoptotic functions [91].
The nerve growth factor (NGF) is a neurotrophic factor playing an essential role in the development and promotion of survival and function of the CNS [92]. Likewise, Trx has protective effects that enhance the action of nerve growth factor via the regulation of antiapoptotic signaling and Trx's antioxidant activity. NGF induces Trx mRNA and protein levels via cyclic AMP responsive element (CREB) as well the nuclear translocation of Trx. The overexpression of the dominant negative type of Trx expression vector resulted in suppression of NGF-induced neurite outgrowth in PC12 cells, playing a critical regulatory role in NGF-mediated signaling transduction and outgrowth in PC12 cells [93, 94], via ERK [95]. Thus, Trx is a neurotrophic cofactor that augments the effect of NGF on neuronal differentiation and regeneration, showing neurotrophic activity in cholinergic neurons [96]. Trx is beneficial in cases of neurodegenerative disease, promoting neural-cell growth and aiding recovery [94].
Mechanisms by which thioredoxin regulates cell growth include binding to signaling molecules such as ASK-1 and thioredoxin-interacting protein (Txnip). ASK1 activates the c-Jun N terminal kinase (JNK) and p38 MAP kinase pathways and requires tumor necrosis factor (TNF-α) to induce apoptosis. Reduced Trx prevents apoptosis via an inhibitory binding to ASK1, which is lost when Trx is oxidized, which is mentioned later in this review. Trxip is an endogenous regulator of Trx that, with high affinity, binds to Trx and inhibits its ability to reduce sulfhydryl groups via NADPH oxidation and reduces the binding of Trx with ASK1 promoting an ASK1 apoptosis mediated pathway [97]. Evidence has established Trxip as a potent metabolic control protein [98, 99]. Several studies describe the control of the expression of Trxip during different conditions in the brain. Diabetic rat brains showed enhanced levels of Trxip mRNA, while Trx1 protein expression is enhanced after exercise in normal rats but not in diabetic rats [100, 101]. Trxip protein expression is induced in hyperglycemic-ischemic mice brains after middle cerebral artery occlusion [102]. Intravitreal NMDA injection augmented the expression of Trxip in rats, which was accompanied by both the release of inflammatory mediators TNFα and interleukin-1β (IL-1β) via ASK1 and the activation of the proapoptotic p38 MAPK/JNK pathway [103]. Exposure to silver nanoparticles induces the expression of the Trxip gene in different regions of the mouse brain [104]. The activation of ASK1 and the increase of Trxip levels produce apoptosis and neurotoxicity, making the cell more vulnerable to death. Hardingham and Bading [105] demonstrate that synaptic NMDAR activity inactivates Trxip via Forkhead box protein O (FOXO) transcription factor, enhancing Trx activity. NMDA receptor overactivity named “excitotoxicity” increased Ca2+ uptake by the mitochondria inducing ROS production. Thus Trx enhanced activity by NMDA receptor activity could reduce cell vulnerability to oxidative damage [105–107].
6. TS and Alzheimer's Disease
A common feature of Alzheimer's Disease (AD), Parkinson's Disease (PD), and Amyotrophic Lateral Sclerosis (ALS) is the extensive evidence of oxidative stress, which might be responsible for the dysfunction and death of neuronal cells that contribute to the pathogenesis of these diseases [108].
AD is the most common form of adult onset dementia. It is characterized by the presence of interneuronal filamentous inclusions, known as neurofibrillary tangles (NFT), and extracellular senile plaques (SP). Hyperphosphorylated Tau is the major protein involved in NFT. Amyloid beta peptide (Aβ), derived from the amyloid precursor protein, is the major protein in SP and amyloid angiopathy [109].
There is direct evidence that supports the theory of increased oxidative stress in the AD brain: (1) increased brain mercury, iron, and aluminum, capable of stimulating free radical generation, (2) increased lipid peroxidation, (3) increased protein and DNA oxidation, (4) diminished energy metabolism and decreased cytochrome c oxidase, (5) advanced glycation end products, malondialdehyde, carbonyls, peroxynitrite, heme oxygenase 1, and SOD-1 in NFT and SP, and (6) Aβ capability to generate free radicals. Overall, evidence suggests that free radicals are possibly involved in the pathogenesis of neuron death in AD and that the antioxidant systems could have an important role in the prevention and control of AD [110].
In AD brains, the ADF/Trx expression in astrocytes of white matter increased (Table 2) [23], while it was found to decrease in some regions of AD brains, in comparison with the controls [38, 111]. Trx80 is also drastically decreased in AD brains. Trx80 inhibits Aβ aggregation and protects against its toxicity, reducing neuronal vulnerability [29]. In amnestic mild cognitive impairment (AMCI, a transition stage between normal aging and AD), brains examined postmortem were characterized by diminished Trx-1 levels in the hippocampus and cerebellum [112]. Rats exposed to chronic intermittent hypoxia exposure, a reversible cause of cognitive loss in patients with AD [113], show impaired spatial learning and memory that are negatively correlated with Trx protein and ARN levels in the hippocampus [114]. Nevertheless, TrxR activity was increased in the cerebellum and amygdala of AD brains, suggesting that TrxR activities increase, perhaps as a compensatory mechanism in the face of increased oxidative stress that is limited by the substrate Trx, and could contribute to the general increase in oxidative stress and subsequent neurodegeneration seen in AD [38].
Table 2.
TS and CNS disorders.
| Disorder | Cell type studied | TS expression | Reference |
|---|---|---|---|
| AD | AD human brain | ↑ ADF/Trx (p, mRNA) astrocytes in white matter | [23] |
| ↓ Trx1 (p) amygdala, hippocampus, and frontal cortex | [38, 111] | ||
|
↓ Trx80 (p) neurons of hippocampus and cortex ↓ Trx80 (p) in CSF |
[29] | ||
| AMCI human brain | ↑ TR (a) hippocampus and cerebellum | [112] | |
| SH-SY5Y cells exposed to Aβ | ↑ Trx1 oxidized | [111] | |
|
| |||
| PD | PC12 cells exposed to MPP+ |
↓ Trx1 ↓ Trx2 ↑ Oxidized Trx1 Trx1-SS ↑ Oxidized Trx2-SS |
[132] |
| SK-DAT cells exposed to paraquat | Oxidizes Trx1 | [131] | |
| SK-DAT cells exposed to rotenone and MPP+ | Oxidizes Trx2 | ||
| SH-SY5Y cells exposed to paraquat | Oxidizes Trx2 | [133] | |
| SH-SY5Y cells exposed to Maneb | ↑ TR1 (mRNA) | ||
|
| |||
| HD | HD patients |
↓ Trx plasma and erythrocytes ↓ TR plasma and erythrocytes |
[142] |
|
| |||
| Schizophrenia | First episode psychosis Long-term schizophrenic |
↑ Trx serum or plasma levels ↑ Trx serum or plasma levels |
[144–146] |
|
| |||
| ALS | Spinal cord of ALS | ↑ Trx (mRNA) | [148] |
|
| |||
| FALS | FALS erythrocytes stable form of mutant SOD-1 | ↑ Trx (p) | [147] |
(p): protein expression; (a): activity; ↑: upregulation; ↓: downregulation.
In vitro studies demonstrated augmented levels of oxidized Trx1 and an increase in the levels of apoptosis in SH-SY5Y cells exposed to Aβ [111]. Reduced Trx is a negative regulator of apoptosis via ASK1 [115]. Studies also show that ASK1 participates in Aβ induced neuronal cell death [116]. The overexpression of Trx1 protects SH-SY5Y cells against Aβ [111]. Furthermore, Trx and TrxR treatments protect primary hippocampal cultures from Aβ toxicity, acting as radical scavenger that inhibits the neuronal injury induced by Aβ [38]. Aβ has a critical methionine residue at position 35 [117]. The reversible product of methionine oxidation is methionine sulfoxide and can be reduced by methionine sulfoxide reductases based on TrxR regulation, while the irreversible oxidation to methionine sulfone is rare and only takes place in the presence of strong oxidants [118]. Methionine sulfoxide modulates oxidative stress and the neurotoxic properties of Aβ, and methionine sulfoxide reductase activity is reduced in the AD brain [119]. Aβ is related to the pathogenesis of other disorders like macular degeneration and glaucoma in mice via the impairment of the TS [120]. Aβ modifications depend on the TS, and the diminished levels of this protein system make the cell more vulnerable to neurotoxic Aβ.
7. TS and Parkinson's Disease
Idiopathic PD is characterized clinically by tremor, rigidity, bradykinesia, and posture instability [121]. PD is diagnosed pathologically by the loss of neurons in the substantia nigra pars compacta of the midbrain, in association with the widespread occurrence of Lewy bodies (intracytoplasmic filamentous aggregates of α-synuclein present in neurons and axons) [122]. Oxidative stress is present in PD, probably due to factors such as increased iron levels, low GSH levels and the impairment of mitochondrial complex I function in the substantia nigra [123].
In patients with sporadic PD, oxidative forms of DJ-1 protein were found [124]. DJ-1 acts as an antioxidant and transcription factor, having been observed in studies as protecting the culture cells and substantia nigra of mice from oxidative stress by inducing Trx1 expression via the transcription factor Nrf2 [125]. Nrf2 transcription factor is related to the expression of antioxidant and detoxifying enzymes, including Trx and TrxR [126]. α-Synuclein inclusions are common in PD, where its methionines and tyrosines are susceptible to oxidation [127]. The oxidation of synuclein methionines stabilizes soluble oligomers, while hetero-oligomers composed of synuclein and oxidized synuclein could have a toxic impact on the cellular environment [128].
Paraquat, MPP+, and rotenone are chemical compounds that mimic PD in animals and exert their toxic actions through the inhibition of mitochondrial complex I, inducing dopaminergic degeneration, as found in rodents. These compounds have been used for the study of neurotoxic mechanisms in PD [129, 130]. Ramachandiran et al. (2007) reported that, in SK-DAT cells, a different mechanism of cell damage operates in the TS, in which paraquat oxidizes Trx1 while rotenone and MPP+ oxidize Trx2. Chen et al. (2010) reported that in PC12 cells exposed to MPP+ decreased expressions of Trx1 and Trx2, although MPP+ decreased the expressions of both Trxs, the ratio of oxidized versus reduced Trx1 and Trx2 was relatively increased. This could explain how each toxin works at different levels within the cell, with rotenone and MPP+ working at a mitochondrial level and Paraquat at a cytosol level in relation to the TS [131, 132]. Another CNS toxin used in PD animal models is the fungicide Maneb, which is a mitochondrial complex III inhibitor. Roede et al. (2011) probed Paraquat and Maneb in SH-SY5Y neuroblastoma cells, finding that Paraquat oxidizes Trx2, whereas Maneb induces the expression of TrxR1, which correlated with the abundant nucleus increase of the transcription factor Nrf2 [133]. Consistently, studies have shown that Trx also protects both SH-SY5Y and PC12 cells against the severe oxidative stress and damage caused by the parkinsonism-producing neurotoxin MPP+ [81, 85, 134]. In mice exposed to MPP+, the activation of both ASK1 and its downstream target JNK was observed, which implicates Trx in the ASK1-mediated redox signaling in the pathogenesis of PD [135, 136].
Mitochondria are the major source of ROS, which are implicated in the pathogenesis of neurodegenerative diseases such as PD [137]. The TS has a significant role in H2O2 detoxification and the consequent cell death in dopaminergic cells. In dopaminergic cells exposed to 6-hydroxydopamine and paraquat, the inhibition of TrxR, induced mitochondrial dysfunction, increased H2O2 levels and cell death through oxidative stress [138]. Studies of the nigral DA cell line after H2O2 using microarray analysis to identify several groups of genes regulated by oxidative stress and related to functional mitochondrial complex I molecules, exocytosis, membrane trafficking, and Trx1 [139].
8. Other Neurodegenerative Diseases
Huntington's disease (HD) is a neurodegenerative disorder, most of whose clinical features can be attributed to CNS neurodegeneration, with up to 95% loss of GABAergic neurons from striatum [140]. Oxidative stress has been proposed as either a causative event or as a secondary constituent of the cell death cascade in HD [141]. The reported reduction of Trx1 and TrxR1 in the plasma and erythrocytes in blood samples from HD patients [142] evidenced an oxidative stress peripheral response to this neurodegeneration. Schizophrenia has a range of cognitive deficits that may involve oxidative stress and possibly contribute to cognitive deficits during aging and in neurodegenerative disorders [143]. Various studies have shown increased levels of Trx in plasma and serum in first episode schizophrenia patients and enhanced Trx levels in the plasma of long-term schizophrenic patients [144–146].
Postmortem of spinal cords presenting amyotrophic lateral sclerosis (ALS) and the erythrocytes of familial amyotrophic lateral sclerosis (FALS) with stable forms of mutant SOD-1 proteins show that Trx genes and protein expression are upregulated [147, 148]. Both studies suggest the involvement of Trx in the etiology and progression of the disease.
9. Concluding Remarks
Evidence shows that the presence of TS proteins is differential in the brain. Since the activity of Trx and TrxR is related to the activation of genes, the cellular cycle, and, especially, cell protection and survival, this differential expression suggests that some brain regions have different requirements for TS proteins for cell functions or against ROS damage. We have revised the modulation of the TS in different animal models, discussing the various mechanisms activating the TS and the mechanisms through which it exercises its functions. These studies demonstrate that the upregulation of TS proteins is accompanied by cell protection against damage, while the downregulation makes cells more vulnerable to death. Research in postmortem brains from different neurodegenerative disorders shows a differential modulating pattern in these disorders. It may depend on disease's stage, which makes the TS a therapeutic target for the treatment and retardation of several neurodegenerative processes. The role on antioxidant functions is important but even more than the antioxidant activity; TS proteins by their redox properties modulate the function and expression of other proteins, including different transcription factors essential for the development and for the control of cell survival or death. Elucidation of specific functions and mechanism of regulation of TS is required in different brain cell types. The role of Trx secretion and the functions as a brain cocytokine and chemokine is needed as well; this will be helpful for the study in pathogenesis of different neurodegenerative diseases.
Acknowledgments
This work was partially supported by a Grant from CONACYT no. 102287. This study was performed in partial fulfillment of the requirements for the Doctorado en Ciencias Biológicas for Daniela Silva Adaya at the Universidad Nacional Autónoma de México.
Conflict of Interests
The authors declare that there is no conflict of interests.
References
- 1.Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV. isolation and characterization of thioredoxin, the hydrogen donor from escherichia coli B. The Journal of Biological Chemistry. 1964;239:3436–3444. [PubMed] [Google Scholar]
- 2.Chae HZ, Kim HJ, Kang SW, Rhee SG. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Research and Clinical Practice. 1999;45(2-3):101–112. doi: 10.1016/s0168-8227(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 3.Lillig CH, Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxidants and Redox Signaling. 2007;9(1):25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 4.Burke-Gaffney A, Callister MEJ, Nakamura H. Thioredoxin: friend or foe in human disease? Trends in Pharmacological Sciences. 2005;26(8):398–404. doi: 10.1016/j.tips.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Arnér ESJ, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. European Journal of Biochemistry. 2000;267(20):6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 6.Pekkari K, Holmgren A. Truncated thioredoxin: physiological functions and mechanism. Antioxidants and Redox Signaling. 2004;6(1):53–61. doi: 10.1089/152308604771978345. [DOI] [PubMed] [Google Scholar]
- 7.Carilho Torrao RB, Dias IH, Bennett SJ, Dunston CR, Griffiths HR. Healthy ageing and depletion of intracellular glutathione influences T cell membrane thioredoxin-1 levels and cytokine secretion. Chemistry Central Journal. 2013;7(1):p. 150. doi: 10.1186/1752-153X-7-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordberg J, Arnér ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology and Medicine. 2001;31(11):1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 9.Zeng XS, Jia JJ, Kwon Y, Wang SD, Bai J. The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease. Free Radical Biology & Medicine. 2013;67:10–18. doi: 10.1016/j.freeradbiomed.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Holmgren A, Luthman M. Tissue distribution and subcellular localization of bovine thioredoxin determined by radioimmunoassay. Biochemistry. 1978;17(19):4071–4077. doi: 10.1021/bi00612a031. [DOI] [PubMed] [Google Scholar]
- 11.Soerensen J, Jakupoglu C, Beck H, et al. The role of thioredoxin reductases in brain development. PLoS ONE. 2008;3(3) doi: 10.1371/journal.pone.0001813.e1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jornstedt MB, Kumar S, Holmgren A. Selenite and selenodiglutathione: reactions with thioredoxin systems. Methods in Enzymology. 1995;252:209–219. doi: 10.1016/0076-6879(95)52024-4. [DOI] [PubMed] [Google Scholar]
- 13.Zhong L, Arnér ESJ, Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(11):5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundstrom J, Holmgren A. Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. Journal of Biological Chemistry. 1990;265(16):9114–9120. [PubMed] [Google Scholar]
- 15.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascotbate by the selenoenzyme thioredoxin reductase. Journal of Biological Chemistry. 1997;272(36):22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 16.May JM, Cobb CE, Mendiratta S, Hill KE, Burk RF. Reduction of the ascorbyl free radical to ascorbate by thioredoxin reductase. Journal of Biological Chemistry. 1998;273(36):23039–23045. doi: 10.1074/jbc.273.36.23039. [DOI] [PubMed] [Google Scholar]
- 17.Arnér ESJ, Nordberg J, Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochemical and Biophysical Research Communications. 1996;225(1):268–274. doi: 10.1006/bbrc.1996.1165. [DOI] [PubMed] [Google Scholar]
- 18.Xia L, Björnstedt M, Nordman T, Eriksson LC, Olsson JM. Reduction of ubiquinone by lipoamide dehydrogenase: an antioxidant regenerating pathway. European Journal of Biochemistry. 2001;268(5):1486–1490. doi: 10.1046/j.1432-1327.2001.02013.x. [DOI] [PubMed] [Google Scholar]
- 19.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain: metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. European Journal of Biochemistry. 2000;267(16):4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 20.Rozell B, Hansson H-A, Luthman M, Holmgren A. Immunohistochemical localization of thioredoxin and thioredoxin reductase in adult rats. European Journal of Cell Biology. 1985;38(1):79–86. [PubMed] [Google Scholar]
- 21.Stemme S, Hansson H-A, Holmgren A, Rozell B. Axoplasmic transport of thioredoxin and thioredoxin reductase in rat sciatic nerve. Brain Research. 1985;359(1-2):140–146. doi: 10.1016/0006-8993(85)91421-0. [DOI] [PubMed] [Google Scholar]
- 22.Tomimoto H, Akiguchi I, Wakita H, Kimura J, Hori K, Yodoi J. Astroglial expression of ATL-derived factor, a human thioredoxin homologue, in the gerbil brain after transient global ischemia. Brain Research. 1993;625(1):1–8. doi: 10.1016/0006-8993(93)90130-f. [DOI] [PubMed] [Google Scholar]
- 23.Asahina M, Yamada T, Yoshiyama Y, Yodoi J. Expression of adult T cell leukemia-derived factor in human brain and peripheral nerve tissues. Dementia and Geriatric Cognitive Disorders. 1998;9(4):181–185. doi: 10.1159/000017044. [DOI] [PubMed] [Google Scholar]
- 24.Godoy JR, Funke M, Ackermann W, et al. Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse. Biochimica et Biophysica Acta. 2011;1810(1):2–92. doi: 10.1016/j.bbagen.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Lippoldt A, Padilla CA, Gerst H, et al. Localization of thioredoxin in the rat brain and functional implications. Journal of Neuroscience. 1995;15(10):6747–6756. doi: 10.1523/JNEUROSCI.15-10-06747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aon-Bertolino ML, Romero JI, Galeano P, et al. Thioredoxin and glutaredoxin system proteins-immunolocalization in the rat central nervous system. Biochimica et Biophysica Acta. 2011;1810(1):93–110. doi: 10.1016/j.bbagen.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 27.Rybnikova E, Damdimopoulos AE, Gustafsson J-Å, Spyrou G, Pelto-Huikko M. Expression of novel antioxidant thioredoxin-2 in the rat brain. European Journal of Neuroscience. 2000;12(5):1669–1678. doi: 10.1046/j.1460-9568.2000.00059.x. [DOI] [PubMed] [Google Scholar]
- 28.Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. Journal of Biological Chemistry. 1992;267(34):24161–24164. [PubMed] [Google Scholar]
- 29.Gil-Bea F, Akterin S, Persson T, et al. Thioredoxin-80 is a product of alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer's disease brain. EMBO Molecular Medicine. 2012;4(10):1097–1111. doi: 10.1002/emmm.201201462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pekkari K, Goodarzi MT, Scheynius A, Holmgren A, Avila-Cariño J. Truncated thioredoxin (Trx80) induces differentiation of human CD14 + monocytes into a novel cell type (TAMs) via activation of the MAP kinases p38, ERK, and JNK. Blood. 2005;105(4):1598–1605. doi: 10.1182/blood-2004-04-1577. [DOI] [PubMed] [Google Scholar]
- 31.Wakasugi N, Tagaya Y, Wakasugi H, et al. Adult T-cell leukemia-derived factor/thioredoxin, produced by both human T-lymphotropic virus type I- and Epstein-Barr virus-transformed lymphocytes, acts as an autocrine growth factor and synergizes with interleukin 1 and interleukin 2. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(21):8282–8286. doi: 10.1073/pnas.87.21.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schubert D, Herrera F, Cumming R, et al. Neural cells secrete a unique repertoire of proteins. Journal of Neurochemistry. 2009;109(2):427–435. doi: 10.1111/j.1471-4159.2009.05968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LoPachin RM, Barber DS. Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Toxicological Sciences. 2006;94(2):240–255. doi: 10.1093/toxsci/kfl066. [DOI] [PubMed] [Google Scholar]
- 34.Hori K, Katayama M, Sato N, Ishii K, Waga S, Yodoi J. Neuroprotection by glial cells through adult T cell leukemia-derived factor/human thioredoxin (ADF/TRX) Brain Research. 1994;652(2):304–310. doi: 10.1016/0006-8993(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 35.Arnér ESJ. Focus on mammalian thioredoxin reductases—important selenoproteins with versatile functions. Biochimica et Biophysica Acta. 2009;1790(6):495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Bindoli A, Rigobello MP. Principles in: from chemistry to functional significance. Redox SignalingAntioxidants & Redox Signaling. 2013;18(13):1557–1593. doi: 10.1089/ars.2012.4655. [DOI] [PubMed] [Google Scholar]
- 37.Hirota K, Nakamura H, Masutani H, Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Annals of the New York Academy of Sciences. 2002;957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 38.Lovell MA, Xie C, Gabbita SP, Markesbery WR. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radical Biology and Medicine. 2000;28(3):418–427. doi: 10.1016/s0891-5849(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 39.Takagi Y, Horikawa F, Nozaki K, Sugino T, Hashimoto N, Yodoi J. Expression and distribution of redox regulatory protein, thioredoxin during transient focal brain ischemia in the rat. Neuroscience Letters. 1998;251(1):25–28. doi: 10.1016/s0304-3940(98)00492-3. [DOI] [PubMed] [Google Scholar]
- 40.Takagi Y, Mitsui A, Nishiyama A, et al. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):4131–4136. doi: 10.1073/pnas.96.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mansur K, Iwahashi Y, Kiryu-Seo S, et al. Up-regulation of thioredoxin expression in motor neurons after nerve injury. Molecular Brain Research. 1998;62(1):86–91. doi: 10.1016/s0169-328x(98)00244-7. [DOI] [PubMed] [Google Scholar]
- 42.Hattori I, Takagi Y, Nakamura H, et al. Intravenous administration of thioredoxin decreases brain damage following transient focal cerebral ischemia in mice. Antioxidants and Redox Signaling. 2004;6(1):81–87. doi: 10.1089/152308604771978372. [DOI] [PubMed] [Google Scholar]
- 43.Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Yodoi J. Expression and distribution of redox regulatory protein, thioredoxin after metabolic impairment by 3-nitropropionic acid in rat brain. Neuroscience Letters. 1999;275(2):145–148. doi: 10.1016/s0304-3940(99)00763-6. [DOI] [PubMed] [Google Scholar]
- 44.Pitten F-A, Kramer A, Herrmann K, Bremer J, Koch S. Formaldehyde neurotoxicity in animal experiments. Pathology Research and Practice. 2000;196(3):193–198. doi: 10.1016/S0344-0338(00)80100-4. [DOI] [PubMed] [Google Scholar]
- 45.Luo F-C, Zhou J, Lv T, et al. Induction of endoplasmic reticulum stress and the modulation of thioredoxin-1 in formaldehyde-induced neurotoxicity. NeuroToxicology. 2012;33(3):290–298. doi: 10.1016/j.neuro.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Smith HS. The role of genomic oxidative-reductive balance as predictor of complex regional pain syndrome development: a novel theory. Pain Physician. 2010;13(1):79–90. [PubMed] [Google Scholar]
- 47.Luo FC, Feng YM, Zhao L, et al. Thioredoxin-1 expression regulated by morphine in SH-SY5Y cells. Neuroscience Letters. 2012;523(1):50–55. doi: 10.1016/j.neulet.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 48.Denny Joseph KM, Muralidhara M. Fish oil prophylaxis attenuates rotenone-induced oxidative impairments and mitochondrial dysfunctions in rat brain. Food and Chemical Toxicology. 2012;50(5):1529–1537. doi: 10.1016/j.fct.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 49.Eftekharpour E, Holmgren A, Juurlink BH. Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia. 2000;31(3):241–248. doi: 10.1002/1098-1136(200009)31:3<241::aid-glia50>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Lee SY, Andoh T, Murphy DL, Chiueh CC. 17beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. The FASEB Journal. 2003;17(8):947–948. doi: 10.1096/fj.02-0807fje. [DOI] [PubMed] [Google Scholar]
- 51.Bardullas U, Limón-Pacheco JH, Giordano M, Carrizales L, Mendoza-Trejo MS, Rodríguez VM. Chronic low-level arsenic exposure causes gender-specific alterations in locomotor activity, dopaminergic systems, and thioredoxin expression in mice. Toxicology and Applied Pharmacology. 2009;239(2):169–177. doi: 10.1016/j.taap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Heimfarth L, Loureiro SO, Reis KP, et al. Diphenyl ditelluride induces hypophosphorylation of intermediate filaments through modulation of DARPP-32-dependent pathways in cerebral cortex of young rats. Archives of Toxicology. 2012;86(2):217–230. doi: 10.1007/s00204-011-0746-6. [DOI] [PubMed] [Google Scholar]
- 53.Comparsi B, Meinerz DF, Franco JL, et al. Diphenyl ditelluride targets brain selenoproteins in vivo: inhibition of cerebral thioredoxin reductase and glutathione peroxidase in mice after acute exposure. Molecular and Cellular Biochemistry. 2012;370(1-2):173–182. doi: 10.1007/s11010-012-1408-6. [DOI] [PubMed] [Google Scholar]
- 54.Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Critical Reviews in Toxicology. 2006;36(8):609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 55.Branco V, Canário J, Lu J, Holmgren A, Carvalho C. Mercury and selenium interaction in vivo: effects on thioredoxin reductase and glutathione peroxidase. Free Radical Biology and Medicine. 2012;52(4):781–793. doi: 10.1016/j.freeradbiomed.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 56.Patenaude A, Murthy MRV, Mirault M-E. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cellular and Molecular Life Sciences. 2005;62(10):1063–1080. doi: 10.1007/s00018-005-4541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka N, Ikeda Y, Ohta Y, et al. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Research. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 58.Tanito M, Agbaga M-P, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radical Biology and Medicine. 2007;42(12):1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 59.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Letters. 2003;546(2-3):181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- 60.Maldonado PD, Pérez-De la Cruz V, Torres-Ramos M, et al. Selenium-induced antioxidant protection recruits modulation of thioredoxin reductase during excitotoxic/pro-oxidant events in the rat striatum. Neurochemistry International. 2012;61(2):195–206. doi: 10.1016/j.neuint.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 61.Chen T-Y, Tsai K-L, Lee T-Y, Chiueh CC, Lee W-S, Hsu C. Sex-specific role of thioredoxin in neuroprotection against iron-induced brain injury conferred by estradiol. Stroke. 2010;41(1):160–165. doi: 10.1161/STROKEAHA.109.562850. [DOI] [PubMed] [Google Scholar]
- 62.Zhao P, Huang Z-N, Chen G, Cheng J-S. Electro-acupuncture attenuates nitric oxide release from rat striatum after transient middle cerebral artery occlusion. Acupuncture and Electro-Therapeutics Research. 2000;25(2):101–107. doi: 10.3727/036012900816356163. [DOI] [PubMed] [Google Scholar]
- 63.Siu FKW, Lo SCL, Leung MCP. Electro-acupuncture potentiates the disulphide-reducing activities of thioredoxin system by increasing thioredoxin expression in ischemia-reperfused rat brains. Life Sciences. 2005;77(4):386–399. doi: 10.1016/j.lfs.2004.10.069. [DOI] [PubMed] [Google Scholar]
- 64.Andoh T, Boon Chock P, Murphy DL, Chiueh CC. Role of the redox protein thioredoxin in cytoprotective mechanism evoked by (-)-deprenyl. Molecular Pharmacology. 2005;68(5):1408–1414. doi: 10.1124/mol.105.012302. [DOI] [PubMed] [Google Scholar]
- 65.Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Current Opinion in Cell Biology. 2001;13(1):41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- 66.Landino LM, Skreslet TE, Alston JA. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: modulation of microtubule assembly kinetics by the thioredoxin reductase system. Journal of Biological Chemistry. 2004;279(33):35101–35105. doi: 10.1074/jbc.M405471200. [DOI] [PubMed] [Google Scholar]
- 67.Hawkins HC, Blackburn EC, Freedman RB. Comparison of the activities of protein disulphide-isomerase and thioredoxin in catalysing disulphide isomerization in a protein substrate. Biochemical Journal. 1991;275, part 2:349–353. doi: 10.1042/bj2750349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mitsui A, Hirakawa T, Yodoi J. Reactive oxygen-reducing and protein-refolding activities of adult T cell leukemia-derived factor/human thioredoxin. Biochemical and Biophysical Research Communications. 1992;186(3):1220–1226. doi: 10.1016/s0006-291x(05)81536-0. [DOI] [PubMed] [Google Scholar]
- 69.Takagi Y, Hattori I, Nozaki K, et al. Excitotoxic hippocampal injury is attenuated in thioredoxin transgenic mice. Journal of Cerebral Blood Flow and Metabolism. 2000;20(5):829–833. doi: 10.1097/00004647-200005000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Kong L, Tanito M, Huang Z, et al. Delay of photoreceptor degeneration in tubby mouse by sulforaphane. Journal of Neurochemistry. 2007;101(4):1041–1052. doi: 10.1111/j.1471-4159.2007.04481.x. [DOI] [PubMed] [Google Scholar]
- 71.Kong L, Zhou X, Li F, Yodoi J, McGinnis J, Cao W. Neuroprotective effect of overexpression of thioredoxin on photoreceptor degeneration in Tubby mice. Neurobiology of Disease. 2010;38(3):446–455. doi: 10.1016/j.nbd.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bai J, Nakamura H, Hattori I, Tanito M, Yodoi J. Thioredoxin suppresses 1-methyl-4-phenylpyridinium-induced neurotoxicity in rat PC12 cells. Neuroscience Letters. 2002;321(1-2):81–84. doi: 10.1016/s0304-3940(02)00058-7. [DOI] [PubMed] [Google Scholar]
- 73.Hee SB, Eun WC, Jin SK, et al. Thioredoxin overexpression in HT-1080 cells induced cellular senescence and sensitization to gamma radiation. FEBS Letters. 2005;579(19):4055–4062. doi: 10.1016/j.febslet.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 74.Munemasa Y, Seok HK, Jae HA, Kwong JMK, Caprioli J, Piri N. Protective effect of thioredoxins 1 and 2 in retinal ganglion cells after optic nerve transection and oxidative stress. Investigative Ophthalmology and Visual Science. 2008;49(8):3535–3543. doi: 10.1167/iovs.08-1716. [DOI] [PubMed] [Google Scholar]
- 75.Chen Y, Yu M, Jones DP, Greenamyre JT, Cai J. Protection against oxidant-induced apoptosis by mitochondrial thioredoxin in SH-SY5Y neuroblastoma cells. Toxicology and Applied Pharmacology. 2006;216(2):256–262. doi: 10.1016/j.taap.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 76.Nakamura T, Nakamura H, Hoshino T, Ueda S, Wada H, Yodoi J. Redox regulation of lung inflammation by thioredoxin. Antioxidants and Redox Signaling. 2005;7(1-2):60–71. doi: 10.1089/ars.2005.7.60. [DOI] [PubMed] [Google Scholar]
- 77.Ma YH, Su N, Chao XD, et al. Thioredoxin-1 attenuates post-ischemic neuronal apoptosis via reducing oxidative/nitrative stress. Neurochemistry International. 2012;60(5):475–483. doi: 10.1016/j.neuint.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 78.Zhou F, Liu PP, Ying GY, Zhu XD, Shen H, Chen G. Effects of thioredoxin-1 on neurogenesis after brain ischemia/reperfusion injury. CNS Neuroscience & Therapeutics. 2013;19(3):204–205. doi: 10.1111/cns.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stroev SA, Gluschenko TS, Tjulkova EI, et al. Preconditioning enhances the expression of mitochondrial antioxidant thioredoxin-2 in the forebrain of rats exposed to severe hypobaric hypoxia. Journal of Neuroscience Research. 2004;78(4):563–569. doi: 10.1002/jnr.20282. [DOI] [PubMed] [Google Scholar]
- 80.Stroev SA, Tjulkova EI, Gluschenko TS, Rybnikova EA, Samoilov MO, Pelto-Huikko M. The augmentation of brain thioredoxin-1 expression after severe hypobaric hypoxia by the preconditioning in rats. Neuroscience Letters. 2004;370(2-3):224–229. doi: 10.1016/j.neulet.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 81.Andoh T, Boon Chock P, Chiueh CC. The roles of thioredoxin in protection against oxidative stress-induced apoptosis in SH-SY5Y cells. Journal of Biological Chemistry. 2002;277(12):9655–9660. doi: 10.1074/jbc.M110701200. [DOI] [PubMed] [Google Scholar]
- 82.Kudin AP, Augustynek B, Lehmann AK, Kovács R, Kunz WS. The contribution of thioredoxin-2 reductase and glutathione peroxidase to H2O2 detoxification of rat brain mitochondria. Biochimica et Biophysica Acta. 2012;1817(10):1901–1906. doi: 10.1016/j.bbabio.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 83.Uhlig S, Wendel A. The physiological consequences of glutathione variations. Life Sciences. 1992;51(14):1083–1094. doi: 10.1016/0024-3205(92)90509-n. [DOI] [PubMed] [Google Scholar]
- 84.Das KC, White CW. Detection of thioredoxin in human serum and biological samples using a sensitive sandwich ELISA with digoxigenin-labeled antibody. Journal of Immunological Methods. 1998;211(1-2):9–20. doi: 10.1016/s0022-1759(97)00163-4. [DOI] [PubMed] [Google Scholar]
- 85.Andoh T, Chiueh CC, Chock PB. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. Journal of Biological Chemistry. 2003;278(2):885–890. doi: 10.1074/jbc.M209914200. [DOI] [PubMed] [Google Scholar]
- 86.Hwang IK, Yoo K-Y, Kim DW, et al. Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radical Biology and Medicine. 2010;48(9):1242–1251. doi: 10.1016/j.freeradbiomed.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 87.Gao J, Zhu Z-R, Ding H-Q, Qian Z, Zhu L, Ke Y. Vulnerability of neurons with mitochondrial dysfunction to oxidative stress is associated with down-regulation of thioredoxin. Neurochemistry International. 2007;50(2):379–385. doi: 10.1016/j.neuint.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 88.Matsui M, Oshima M, Oshima H, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Developmental Biology. 1996;178(1):179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 89.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. The EMBO Journal. 2002;21(7):1695–1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Molecular and Cellular Biology. 2003;23(3):916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxidants and Redox Signaling. 2006;8(11-12):2143–2151. doi: 10.1089/ars.2006.8.2143. [DOI] [PubMed] [Google Scholar]
- 92.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annual Review of Neuroscience. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bai J, Nakamura H, Kwon Y-W, et al. Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. Journal of Neuroscience. 2003;23(2):503–509. doi: 10.1523/JNEUROSCI.23-02-00503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Masutani H, Bai J, Kim Y-C, Yodoi J. Thioredoxin as a neurotrophic cofactor and an important regulator of neuroprotection. Molecular Neurobiology. 2004;29(3):229–242. doi: 10.1385/MN:29:3:229. [DOI] [PubMed] [Google Scholar]
- 95.Impey S, Obrietan K, Wong ST, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21(4):869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 96.Endoh M, Kunishita T, Tabita T. Thioredoxin from activated macrophages as a trophic factor for central cholinergic neurons in vitro. Biochemical and Biophysical Research Communications. 1993;192(2):760–765. doi: 10.1006/bbrc.1993.1479. [DOI] [PubMed] [Google Scholar]
- 97.Hui TY, Sheth SS, Diffley JM, et al. Mice lacking thioredoxin-interacting protein provide evidence linking cellular redox state to appropriate response to nutritional signals. Journal of Biological Chemistry. 2004;279(23):24387–24393. doi: 10.1074/jbc.M401280200. [DOI] [PubMed] [Google Scholar]
- 98.Bodnar JS, Chatterjee A, Castellani LW, et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nature Genetics. 2002;30(1):110–116. doi: 10.1038/ng811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oka S-I, Liu W, Masutani H, et al. Impaired fatty acid utilization in thioredoxin binding protein-2 (TBP-2)-deficient mice: a unique animal model of Reye syndrome. The FASEB Journal. 2006;20(1):121–123. doi: 10.1096/fj.05-4439fje. [DOI] [PubMed] [Google Scholar]
- 100.Lappalainen Z, Lappalainen J, Oksala NKJ, et al. Diabetes impairs exercise training-associated thioredoxin response and glutathione status in rat brain. Journal of Applied Physiology. 2009;106(2):461–467. doi: 10.1152/japplphysiol.91252.2008. [DOI] [PubMed] [Google Scholar]
- 101.Nishiyama A, Masutani H, Nakamura H, Nishinaka Y, Yodoi J. Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life. 2001;52(1-2):29–33. doi: 10.1080/15216540252774739. [DOI] [PubMed] [Google Scholar]
- 102.Kim GS, Jung JE, Narasimhan P, Sakata H, Chan PH. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiology of Disease. 2012;46(2):440–449. doi: 10.1016/j.nbd.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Al-Gayyar MMH, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Thioredoxin interacting protein is a novel mediator of retinal inflammation and neurotoxicity. British Journal of Pharmacology. 2011;164(1):170–180. doi: 10.1111/j.1476-5381.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rahman MF, Wang J, Patterson TA, et al. Expression of genes related to oxidative stress in the mouse brain after exposure to silver-25 nanoparticles. Toxicology Letters. 2009;187(1):15–21. doi: 10.1016/j.toxlet.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 105.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Reviews Neuroscience. 2010;11(10):682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochemical Research. 2003;28(10):1563–1574. doi: 10.1023/a:1025682611389. [DOI] [PubMed] [Google Scholar]
- 107.Papadia S, Soriano FX, Léveillé F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nature Neuroscience. 2008;11(4):476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidatives stress. Nature Reviews Drug Discovery. 2004;3(3):205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 109.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Archives of Neurology. 1985;42(11):1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 110.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radical Biology and Medicine. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 111.Akterin S, Cowburn RF, Miranda-Vizuete A, et al. Involvement of glutaredoxin-1 and thioredoxin-1 in β-amyloid toxicity and Alzheimer’s disease. Cell Death and Differentiation. 2006;13(9):1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- 112.di Domenico F, Sultana R, Tiu GF, et al. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Research. 2010;1333:72–81. doi: 10.1016/j.brainres.2010.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ancoli-Israel S, Palmer BW, Cooke JR, et al. Cognitive effects of treating obstructive sleep apnea in Alzheimer’s disease: a randomized controlled study. Journal of the American Geriatrics Society. 2008;56(11):2076–2081. doi: 10.1111/j.1532-5415.2008.01934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang XH, Liu HG, Liu X, Chen JN. Thioredoxin and impaired spatial learning and memory in the rats exposed to intermittent hypoxia. Chinese Medical Journal. 2012;125(17):3074–3080. [PubMed] [Google Scholar]
- 115.Song JJ, Lee YJ. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of appptosis signal-regulating kinase 1. Biochemical Journal. 2003;373, part 3:845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kadowaki H, Nishitoh H, Urano F, et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death and Differentiation. 2005;12(1):19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- 117.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochimica et Biophysica Acta. 2005;1703(2):149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 118.Sun Q-A, Wu Y, Zappacosta F, et al. Redox regulation of cell signaling by selenocysteine in mammalian thioredoxin reductases. Journal of Biological Chemistry. 1999;274(35):24522–24530. doi: 10.1074/jbc.274.35.24522. [DOI] [PubMed] [Google Scholar]
- 119.Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. Journal of Neurochemistry. 1999;73(4):1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 120.Lamoke F, Ripandelli G, Webster S, et al. Loss of thioredoxin function in retinas of mice overexpressing amyloid beta. Free Radical Biology & Medicine. 2012;53(3):577–588. doi: 10.1016/j.freeradbiomed.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 121.Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. 1967. Neurology. 2001;57(10, supplement3):S11–S26. [PubMed] [Google Scholar]
- 122.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Archives of Neurology. 1999;56(1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 123.Jenner P. Oxidative stress and Parkinson's disease. Handbook of Clinical Neurology. 2007;83:507–520. doi: 10.1016/S0072-9752(07)83024-7. [DOI] [PubMed] [Google Scholar]
- 124.Choi J, Sullards MC, Olzmann JA, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. Journal of Biological Chemistry. 2006;281(16):10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Im JY, Lee KW, Woo JM, Junn E, Mouradian MM. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Human Molecular Genetics. 2012;21(13):3013–3024. doi: 10.1093/hmg/dds131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(41):15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chavarria C, Souza JM. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Archives of Biochemistry and Biophysics. 2013;533(1-2):25–32. doi: 10.1016/j.abb.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 128.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human α-synuclein in vitro. FEBS Letters. 2002;517(1–3):239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 129.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature Neuroscience. 2000;3(12):1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 130.Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson’s disease? Brain Research. 2000;873(2):225–234. doi: 10.1016/s0006-8993(00)02496-3. [DOI] [PubMed] [Google Scholar]
- 131.Ramachandiran S, Hansen JM, Jones DP, Richardson J.R. JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicological Sciences. 2007;95(1):163–171. doi: 10.1093/toxsci/kfl125. [DOI] [PubMed] [Google Scholar]
- 132.Chen VTK, Huang C-L, Lee Y-C, Liao W-C, Huang N-K. The roles of the thioredoxin system and peroxiredoxins in 1-methyl-4-phenyl-pyridinium ion-induced cytotoxicity in rat pheochromocytoma cells. Toxicology in Vitro. 2010;24(6):1577–1583. doi: 10.1016/j.tiv.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 133.Roede JR, Hansen JM, Go Y-M, Jones DP. Maneb and paraquat-mediated neurotoxicity: involvement of peroxiredoxin/thioredoxin system. Toxicological Sciences. 2011;121(2):368–375. doi: 10.1093/toxsci/kfr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Estévez AG, Spear N, Anthony Thompson J, et al. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. Journal of Neuroscience. 1998;18(10):3708–3714. doi: 10.1523/JNEUROSCI.18-10-03708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fujino G, Noguchi T, Takeda K, Ichijo H. Thioredoxin and protein kinases in redox signaling. Seminars in Cancer Biology. 2006;16(6):427–435. doi: 10.1016/j.semcancer.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 136.Karunakaran S, Diwakar L, Saeed U, et al. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson’s disease: protection by α-lipoic acid. The FASEB Journal. 2007;21(9):2226–2236. doi: 10.1096/fj.06-7580com. [DOI] [PubMed] [Google Scholar]
- 137.Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology. 1996;47(6, supplement 3):S161–S170. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
- 138.Lopert P, Day BJ, Patel M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0050683.e50683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yoo MS, Chun HS, Son JJ, et al. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: correlation with the known pathology in Parkinson’s disease. Molecular Brain Research. 2003;110(1):76–84. doi: 10.1016/s0169-328x(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 140.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. The Lancet Neurology. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 141.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathology. 1999;9(1):147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sánchez-López F, Tasset I, Agüera E, et al. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington's disease. Journal of Neurology Research. 2012;34(7):721–724. doi: 10.1179/1743132812Y.0000000073. [DOI] [PubMed] [Google Scholar]
- 143.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. The Lancet Neurology. 2011;10(2):187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang XY, Chen DC, Xiu MH, et al. The novel oxidative stress marker thioredoxin is increased in first-episode schizophrenic patients. Schizophrenia Research. 2009;113(2-3):151–157. doi: 10.1016/j.schres.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 145.Owe-Larsson B, Ekdahl K, Edbom T, et al. Increased plasma levels of thioredoxin-1 in patients with first episode psychosis and long-term schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(4):1117–1121. doi: 10.1016/j.pnpbp.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 146.Zhang XY, Chen da C, Xiu MH, et al. Thioredoxin, a novel oxidative stress marker and cognitive performance in chronic and medicated schizophrenia versus healthy controls. Schizophrenia Research. 2013;143(2-3):301–306. doi: 10.1016/j.schres.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 147.Ogawa Y, Kosaka H, Nakanishi T, et al. Stability of mutant superoxide dismutase-1 associated with familial amyotrophic lateral sclerosis determines the manner of copper release and induction of thioredoxin in erythrocytes. Biochemical and Biophysical Research Communications. 1997;241(2):251–257. doi: 10.1006/bbrc.1997.7804. [DOI] [PubMed] [Google Scholar]
- 148.Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. Journal of Neurochemistry. 2001;77(1):132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]