

Highlights
-
•
The bacterial metabolic pathway of trans-3-hydroxy-l-proline (T3LHyp) has been identified.
-
•
Azospirillum brasilense can grow on T3LHyp as a sole carbon source.
-
•
The T3LHyp pathway comprises T3LHyp dehydratase and Δ1-pyrroline-2-carboxylate reductase.
-
•
This pathway is similar to one found in mammals.
Abbreviations: l-Hyp, hydroxy-l-proline; T4LHyp, trans-4-hydroxy-l-proline; T3LHyp, trans-3-hydroxy-l-proline; C4LHyp, cis-4-hydroxy-l-proline; C4DHyp, cis-4-hydroxy-d-proline; Pyr4RH2C, Δ1-pyrroline-4R-hydroxy-2-carboxylate; C4DHypDH, C4DHyp dehydrogenase; Pyr2C, Δ1-pyrroline-2-carboxylate; Pip2C, Δ1-piperidine-2-carboxylate; OCD, ornithine cyclodeaminase; LCD, l-lysine cyclodeaminase
Keywords: Hydroxyproline, trans-3-Hydroxy-l-proline metabolism, trans-3-Hydroxy-l-proline dehydratase, Δ1-Pyrroline-2-carboxylate reductase, Convergent evolution of enzyme
Abstract
trans-4-Hydroxy-l-proline (T4LHyp) and trans-3-hydroxy-l-proline (T3LHyp) occur mainly in collagen. A few bacteria can convert T4LHyp to α-ketoglutarate, and we previously revealed a hypothetical pathway consisting of four enzymes at the molecular level (J Biol Chem (2007) 282, 6685–6695; J Biol Chem (2012) 287, 32674–32688). Here, we first found that Azospirillum brasilense has the ability to grow not only on T4LHyp but also T3LHyp as a sole carbon source. In A. brasilense cells, T3LHyp dehydratase and NAD(P)H-dependent Δ1-pyrroline-2-carboxylate (Pyr2C) reductase activities were induced by T3LHyp (and d-proline and d-lysine) but not T4LHyp, and no effect of T3LHyp was observed on the expression of T4LHyp metabolizing enzymes: a hypothetical pathway of T3LHyp → Pyr2C → l-proline was proposed. Bacterial T3LHyp dehydratase, encoded to LhpH gene, was homologous with the mammalian enzyme. On the other hand, Pyr2C reductase encoded to LhpI gene was a novel member of ornithine cyclodeaminase/μ-crystallin superfamily, differing from known bacterial protein. Furthermore, the LhpI enzymes of A. brasilense and another bacterium showed several different properties, including substrate and coenzyme specificities. T3LHyp was converted to proline by the purified LhpH and LhpI proteins. Furthermore, disruption of LhpI gene from A. brasilense led to loss of growth on T3LHyp, d-proline and d-lysine, indicating that this gene has dual metabolic functions as a reductase for Pyr2C and Δ1-piperidine-2-carboxylate in these pathways, and that the T3LHyp pathway is not linked to T4LHyp and l-proline metabolism.
1. Introduction
Hydroxy-l-proline (l-Hyp) has been found in certain proteins, in particular collagen, and in some peptide antibiotics. In mammalian systems, l-proline residue is post-translationally hydroxylated to trans-4-hydroxy-l-proline (T4LHyp) or trans-3-hydroxy-l-proline (T3LHyp) by prolyl 4-hydroxylase (EC 1.14.11.2) and propyl 3-hydroxylase (EC 1.14.11.7), respectively [1]. Additionally, it is known that a few bacterial enzymes directly hydroxylate free l-proline to T4LHyp [2], cis-4-hydroxy-l-proline (C4LHyp) [3] or cis-3-hydroxy-l-proline (C3LHyp) [4]. Among several stereoisomers of l-Hyp, T4LHyp is the most common in nature. In mammals, T4LHyp is converted to pyruvate and glyoxylate via three intermediates, Δ1-pyrroline-3-hydroxy-5-carboxylate (Pyr3H5C), 4-hydroxyglutamate, and 4-hydroxy-3-oxo-glutarate (HOG), by four mitochondrial enzymes as follows: T4LHyp oxidase, Pyr3H5C dehydrogenase (EC 1.5.1.12), aspartate aminotransferase (EC 2.6.1.23), and HOG aldolase (EC 4.1.3.16) [5]. On the other hand, over 50 years after the discovery of bacteria capable of growing on T4LHyp as a sole carbon source, the enzymes (genes) involved in the hypothetical degradation were recently understood at the molecular level [6,7]. In contrast to mammalians, bacteria metabolize T4LHyp to α-ketoglutarate through four enzymatic steps (Fig. 1A). First, T4LHyp epimerase (EC 5.1.1.8; encoded by LhpA) catalyzes the isomerization of T4LHyp to cis-4-hydroxy-d-proline (C4DHyp), and this is then oxidized to Δ1-pyrroline-4R-hydroxy-2-carboxylate (Pyr4RH2C) by C4DHyp dehydrogenase (C4DHypDH; EC 1.4.99.-). Pyr4RH2C is converted to α-ketoglutaric semialdehyde (αKGSA) by Pyr4RH2C deaminase (EC 3.5.4.22; LhpC) and, in the fourth step, αKGSA is oxidized to α-ketoglutarate by the enzyme αKGSA dehydrogenase (EC 1.2.1.26; LhpG). Interestingly, there are two types of C4DHypDHs: Pseudomonas aeruginosa and Azospirillum brasilense, α4β4γ4-type enzyme encoded by LhpB (encoding to β-subunit), LhpE (α-subunit) and LhpF genes (γ-subunit); Pseudomonas putida; homodimeric-type enzyme encoded by LhpB gene. This finding strongly suggests that the T4LHyp pathway clearly evolved convergently in bacteria. LhpABCEFG genes are often clustered together with gene(s) encoding putative amino acid transporter on bacterial genomes (referred to as T4LHyp gene cluster) (Fig. 1D).
Fig. 1.
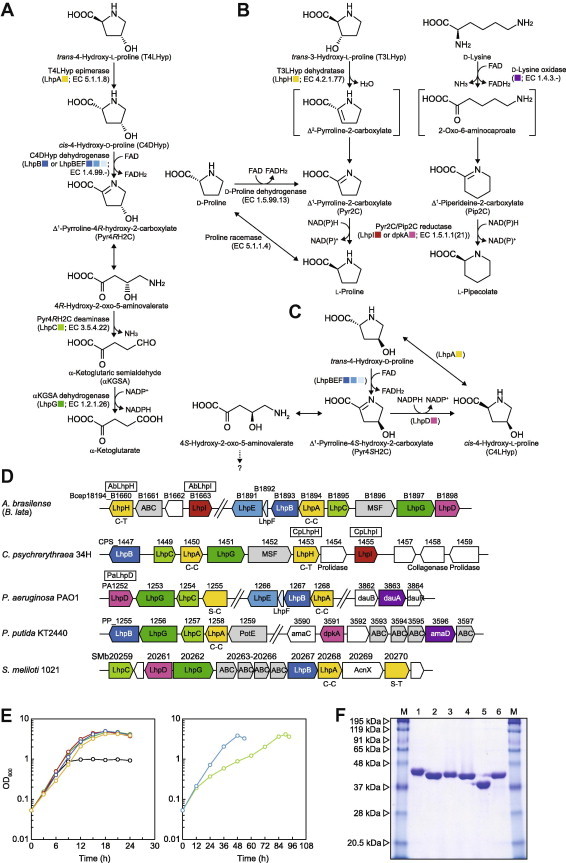
(A) Bacterial pathway of T4LHyp metabolism. Homologous genes are indicated in the same color and correspond to B–D. (B) Novel T3LHyp pathway and the metabolic networks with d-lysine and d-proline. (C) Hypothetical pathway of C4LHyp metabolism. (D) Schematic gene clusters related to T4LHyp and/or T3LHyp metabolism of bacteria. Gene cluster of A. brasilense was assumed from the genome sequence of B. lata (see text). Putative genes in the box were purified and characterized in this study (see F). C-C and C-T indicate a pair of catalytic amino acid residues of proline racemase superfamily enzymes (see Fig. S3A). Gray putative genes are sequentially similar to other amino acid transporters. (E) Growth curves of A. brasilense on glucose (black), l-proline (green), d-proline (light-green), T4LHyp (red), C4DHyp (blue), T3LHyp (yellow) and d-lysine (aqua) as a sole carbon source (30 mM). (F) Purification of recombinant His6 tag proteins. Five micrograms of each of purified protein were applied to 12% (w/v) gel. M: marker proteins. Lane 1, C14orf149; lane 2, AbLhpH; lane 3, CpLhpH; lane 4, PaLhpD; lane 5, AbLhpI; lane 6, CpLhpI. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In the case of T3LHyp, small amounts (one amino acid per 1000 amino acids) have been shown to occur in different types of collagen, including collagen I, II and III, but it was found to be particularly abundant in collagen IV (as much as 10% of the total l-Hyp content). Although degradation by organism(s) is poorly understood, Visser et al. [8] recently reported that a human C14orf149 protein catalyzes the dehydration of T3LHyp to Δ1-pyrroline-2-carboxylate (Pyr2C) via a putative Δ2-pyrroline-2-carboxylate intermediate, although the mechanism of the ability to metabolize T3LHyp by human cells is unclear. Interestingly, in spite of different reactions, the T3LHyp dehydratase (EC 4.2.1.77) belongs to the proline racemase superfamily, in which the archetypical proline racemase (EC 5.1.1.4, Ref. [9]) and T4LHyp epimerase [10] are also contained (see Fig. S3B). It had been believed that the T3LHyp dehydratase is found only in animals and fungi [8].
Although the metabolic fate of Pyr2C may be conversion to l-proline by NAD(P)H-dependent reductase (Fig. 1B), the corresponding gene has not yet been identified. On the other hand, reductase for Pyr2C from bacteria has been already studied. In Pseudomonas strains including P. putida [11] and Pseudomonas syringae [12], d-lysine is metabolized to α-ketoadipate through the so-called “l-pipecolate pathway”, in which dpkA protein catalyzes the second step (conversion of Δ1-piperidine-2-carboxylate (Pip2C) to l-pipecolate) as a Pip2C reductase (EC 1.5.1.21) (Fig. 1B). Indeed, this gene also functions as a Pyr2C reductase (EC 1.5.1.1) involved in (hypothetical) d-proline metabolism, in which Pyr2C may be produced from d-proline by (an unknown) d-amino acid oxidase [11]. The bifunctional Pyr2C/Pip2C reductase belongs to a novel NAD(P)H-dependent malate/l-lactate dehydrogenase (MDH/LDH) superfamily with no sequence homology to “orthodox” MDH/LDH, and shows strict NADPH dependence. It is unclear whether the dpkA(-like) protein functions as Pyr2C reductase in T3LHyp metabolism, because there is no homolog on mammalian genomes, and P. putida cannot metabolize T3LHyp (see in text).
In this study, we first identified that A. brasilense, previously known as a T4LHyp-metabolizing bacterium [6], has the ability to grow on T3LHyp as a sole carbon source, and that the metabolic pathway actually contains T3LHyp dehydratase and Pyr2C reductase, as proposed in mammals. Interestingly, Pyr2C reductase is a novel member of the ornithine cyclodeaminase/μ-crystallin superfamily, different from known dpkA protein, and there are several significant differences in the enzymatic properties between A. brasilense and another bacteria: substrate and coenzyme specificities. Metabolic networks among T3LHyp, T4LHyp, d-proline and d-lysine are also discussed.
2. Results
2.1. Hypothetical metabolic pathway of T3LHyp in A. brasilense
First, we found that among three bacteria capable of metabolizing T4LHyp, only A. brasilense can grow on T3LHyp as a sole carbon source, not P. putida and P. aeruginosa; to our knowledge, this is the first report of T3LHyp metabolism by an organism(s) (Figs. 1E and S1). Next, we estimated whether the T4LHyp pathway is related to T3LHyp metabolism because of its structural similarity. However, all four enzymes involved in T4LHyp metabolism were induced only by T4LHyp (and C4DHyp), not by T3LHyp (Fig. 2A). On the other hand, significant activities of T3LHyp dehydratase and Pyr2C reductase with dual specificity between NADPH and NADH were found in cell-free extract prepared from A. brasilense cells grown not only on T3LHyp but also d-proline and d-lysine. Unexpectedly, although T4LHyp and C4DHyp also induced Pyr2C reductase, enzyme activity was clearly NADPH dependent. These results indicated that T3LHyp dehydratase and Pyr2C reductase are actually involved in the hypothetical T3LHyp pathway not only of mammalians but also bacteria, and that there are Pyr2C reductase isozymes with different inductivity by carbon sources and coenzyme specificity.
Fig. 2.
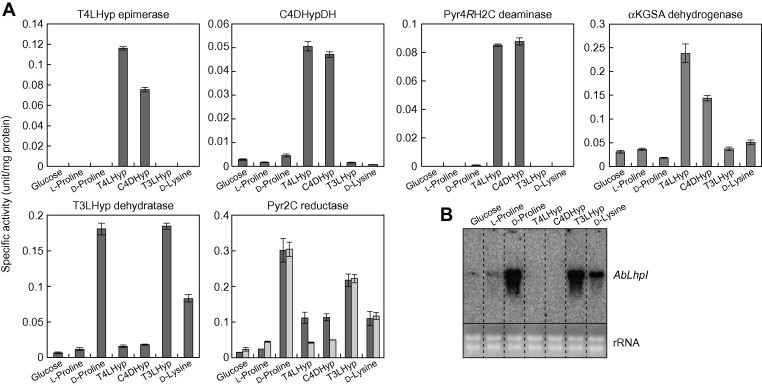
(A) Enzyme activities of cell-free extracts prepared from A. brasilense cells grown on several carbon sources. Values are the means ± SD, n = 2. Light- and dark-gray bars in Pyr2C reductase indicate NADPH- and NADH-dependent activities, respectively. (B) The transcriptional effect of carbon source on AbLhpI gene. Total RNAs (4 μg per lane) were isolated from the A. brasiliense cells grown on the indicated carbon sources.
2.2. Candidates of T3LHyp dehydratase and Pyr2C reductase genes
Although the genome sequence of A. brasilense is unavailable, nucleotide sequences of several genes from this bacterium show very high similarity (>∼98%) to those of Burkholderia lata, which was formerly described as Burkholderia sp. 183 [6]. Therefore, a homology search using the Protein-BLAST program was carried out against the genome sequence of B. lata using C14orf149 (T3LHyp dehydratase) as the probe protein sequence, although it had been believed that only animals and fungi possess this enzyme, not bacteria [8]. Among two homologous proteins (genes) annotated as putative proline racemases, Bcep18194_B1894 and Bcep18194_B1660 with sequence similarities of 29% and 44% to C14orf149, respectively, the former corresponded to T4LHyp epimerase (LhpA) (Fig. 1D), whereas the latter possessed two specific active sites for T3LHyp dehydratase (see below; Ref. [8]) (referred to as LhpH) (Fig. S3A). Therefore, we thought that the LhpH gene was the first candidate for a T3LHyp dehydratase.
B. lata (probably also A. brasilense) possessed one homologous protein (gene) to dpkA from P. putida (∼40% identity; PP_3591). However, this gene (Bcep18194_B1898; referred to as LhpD) was contained within the T4LHyp gene cluster (Fig. 1D), which was up-regulated only by T4LHyp, not T3LHyp, as described above (Fig. 2A). On the other hand, further bioinformatics analysis revealed that a (putative) LhpH gene from other bacteria such as Colwellia psychrerythraea 34H (CPS_1453) is located within the T4LHyp gene cluster together with one function-unknown protein (gene) annotated as a ornithine cyclodeaminase (OCD; EC 4.3.1.12) (referred to as LhpI), instead of LhpD gene (Fig. 1D). A gene homologous to CpLhpI gene was also found within the flanking region of AbLhpH gene, and the enzyme reaction by OCD contained Pyr2C as an intermediate (see Fig. 5C). Based on these analysis, we selected LhpD and/or LhpI gene as candidates for Pyr2C reductase.
Fig. 5.
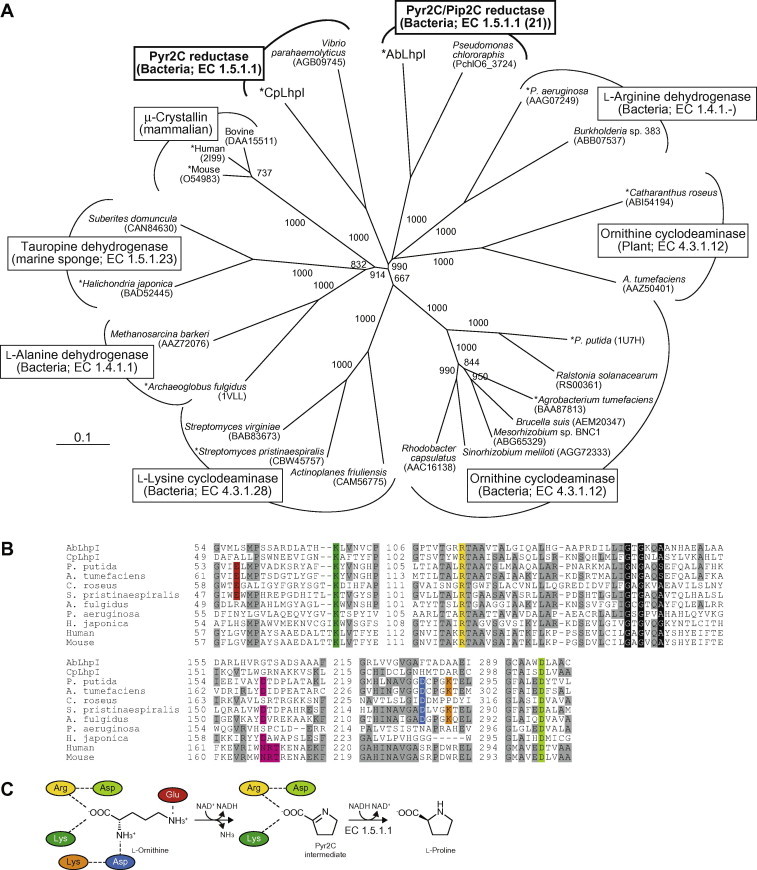
(A) Phylogenetic tree of OCD/μ-crystallin protein family. The number on each branch indicates the bootstrap value. Proteins with asterisks were used for B. (B) Partial multiple sequence alignment of deduced amino acid sequences of Pyr2C reductases. Binding sites for a carboxyl group of substrate, lysine, arginine and aspartate, are shaded in green, yellow and light-green, respectively. Aspartate and glutamate residues, related to the cyclization by OCD and LCD, are shaded in red and blue, respectively. Amino acid residues interacting with 2′- and 3′-functional groups in the ribosyl moiety of NAD+(H) and NADP+(H) are shaded in pink. Coenzyme-binding motif of Rossmann-fold are shown as white letters in black boxes. Gray-shaded letters indicate highly conserved amino acid residues. (C) Schematic diagram showing the interactions of l-ornithine, Pyr2C, and nearby residues in OCD. Color of residues correspond to B. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.3. Preparation of recombinant His6-tag proteins
After cloning all target genes into the vector pETDuet-1, the recombinant enzymes with attached His6-tags at their N-termini were expressed in Escherichia coli and purified with an Ni2+-chelating affinity column (PaLhpD was characterized instead of AbLhpD because of its successful expression in E. coli cells, and there is 59.8% identity between the proteins) (Fig. 1F). Apparent molecular masses of AbLhpH, CpLhpH, C14orf149, PaLhpD, AbLhpI, and CpLhpI, estimated by SDS–PAGE, were 40 (37,805.87), 40 (40,437.15), 44 (39,618.72), 40 (37,164.78), 37 (33,312.46), and 40 (36,181.76) kDa (values in parentheses indicate the calculated molecular mass of the enzyme with His6-tag), and those estimated by analytic gel filtration were 87, 98, 70, 62, 78 and 81 kDa, respectively (Fig. S2). Therefore, all of these enzymes appear to be dimeric.
2.4. Characterization of LhpH protein as bacterial T3LHyp dehydratase
Potential T3LHyp dehydratase activity in LhpH proteins were assayed by the colorimetric method based on the reaction of 2-aminobenzaldehyde with Pyr2C [8], described in Section 4. Specific activities with T3LHyp of AbLhpH, CpLhpH, and C14orf149 (as a reference) were 19.8, 19.3, and 7.22 U mg protein−1, respectively. Optimum pH values was also determined by this method: common range of pH 8.0–9.5 (data not shown). This indicated that the LhpH gene encodes T3LHyp dehydratase (first example for bacteria), and that the biochemical properties are similar to the mammalian enzyme C14orf149.
2.5. Characterization of LhpD as Pyr2C reductase
The purified PaLhpD showed similar reductase activities for both Pyr2C and Pip2C in the presence of NADPH but not NADH: 42.3 and 32.8 U mg protein−1, respectively (Table 1). Although both l-proline and l-pipecolate underwent NADP+-dependent oxidization, their kcat/Km values were ∼500 and ∼350-fold lower than those with Pyr2C and Pip2C, respectively (Table 2). Namely, the reaction equilibrium favors the direction toward NADPH-dependent reduction. Optimum pH values in reduction and oxidization using Pyr2C and l-proline were pH 7.0 and pH 10.0, respectively (Fig. 3A). These substrate and coenzyme specificities also corresponded to zymogram staining analysis (Fig. 3C). Overall, PaLhpD (probably also AbLhpD) showed similar enzymatic properties to dpkA [11,12]. Furthermore, it was likely that based on coenzyme specificity, LhpD corresponds to an enzyme induced by T4LHyp (and C4DHyp) in A. brasilense cells (Fig. 2A).
Table 1.
Kinetic parameters of AbLhpI, CpLhpI, and PaLhpD in the forward direction.
| Enzymes | Substrates | pHa | Coenzymes | Specific activity (units/mg protein) | Km (mM) | kcat (min−1) | kcat/Kmb (min−1 mM−1) |
|---|---|---|---|---|---|---|---|
| AbLhpI | Pyr2C | 6.5 | NADPH | 584 | 0.628 ± 0.045 | 31400 ± 1300 | 50100 ± 1400 |
| NADH | 600 | 0.837 ± 0.080 | 36900 ± 2400 | 44100 ± 1400 | |||
| Pip2C | NADPH | 220 | 0.474 ± 0.050 | 9950 ± 610 | 21100 ± 1010 | ||
| NADH | 177 | 0.600 ± 0.005 | 9690 ± 380 | 16200 ± 680 | |||
| Pyr4SH2C | NADPH | 12.0 | 0.491 ± 0.063 | 621 ± 58 | 1270 ± 45 | ||
| CpLhpI | Pyr2C | NADPH | 30.2 | 5.90 ± 0.36 | 7470 ± 470 | 1270 ± 2 | |
| NADH | 4.76 | 2.79 ± 0.42 | 621 ± 34 | 225 ± 24 | |||
| Pip2C | NADPH | 0.291 | 7.26 ± 1.22 | 82.9 ± 13.7 | 11.4 ± 0.2 | ||
| NADH | 0.0464 | 2.10 ± 0.17 | 5.27 ± 0.24 | 2.51 ± 0.09 | |||
| Pyr4SH2C | NADPH | 0.174 | 8.28 ± 2.45 | 61.3 ± 17.2 | 7.43 ± 0.14 | ||
| PaLhpD | Pyr2C | 7.0 | NADPH | 42.3 | 0.447 ± 0.040 | 2500 ± 120 | 5600 ± 220 |
| Pip2C | 38.3 | 1.57 ± 0.3 | 2120 ± 290 | 1350 ± 68 | |||
| Pyr4SH2C | 12.1 | 0.835 ± 0.109 | 868 ± 71 | 1043 ± 48 |
pH of potassium phosphate buffer for assay.
Illustrated in Fig. 3D.
Table 2.
Kinetic parameters of AbLhpI, CpLhpI and PaLhpD in the reverse direction.
| Enzymes | Substrates | pHa | Coenzymes | Specific activity (units/mg protein) | Km (mM) | kcat (min−1) | kcat/Kmb (min−1 mM−1) |
|---|---|---|---|---|---|---|---|
| AbLhpI | l-Proline | 10.5 | NADP+ | 5.18 | 3.63 ± 0.31 | 235 ± 7 | 65.0 ± 3.7 |
| NAD+ | 5.50 | 3.99 ± 0.18 | 254 ± 3 | 63.6 ± 2.1 | |||
| l-Pipecolate | NADP+ | 2.92 | 6.54 ± 0.96 | 150 ± 14 | 23.0 ± 1.2 | ||
| NAD+ | 2.72 | 14.8 ± 2.1 | 222 ± 24 | 15.0 ± 0.5 | |||
| CpLhpI | l-Proline | NADP+ | 0.630 | 18.3 ± 2.4 | 65.3 ± 6.9 | 3.57 ± 0.09 | |
| NAD+ | 0.00451 | 18.8 ± 3.8 | 0.415 ± 0.077 | 0.0222 ± 0.0004 | |||
| l-Pipecolate | NADP+ | 0.00435 | 80.1 ± 2.8 | 1.28 ± 0.04 | 0.016 ± 0.001 | ||
| NAD+ | 0.000457 | N.D.c | N.D. | N.D. | |||
| PaLhpD | l-Proline | 10.0 | NADP+ | 1.98 | 18.5 ± 2.1 | 205 ± 18 | 11.1 ± 0.3 |
| l-Pipecolate | 0.679 | 34.8 ± 4.0 | 135 ± 14 | 3.88 ± 0.05 | |||
| T3LHyp | 0.490 | 132 ± 26 | 272 ± 53 | 2.07 ± 0.01 | |||
pH of glycine–NaOH buffer for assay.
Illustrated in Fig. 3E.
Not determined due to trace activity.
Fig. 3.
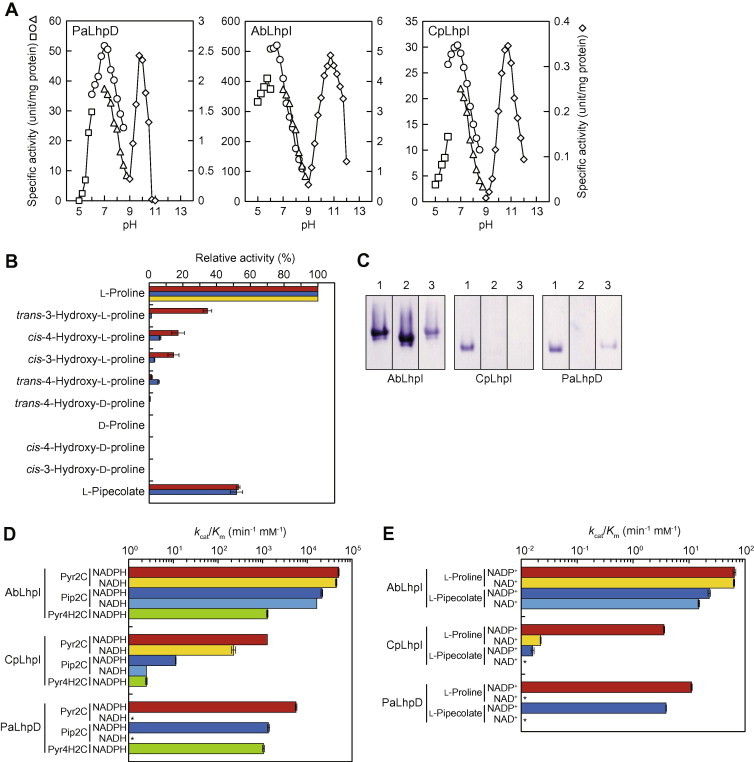
Enzymatic properties of Pyr2C reductase. (A) Effect of pH on the activity. Left panel, PaLhpD; middle panel, AbLhpI; right panel, CpLhpI. 50 mM acetate-NaOH (pH 4.0–6.0) (square), 50 mM potassium phosphate (pH 6.0–8.5) (circle), 50 mM Tris–HCl (pH 7.0–9.0) (triangle) for the reduction of Pyr2C (left axis), and 50 mM glycine–NaOH (diamond) for the oxidization of l-proline (right axis), instead of 50 mM Tris-HCl (pH 8.0) under standard assay conditions. (B) Substrate specificity of NADP+-dependent oxidization reaction. PaLhpD, red bar; AbLhpI, blue bar; CpLhpI, yellow bar. The assay was performed with standard assay solution containing the indicated substrate (10 mM). Relative values were expressed as percents of the values obtained in l-proline (means ± SD, n = 3). (C) Zymogram staining. Five micrograms of each of purified protein (left panel, AbLhpI; middle panel, CpLhpI; right panel, PaLhpD) was applied on 10% (w/v) gel. After electrophoresis, the gel was soaked in staining solution in the presence of substrates and coenzymes as follows (each of 10 mM): lane 1, l-proline and NADP+; lane 2, l-proline and NAD+; lane 3, l-pipecolate and NADP+. (D) Catalytic efficiency (kcat/Km) of NADPH-dependent reduction (left panel) and NADP+-dependent oxidization reactions (right panel). Asterisks indicate no (or trace) activity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.6. Characterization of LhpI as novel Pyr2C reductase
First, the LhpI proteins were assayed for OCD activity, with none detected (data not shown). Alternatively, when Pyr2C and NADPH was used as the substrate and coenzyme, respectively, significant reduction of activity was observed. Furthermore, l-proline was the active substrate for the NADP+-dependent oxidization reaction, and ratios of Pyr2C to l-proline in kcat/Km were 771 and 356 for AbLhpI and CpLhpI, respectively (Tables 1 and 2), suggesting the preference of the reaction equilibrium to the direction toward NADPH-dependent reduction. Their optimum pH values in reduction and oxidization using Pyr2C and l-proline were pH 6.5 and pH 10.5, respectively (Fig. 3A). These properties as a Pyr2C reductase were similar to PaLhpD.
On the other hand, there were also several significant differences in enzymatic properties between AbLhpI and CpLhpI (and PaLhpD). First, the kcat/Km value for Pyr2C of AbLhpI (50,100 min−1 mM−1) was ∼350- and 8.9-fold higher than those of CpLhpI and PaLhpD, respectively, commonly caused by the higher kcat values. Second, there was no preference for coenzyme utilization between NADPH and NADH in AbLhpI, whereas the kcat/Km value for Pyr2C of CpLhpI in the presence of NADH was 5.6-fold lower than that in the presence of NADPH, mainly caused by the 12-fold lower kcat value. Much higher preference for coenzymes was found in the oxidization of l-proline: the ratio of NADPH to NADH in kcat/Km value was 161. Third, AbLhpI could utilize Pip2C (and l-pipecolate) in almost the same manner as Pyr2C (l-proline). On the other hand, kcat/Km values for Pip2C and l-pipecolate of CpLhpI (in the presence of NADP+(H)) were 111- and 223-fold lower than those for Pyr2C and l-proline, mainly caused by 90- and 51-fold lower kcat values, respectively. To estimate more detailed substrate specificity, several proline analog substrates (because of easy preparation) were used for the NADP+-dependent oxidization reaction, in addition of l-proline and l-pipecolate. Only PaLhpD showed 32.8%, 14.4% and 12.2% activity for T3LHyp, cis-4-hydroxy-l-proline (C4LHyp) and cis-3-hydroxy-l-proline (C3LHyp) as a percent of l-proline, respectively (Fig. 3B). Among the corresponding substrates for the NADPH-dependent reduction reaction, we successfully synthesized Δ1-pyrroline-4S-hydroxy-2-carboxylate (Pyr4SH2C) for C4LHyp. The ratio of Pyr2C to Pyr4SH2C of PaLhpD, AbLhpI and CpLhpI (in the presence of NADPH) was 5.4, 39.4 and 171, respectively, raising the possibility that PaLhpD can utilize Pyr4SH2C as a physiological substrate (see Section 3). Such different properties among these three enzymes also corresponded to zymogram staining analysis (Fig. 3C). These results suggested that AbLhpI and CpLhpI are bifunctional NAD(P)H-dependent Pyr2C/Pip2C reductase and monofunctional NADPH-preference Pyr2C reductase, respectively, and that Pyr2C reductase activities induced by T3LHyp, d-proline and d-lysine in A. brasilense cells may be derived from LhpI but not LhpD (Fig. 2A).
In HPLC analysis (Fig. 4), a peak corresponding to T3LHyp was completely eliminated by incubation with purified AbLhpH (or CpLhpH), probably caused by no reaction of Pyr2C with labeling reagent for amino acid analysis. On the other hand, when T3LHyp was incubated together with purified AbLhpH and AbLhpI (or CpLhpH and CpLhpI) in the presence of NADPH, a novel peak corresponding to (l-)proline appeared. This suggested that T3LHyp is converted to l-proline by continuous reactions with LhpH and LhpI under physiologically neutral conditions (pH 7.0).
Fig. 4.
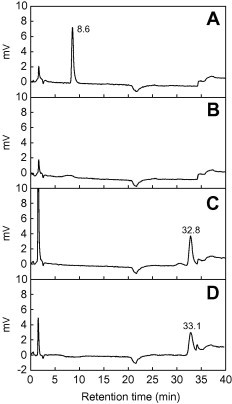
HPLC analysis of the reaction products from T3LHyp by AbLhpH and AbLhpI. (A) Authentic T3LHyp. (B) Reaction product by AbLhpH. (C) Reaction product by AbLhpH and AbLhpI in presence of NADPH. (D) Authentic l-proline. Similar results were obtained when CpLhpH and CpLhpI were used as alternatives (data not shown).
2.7. Amino acid sequence analysis of LhpI
As expected from preliminary annotation, LhpI belongs to the OCD/μ-crystallin superfamily including the archetype OCD [13], μ-crystallin [14], l-alanine dehydrogenase (EC 1.4.1.1) [15], l-arginine dehydrogenase [16], l-lysine cyclodeaminase (LCD; EC 4.3.1.28) [17] and tauropine dehydrogenase (EC 1.5.1.23) [18] (Fig. 5A). On the other hand, phylogenetic analysis revealed that AbLhpI and CpLhpI have poor relationship not only to any subclasses of the other members but also each other. Putative amino acid sequences of the LhpI proteins contained essential amino acid residues for coenzyme binding (Rossmann-fold motif consisting of Gly-X-Gly-X2-[Ala/Ser], where X indicates any amino acid) and the catalytic triad for binding to a carboxyl group of substrate (Lys-Arg-Asp): Gly137-Thr-Gly-Lys-Gln-Ala142 and Lys70-Arg113-Asp294 in AbLhpI, respectively (Fig. 5B and C). On the other hand, putative amino acid residues, responsible for discrimination between NAD+(H) and NADP+(H), were different not only from other OCD/μ-crystallin members but also within LhpI proteins: AbLhpI, Gly162-Thr163; CpLhpI, Gly158-Arg159.
2.8. Gene regulation and disruptant analysis
It is likely that AbLhpH and AbLhpI genes are clustered together with the putative amino acid transporter gene on the genome of A. brasilense (Fig. 1D). Northern blot analysis revealed that AbLhpI gene was induced by T3LHyp, d-proline and d-lysine, but not T4LHyp (and C4DHyp) (Fig. 2B). To further estimate the physiological roles of LhpI gene, we carried out gene disruption experiments by introducing a kanamycin-resistant gene (Kmr) into AbLhpI gene. The obtained AbLhpI− mutant strain was distinct from the wild-type strain in that T3LHyp, d-proline and d-lysine did not support growth as a sole carbon source. On the other hand, there was no difference in growth on other carbon sources, including T4LHyp, between the two strains. Although we did not analyze AbLhpD in this study, it has been reported that disruption of LhpD gene from Sinorhizobium meliloti had no effect on growth on T4LHyp, in spite of transcriptional induction by T4LHyp (Fig. 1D) [19]. Overall, these results suggested clearly that LhpI gene is a Pyr2C/Pip2C reductase involved in T3LHyp, d-proline and d-lysine metabolism, and that LhpD gene is not related directly to T3LHyp and T4LHyp metabolism (see below).
3. Discussion
In this study, we identified T3LHyp pathway consisting of T3LHyp dehydratase and Pyr2C reductase of bacteria. A similar T3LHyp metabolic pathway may exist in mammals [8], however, this contrasts with T4LHyp metabolic pathways as there are complete different in bacteria and mammals [5–7].
On the basis of two specific residues at the active sites, proline racemase-like enzymes have been classified into three types: Cys-Cys type (proline racemase and T4LHyp epimerase); Cys-Thr type (T3LHyp dehydratase); Ser-Cys type (function-unknown) (Fig. S3A). Interestingly, a mutant enzyme of T3LHyp dehydratase (C14orf149) with a substitution of threonine to cysteine shows “T3LHyp epimerase” activity [8]. In the phylogenetic tree (Fig. S3B), T4LHyp epimerase and T3LHyp dehydratase in the same bacteria (LhpA and LhpH, respectively) belong to different subfamilies, indicating that dehydratase activity with T3LHyp in this superfamily was acquired once at an early evolutional stage.
In mammalians, Pyr2C is one of the substrates for ketimine reductase (EC 1.5.1.25), in which Pip2C and several ketimine compounds with neurological functions are also contained [20]. It was recently reported that μ-crystallin, belonging to the same protein family as LhpI, has ketimine reductase activity [21]: this protein was previously known as an NADPH-dependent thyroid hormone-binding protein without enzymatic function [14]. However, the purified μ-crystallin accounted for only 0.19% of total enzyme activity measured in a cell-free extract of the lamb forebrain, and enzyme activity was optimal at acidic pH 4.5–5.0, in contrast with neutral pH of LhpI (Fig. 3A): it is doubted that this protein physiologically functions as the major ketimine reductase (and also Pyr2C reductase involved in T3LHyp metabolism) [22]. Furthermore, in spite of reaction similarity, bacterial LhpI proteins form a distinct subfamily from μ-crystallin in the phylogenetic tree (Fig. 5A). Similar phenomena were also found in OCDs of bacteria and plants [13,23]. Based on these insights, we assumed that these subfamilies appeared by the independent acquisition of substrate specificity for Pyr2C (and/or Pip2C) rather than divergence from a common ancestor: convergent evolution. Namely, it is still unclear whether T3LHyp pathways of bacteria and mammalian evolved from the same ancestor(s).
OCD and LCD catalyze unusual cyclization to convert l-ornithine and l-lysine to l-proline and l-pipecolate via Pyr2C and Pip2C intermediates, with the release of ammonia, respectively (Fig. 5C; Refs. [13,17]). Homologous aspartate and glutamate residues, related to cyclization [24], are not found in LhpI (and other OCD/μ-crystallin members), confirming only Pyr2C (and/or Pip2C) reductase activity. In OCD (from P. putida), Asp161 forms hydrogen bonds with the 2′- and 3′-hydroxyl groups of the NADH ribose moiety [13,24] (Fig. 5B). On the other hand, in μ-crystallin (from human), the 2′-phosphate group of the NADPH ribose moiety interacts with side-chains of Asn168 (equivalent to Asp161 in OCD), Arg169 and Thr170, in which the enzyme favors binding to NADP+(H) with negative charge [14]. Such a tendency has been observed in other many dinucleotide-binding domains [25]. Both AbLhpI and CpLhpI possess no homologous Asp residue to Asp161 in OCD (substitution to Gly), and the homologous Arg residue to Arg169 in μ-crystallin is found only in CpLhpI. Site-directed mutagenetic study is in progress to assess the unique coenzyme specificity of LhpI at the molecular level.
No homologous gene with known metabolic genes (proteins) involved in the l-pipecolate pathway of d-lysine has been found on the genome of C. psychrerythraea, confirming that CpLhpI shows no significant Pip2C reductase activity (Tables 1 and 2): this bacteria may probably possess no ability to metabolize d-lysine. Such different substrate specificity from AbLhpI may be related to their distant phylogenetic relationship (only 21.8% identity) (Fig. 5A): there is a possibility of convergent evolution even within bacterial LhpI proteins, similar to C4DHypDH proteins in T4LHyp metabolism [6].
One of the most interesting points in this study is that in A. brasilense, LhpI (but not LhpD), functions as a Pyr2C/Pip2C reductase not only in T3LHyp but also d-proline and d-lysine metabolism (Fig. 1B), which are good examples of convergent evolution. Indeed, the OCD/μ-crystallin superfamily (but not MDH/LDH superfamily including dpkA) contains several enzymes related to proline, lysine and arginine metabolism (Fig. S4). Since the final product of T3LHyp metabolism is l-proline, these pathways might have evolved by duplication and divergence of a common ancestor. Then, what is the physiological role of LhpD? This enzyme can efficiently utilize Pyr4SH2C (and C4LHyp) as a substrate (Table 1 and Fig. 3B, D and E). Furthermore, we recently found that C4LHyp and T4DHyp are also substrates of T4LHyp epimerase and C4DHypDH, respectively, by which C4LHyp is converted to T4DHyp (via Pyr4SH2C) (Fig. 1C) [26]. Such l- to d-epimerization (racemization) of amino acids consisting of two distinct FAD- and NAD(P)+-dependent dehydrogenase (oxidase) is also found in arginine, lysine and proline metabolism from bacteria (Fig. S4) [16]. Therefore, it is likely that LhpD is involved in the degradation of C4LHyp, a compound which is generated by the hydroxylation of free l-proline by bacteria [3].
A. brasilense possesses two separated gene clusters for T4LHyp and T3LHyp metabolism (Fig. 1D), which are up-regulated only by each carbon source (Fig. 2). These properties may be suitable for the metabolism of T4LHyp and T3LHyp (and C4LHyp) produced by direct hydroxylation of free l-proline, as described in Section 1: in fact, the hydroxylase is often found in soil bacteria that fix nitrogen (so-called rhizobia; Ref. [3]), similar to A. brasilense. In contrast, the homologous gene clusters of C. psychrerythraea, a marine bacteria, are combined (Fig. 1D): this gene cluster may be induced by both T4LHyp and T3LHyp. If this hypothesis is true, it is likely that gene regulation is advantageous for the utilization of much marine collagen, because of the co-production of T4LHyp and T3LHyp.
Since post-translational hydroxylation of l-proline residues is almost specific to collagen protein, l-Hyp(s) provides an important marker to directly measure collagen content in several sample types, including foods and tissue fibrosis. Furthermore, l-Hyp(s) in urine and serum has been focused on as a significant biomarker for bone resorption and many human diseases (urinal T3LHyp for cancer; Ref. [27]). On the other hand, the most popular HPLC method for determination of l-Hyp(s) is time-consuming and requires expensive and large apparatus and organic solvent, and l-Hyp(s) (and l-proline), a cyclic imino acid, cannot react with a general labeling reagent for amino acids. Alternatively, an enzymatic method for the T3LHyp dehydratase assay, described in Section 4, would be helpful for conventional detection of T3LHyp in in vivo samples. This line of study is in progress in our laboratory [28].
4. Experimental procedures
4.1. Materials
T3LHyp was purchased from Kanto Chemical (Tokyo, Japan). C4LHyp, l-pipecolate, and d-pipecolate were obtained from Tokyo Chemical Industry (Tokyo, Japan). T4DHyp and C3LHyp were from Sigma Aldrich (USA). T4LHyp, C4DHyp, l-proline, and d-proline were from Wako Pure Chemical Industries (Osaka, Japan).
4.2. General procedures
Basic recombinant DNA techniques were performed as described by Sambrook et al. [29]. Bacterial genomic DNA was prepared using a DNeasy Tissue Kit (Qiagen). PCR was carried out using a GeneAmp PCR System 2700 (Applied Biosystems) for 30 cycles in 50 μL reaction mixture containing 1 U of KOD FX DNA polymerase (TOYOBO), appropriate primers (15 pmol) and template DNA under the following conditions: denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s and extension at 68 °C for time periods calculated at an extension rate of 1 kbp min−1. DNA sequencing was carried out using the BigDye Cycle Sequencing Kit ver.3.1 (Applied Biosystems) and appropriate primers with the Genetic Analyzer 3130 (Applied Biosystems). Protein concentrations were determined by the method of Lowry et al. [30] with bovine serum albumin as the standard. SDS–PAGE was performed as described by Laemmli [31]. Amino acids were identified using an amino acid analyzer (L-8900; Hitachi, Tokyo, Japan) using commercial standards (Wako).
4.3. Substrates
Pyr2C was enzymatically synthesized from T3LHyp with C14orf149. The reaction mixture (10 mL) consisted of 50 mM potassium phosphate buffer (pH 7.0) and 10 mM T3LHyp. After the addition of ∼20 mg of purified C14orf149, the mixture was left at 30 °C overnight. For enzymatic synthesis of Pip2C and Pyr4SH2C, a reaction mixture (10 mL) consisting of 50 mM Tris-HCl buffer (pH 9.0), 10 mM d-pipecolate (for Pip2C) or T4DHyp (for Pyr4SH2C), 0.02 mM phenazine methosulfate (PMS), and ∼20 mg purified AbLhpBEF (C4DHypDH) was incubated with shaking at 30 °C overnight in the dark. Pyr2C, Pip2C, and Pyr4SH2C in each reaction mixture were purified using a Dowex 1 × 2 Cl− form (100–200 mesh) resin column, described previously [11].
4.4. Bacterial strain, culture conditions and preparation of cell-free extracts
A. brasilense ATCC29145 and P. aeruginosa PAO1 were cultured aerobically with vigorous shaking at 30 °C in minimal medium [7] supplemented with 30 mM carbon source. C. psychrerythraea 34H was grown at 8 °C in Marine Broth (Difco 2216). The grown cells were harvested by centrifugation at 30,000g for 20 min, suspended in 50 mM Tris-HCl (pH 8.0), and disrupted by sonication for 20 min at appropriate intervals on ice using Ultra Sonic Disruptor Model UR-200P (TOMY SEIKO Co., Ltd., Tokyo, Japan) and then centrifuged at 108,000g for 20 min at 4 °C to obtain cell-free extracts.
4.5. Plasmid construction for expression of recombinant proteins
Primer sequences used in this study are shown in Table S1. In this report, the prefixes Ab (A. brasilense), Pa (P. aeruginosa), Cp (C. psychrerythraea) and Pp (P. putida) have been added to gene symbols or protein designations when required for clarity. PaLhpD (PA1252), CpLhpH (CPS_1453), and CpLhpI genes (CPS_1455) were amplified by PCR using primers containing appropriate restriction enzyme sites at the 5′- and 3′-ends and genome DNA of P. aeruginosa or C. psychrerythraea as a template. C14orf149 gene was obtained from Human cDNA clone AK058165 (NITE Biological Resource Center (NBRC), Chiba, Japan). AbLhpH and AbLhpI genes were amplified by PCR using primers designed from putative proline racemase (Bcep18194_B1660) and OCD genes (Bcep18194_B1663) from B. lata and genome DNA of A. brasilense as a template, and the amplified products were sequenced. The nucleotide sequences of AbLhpH and AbLhpI genes were submitted to GenBank with accession numbers GenBank: AB894494 and GenBank: AB894495, respectively.
Each amplified DNA fragment was introduced into BamHI-HindIII sites (for PaLhpD, AbLhpH, AbLhpI and CpLhpI genes) or BamHI-PstI (for C14orf149 and CpLhpH genes) in pETDuet-1 (Novagen), a plasmid vector for conferring N-terminal His6-tag on expressed proteins, to obtain pET/PaLhpD, pET/AbLhpH, pET/AbLhpI, pET/CpLhpH, pET/CpLhpI, and pET/C14orf149. The five former and pET/C14orf149 were transformed into E. coli strains BL21(DE3) and BL21(DE3)-RIL (Novagen), respectively.
4.6. Expression and purification of His6-tagged recombinant proteins
E. coli harboring the expression plasmid for His6-tagged proteins was grown at 37 °C to a turbidity of 0.6 at 600 nm in Super broth medium (pH 7.0, 12 g tryptone, 24 g yeast extract, 5 mL glycerol, 3.81 g KH2PO4, and 12.5 g K2HPO4 per liter) containing 50 mg/liter ampicillin. After the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), the culture was further grown for 6 h at 37 °C (for C14orf149 and AbLhpH) or for 18 h at 18 °C (for PaLhpD, AbLhpI, CpLhpH and CpLhpI) to induce the expression of His6-tagged protein. Cells were harvested and resuspended in Buffer A (50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl and 10 mM imidazole). The cells were then disrupted by sonication, and the solution was centrifuged. The supernatant was loaded onto a Ni-NTA Superflow column (Qiagen) equilibrated with Buffer A linked to the BioAssist eZ system (TOSOH). The column was washed with Buffer B (50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl, 10% (v/v) glycerol, and 50 mM imidazole). The enzymes were then eluted with Buffer C (pH 8.0, Buffer B containing 250 mM imidazole instead of 50 mM imidazole), concentrated by ultrafiltration with Centriplus YM-30 (Millipore), dialyzed against 50 mM Tris–HCl buffer (pH 8.0) containing 50% (v/v) glycerol, and stored at −35 °C until use.
The native molecular mass of recombinant proteins was estimated by gel filtration, which was carried out using HPLC with a Multi-Station LC-8020 model II system (TOSOH) at a flow rate of 1 mL min−1. The purified enzyme (∼10 mg mL−1) was loaded onto a TSKgel G3000SWXL column (TOSOH) equilibrated with 50 mM Tris–HCl buffer (pH 8.0). A high molecular weight gel filtration calibration kit (GE Healthcare) was used as a molecular marker.
4.7. Enzyme assay
All enzyme assays were performed at 30 °C.
T3LHyp dehydratase was assayed spectrophotometrically in the coupling system with PaLhpD (NADPH-dependent Pyr2C reductase) using a Shimadzu UV-1800 spectrophotometer (Shimadzu GLC Ltd., Tokyo, Japan). The reaction mixture consisted of 50 mM Tris–HCl (pH 8.0), 0.15 mM NADPH and 10 μg purified PaLhpD. The reaction was started by the addition of 100 mM T3LHyp (100 μL) with a final reaction volume of 1 mL. One unit of enzyme activity refers to 1 μmol NADPH produced/min. Km and kcat values were calculated by a Lineweaver–Burk plot. The enzyme was alternatively assayed by the colorimetric method based on the reaction of 2-aminobenzaldehyde with Pyr2C, which yields a yellow reaction product [8]. This method was used for the determination of optimum pH for the activity.
Pyr2C/Pip2C reductase was assayed routinely in the direction of Pyr2C reduction by measuring the oxidization of NAD(P)H at 340 nm. The standard assay mixture contained 10 mM Pyr2C (or Pip2C) in 50 mM potassium phosphate (pH 6.5 for AbLhpI and CpLhpI, or pH 7.0 for PaLhpD) buffer. The reactions were started by the addition of 100 μL of a 1.5 mM NAD(P)H solution to a final volume of 1 mL. To assay the reverse reaction, the reaction mixture consisted of 50 mM Glycine–NaOH (pH 10.0 for PaLhpD or pH 10.5 for AbLhpI and CpLhpI) and 10 mM l-proline (or l-pipecolate). The reaction was started by the addition of 15 mM NAD(P)+ (100 μL) with a final reaction volume of 1 mL. One unit of enzyme activity refers to 1 μmol NAD(P)H produced/min. Potential OCD activity in LhpI was assayed by the method described previously [23].
T4LHyp epimerase, C4DHypDH, Pyr4RH2C deaminase, and αKGSA dehydrogenase, involved in T4LHyp metabolism, were assayed by the method described previously [7]. If necessary, recombinant AbLhpBEF [26] was used as a coupling enzyme of the epimerase assay as C4DHypDH, instead of recombinant PaLhpBEF.
4.8. Reaction product analysis
Purified AbLhpH and AbLhpI or CpLhpH and CpLhpI (each 10 μg) were added to 50 mM Tris–HCl buffer (pH 8.0) containing 10 mM T3LHyp and 0.15 mM NADPH (1 mL). After incubation at 30 °C overnight, each enzyme product was then analyzed by Hitachi L-8900 amino acid analyzer (Tokyo, Japan), using ion exchange chromatography followed by post-column derivatization with ninhydrin. Retention times of T3LHyp and l-proline (potential product) were appropriately 8.6 and 33 min, respectively.
4.9. Zymogram staining analysis
Purified PaLhpD, AbLhpI and CpLhpI were separated at 4 °C on non-denaturing PAGE with 10% (w/v) gel, which was performed by omitting SDS and 2-mercaptoethanol from the solution used in SDS–PAGE. The gels were then soaked in 10 mL staining solution consisting of 50 mM Glycine–NaOH (pH 10), 0.25 mM nitroblue tetrazolium (NBT), 0.06 mM PMS, 10 mM substrate (l-proline or l-pipecolate), and 15 mM NAD(P)+ at room temperature for 15 min in the dark. Dehydrogenase activity appeared as a dark blue band.
4.10. Northern blot analysis
A. brasilense cells were cultured at 30 °C to the mid-log phase (OD600 = 0.6–0.8) in minimal medium containing 30 mM carbon source, and harvested by centrifugation. Total RNA preparation was isolated using the SV Total RNA Isolation Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The isolated RNA (4 μg) was subjected to electrophoresis on 1.2% (w/v) agarose gel containing 0.66 M formaldehyde, and blotted to Hybond-N (GE Healthcare) by capillary transfer using 10× SSC as a transfer buffer (1× SSC is 15 mM sodium citrate (pH 7.0), and 0.15 M NaCl). The blotted filter was cross-linked in a UV cross-linker CX-2000 (Ultra-Violet Products, Ltd.). A double-stranded probe DNA was labeled with digoxigenin-11-dUTP and hybridized using a DIG-High Prime DNA labeling and detection starter kit (Roche Applied Science). Membrane was visualized using a nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate reagent detection system (Roche Applied Science).
4.11. Target disruption of AbLhpI gene
The Tn5-derived SacII 1.3-kbp kanamycin resistance (Kmr) cassette was amplified by PCR using pUC4K (GE Healthcare) as a template and two primers P13 and P14 (Table S1), and inserted into the single SacII site in the coding sequence of AbLhpI gene of pET/AbLhpI to yield pLhpI::Km. To introduce the restriction site for MfeI at the 5′- and 3′-end of the DNA fragment containing the Kmr gene in the AbLhpI gene, PCR was carried out using pLhpI::Km as a template and two primers P15 and P16. The 2.2-kbp MfeI DNA fragment was subcloned into EcoRI site in a chloramphenicol resistance (Cmr) cassette of the suicide vector pSUP202 [32] to yield pSUP/LhpI::Km. E. coli S17-1 [32] was transformed with pSUP/LhpI::Km, and then the transformant was further mobilized to A. brasilense by biparental mating. The transconjugants were selected on a minimal medium agar plate supplemented with 5 g sodium malate and 25 μg kanamycin per liter using Kmr (the presence of Kmr cassette) and TcS (loss of pSUP202) phenotypes. The construction was confirmed by genomic PCR.
4.12. Amino acid sequence alignment and phylogenetic analysis
Protein sequences were analyzed using the Protein-BLAST and Clustal W program distributed by DDBJ (DNA Data Bank of Japan) (www.ddbj.nig.ac.jp). The phylogenetic tree was produced using the TreeView 1.6.1. program.
5. Database
Nucleotide sequence data are available in the DDBJ/EMBL/GenBank databases under the accession number(s) GenBank: AB894494 and GenBank: AB894495 for T3LHyp dehydratase and Pyr2C reductase genes from A. brasilense.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan (25440049) (to S.W.), the A-STEP feasibility study program (AS242Z00554M) from the Japan Science and Technology Agency (JST) (to S.W.), and Hokuto Foundation for Bioscience (to S.W.). We thank Prof. Miyuki Kawano-Kawada (Integrated Center for Sciences (INCS), Ehime University) for amino acid analysis. Our thanks are extended especially to Profs. Hiroshi Takagi and Iwao Ohtsu, and Dr. Yusuke Kawano (Nara Institute of Technology) for invaluable advice.
Appendix A. Supplementary data
Th PDF file contains Supplementary Figs. S1–S4 and Table S1.
References
- 1.Adams E., Frank L. Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 2.Shibasaki T., Mori H., Chiba S., Ozaki A. Microbial proline 4-hydroxylase screening and gene cloning. Appl. Environ. Microbiol. 1999;65:4028–4031. doi: 10.1128/aem.65.9.4028-4031.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hara R., Kino K. Characterization of novel 2-oxoglutarate dependent dioxygenases converting l-proline to cis-4-hydroxy-l-proline. Biochem. Biophys. Res. Commun. 2009;379:882–886. doi: 10.1016/j.bbrc.2008.12.158. [DOI] [PubMed] [Google Scholar]
- 4.Mori H., Shibasaki T., Yano K., Ozaki A. Purification and cloning of a proline 3-hydroxylase, a novel enzyme which hydroxylates free l-proline to cis-3-hydroxy-l-proline. J. Bacteriol. 1997;179:5677–5683. doi: 10.1128/jb.179.18.5677-5683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu G., Bazer F.W., Burghardt R.C., Johnson G.A., Kim S.W., Knabe D.A., Li P., Li X., McKnight J.R., Satterfield M.C., Spencer T.E. Proline and hydroxyproline metabolism: implications for animal and human nutrition. Amino Acids. 2011;40:1053–1063. doi: 10.1007/s00726-010-0715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe S., Yamada M., Ohtsu I., Makino K. α-Ketoglutaric semialdehyde dehydrogenase isozymes involved in metabolic pathways of d-glucarate, d-galactarate and hydroxy-l-proline: molecular and metabolic convergent evolution. J. Biol. Chem. 2007;282:6685–6695. doi: 10.1074/jbc.M611057200. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe S., Morimoto D., Fukumori F., Shinomiya H., Nishiwaki H., Kawano-Kawada M., Sasai Y., Tozawa Y., Watanabe Y. Identification and characterization of d-hydroxyproline dehydrogenase and Δ1-pyrroline-4-hydroxy-2-carboxylate deaminase involved in novel l-hydroxyproline metabolism of bacteria: metabolic convergent evolution. J. Biol. Chem. 2012;287:32674–32688. doi: 10.1074/jbc.M112.374272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visser W.F., Verhoeven-Duif N.M., de Koning T.J. Identification of a human trans-3-hydroxy-l-proline dehydratase, the first characterized member of a novel family of proline racemase-like enzymes. J. Biol. Chem. 2012;287:21654–21662. doi: 10.1074/jbc.M112.363218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buschiazzo A., Goytia M., Schaeffer F., Degrave W., Shepard W., Grégoire C., Chamond N., Cosson A., Berneman A., Coatnoan N., Alzari P.M., Minoprio P. Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc Natl Acad Sci U S A. 2006;103:1705–1710. doi: 10.1073/pnas.0509010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goytia M., Chamond N., Cosson A., Coatnoan N., Hermant D., Berneman A., Minoprio P. Molecular and structural discrimination of proline racemase and hydroxyproline-2-epimerase from nosocomial and bacterial pathogens. PLoS One. 2007;2:e885. doi: 10.1371/journal.pone.0000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muramatsu H., Mihara H., Kakutani R., Yasuda M., Ueda M., Kurihara T., Esaki N. The putative malate/lactate dehydrogenase from Pseudomonas putida is an NADPH-dependent Δ1-piperideine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase involved in the catabolism of d-lysine and d-proline. J. Biol. Chem. 2005;280:5329–5335. doi: 10.1074/jbc.M411918200. [DOI] [PubMed] [Google Scholar]
- 12.Goto M., Muramatsu H., Mihara H., Kurihara T., Esaki N., Omi R., Miyahara I., Hirotsu K. Crystal structures of Δ1-piperideine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase belonging to a new family of NAD(P)H-dependent oxidoreductases: conformational change, substrate recognition, and stereochemistry of the reaction. J. Biol. Chem. 2005;280:40875–40884. doi: 10.1074/jbc.M507399200. [DOI] [PubMed] [Google Scholar]
- 13.Goodman J.L., Wang S., Alam S., Ruzicka F.J., Frey P.A., Wedekind J.E. Ornithine cyclodeaminase: structure, mechanism of action, and implications for the mu-crystallin family. Biochemistry. 2004;43:13883–13891. doi: 10.1021/bi048207i. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Z., Sun L., He J., Gong W. Crystal structure of human μ-crystallin complexed with NADPH. Protein Sci. 2007;16:329–335. doi: 10.1110/ps.062556907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher D.T., Monbouquette H.G., Schröder I., Robinson H., Holden M.J., Smith N.N. Structure of alanine dehydrogenase from Archaeoglobus: active site analysis and relation to bacterial cyclodeaminases and mammalian mu crystallin. J. Mol. Biol. 2004;342:119–130. doi: 10.1016/j.jmb.2004.06.090. [DOI] [PubMed] [Google Scholar]
- 16.Li C., Lu C.D. Arginine racemization by coupled catabolic and anabolic dehydrogenases. Proc Natl Acad Sci U S A. 2009;106:906–911. doi: 10.1073/pnas.0808269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatto G.J., Jr., Boyne M.T., 2nd, Kelleher N.L., Walsh C.T. Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J. Am. Chem. Soc. 2006;128:3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]
- 18.Kan-No N., Matsu-Ura H., Jikihara S., Yamamoto T., Endo N., Moriyama S., Nagahisa E., Sato M. Tauropine dehydrogenase from the marine sponge Halichondria japonica is a homolog of ornithine cyclodeaminase/mu-crystallin. Comp. Biochem. Physiol. 2005;B141:331–339. doi: 10.1016/j.cbpc.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 19.White C.E., Gavina J.M., Morton R., Britz-McKibbin P., Finan T.M. Control of hydroxyproline catabolism in Sinorhizobium meliloti. Mol. Microbiol. 2012;85:1133–1147. doi: 10.1111/j.1365-2958.2012.08164.x. [DOI] [PubMed] [Google Scholar]
- 20.Nardini M., Ricci G., Caccuri A.M., Solinas S.P., Vesci L., Cavallini D. Purification and characterization of a ketimine-reducing enzyme. Eur. J. Biochem. 1988;173:689–694. doi: 10.1111/j.1432-1033.1988.tb14053.x. [DOI] [PubMed] [Google Scholar]
- 21.Hallen A., Cooper A.J., Jamie J.F., Haynes P.A., Willows R.D. Mammalian forebrain ketimine reductase identified as μ-crystallin; potential regulation by thyroid hormones. J. Neurochem. 2011;118:379–387. doi: 10.1111/j.1471-4159.2011.07220.x. [DOI] [PubMed] [Google Scholar]
- 22.Reed P.W., Bloch R.J. Crystallin-gazing: unveiling enzymatic activity. J. Neurochem. 2011;118:315–316. doi: 10.1111/j.1471-4159.2011.07249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trovato M., Maras B., Linhares F., Costantino P. The plant oncogene rolD encodes a functional ornithine cyclodeaminase. Proc Natl Acad Sci U S A. 2001;98:13449–13453. doi: 10.1073/pnas.231320398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ion B.F., Bushnell E.A., Luna P.D., Gauld J.W. A molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) study on ornithine cyclodeaminase (OCD): a tale of two iminiums. Int. J. Mol. Sci. 2012;13:12994–13011. doi: 10.3390/ijms131012994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Branden C., Tooze J. Garland Publishing; New York: 1991. Introduction to Protein Structure. pp. 141–159. [Google Scholar]
- 26.Watanabe, S., Morimoto, D. and Sasai, Y. (2013) Japanese Patent Application No. JP2013-95772.
- 27.Saito J., Imamura Y., Itoh J., Matsuyama S., Maruta A., Hayashi T., Sato A., Wada N., Kashiwazaki K., Inagaki Y., Watanabe T., Kitagawa Y., Okazaki I. ELISA measurement for urinary 3-hydroxyproline-containing peptides and its preliminary application to healthy persons and cancer patients. Anticancer Res. 2010;30:1007–1014. [PubMed] [Google Scholar]
- 28.Watanabe, S. and Tanimoto, Y. (2013) Japanese Patent Application No. JP2013-186647.
- 29.Sambrook J., Fritsch E.F., Maniatis T. 3rd ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 30.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 31.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Simon R., Priefer U., Puhler A. A broad range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1983;1:789–791. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Th PDF file contains Supplementary Figs. S1–S4 and Table S1.