

Abstract
Single-molecule (SM) fluorescence methods have been increasingly instrumental in our current understanding of a number of key aspects of protein folding and aggregation landscapes over the past decade. With the advantage of a model free approach and the power of probing multiple subpopulations and stochastic dynamics directly in a heterogeneous structural ensemble, SM methods have emerged as a principle technique for studying complex systems such as intrinsically disordered proteins (IDPs), globular proteins in the unfolded basin and during folding, and early steps of protein aggregation in amyloidogenesis. This review highlights the application of these methods in investigating the free energy landscapes, folding properties and dynamics of individual protein molecules and their complexes, with an emphasis on inherently flexible systems such as IDPs.
1. Introduction
Proteins play critical roles in all forms of life. Folding landscapes of proteins encode essential information that leads to proper operation of cells and organisms. Over the past several decades, experiment and theory have consistently revealed new complexity on these landscapes that is intimately tied to optimal protein function in biology1–11. Not surprisingly, it is now widely known that a diverse array of diseases, including neurodegeneration and cancer, are linked to dysfunctional folding states of proteins. In fact, the direct link between alteration in protein folding landscapes and human diseases is highlighted by amyloidogenesis12. This represents the largest group of “protein conformational” diseases, where the pathology is marked by the formation of highly ordered, insoluble protein filaments known as amyloid fibrils from soluble functional proteins in a yet unknown mechanism.
Research on the protein folding problem began more than 50 years ago with a basic quest for understanding how the primary sequence of a polypeptide chain codes for a unique native, folded 3D structure of a protein, and what the underlying physical forces are that guide folding processes to be extremely fast as compared with a random search 13, 14. The well-defined 3D structure of a protein was perceived as tightly coupled with the function of the protein. However, over the last decade, the field of protein science has recognized the existence of a new class of proteins which challenged this classical ‘structure-function’ paradigm. An increasing number of studies pointed out that the prerequisite of well-defined secondary and tertiary structures in the native state does not hold true for these proteins to be functional15–17. In fact, the benchmark of these highly dynamic proteins, often termed as intrinsically disordered proteins (IDPs), is the lack of any significant structural elements in their native conformations. It is now known that ~ 30% proteins in human proteome contain a substantial degree of intrinsic disorder. IDPs are involved in important cellular functions such as signaling and regulation, and dysfunction of many are associated with human amyloidosis, neurodegeneration18 and cancer. Important members of this class of proteins include p53, Aβ, α–synuclein and prion proteins to name a few. One unique feature of these proteins is that they are able to interact with a diverse array of binding partners such as proteins, nucleic acids, and membranes, which results in coupled binding and folding events19–21 via a complex free energy landscape.
A significant body of basic science research over the years bridging the fields of physics, chemistry and biology has evolved our understanding of the process of protein folding, misfolding and aggregation. A wealth of structural biology tools such as X-ray crystallography, NMR spectroscopy, solution X-ray scattering and a variety of ensemble spectroscopic methods have been extensively employed for the investigation of folding pathways of proteins, characterization of the intermediates involved and elucidating properties of folded and unfolded conformational states, which were appropriately aided by a surge of theoretical studies and computational modeling. However, protein folding landscapes are complex, and their ruggedness encodes features such as more than one local minimum, dynamic conformations which interconvert among each other, degenerate states in folded and unfolded basins, intrinsically disordered native conformations, and existence of multiple folding pathways. Many of these properties are almost impossible to directly access in detail by ensemble methods due to their inherent limitation of averaging out many aspects of heterogeneity and dynamics. Single-molecule methodologies offer the power of watching one molecule at a time over a broad range of dynamical fluctuations22, thus accessing their properties directly. Moreover, ensemble methods are limited in studies of IDPs, which have emerged as the proteins without stable structural elements and with a shallow folding landscape. These proteins are also capable of adopting multiple conformations via interaction with their binding partners, sometimes simultaneously, complexity which may be normally obscured due to ensemble averaging. Thus far, SM optical methods have been applied extensively in the study of protein folding after their first introduction to the problem in the late 90s. Here, we describe unique insights and new findings in the study of protein folding, misfolding and aggregation pathways using single-molecule fluorescence methods highlighting the immense potential of these methods with a special emphasis on studies focused on IDPs.
2. Single-Molecule (SM)Fluorescence Methods
Studies of individual molecules have advantages over ensemble measurements, as hidden conformational states can be detected and ensemble averaging can be avoided for inherently heterogeneous systems like IDPs23. Several single-molecule (SM) fluorescence methods have been useful in gaining critical understanding of protein folding and misfolding and the mechanism of amyloid formation21, 24–28 (see Fig. 1). These methods are often used in combination with each other to get complimentary information. For example, single-molecule Förster (Fluorescence) Resonance Energy Transfer (smFRET) can provide insights about conformational states and stochastic transitions of proteins, and reveals if more than one conformational state coexists21. Intermediates in the folding pathway induced by small molecules, osmolytes, salts, chaperone proteins, etc., can be detected as well21, 24. Fluorescence correlation spectroscopy (FCS) and FRET-correlation studies reports on rapid conformational dynamics of proteins21, 29. Fluorescence coincidence analysis gives us information about the aggregation states of the proteins as well as kinetics of the underlying process30, 31. Fluorescence microscopy in conjunction with FRET imaging techniques is also particularly useful for in vivo studies of amyloidogensis32. Below, we briefly outline the experimental details of some of the key fluorescence methods.
Figure-1.
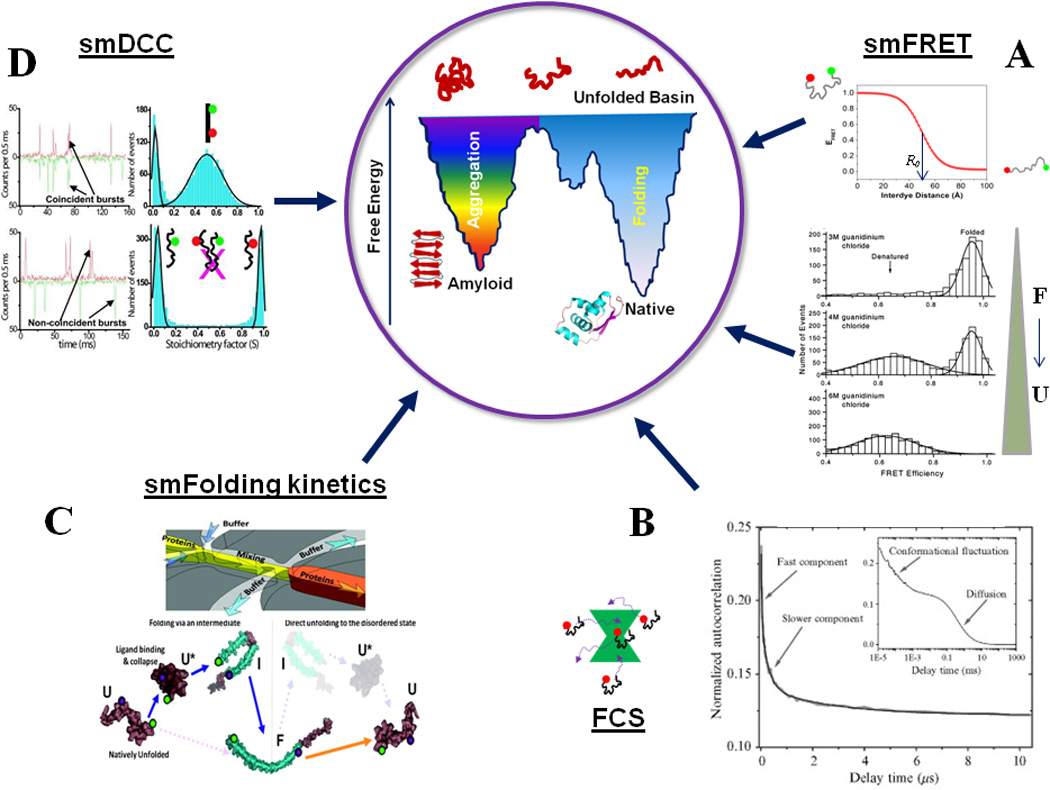
An overview of single-molecule fluorescence methods for protein folding studies. (A) smFRET can detect individual subpopulations of protein conformations in a heterogeneous structural ensemble by means of FRET efficiency, which is a function of inter-dye distance (Eqn. 1). smFRET histograms showing denaturant induced unfolding of a two-state folder, CI2. Adapted from Deniz et al., Proc. Natl. Acad. Sci. USA (2000), 97:5179 50. (B) FCS can be employed to detect slow and fast conformational dynamics of proteins: shown here are the autocorrelation curves for the NM domain of Sup35. Adapted from Mukhopadhyay et al. Proc. Natl. Acad. Sci., USA (2007) 104:2649 29. (C) Shows a microfluidic mixer which can be coupled with smFRET detection to study single-molecule folding and unfolding pathways of proteins. The bottom shows a schematic of the observed pathways for α-synuclein. Adapted from Gambin et al., Nature Methods (2011) 8:239 102. (D) Shows the application of smDCC for directly detecting lack of aggregation of the NM domain of Sup35 under experimental conditions. Adapted from Mukhopadhyay et al., Proc. Natl. Acad. Sci. USA (2007) 104:2649 29. Aggregation would be signaled by coincident bursts, which could be analyzed for further quantification. The concepts behind the central cartoon showing a double-funnel for protein folding and aggregation are discussed in 146. The native state protein structure at the bottom of the folding funnel is a cartoon for MJ0366 (pdb ID: 2EFV).
2.1. Single-Molecule Fluorescence/Förster Resonance Energy Transfer (smFRET)
Förster (also called Fluorescence) Resonance Energy Transfer (FRET) is a process where singlet excitation energy of the donor is transferred to the acceptor in a nonradiative way by coupling of their respective dipoles. The efficiency of FRET (EFRET) is determined by the following relation:
| (1) |
where r is the inter-chromophore distance, and R0 is called the Förster distance, which is a property of the donor-acceptor pair. R0 is defined as the inter-dye distance where EFRET is 0.50 (Fig-1). For commonly used dye pairs, EFRET shows strong distance dependence in 30–70Å range and is widely used for studying conformational states and fluctuations in proteins and nucleic acids.
smFRET experiments can be carried out either on molecules freely diffusing in solution, or on surface immobilized molecules33. With the latter format21, 27, 29, 34, long time trajectories of conformational fluctuations of single molecules of proteins and DNA can be measured, which is useful in many cases to derive their equilibrium and kinetic properties. However, covalent interactions with surfaces may alter conformational properties of the biomolecules and could result in artifacts in some cases. This problem has recently been addressed by researchers by encapsulating protein molecules inside lipid vesicles35–37 which are surface-tethered, to minimize such perturbations due to direct interactions. On the other hand, smFRET measurements on freely diffusing molecules provide the opportunity to watch large numbers of individual molecules one at a time, revealing for example population distributions of different conformational states. This method has advantages over surface immobilized methods for studying IDPs since these proteins have a relatively shallow free energy surface which could be especially prone to be affected by surface interactions. Indeed, recent single-molecule fluorescence studies on IDPs and other amyloidogenic proteins were mostly done on freely diffusing molecules.
In a nutshell, a typical experimental set up for smFRET detection on freely diffusing molecules23, 38 consists of laser excitation of donor fluorophores attached to the biomolecules of interest (≤ 50 pM) with the aid of a high numerical aperture objective, followed by collection of the fluorescence signal (both donor and acceptor) via the same objective through a sub-fL detection volume (achieved by using a pinhole of 30–100 µm). The fluorescence signal of donor and acceptor are then optically separated after the excitation signal is filtered from the emission signal using a proper dichroic mirror. Bursts of photons for donor and the acceptor are subsequently recorded using highly sensitive avalanche photodiode detectors (APDs), separated from the background noise using a threshold, and the FRET efficiency is determined in a ratiometric way using the following relation:
| (2) |
Here, IA and ID are the photon counts in the acceptor and the donor channels, respectively, while γ takes care of the unequal quantum yields and detection efficiencies (if any) of the donor and acceptors. The data are plotted as histograms which reveal populations of different conformational states and can be used to detect changes in conformational properties and fluctuations during protein folding, conformational transitions, protein-protein, protein-membrane, or protein-small molecule interactions. Care should be taken for correct estimation of γ for more accurately quantifying the EFRET values in terms of inter-fluorophore distances and interpreting relatively small changes in the FRET efficiencies. Furthermore, a value of 2/3 (for the case of dynamic averaging) is usually used for the orientation factor, κ2, which contributes towards the Förster distance (R0). For quantitative distance measurements, this should be validated and further analyzed 39. These issues have been discussed in more detail by Ferreon et al.21 and references therein. For more accurate detection of conformational states with low EFRET, a more advanced smFRET technique, known as alternating laser excitation-FRET or ALEX-FRET can be used40, 41.
As noted above, the other common way of smFRET detection involves surface-immobilized biomolecules and total internal reflection fluorescence microscopy (TIRF). In this technique, the donor molecules on the immobilized sample are excited by an evanescent field (stretches a few hundred nm from the surface into the sample) created by the excitation laser. The fluorescence signal collection in the donor and acceptor channels is done in a similar fashion as discussed for freely diffusing molecules, and the detection of the emission signals from several of molecules are done simultaneously in parallel, usually using an advanced EM-CCD camera. The data analysis involves processing of raw composite image files into time trajectories for a large set of single-molecules, and the FRET efficiencies (equation-2) are estimated using the similar protocol as described above33.
2.2. Single-Molecule Dual Color Coincidence Measurements (smDCC)
The interaction between two or more different protein molecules to form higher molecular assemblies and protein aggregates can be detected using dual color coincidence measurements29, 30, 42. In this method, two different batches of protein molecules are singly-labeled with two separate fluorophores. The experimental format follows the same principle as smFRET measurements, the only difference being simultaneous excitation of both the fluorophores which need not participate in FRET. The data collection and the dual-channel detection of the emission signal follow the same protocol as well. The data processing involves calculation of a stoichiometry factor, S = (IR/(IR+IB)) (IB/R = photon counts in the blue/red channel), after correcting for the cross-talk between two channels, and eliminating the background noise by setting a threshold, and is plotted as a histogram. The excitation laser powers can be set in such a way that the value of S = 0.5 for a coincidence “control” sample (e.g., a DNA control sample)29. Furthermore, information regarding the relative size of oligomeric species can be extracted by means of analyzing average photon counts in the red and blue channels relative to the bursts of a monomer30. This method has been successfully applied to confirm lack of aggregation in an amyloidogenic IDP in the experimental conditions by distinguishing oligomers from monomeric protein molecules29, and to gain direct insights into the pathways of amyloidogenesis by its use in conjunction with smFRET30, 31.
2.3. Fluorescence Correlation Spectroscopy (FCS)
Fluorescence correlation measurements provide important information about conformational fluctuations and associated time-scales for proteins and other biomolecules43. Typically, fluorescence correlation analysis is done by recording fluctuations in the fluorescence signal from a given set of molecules which underlie specific molecular event(s) with characteristic timescale(s) associated with them, and fitting the autocorrelation curve using standard functions43, 44. An experimental set up similar to that for diffusion smFRET measurements described above can be used for FCS measurements and it is often used in conjunction with FRET measurements, called FRET-FCS. The method generally requires proteins to be singly labeled. However, dual labeling is required for more advanced type of measurements such as FRET-FCS. Quantitative information regarding the diffusion coefficients, the size of biomolecules as well as biomolecular association45 can be obtained from autocorrelation analysis. For example, for intrinsically disordered, amyloidogenic proteins such as α-synuclein, yeast prion and polyQ, critical information regarding the timescale of the conformational dynamics, diffusion coefficients and molecular dimensions have been extracted from correlation analysis21, 29, 44.
2.4. Site-specific Labeling Strategies
SM fluorescence based methods require labeling the protein under investigation with fluorescent dyes in one or more specific sites. Site-specific protein labeling can be achieved by taking advantage of appropriate chemistries. For example, one of the most common protein labeling strategies is to react cysteine(s) in the protein of interest with a thiol-reactive derivative (usually a maleimide) of a dye of choice. For proteins without any cysteine residues, site-directed introduction of cysteines without perturbation of the structure and function of the proteins is a common way to covalently attach fluorescent dyes23. A similar but less efficient way is to use amine-reactive dyes which can react with side chains of lysines and the N-terminus of a protein. However, this method is not very popular due to low labeling specificity and high frequency of natural occurrence of lysines compared to cysteines23. One way of increasing labeling specificity is the site-specific introduction of non-natural amino acids46 with different functional moieties such as a ketone, and use of orthogonal chemistry to produce dual-labeled protein with one dye being thiol-reactive and the other being ketone reactive (e.g., an alkoxyamine derivative for reaction with a ketone). A study47 successfully demonstrated this technique for T4 lysozyme (T4L) where the protein was dual labeled in one pot highlighting the compatibility of the reaction conditions. The other method of achieving site-specific dual labeling is native chemical ligation (NCL)48–50, for example by ligation of two different fragments of the protein of interest combined with labeling prior to (and post) ligation.
3. Globular Protein Folding Studies
3.1. Early Work
In 2000, Deniz et al. used smFRET to directly demonstrate a two-state transition during the denaturant induced unfolding of a small protein, chymotrypsin inhibitor 2 (CI2), under freely diffusing solution conditions 50. Subpopulations of folded and unfolded states were measured at various concentrations of denaturant, and the observed two-state behavior directly validated the conclusions of previous ensemble studies. In 2002, Schuler et al. used smFRET to characterize the folding free-energy landscape of a cold shock protein, CspTm51. From analysis of their data, the authors provided an estimation of the bounds to the energy barrier in the free-energy surface - the so called “pre-exponential factor” for protein folding – from the reconfiguration time of polypeptide chain. In a related work on the same protein, Lipman et al. then measured folding kinetics at the SM level, by coupling a rapid microfluidic mixer with their smFRET set up52. The rate of SM folding of CspTm, measured from the time dependent evolution of folded and unfolded subpopulation, was found to be in good agreement with the ensemble kinetics data. Moreover, a rapid collapse of the unfolded state prior to folding was observed 51. Subsequently, Rhoades et al. demonstrated the existence of multiple folding pathways for the protein adenylate kinase35, previously suggested as a multi-state folder. This study was carried out by an improved version of surface immobilization, where individual protein molecules were confined inside synthetic vesicles, thus minimizing artificial surface interactions, and providing insights about the ruggedness of protein-folding landscape. These early studies along with others53 showed the unprecedented power of SM optical methods in the folding field, and paved the path for a surge of SM fluorescence studies in protein folding in the years to follow.
3.2. New Frontiers in Protein Folding
3.2.1. Characterization of the unfolded conformation
Understanding unfolded chain properties is important to evaluate the role of intrinsic dynamics of the polypeptide chain, local residual structural elements and nature of non-covalent interactions in the folding properties of individual protein molecules. Early studies from Nienhaus lab on RNase H54, 55, by Sherman et al.56 on protein L, and by the Eaton group57 on protein L and CspTm, have demonstrated that smFRET measurements, in conjunction with theoretical modeling and molecular simulations, can be used quantitatively to access the structural and dynamical features of unfolded states of proteins. A non-specific collapse of the polypeptide chain was identified at low denaturant condition by these researchers, and an overall non-sigmoidal increase in the radius of gyration in the unfolded basin with denaturant concentration was observed. The smFRET experiments on RNase H were performed on surface immobilized molecules by means of a strategy which ensured negligible spurious interactions of the folded and unfolded polypeptide chains with the surface, and highlights the strength of this approach for probing the long time trajectories of protein folding dynamics. The experimental findings which suggested the existence of “continuum-of-states” in the unfolded basin55, 58 were supported by molecular simulations57, but did not agree well with previously reported time resolved SAXS data59 at low denaturant concentration. It was concluded that the origin of this discrepancy might be a direct manifestation of higher protein concentrations used in the SAXS measurement57. A recent study using SAXS on protein L, however, failed to reproduce the chain collapse under similar conditions as smFRET study was done60. While further investigations using multiple probes will be necessary to resolve this issue, it is worth mentioning here that independent studies using both single molecule and ensemble fluorescence methods from different laboratories on a number of unrelated proteins, both globular and natively disordered in nature, unequivocally suggested the occurrence of unfolded protein chain collapse under low denaturant concentration21, 29, 50, 52, 56, 57, 61.
smFRET studies on the unfolded state of SNase also showed similar characteristics as described above62. Furthermore, comparing the folding characteristics of the β-subdomain and overall protein, the authors suggested a “subdomain-specific collapse” leading to a subpopulation of unfolded states with native-like structures and folding intermediates during early events of SNase folding62.
A related study by a different approach, FCS, on disordered, monomeric polyQ proteins by Pappu and colleagues44 revealed that an ensemble of collapsed structures in aqueous environment are preferred by the protein, a surprising observation given the lack of hydrophobic residues in these proteins. These results provided a molecular basis for polyQ aggregation in aqueous solution, described as a poor-solvent for the protein, based on a polymer description of the polypeptide chain63. As noted below, work by Mukhopadhyay et al. subsequently demonstrated existence of such a heterogeneous collapsed ensemble for the NM domain of the yeast prion protein Sup3529. More recent studies from the Schuler group on the effect of temperature on the dimension of the unfolded state revealed some interesting aspects64. Using CspTm, they have shown that the unfolded polypeptide chain undergoes a compaction with increased temperature, which cannot be explained on the basis of a simple homopolymer model of the polypeptide chain. Using highly charged intrinsically disordered protein prothymosin α, the authors show that “hydrophobic collapse” cannot account for such an effect alone, and the effect of other non-covalent interactions such as hydrogen bonding was highlighted64 in the context of residual secondary structure in the temperature induced collapse of unfolded state. In a separate study, the same group investigated the effect of overall polypeptide chain composition on the size of the natively unfolded state of proteins65, which revealed that charge-charge interactions can be a determining factor of the dimensions in such cases under physiological buffer conditions. The above works showed that intrinsically disordered proteins encode features both similar and distinct from the denatured states of globular proteins. Additionally, these results are believed to have important implications on the roles of disordered states in molecular recognition and protein-protein interactions.
3.2.2. Novel Insights into the Thermodynamics and Kinetics of Protein Folding
smFRET experiments are well suited for identifying discrete conformational switching in protein molecules, which are thought to be important for their biological functions. For example, Gambin et al. showed that specific mutations in the homo-dimeric RNA-binding protein ROP leads to a heterogeneous mixture of two native structures26, designated as “anti” (functionally active form) and “syn” (functionally inactive form). Direct observations of complex dynamics of folding landscapes of large, multimeric and multidomain proteins have also been demonstrated recently by smFRET experiments. For example, information about multiple conformations in the full length, tetrameric, tumor suppressor transcription factor p53 were reported by Huang et al.66, while the role of sequence specificity in folding and misfolding of multidomain proteins was shown using tandem constructs of the well-characterized Ig domains of the muscle protein titin as a successful evolutionary mechanism to minimize protein misfolding in multidomain proteins67. A recent study on the protein adenylate kinase (encapsulated into lipid vesicles which were immobilized on a glass surface) by Prichi et al.68 highlights how a combination of smFRET experiments and statistical analysis using hidden Markov modeling can be used to elucidate protein folding landscapes. Interestingly, they observed the occurrence of multiple folding intermediates, the relative population of which are tunable by varying denaturant concentration, and multiple coexisting folding pathways as previously suggested by Rhoades et al.35.
smFRET provides a convenient way to probe the unique behaviors of one-state folders – the so called “downhill” or barrier-less protein folding as predicted by energy landscape theory69. Since downhill folding occurs at the “folding speed limit” (~106 s−1)70, 71, it poses a challenge to the limits of single-molecule detection efficiency, and hence such measurements are difficult. An interesting example is the protein BBL, where single-molecule experiments have raised questions about whether it is or is not it folds via a downhill mechanism72,73–78,79 While further investigations of its complex folding landscape are needed to fully understand the folding mechanism of BBL, these studies nevertheless begin to highlight the power of smFRET methods in studying one state folders.
smFRET experiments have been helpful in elucidating the complex physics underlying protein folding kinetics in recent years. Schuler and colleagues have employed ns-FCS in combination with smFRET to quantify the role of internal friction – a measure of the ruggedness of the folding landscape – on the relaxation dynamics of unfolded state and provided valuable insights about the role of primary sequence on internal friction80 (Fig. 2). Interestingly, the same group recently reported that the internal friction is specifically localized and has pronounced effects on the early transition state formation rather than the late transition states in the spectrin folding landscape81 (Fig. 2), which is consistent with a nucleation-condensation mechanism of protein folding82. The role of such localized internal friction in determining the transition path times will be an important next goal for smFRET experiments. Recently, Eaton and coworkers have demonstrated how protein folding and unfolding rate coefficients can be obtained for fast folders by combining experimentally measured single-photon trajectories, smFRET histograms and statistical analysis83. Further studies from the same group were successful in determining transition path times for model two-state folders with very different folding rates84: formin binding protein WW domain (folds in sub-millisecond) and GB1 (folds in seconds) (Fig. 3), using a similar approach. While the transition path time for WW is ~ 2 µs, the same for GB1 is ~ 10µs. Their work showed that though there is a remarkable difference in the rates of folding between these two proteins, the average time taken for a successful one-way transition from the unfolded basin to the folded basin is similar. These types of studies show promise for a more complete description of protein folding landscapes in the future.
Figure-2.
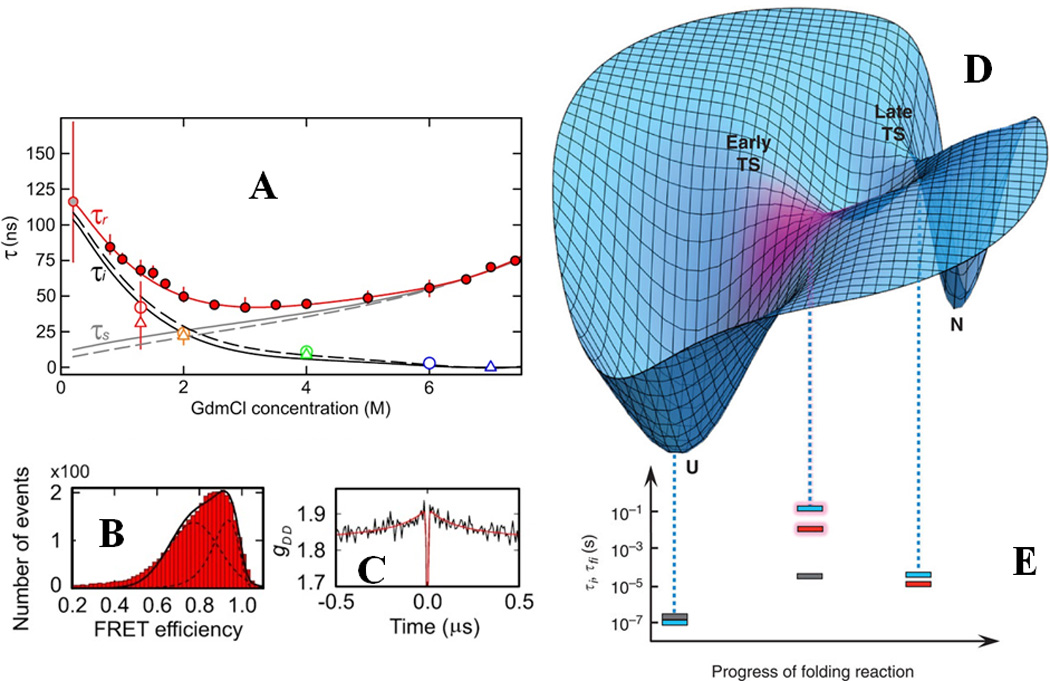
(A) Quantitative determination of internal friction in the unfolded state of Csp using smFRET (B) and nanosecond FCS (C). The red filled circles in (A) denote experimentally determined reconfiguration time (τr) for end-labeled Csp as a function of guanidinium concentrations under equilibrium conditions, while grey filled circles denote the same observable measured using a microfluidic mixer. The red line is a polynomial fit of the data. τi and τs are the reconfiguration timescales independent and dependent on solvent viscosity, respectively. The symbols represent experimental values for τi and solid and dashed lines are derived from a Rouse model with internal friction. (D) Cartoon depicting the internal friction being localized at the early TS during spectrin folding as shown by magenta. (E) The time constants which are solvent-viscosity independent are indicated by dotted lines as a function of the reaction coordinate: grey for R15 domain, blue for R16 domain and red for R17 domain. The pronounced effect of internal friction on the early TS for R16 and R17 are shown by shaded bars. (A)-(C) are adapted by permission from Soranno et al., Proc. Natl. Acad. Sci. USA (2012) 109:17800 80 (copyright (2012) National Academy of Science, U.S.A.), (D) and (E) are adapted from Borgia et al., Nat. Comm. (2012) 3:1195 by permission from Macmillan Publishers Ltd: [Nature Communication]81, copyright (2012).
Figure-3.
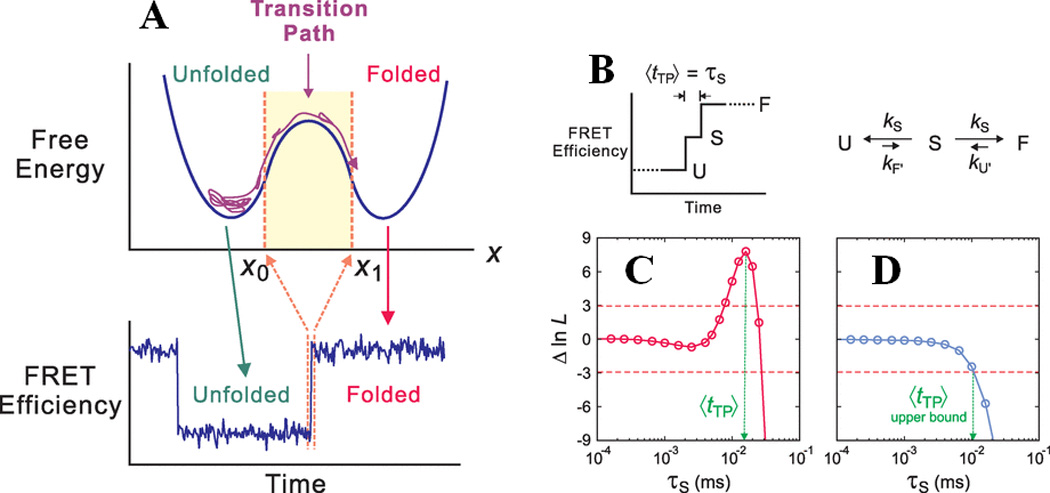
Determination of the transition path time for protein folding using smFRET. Shown here are (A) a one dimensional free energy diagram highlighting the possible conformational sampling of the unfolded polypeptide chains before it can successfully make a transition from x0 to x1 along the reaction coordinates. This is termed the transition path, and the associated time can be measured by monitoring the FRET transition time from folded to unfolded states, as shown at the bottom. (B) A kinetic model for determining the average transition path time, <tTP>, which is equal to the lifetime (τs) of a “virtual intermediate state”, for a two-state folder. (C) and (D) show plots of the difference of log likelihood between a pathway where τs has a finite value, and a pathway where τs = 0, as a function of τs for WW and GB1, respectively. The positive peak value in WW represents <tTP>, whereas the intersection of the function with the red dashed line at -3 gives the upper bound of <tTP> for GB1. The figure is adapted from Chung et al., Science (2012) 335:981 84 and reprinted with permission from AAAS.
3.2.3. Chaperone-mediated Protein Folding
Protein folding machines are critical to ensure correct folding of nascent polypeptide chains as well as rescuing kinetically trapped states in the crowded cellular environment. Recent smFRET studies highlighted some key aspects of the folding of individual protein molecules in the “nano-cage” of GroEL/GroES chaperonin system which is the best characterized folding machine. Using rhodanese as a model substrate, Hillger et al.85 demonstrated that GroEL binding to the substrate leads to substantial broadening of the smFRET histograms, suggesting a heterogeneous ensemble with a folding intermediate containing a native-like topology. The nature of this folding intermediate was found to be independent of the properties of the unfolded ensemble, thus suggesting an on-pathway species during folding of rhodanese. Subsequent addition of GroES and ATP led to the formation of the well-defined folded substrate. Furthermore, in a recent report it was shown that the rate of N-terminal domain folding of the same substrate does not alter significantly, while the C-terminal domain folds much slowly in the chaperonin cage86. Rapid microfluidic-chip based kinetic measurements highlighted the increased inter-molecular friction as the underlying mechanism of slower configurational dynamics of C-terminal domain during folding. An smFRET-based study by Sharma et al.87 on the GroEL/GroES assisted MBP folding probed polypeptide chain dynamics along a complex folding pathway. These and other studies88 indicate that confinement of the polypeptide chain in a narrow chaperonin cavity ensures minimal non-native contact formation, and reduces the chances for off-pathway products leading to misfolded states and protein aggregation.
4. Exploring the Structure and Dynamics of Intrinsically Disordered Proteins (IDPs)
A combination of fluorescence methods, especially at the single-molecule level, has been useful to conduct a number of studies probing amyloidogenic IDPs in recent years. Here we aim to provide some critical case studies with a broad overview for other IDPs. Perhaps the most extensively studied system is the Parkinson’s disease associated protein α–synuclein, which will be discussed first. Then, we will discuss the NM domain of the yeast prion protein Sup35, which is believed to form functional amyloids. Finally, we briefly mention some recent studies of a couple of other important IDPs, islet amyloid polypeptide (IAPP) and tau protein. We note that some of these studies demonstrate the unique applicability of SM fluorescence methods for studying aggregation-prone species which are generally inaccessible by other methods89.
4.1. α–synuclein
The Deniz laboratory has done some of the early single-molecule fluorescence studies21, 23, 25 on the presynaptic neuronal protein α–synuclein, which is natively unstructured. It was shown that α–synuclein assumes a disordered state under native conditions, and this state undergoes a non-cooperative transition towards a more expanded state as a function of denaturant concentration23. The smaller dimension of the native state of α–synuclein and other IDPs, compared to fully-expanded random coil structure of the globular proteins is suggested to be determined by the overall composition of the charged amino acids and might have important implications in the biological function and pathological roles of these proteins65, 90. Interestingly, in the same line, a recent smFRET study from Deniz group24 suggested that osmolytes, such as TMAO, leads to gradual compaction in the dimension, rather than cooperative folding of α–synuclein, unlike for globular proteins.
One of the interesting features of IDPs is the potential for populating multiple folded structures when they interact with their biological partners. Along these lines, α–synuclein shows a transition to an α–helix-rich state via interactions with membranes91. Hence, a next set of single-molecule studies was aimed to study the detailed folding behavior of α–synuclein in presence of membranes. SDS was used as a well-studied and tunable model for negatively charged lipids and membranes for several of those studies. The protein was dual-labeled at key positions with Alexa488 (D) and Alexa594 (A) dyes via site-specifically introduced cysteines. The smFRET data directly revealed a complex multi-state folding landscape of the protein21, as multiple conformations were observed as a function of SDS concentration (Fig. 4A and B). Initially, the natively unfolded state of α–synuclein (U state, EFRET = 0.4) undergoes a transition to a compact high-EFRET state, which then switches to an extended low EFRET state at higher SDS concentration. Interestingly, further increase in SDS concentration led to additional switching of the protein back and forth into similar high and low-EFRET states. Combining these data with helicity measurements by CD91 and information from earlier NMR92 and EPR93 structural studies, the two states were assigned to “hairpin” or “broken-helix” (I) and extended helical (F) forms of the protein, as shown in Figure-4A and B. The different conformational states of the protein showed fluctuations in the ns-µs timescale as probed by FRET based fluorescence correlation measurements (FRET-FCS). Interesting enough, though the I and Im states have very similar structural features and EFRET values, they showed different dynamical behavior (Fig. 4A). Furthermore, the ALEX-smFRET method was applied to confirm the occurrence of the low-FRET F state, which happened to be the predominant conformation of the protein in the presence of phospholipid membranes (Fig. 4C), as well as high-FRET conformations of membrane-bound α–synuclein. These results have potential implications for the biological function and dysfunction as well as aggregation of α–synuclein under physiological conditions, since it has been shown that low amount of SDS94, organic solvents such as TFE95 and some phospholipids96–99 accelerated or otherwise modulated aggregation and fibrillation of the protein, suggesting one or more of these helical conformations are on-pathway for amyloid fibril formation. Single-molecule and ESR studies from other laboratories have also provided important information about aspects of these α–synuclein interactions 34, 100, 101. In addition, in a follow up work on the Parkinson’s disease associated A30P mutant25, it was demonstrated that the mutant protein had substantially altered folding behavior as the F state was disfavored (Fig. 4D). This result has important implications for understanding the molecular basis of misfunction in this mutant.
Figure-4.
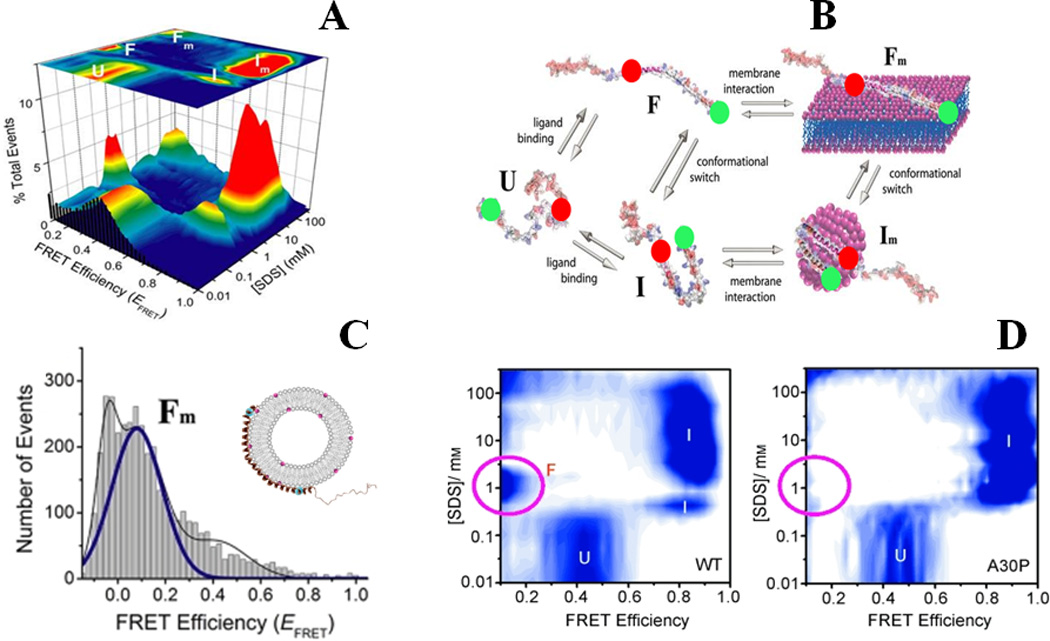
Binding-coupled-folding of α-synuclein: (A) and (B) show multistate conformational sampling of synuclein as revealed during SDS-binding of the protein probed by smFRET studies. smFRET histograms are shown in (A), and a schematic describing different states of α-synuclein is shown in (B). Red and green spheres in (B) represent donor and acceptor dyes. (C) Shows an extended helical conformation, the Fm state, when the protein is bound to phospholipid SUVs (POPA:POPC = 1:1). The disease-linked A30P mutation alters the protein-folding landscape substantially, as observed in (D) by a largely absent F state. (A), (B) and (C) are adapted from Ferreon et al., Proc. Natl. Acad. Sci. (2009) 106:5645 21, and (D) is adapted from Ferreon, Moran et al., Angew. Chem. Int. Ed. (2010) 49:3469 25.
In a more advanced study102 combining microfluidics with smFRET, the kinetics of conformational switching of α–synuclein via interaction with SDS was probed. Initial interactions with SDS showed a rapid transition of the U state via increasingly compact protein forms to a hairpin-like state (in ~ 1 ms), and then relatively slow transition to an extended helical state (in ~ 8 ms).
The effect of oxidative modifications, such as nitration of tyrosines, on the conformational landscape of α–synuclein, has been probed recently by the Rhoades group using smFRET measurements 103. It was shown that nitration of tyrosines in the N-terminal region of the protein results in direct perturbations in membrane binding, while nitration in the C-terminal region results in a more subtle change in the interaction with membranes via an allosteric effect103. This result has potential in identifying regions in the α–synuclein structure which control its interactions with biological partners via distinct mechanisms, and hence contribute in aggregation and dysfunction.
The aggregation properties of the protein have been subjected to smFRET analysis by the Rhoades group by exposing the protein under aggregation-favoring conditions, such as lower pH, and small-molecule inducer of the α–synuclein aggregation27. The highly charged C-terminal tail was observed to assume a collapsed but disordered conformation at low pH without any significant structural changes in the other regions of the protein. However, binding of polyions such as heparin and spermine did not alter the structural ensemble of the protein. Thus, the conformational changes at low pH and charge-screening effect by aggregation-inducers are attributed to be the underlying mechanism of the protein aggregation, indicating more than one mechanistic pathway could be operative.
4.2. Yeast Prion Protein Sup35
The application of smFRET for studies of IDPs was initiated on the yeast prion protein Sup35, a protein that has been ascribed to the class of functional amyloids. The prion-determining region (known as the NM segment) of this protein is composed of a mostly disordered N-terminus, known as the amyloid core region, and a middle region (M) which is highly charged104. smFRET measurements on a protein FRET-labeled across the amyloid core region showed that the native state of the protein is unstructured yet compact. The protein showed a non-sigmoidal transition to an expanded state when titrated with denaturant. Dual-color coincidence measurements were able to clearly confirm that the monomeric state was being studied. Moreover, Sup35 NM showed an ensemble of structures in the native state which are rapidly fluctuating in the nanosecond to sub-microsecond time scale as probed by FCS studies. The study overall showed that the native monomer has structural features some shared (disordered, rapidly fluctuations) and others distinctly different (compaction) from the unfolded states of globular proteins. These results29 were suggestive of an interesting mechanism for amyloid formation, via initial formation of less-ordered oligomers105.
4.3. Islet Amyloid Polypeptide
The Islet amyloid polypeptide (IAPP) is a small (37 AA), intrinsically disordered hormone which is associated with type II diabetes106. Similar to the protein α–synuclein, IAPP interacts with anionic membranes to assume helical structural ensemble, which also results in much faster amyloidogenesis107. A recent study from the Rhoades lab108 probed the structure of rat IAPP when bound to the membrane using smFRET experiments and Rosetta simulations. They showed that the membrane-bound form of the protein is an anti-parallel helical dimer. This result has important implications as significant structural rearrangement is anticipated from such a species to form amyloid fibrils with parallel orientation, thus providing insight about possible strategies of therapeutic intervention for the disease.
4.4. Tau Protein
Tau is another important IDP which is associated with tauopathies which include Alzheimer’s and many other neudegenerative diseases109, 110. In an ensemble FRET study111, Tau was shown to adopt a compact fold with long range contacts between the N- and C-termini as well as with micro-tubule binding domain (MTBD) even in the globally disordered native state. Based on an examination of several FRET constructs distributed throughout the protein structure, the authors proposed a “paper-clip” like structure for the protein. In a recent study on Tau112, the Rhoades group identified features of the full-length protein as well individual domains using smFRET. Most interestingly, Tau binding to the aggregation-inducer heparin showed a cooperative transition to a distinct conformation, unlike many other IDPs as discussed here. There is an overall increase in the protein dimension, and in contrast, the micro-tubule binding domain (MTBD), which forms the core of the fibrils and the paired helical filaments, showed a two-state transition to a compact conformation due to heparin binding. These results about the conformational states of Tau under aggregation-prone conditions provide insights for understanding the mechanism of amyloidogenesis and tauopathy.
5. Probing Protein Aggregation and Amyloid Formation
Fluorescence methods have been useful for studying protein aggregation and amyloid formation both in vitro and in cells. The well-known amyloid-reporter dye Thioflavin-T (ThT) has been used extensively after it was first introduced in 1959113, for monitoring amyloidogenesis both in vitro and in vivo. Moreover, fusion of the protein of interest with fluorescent proteins provides a convenient way of watching protein aggregation in live cells114, 115, and monitoring homologous and/or heterologous protein-protein interactions by FRET between a donor and an acceptor fluorescent protein116. FLIM32, a fluorescence lifetime-based in vivo microscopic method, is another popular and useful method for studying protein-aggregation. On the other hand, ensemble fluorescence techniques like ones utilizing fluorescence properties of biarsenical reagents attached to a tetra-cysteine moiety117 have been useful in gaining insights about mechanistic aspects of amyloidogenesis. Recently, SM techniques, such as dual-color coincidence measurements in conjunction with smFRET30, 31, have been used to provide new insights about in vitro amyloidogenesis kinetics.
5.1. In Vitro Amyloidogenesis
The Klenerman group has used dual-color coincidence measurements30, 42 to study the kinetics and oligomerization of amyloidogenic proteins. Initial studies on SH3-PI3 system30 demonstrated the aggregation kinetics of the proteins at the single-particle level. The protein was observed to populate “small” oligomers very rapidly under the experimental conditions (low pH and low salt), which then grow to larger oligomers relatively slowly (total 2–4 hrs reaction time). Both of these on-pathway oligomers were shown to be dissociable into monomeric protein simply by dilution. However, after 48 hrs, these oligomers convert to stable “amyloid-prone” oligomers, which then proliferate to form amyloid fibrils. A similar method was also applied for Aβ aggregation and its interactions with the chaperone protein clusterin, to demonstrate a protective role of clusterin118. A more recent study31 on α–synuclein using coincidence measurements, in conjunction with smFRET, revealed heterogeneous oligomeric distributions in α–synuclein aggregation pathway. A kinetic model emerged from the detailed analysis of the SM data, where a slow conversion of initial oligomers into more stable and toxic aggregates was identified as the critial step in the pathway. In a separate study on IAPP, the aggregation process and membrane disruption by the oligomers were also observed to follow at least a two state mechanism119.
The amyloid formation pathway for functional amyloids, such as the yeast prion protein Sup35-NM, has recently been highlighted using a combination of fluorescence quenching and anisotropy measurements as well as antibodies reactive to specific oligomeric conformations105. Multiple oligomeric species were identified to be populated the amyloid assembly reaction. The conformational conversion of these oligomers into a more compact form were directly probed using ensemble fluorescence quenching of Cy3B/Cy5 dye by tyrosines and single-molecule anisotropy measurements. The “less-compact” early oligomers were observed to be toxic for the cells just like the disease-linked amyloids. However, for functional amyoids, the on-pathway toxic oligomers are believed to rapidly mature and form fibrils in a distinctly different mechanism (Fig. 5), compared to non-functional amyloids.
Figure-5.
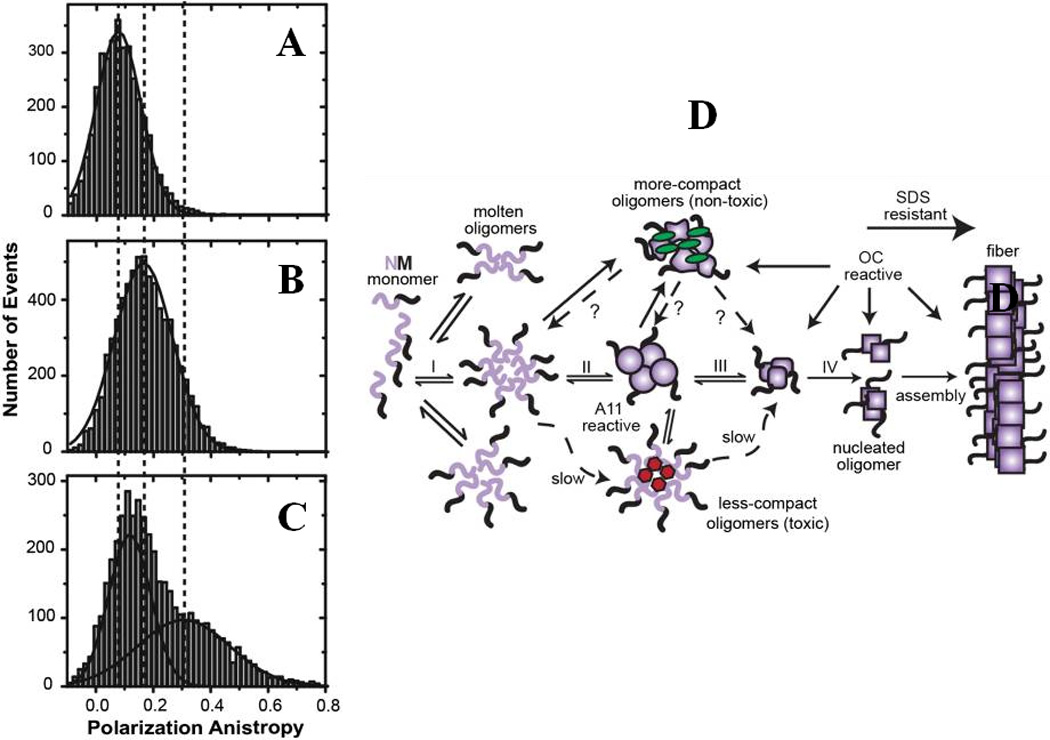
Single-molecule (SM) study of amyloidogenesis for Sup35-NM: (A-C) SM anisotropy measurements revealed progressive conversion of the monomeric protein to the matured oligomers through intermediate oligomeric species. (D) Proposed amyloid pathway for the Sup35-NM. The figure is adapted from Krishnan et al., Proc. Natl. Acad. Sci., USA, (2012) 109:11172 105.
6. Single-molecule Protein Folding: What is Next?
We expect the field to make hand-in-hand advances in the complexity of folding problem investigated and the quality of single-molecule methods developed and used.
Fluorescence techniques, especially in the single-molecule and imaging field, will continue to evolve and be necessary for probing many complex, elusive features of protein folding landscapes and underlying mechanisms of amyloidogensis to address health issues. A significant improvement in SM methodology is attained by combining microfluidics with smFRET measurements52, 102, 120–123 which resulted in improved detection efficiency, less photobleaching, increased brightness of fluorephores and addressing “out-of-equilibrium” biophysics of protein folding. To overcome complex features of analyzing smFRET data and extract quantitative structural information with reduced errors, multi-parameter fluorescence detection has many benefits124. We expect a broader scope of such methodologies with improved detection efficiencies leading to a superior detection of ultra-fast biomolecular dynamics in the µs time scale and faster in the future. This will be necessary, for example, to obtain a quantitative description of the folding properties of proteins which fold near the folding speed limit69, such as BBL, thus revealing key details of their folding landscapes. Additionally, rapid and high-resolution analysis will permit better exploration of folding landscapes, for example for further exploration of the transition region experimentally84. These types of studies are particularly exciting to look forward to, since they can in principle directly report on roughness predicted from energy landscape theory69. Another important focus will be in developing efficient and general ways for performing three- and multi-color FRET measurements125, 126. These experiments will be important and necessary in understanding simultaneous and correlated conformational changes in proteins, especially larger, mutidomain protein complexes as well as protein-protein interactions. Preliminary studies have already shown how insightful such measurements can be at the single-molecule level126, 127. A big challenge for a multi-color FRET experiment is to site-specifically labeling the protein of interest at least at three different positions, and thus orthogonal labeling approaches are essential, for example by use of non-natural amino acids47 for orthogonal chemistry or by other approaches23, 128.
One other important future direction of single-molecule protein folding research will be focused on studying the behaviors of proteins under cellular conditions, since cellular factors, such as crowding and molecular interactions, are important determinants of folding and aggregation properties of proteins which are difficult to probe by simple two-component in vitro systems. Recent methodological advances in studying protein folding dynamics inside living cells129, 130 by combining T-jump and FRET imaging provide an excellent step in one path for such studies in future, preferably at the single-molecule level. Moreover, recent advances in co-translational protein folding131 studies offer a complex, yet attractive system to explore the effects of key components associated with cellular protein folding. Further improvement in imaging techniques and dye photophysics will also be helpful to improve single-molecule studies on in vivo protein folding and amyloidosis.
Improvement of data analysis methods and use of computer simulations in conjunction with experimental tools will be necessary for understanding complex behaviors of individual molecules26, 132. The potential impact of computer simulation in the field of single-molecule protein folding has been clearly highlighted by several important studies recently. Examples include, but certainly are not limited to, studies by Best et al. on the dependence of the diffusion coefficients on the folding coordinate133, by Nath et al. in characterizing conformational ensemble of IDPs134, and by Voelz et al. in demonstrating the attainment of structure of the unfolded state which marks the early phases of the folding of acyl-coenzyme A binding protein135.
Complex aspects of kinetics and thermodynamics of protein folding have started emerging from smFRET studies. These include recent reports of the role and localization of internal friction during protein folding, measurements of transition-path times, and characterization of folding intermediates of an IDP, α-synuclein, during binding-induced-folding with model membranes. A recent report136 on an oncogenic IDP, E1A, used smFRET to reveal complex layers of allostery modulation that could tune the populations and thus corresponding functions of various binding states of the protein (Fig. 6). Such findings highlight the potential of smFRET methods for shedding light on this complex area of research. Also, single-molecule methods have the obvious advantage over ensemble methods for studying aggregation-prone proteins under native conditions (as demonstrated for E1A) since the required protein concentration is usually less than nM. Thus, application of single-molecule methods will be significant in studying the structural features of aggregation-prone systems in understanding the mechanism of formation of various forms of protein aggregates and their connections with diseases. On the other hand, smFRET will also be important to dissect unique topological features of recently discovered protein topologies such as knotted conformations137, 138, as well as probing the thermodynamics and kinetics associated with complex landscapes of protein knotting. Similarly, single-molecule spectroscopy will be critical to study another important family of proteins, membrane proteins. Several studies by force spectroscopy have shown promise139, 140 in understanding the dynamics of the folding landscapes of membrane proteins. It is worthy of mention here that recent advances in single-molecule force spectroscopy have been key in revealing novel complexities in protein folding pathways. For example, a study from Marqusee group reported on a new level of complexities during force-induced unfolding of a two-state folder, src-SH3141, while Stigler et al. have shown the existence of a complex network of multiple folding pathways en route to folding of individual protein molecules of calmodulin142 (Fig. 7). The latter study also presents a successful combination of single-molecule force spectroscopy and molecular simulation, an approach that has been proven to be powerful in the context of smFRET as discussed here. In addition, high-speed AFM is a relatively recent development, and has been applied to initial study of an IDP143. A future experimental approach may constitute a successful combination of smFRET with single-molecule force methods144, 145 along with molecular simulations in studying these complex systems to understand the behavior of their structural ensembles that underlies function, folding properties, lipid–protein interactions as well as for screening potent drug-like molecules as modulators of the functions of membrane proteins. We envision that improvements in methodology as described above will be critical in achieving new frontiers of protein folding research, especially in understanding complex, biologically-relevant interactions involving multiple partners, structurally heterogeneous systems, oligomeric and multi domain, large protein complexes, and the effect of environmental factors in protein folding, misfolding and aggregation.
Figure-6.
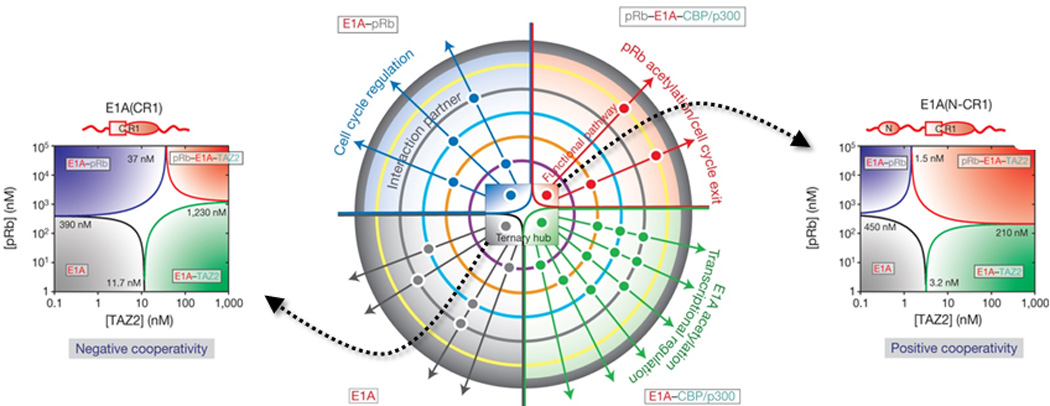
Single-molecule observation of allosteric regulation of protein–protein interactions by intrinsic disorder: The center shows a schematic of functional regulation by modulation of an E1A-CBP/p300-pRb ternary hub (shown by a central phase diagram). The four color-coded quadrants represent four different states: E1A (grey), E1A-pRb (blue), E1A-CBP/p300 (green), and ternary complex (red). Additional binding partners are indicated using color-coded concentric circles, and positive interactions using dots. Corresponding ternary phase diagrams of pRb-TAZ2 interacting with different constructs of E1A reveal a striking switchable cooperativity, likely a key contributor to functional regulation, are also indicated in the figure (Right - negative cooperativity; Left - positive cooperativity). The figure is adapted from Ferreon et al., Nature (2013) 498, 390–394 136.
Figure-7.
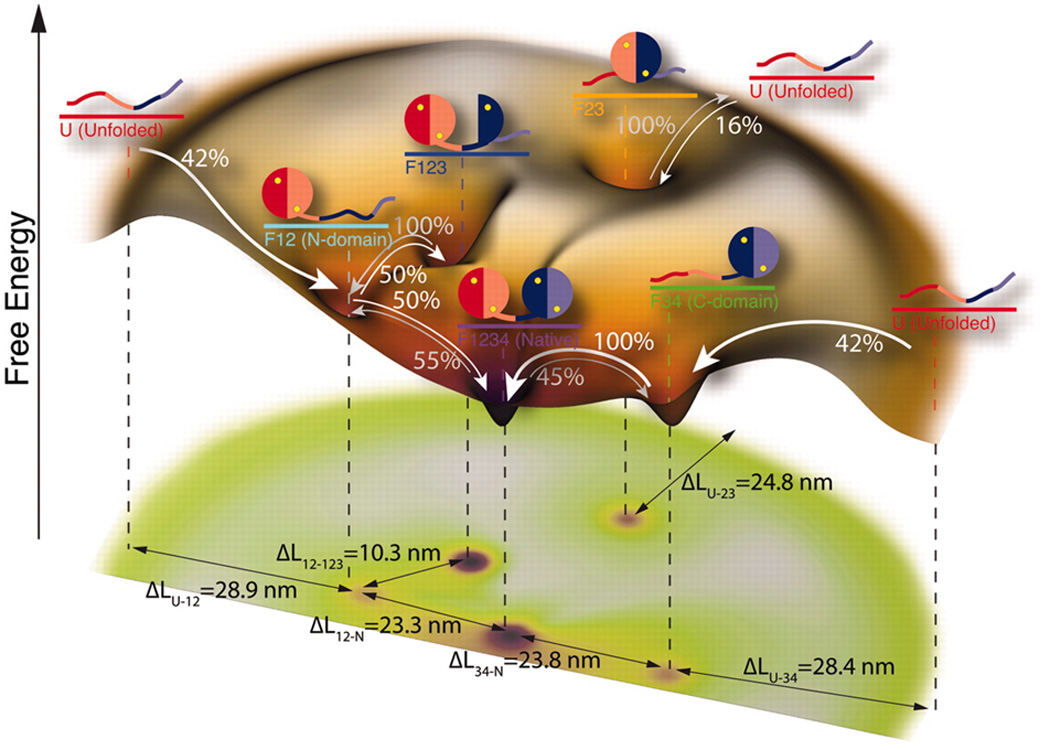
Folding landscape of a single calmodulin molecule as revealed by single-molecule force spectroscopy in conjunction with hidden Markov modeling: four intermediate states were observed in this complex folding network and the observed transitions among the states are indicated by arrows. Respective differences in contour lengths (ΔL) are also denoted in the figure. The figure is adapted from Stigler et al., Science (2011) 334, 512–516 142 and reprinted with permission from AAAS.
7. Concluding Remarks
The complex process of protein folding involves several aspects which are better resolved by advanced single-molecule analysis. Throughout this review, we intended to give the reader an understanding of the utility of single-molecule fluorescence methods for studying the process of protein folding and misfolding. One highlight was IDPs which are an emerging class of proteins with key regulatory functions and that are difficult to probe using ensemble methods. The power of SM-fluorescence techniques is vividly demonstrated in a diverse array of studies spanning conformational biophysics of the protein to imaging protein aggregation kinetics inside living cells. SM-fluorescence methods will continue to improve, with the advent of new techniques providing novel dimensions in protein folding research.
Acknowledgements
We thank members of the Deniz Lab for their contributions to some of the work discussed in this review, and gratefully acknowledge support from the National Science Foundation, USA (MCB 1121959 to A.A.D.) and the NIGMS, National Institutes of Health, USA (GM RO1 066833 to A.A.D.).
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Leopold PE, Montal M, Onuchic JN. Protein folding funnels: a kinetic approach to the sequence-structure relationship. Proc Natl Acad Sci U S A. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dill KA, Ozkan SB, Shell MS, Weikl TR. The protein folding problem. Annu Rev Biophys. 2008;37:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fersht AR. From the first protein structures to our current knowledge of protein folding: delights and scepticisms. Nat Rev Mol Cell Biol. 2008;9:650–654. doi: 10.1038/nrm2446. [DOI] [PubMed] [Google Scholar]
- 5.Lindorff-Larsen K, Rogen P, Paci E, Vendruscolo M, Dobson CM. Protein folding and the organization of the protein topology universe. Trends Biochem Sci. 2005;30:13–19. doi: 10.1016/j.tibs.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Thirumalai D, O'Brien EP, Morrison G, Hyeon C. Theoretical perspectives on protein folding. Annu Rev Biophys. 2010;39:159–183. doi: 10.1146/annurev-biophys-051309-103835. [DOI] [PubMed] [Google Scholar]
- 7.Bartlett AI, Radford SE. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat Struct Mol Biol. 2009;16:582–588. doi: 10.1038/nsmb.1592. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- 9.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 10.Thirumalai D, Liu Z, O'Brien EP, Reddy G. Protein folding: from theory to practice. Curr Opin Struct Biol. 2013;23:22–29. doi: 10.1016/j.sbi.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 12.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 13.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 14.Dill KA, MacCallum JL. The protein-folding problem 50 years on. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 15.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 16.Dunker AK, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 17.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 18.Uversky VN. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res. 2008;5:260–287. doi: 10.2174/156720508784533312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 20.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci U S A. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuler B, Hofmann H. Single-molecule spectroscopy of protein folding dynamics--expanding scope and timescales. Curr Opin Struct Biol. 2013;23:36–47. doi: 10.1016/j.sbi.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Ferreon AC, Moran CR, Gambin Y, Deniz AA. Single-molecule fluorescence studies of intrinsically disordered proteins. Methods Enzymol. 2010;472:179–204. doi: 10.1016/S0076-6879(10)72010-3. [DOI] [PubMed] [Google Scholar]
- 24.Ferreon AC, Moosa MM, Gambin Y, Deniz AA. Counteracting chemical chaperone effects on the single-molecule alpha-synuclein structural landscape. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1201802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreon AC, Moran CR, Ferreon JC, Deniz AA. Alteration of the alpha-synuclein folding landscape by a mutation related to Parkinson's disease. Angew Chem Int Ed Engl. 2010;49:3469–3472. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gambin Y, et al. Direct single-molecule observation of a protein living in two opposed native structures. Proc Natl Acad Sci U S A. 2009;106:10153–10158. doi: 10.1073/pnas.0904461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trexler AJ, Rhoades E. Single molecule characterization of alpha-synuclein in aggregation-prone states. Biophys J. 2010;99:3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci U S A. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orte A, et al. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc Natl Acad Sci U S A. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas AV, Berezovska O, Hyman BT, von Arnim CA. Visualizing interaction of proteins relevant to Alzheimer's disease in intact cells. Methods. 2008;44:299–303. doi: 10.1016/j.ymeth.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veldhuis G, Segers-Nolten I, Ferlemann E, Subramaniam V. Single-molecule FRET reveals structural heterogeneity of SDS-bound alpha-synuclein. Chembiochem. 2009;10:436–439. doi: 10.1002/cbic.200800644. [DOI] [PubMed] [Google Scholar]
- 35.Rhoades E, Gussakovsky E, Haran G. Watching proteins fold one molecule at a time. Proc Natl Acad Sci U S A. 2003;100:3197–3202. doi: 10.1073/pnas.2628068100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okumus B, Wilson TJ, Lilley DM, Ha T. Vesicle encapsulation studies reveal that single molecule ribozyme heterogeneities are intrinsic. Biophys J. 2004;87:2798–2806. doi: 10.1529/biophysj.104.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benitez JJ, et al. Probing transient copper chaperone-Wilson disease protein interactions at the single-molecule level with nanovesicle trapping. J Am Chem Soc. 2008;130:2446–2447. doi: 10.1021/ja7107867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay S, Deniz AA. Fluorescence from diffusing single molecules illuminates biomolecular structure and dynamics. J Fluoresc. 2007;17:775–783. doi: 10.1007/s10895-007-0214-0. [DOI] [PubMed] [Google Scholar]
- 39.Ha T. Single-molecule fluorescence resonance energy transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 40.Kapanidis AN, et al. Alternating-laser excitation of single molecules. Acc Chem Res. 2005;38:523–533. doi: 10.1021/ar0401348. [DOI] [PubMed] [Google Scholar]
- 41.Lee NK, et al. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orte A, Clarke R, Balasubramanian S, Klenerman D. Determination of the fraction and stoichiometry of femtomolar levels of biomolecular complexes in an excess of monomer using single-molecule, two-color coincidence detection. Anal Chem. 2006;78:7707–7715. doi: 10.1021/ac061122y. [DOI] [PubMed] [Google Scholar]
- 43.Bacia K, Schwille P. Fluorescence correlation spectroscopy. Methods Mol Biol. 2007;398:73–84. doi: 10.1007/978-1-59745-513-8_7. [DOI] [PubMed] [Google Scholar]
- 44.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci U S A. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie J, Schultz PG. A chemical toolkit for proteins--an expanded genetic code. Nat Rev Mol Cell Biol. 2006;7:775–782. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 47.Brustad EM, Lemke EA, Schultz PG, Deniz AA. A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer. J Am Chem Soc. 2008;130:17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dirksen A, Dawson PE. Expanding the scope of chemoselective peptide ligations in chemical biology. Curr Opin Chem Biol. 2008;12:760–766. doi: 10.1016/j.cbpa.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Muralidharan V, Muir TW. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 50.Deniz AA, et al. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc Natl Acad Sci U S A. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 52.Lipman EA, Schuler B, Bakajin O, Eaton WA. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 53.Talaga DS, et al. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc Natl Acad Sci U S A. 2000;97:13021–13026. doi: 10.1073/pnas.97.24.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule Forster resonance energy transfer study of protein dynamics under denaturing conditions. Proc Natl Acad Sci U S A. 2005;102:15471–15476. doi: 10.1073/pnas.0507728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule FRET study of denaturant induced unfolding of RNase H. J Mol Biol. 2006;357:313–324. doi: 10.1016/j.jmb.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 56.Sherman E, Haran G. Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci U S A. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci U S A. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker MJ, Marqusee S. The cooperativity of burst phase reactions explored. J Mol Biol. 1999;293:1195–1210. doi: 10.1006/jmbi.1999.3204. [DOI] [PubMed] [Google Scholar]
- 59.Plaxco KW, Millett IS, Segel DJ, Doniach S, Baker D. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat Struct Biol. 1999;6:554–556. doi: 10.1038/9329. [DOI] [PubMed] [Google Scholar]
- 60.Yoo TY, et al. Small-angle X-ray scattering and single-molecule FRET spectroscopy produce highly divergent views of the low-denaturant unfolded state. J Mol Biol. 2012;418:226–236. doi: 10.1016/j.jmb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haran G. How, when and why proteins collapse: the relation to folding. Curr Opin Struct Biol. 2012;22:14–20. doi: 10.1016/j.sbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu P, Meng X, Qu P, Zhao XS, Wang CC. Subdomain-specific collapse of denatured staphylococcal nuclease revealed by single molecule fluorescence resonance energy transfer measurements. J Phys Chem B. 2009;113:12030–12036. doi: 10.1021/jp809825x. [DOI] [PubMed] [Google Scholar]
- 63.Chan HS, Dill KA. Polymer principles in protein structure and stability. Annu Rev Biophys Biophys Chem. 1991;20:447–490. doi: 10.1146/annurev.bb.20.060191.002311. [DOI] [PubMed] [Google Scholar]
- 64.Nettels D, et al. Single-molecule spectroscopy of the temperature-induced collapse of unfolded proteins. Proc Natl Acad Sci U S A. 2009;106:20740–20745. doi: 10.1073/pnas.0900622106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muller-Spath S, et al. From the Cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc Natl Acad Sci U S A. 2010;107:14609–14614. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang F, et al. Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2009;106:20758–20763. doi: 10.1073/pnas.0909644106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borgia MB, et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature. 2011;474:662–665. doi: 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pirchi M, et al. Single-molecule fluorescence spectroscopy maps the folding landscape of a large protein. Nat Commun. 2011;2:493. doi: 10.1038/ncomms1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 70.Yang WY, Gruebele M. Folding at the speed limit. Nature. 2003;423:193–197. doi: 10.1038/nature01609. [DOI] [PubMed] [Google Scholar]
- 71.Li P, Oliva FY, Naganathan AN, Munoz V. Dynamics of one-state downhill protein folding. Proc Natl Acad Sci U S A. 2009;106:103–108. doi: 10.1073/pnas.0802986106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang F, Ying L, Fersht AR. Direct observation of barrier-limited folding of BBL by single-molecule fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2009;106:16239–16244. doi: 10.1073/pnas.0909126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garcia-Mira MM, Sadqi M, Fischer N, Sanchez-Ruiz JM, Munoz V. Experimental identification of downhill protein folding. Science. 2002;298:2191–2195. doi: 10.1126/science.1077809. [DOI] [PubMed] [Google Scholar]
- 74.Oliva FY, Munoz V. A simple thermodynamic test to discriminate between two-state and downhill folding. J Am Chem Soc. 2004;126:8596–8597. doi: 10.1021/ja048309w. [DOI] [PubMed] [Google Scholar]
- 75.Sadqi M, Fushman D, Munoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–321. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 76.Arbely E, Rutherford TJ, Sharpe TD, Ferguson N, Fersht AR. Downhill versus barrier-limited folding of BBL 1: energetic and structural perturbation effects upon protonation of a histidine of unusually low pKa. J Mol Biol. 2009;387:986–992. doi: 10.1016/j.jmb.2008.12.055. [DOI] [PubMed] [Google Scholar]
- 77.Neuweiler H, et al. Downhill versus barrier-limited folding of BBL 2: mechanistic insights from kinetics of folding monitored by independent tryptophan probes. J Mol Biol. 2009;387:975–985. doi: 10.1016/j.jmb.2008.12.056. [DOI] [PubMed] [Google Scholar]
- 78.Settanni G, Fersht AR. Downhill versus barrier-limited folding of BBL 3. Heterogeneity of the native state of the BBL peripheral subunit binding domain and its implications for folding mechanisms. J Mol Biol. 2009;387:993–1001. doi: 10.1016/j.jmb.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 79.Liu J, et al. Exploring one-state downhill protein folding in single molecules. Proc Natl Acad Sci U S A. 2012;109:179–184. doi: 10.1073/pnas.1111164109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soranno A, et al. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc Natl Acad Sci U S A. 2012;109:17800–17806. doi: 10.1073/pnas.1117368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Borgia A, et al. Localizing internal friction along the reaction coordinate of protein folding by combining ensemble and single-molecule fluorescence spectroscopy. Nat Commun. 2012;3:1195. doi: 10.1038/ncomms2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abkevich VI, Gutin AM, Shakhnovich EI. Specific nucleus as the transition state for protein folding: evidence from the lattice model. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 83.Chung HS, et al. Extracting rate coefficients from single-molecule photon trajectories and FRET efficiency histograms for a fast-folding protein. J Phys Chem A. 2011;115:3642–3656. doi: 10.1021/jp1009669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chung HS, McHale K, Louis JM, Eaton WA. Single-molecule fluorescence experiments determine protein folding transition path times. Science. 2012;335:981–984. doi: 10.1126/science.1215768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hillger F, et al. Probing protein-chaperone interactions with single-molecule fluorescence spectroscopy. Angew Chem Int Ed Engl. 2008;47:6184–6188. doi: 10.1002/anie.200800298. [DOI] [PubMed] [Google Scholar]
- 86.Hofmann H, et al. Single-molecule spectroscopy of protein folding in a chaperonin cage. Proc Natl Acad Sci U S A. 2010;107:11793–11798. doi: 10.1073/pnas.1002356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharma S, et al. Monitoring protein conformation along the pathway of chaperonin-assisted folding. Cell. 2008;133:142–153. doi: 10.1016/j.cell.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 88.Chakraborty K, et al. Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell. 2010;142:112–122. doi: 10.1016/j.cell.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 89.Deniz AA, et al. Ratiometric single-molecule studies of freely diffusing biomolecules. Annu Rev Phys Chem. 2001;52:233–253. doi: 10.1146/annurev.physchem.52.1.233. [DOI] [PubMed] [Google Scholar]
- 90.Mao AH, Crick SL, Vitalis A, Chicoine CL, Pappu RV. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc Natl Acad Sci U S A. 2010;107:8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferreon AC, Deniz AA. Alpha-synuclein multistate folding thermodynamics: implications for protein misfolding and aggregation. Biochemistry. 2007;46:4499–4509. doi: 10.1021/bi602461y. [DOI] [PubMed] [Google Scholar]
- 92.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 93.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci U S A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giehm L, Oliveira CL, Christiansen G, Pedersen JS, Otzen DE. SDS-induced fibrillation of alpha-synuclein: an alternative fibrillation pathway. J Mol Biol. 2010;401:115–133. doi: 10.1016/j.jmb.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 95.Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW, Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proc Natl Acad Sci U S A. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 97.Zhu M, Li J, Fink AL. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 98.Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 99.Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during alpha-synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc. 2012;134:5090–5099. doi: 10.1021/ja209019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trexler AJ, Rhoades E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J Am Chem Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gambin Y, et al. Visualizing a one-way protein encounter complex by ultrafast single-molecule mixing. Nat Methods. 2011;8:239–241. doi: 10.1038/nmeth.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: oxidative modifications to alpha-synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011;133:7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 105.Krishnan R, et al. Conserved features of intermediates in amyloid assembly determine their benign or toxic states. Proc Natl Acad Sci U S A. 2012;109:11172–11177. doi: 10.1073/pnas.1209527109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Westermark P, et al. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Williamson JA, Loria JP, Miranker AD. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J Mol Biol. 2009;393:383–396. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nath A, Miranker AD, Rhoades E. A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew Chem Int Ed Engl. 2011;50:10859–10862. doi: 10.1002/anie.201102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol. 2001;13:41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- 110.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 111.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 112.Elbaum-Garfinkle S, Rhoades E. Identification of an Aggregation-Prone Structure of Tau. J Am Chem Soc. 2012 doi: 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol. 1959;68:487–498. [PubMed] [Google Scholar]
- 114.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 115.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 116.Pollitt SK, et al. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron. 2003;40:685–694. doi: 10.1016/s0896-6273(03)00697-4. [DOI] [PubMed] [Google Scholar]
- 117.Luedtke NW, Dexter RJ, Fried DB, Schepartz A. Surveying polypeptide and protein domain conformation and association with FlAsH and ReAsH. Nat Chem Biol. 2007;3:779–784. doi: 10.1038/nchembio.2007.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Narayan P, et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1–40) peptide. Nat Struct Mol Biol. 2012;19:79–83. doi: 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci U S A. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hamadani KM, Weiss S. Nonequilibrium single molecule protein folding in a coaxial mixer. Biophys J. 2008;95:352–365. doi: 10.1529/biophysj.107.127431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pfeil SH, Wickersham CE, Hoffmann A, Lipman EA. A microfluidic mixing system for single-molecule measurements. Rev Sci Instrum. 2009;80:055105. doi: 10.1063/1.3125643. [DOI] [PubMed] [Google Scholar]
- 122.Lemke EA, et al. Microfluidic device for single-molecule experiments with enhanced photostability. J Am Chem Soc. 2009;131:13610–13612. doi: 10.1021/ja9027023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gambin Y, Simonnet C, VanDelinder V, Deniz A, Groisman A. Ultrafast microfluidic mixer with three-dimensional flow focusing for studies of biochemical kinetics. Lab Chip. 2010;10:598–609. doi: 10.1039/b914174j. [DOI] [PubMed] [Google Scholar]
- 124.Sisamakis E, Valeri A, Kalinin S, Rothwell PJ, Seidel CA. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010;475:455–514. doi: 10.1016/S0076-6879(10)75018-7. [DOI] [PubMed] [Google Scholar]
- 125.Gambin Y, Deniz AA. Multicolor single-molecule FRET to explore protein folding and binding. Mol Biosyst. 2010;6:1540–1547. doi: 10.1039/c003024d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clamme JP, Deniz AA. Three-color single-molecule fluorescence resonance energy transfer. Chemphyschem. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 127.Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hejjaoui M, et al. Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies: alpha-synuclein phosphorylation at tyrosine 125. J Am Chem Soc. 2012;134:5196–5210. doi: 10.1021/ja210866j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dhar A, Prigozhin M, Gelman H, Gruebele M. Studying IDP stability and dynamics by fast relaxation imaging in living cells. Methods Mol Biol. 2012;895:101–111. doi: 10.1007/978-1-61779-927-3_8. [DOI] [PubMed] [Google Scholar]
- 130.Ebbinghaus S, Dhar A, McDonald JD, Gruebele M. Protein folding stability and dynamics imaged in a living cell. Nat Methods. 2010;7:319–323. doi: 10.1038/nmeth.1435. [DOI] [PubMed] [Google Scholar]
- 131.Clark PL, Ugrinov KG. Measuring cotranslational folding of nascent polypeptide chains on ribosomes. Methods Enzymol. 2009;466:567–590. doi: 10.1016/S0076-6879(09)66024-9. [DOI] [PubMed] [Google Scholar]
- 132.Gopich IV, Szabo A. Single-molecule FRET with diffusion and conformational dynamics. J Phys Chem B. 2007;111:12925–12932. doi: 10.1021/jp075255e. [DOI] [PubMed] [Google Scholar]
- 133.Best RB, Hummer G. Coordinate-dependent diffusion in protein folding. Proc Natl Acad Sci U S A. 2010;107:1088–1093. doi: 10.1073/pnas.0910390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nath A, et al. The conformational ensembles of alpha-synuclein and tau: combining single-molecule FRET and simulations. Biophys J. 2012;103:1940–1949. doi: 10.1016/j.bpj.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Voelz VA, et al. Slow unfolded-state structuring in Acyl-CoA binding protein folding revealed by simulation and experiment. J Am Chem Soc. 2012;134:12565–12577. doi: 10.1021/ja302528z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sulkowska JI, Noel JK, Onuchic JN. Energy landscape of knotted protein folding. Proc Natl Acad Sci U S A. 2012;109:17783–17788. doi: 10.1073/pnas.1201804109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sulkowska JI, Rawdon EJ, Millett KC, Onuchic JN, Stasiak A. Conservation of complex knotting and slipknotting patterns in proteins. Proc Natl Acad Sci U S A. 2012;109:E1715–1723. doi: 10.1073/pnas.1205918109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Janovjak H, Sapra KT, Kedrov A, Muller DJ. From valleys to ridges: exploring the dynamic energy landscape of single membrane proteins. Chemphyschem. 2008;9:954–966. doi: 10.1002/cphc.200700662. [DOI] [PubMed] [Google Scholar]
- 140.Kedrov A, Janovjak H, Sapra KT, Muller DJ. Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu Rev Biophys Biomol Struct. 2007;36:233–260. doi: 10.1146/annurev.biophys.36.040306.132640. [DOI] [PubMed] [Google Scholar]
- 141.Jagannathan B, Elms PJ, Bustamante C, Marqusee S. Direct observation of a force-induced switch in the anisotropic mechanical unfolding pathway of a protein. Proc Natl Acad Sci U S A. 2012;109:17820–17825. doi: 10.1073/pnas.1201800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stigler J, Ziegler F, Gieseke A, Gebhardt JC, Rief M. The complex folding network of single calmodulin molecules. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 143.Miyagi A, et al. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. Chemphyschem. 2008;9:1859–1866. doi: 10.1002/cphc.200800210. [DOI] [PubMed] [Google Scholar]
- 144.Hohng S, et al. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the holliday junction. Science. 2007;318:279–283. doi: 10.1126/science.1146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tarsa PB, et al. Detecting force-induced molecular transitions with fluorescence resonant energy transfer. Angew Chem Int Ed Engl. 2007;46:1999–2001. doi: 10.1002/anie.200604546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]