

Abstract
Genes and proteins generate molecular circuitry that enables the cell to process information and respond to stimuli. A major challenge is to identify characteristic patterns in this network of interactions that may shed light on basic cellular mechanisms. Previous studies have analyzed aspects of this network, concentrating on either transcription–regulation or protein–protein interactions. Here we search for composite network motifs: characteristic network patterns consisting of both transcription–regulation and protein–protein interactions that recur significantly more often than in random networks. To this end we developed algorithms for detecting motifs in networks with two or more types of interactions and applied them to an integrated data set of protein–protein interactions and transcription regulation in Saccharomyces cerevisiae. We found a two-protein mixed-feedback loop motif, five types of three-protein motifs exhibiting coregulation and complex formation, and many motifs involving four proteins. Virtually all four-protein motifs consisted of combinations of smaller motifs. This study presents a basic framework for detecting the building blocks of networks with multiple types of interactions.
Cellular processes are regulated by interactions between various types of molecules such as proteins, DNA, and metabolites (1–4). Among these, the interactions between proteins and the interactions between transcription factors and their target genes play a prominent role, controlling the activity of proteins and the expression levels of genes. A significant number of such interactions have been revealed recently by means of high-throughput technologies such as yeast two-hybrid (5, 6) and chromatin immunoprecipitation (7–10). By using these data, one can build a network of interactions and thus describe the circuitry responsible for a variety of cellular processes. The analysis of this cellular circuitry is one of the major goals in the postgenomic era.
What are the building blocks of this cellular circuitry? Recent studies have analyzed the structure of the transcriptional networks of Escherichia coli (11) and Saccharomyces cerevisiae (10, 12), consisting solely of interactions between transcription factors and their target genes. These transcriptional networks were shown to be composed, to a large extent, of a small set of network motifs: patterns of interactions that recur in the cellular network significantly more often than in randomized networks (11, 12). Each of these motifs was suggested to perform a specific information-processing role in the network. In parallel, the network of protein–protein interactions (PPIs) in S. cerevisiae has also been studied intensively and shown to consist of clusters of interacting proteins (13–16). Yet, analyzing transcriptional networks and PPI networks separately hides the full complexity of the cellular circuitry, because many processes involve combinations of these two types of interactions.
Here we systematically analyze the cellular circuitry comprising two types of interactions: those between transcription factors and their target genes and those between proteins. To this end, we extended the concept of network motifs to include motifs involving these two types of interactions. We developed algorithms for detecting composite motifs in networks comprising two or more types of connections and apply them here to the cellular network of the yeast S. cerevisiae.
Intriguingly, our analysis revealed a few network motifs involving two or three proteins (by “protein” we refer both to the protein and to the gene encoding it) and several four-protein motifs, virtually all of which consisted of combinations of smaller motifs. These findings suggest that the cellular network consists of small network motifs that can be interpreted as basic building blocks. Particularly, the smaller motifs we revealed were a two-protein motif defining a mixed-feedback loop involving both transcription–regulation interaction (TRI) and PPI and five types of three-protein motifs. Two of these five motifs are purely decoupled motifs of either TRIs or PPIs. The other three motifs present biologically meaningful combinations of the two types of interactions. Altogether the five motifs manifest the tendency of eukaryotic cells toward coregulation and complex formation. This study presents a framework for detecting the building blocks of cellular networks with multiple types of interactions, which can be utilized to analyze any network with more than one type of connection.
Methods
Network Data. Experimentally identified interactions between transcription factors and their target genes in S. cerevisiae were extracted from the SCPD Promoter Database of Saccharomyces cerevisiae (http://cgsigma.cshl.org/jian) (17), the Yeast Proteome Database (www.incyte.com/control/researchproducts/insilico/proteome) (18), and genome-wide experiments that locate binding sites of given transcription factors (7–10). For the latter, we used the experimental thresholds used in the original articles.
Experimentally identified PPIs were extracted from the Database of Interacting Proteins (http://dip.doe-mbi.ucla.edu) (19), Biomolecular Interaction Network Database (http://binddb.org) (20), and Munich Information Center for Protein Sequences database (http://mips.gsf.de/proj/yeast/tables/interaction) (21), all providing manually reviewed lists of interacting proteins, and from high-throughput yeast two-hybrid studies (5, 6). We excluded from the analysis interactions in which one of the pair mates interacts with >50 different proteins to avoid false interactions caused by “sticky” proteins (22). Self-interactions representing autoregulation or protein homodimerization were not included in the analysis.
Data Representation. We based our analysis on network representation of the two types of data (23). A node represents both a protein and the gene encoding it. A PPI is represented by a bidirected edge connecting the interacting proteins. A TRI is an interaction between a transcription factor and its target gene and is represented by a directed edge pointing from the transcription factor to its target gene.
Detecting k-Protein Network Motifs. All connected subnetworks containing k nodes in the interaction network were collated into isomorphic patterns, and the number of times each pattern occurred was counted. If the number of occurrences was at least five and statistically significantly higher than in randomized networks, the pattern was considered as a network motif. The statistical-significance test was performed by generating 1,000 randomized networks (4, 24, 25) and computing the fraction of randomized networks in which the pattern appeared at least as often as in the interaction network. A pattern with P ≤ 0.05 was considered statistically significant.
To generate randomized networks containing two types of edges, we extended the approach of Shen-Orr et al. (11), who considered networks involving one type of connection. There they generated randomized networks with the same network characteristics by preserving the node degrees. For dealing with networks with multiple types of connections we defined two terms:
Extended degree of a node: the number of edges per type that point to/from a node (demonstrated in Fig. 1a). Two nodes have the same extended degree if they have the same number of ingoing and outgoing edges for each edge type.
Edge profile of two nodes: the set of edges connecting the two nodes with the type and direction of each edge detailed (exemplified in Fig. 1a).
Fig. 1.

The randomization procedure. (a) Extended node degree and edge profile. Nodes represent proteins; black, bidirected edges represent PPIs; and red, directed edges represent TRIs. Extended node degrees: a, one PPI, one outgoing TRI, and two ingoing TRIs; b, two PPIs and one outgoing TRI; c, one PPI and one outgoing TRI; d, two PPIs, one outgoing TRI, and two ingoing TRIs. Examples for edge profiles: (a,b), one PPI and one ingoing TRI; (b,a), one PPI and one outgoing TRI; (a,d), one outgoing TRI and one ingoing TRI; (b,d), one PPI. The edge profile of (d,c) is equivalent to that of (a,b). (b) The four-point-switchability condition. If edge profile (s1,t1) = edge profile (s2,t2) and edge profile (s1,t2) = edge profile (s2,t1), then edges can be switched as exemplified. For clarity, each edge color represents a type of edge profile. Note that if (s1,t1; s2,t2) are switchable, then so are (s1,t2;s2,t1), (t1,s1;t2,s2), and (t2,s1;t1,s2). Switch-ability is considered only for cases in which all four nodes are distinct and at least one edge profile is not empty.
The extended degree reflects the local connectivity of a node, and the edge profile provides a local measure of the relation between two nodes. The randomized networks are generated such that both the extended degree of each node and the profile of each edge in the network are retained. We developed an algorithm that generates such randomized networks by an iterative switching of edges. The four-point-switchability condition described in Fig. 1b provides sufficient conditions for the retention of all edge profiles and the extended degrees of all nodes.
For k = 2 a slight change is required, because the preservation of edge profiles implies that all two-node patterns (Fig. 2) remain fixed. For the assessment of two-node patterns, we created 1,000 randomized networks by (i) decoupling the two types of connections to form two separate networks, each representing a single type of connection; (ii) separately randomizing each of the decoupled networks; and (iii) integrating them into a single random network. Also, the statistical significance of the pattern in Fig. 2C was computed analytically by assuming a uniform distribution of TRIs over transcription factor pairs.
Fig. 2.

All possible interaction patterns between two connected proteins. A node represents a gene and its protein product; a red, directed edge represents a TRI; and a black bidirected edge represents a PPI.
Results
The approach underlying the present analysis extends the methodology for motif discovery of Shen-Orr et al. (11), who studied networks comprising a single type of connection. It starts with representing the integrated cellular network as a network in which there is a distinction between TRIs and PPIs. A node in the network represents both a gene and its protein product; a TRI is represented by a directed edge pointing from the transcription factor to its target gene; and a PPI is represented by a bidirected edge connecting the interacting proteins (23). The network can be represented graphically by using edges of two “colors”: directed, red edges representing transcription regulation and bidirected, black edges representing protein interactions. The resulting network is analyzed to find network motifs: patterns of connections involving TRI, PPI, or both that recur in the network significantly more often than in randomized networks (P ≤ 0.05). The randomized networks are generated under the requirement that the extended node degrees and edge profiles (Fig. 1a) are the same as in the original network for all nodes and edges. To generate these randomized networks, we developed a randomization algorithm in which edges of a network are switched if a four-point-switchability condition holds (Fig. 1b). This condition guarantees the retention of extended node degrees and edge profiles as required. For the analysis of patterns consisting of three and four nodes, we generated 1,000 randomized networks by iterative applications of the switchability condition. The analysis of two-node patterns is slightly different, as detailed in Methods.
We applied our approach to network data of the yeast S. cerevisiae. To overcome the noisiness of experimental interaction data collected via high-throughput methods, we generated a stringent data set containing 3,183 interactions between 1,863 proteins: PPIs were included if detected by at least two different experimental studies (different yeast two-hybrid methods were considered as different studies, as in ref. 22). TRIs were included if detected by methods other than genome-wide experiments. The resulting network was denoted as the stringent network. The robustness of the results was confirmed by performing the same analysis on a network containing all 12,413 experimentally identified interactions between 4,651 proteins, denoted the nonstringent network (see Methods for a detailed description of the data). Table 1 lists the number of interactions in the stringent and nonstringent networks.
Table 1. Number of interactions in the stringent and nonstringent networks.
| Network type | No. of PPIs* | No. of TRIs |
|---|---|---|
| Stringent network | 1,832 interactions between 1,385 proteins | 1,351 interactions between 128 transcription factors and 591 target genes |
| Nonstringent network | 6,159 interactions between 3,617 proteins | 6,254 interactions between 160 transcription factors and 2,698 target genes |
This number does not include the 177 and 235 self-interactions that are present in the stringent and the nonstringent data sets, correspondingly.
Next we present the motifs that emerged from the analysis.
Two-Protein Network Motifs. There are five possible two-protein connected patterns (Fig. 2). We assessed the statistical significance of the five patterns by comparing the number of their occurrences in the interaction network to that expected at random (see Methods). We found that only one of the patterns, the mixed-feedback loop comprising one PPI edge and one TRI edge (Fig. 2D), was a motif (P < 0.001 both in the stringent and nonstringent interaction networks). In this motif, protein P regulates gene g at the transcription level, and the product of gene g, protein G, interacts with P at the protein level. The eight two-protein mixed-feedback loops identified in the stringent network included five characterized feedback loops, of which three were positive and two were negative. The pair Gal4–Gal80 presents an example for a negative-feedback loop: Gal4 is a transcription factor that activates genes participating in galactose catabolism, including GAL80. Gal80 binds to Gal4 and, in the absence of galactose, represses its activity (26). A detailed list of mixed-feedback loops as well as detection and analysis of more complex cases of mixed-feedback loops in S. cerevisiae can be found in ref. 23.
Three-Protein Network Motifs. There are 13 possible three-protein connected patterns with a single type of directed interaction (12). When there are two types of interactions, such as TRI and PPI, the number of possible patterns rises to 100. Of the 100 possible three-protein connected patterns, 29 different patterns occurred in the stringent network. Only five of these occurred significantly more often than in random networks (P < 0.001) and thus are network motifs (Table 2). These five motifs were also found to be network motifs in the nonstringent network. We now describe the motifs in descending order of abundance:
Table 2. Three-protein network motifs in the stringent network.
| No. of occurrences
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Stringent network
|
Nonstringent network
|
Comments regarding occurrences in the stringent network | ||||||
| Motif* | Illustration† | N real | N rand ± SD | z score | N real | N rand ± SD | Z score | |
| A. Protein clique | 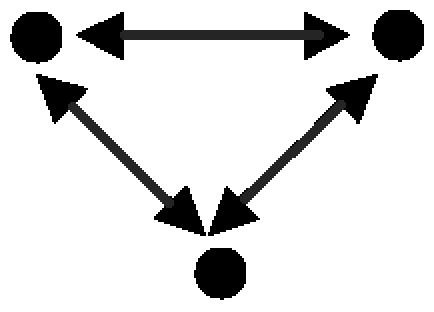 |
1,293 | 14 ± 3.8 | 332.7 | 2,016 | 87 ± 10.8 | 177.9 | 1,198 occurrences in experimentally identified complexes |
| B. Interacting transcription factors that coregulate a third gene | 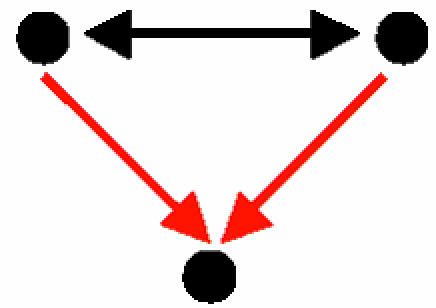 |
243 | 2.4 ± 2.1 | 115.9 | 476 | 9.6 ± 7.8 | 59.7 | 21 pairs of coregulating proteins, most of which act in concert |
| C. Feed-forward loop | 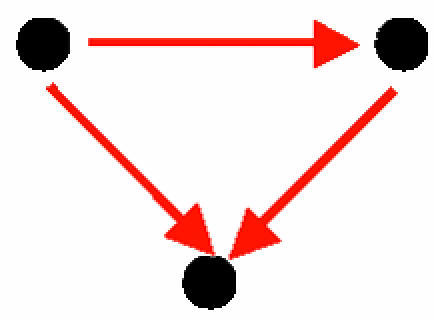 |
83 | 26 ± 6 | 9.5 | 994 | 473 ± 36.7 | 14.2 | Analyzed in refs. 11, 40, and 41 |
| D. Coregulated interacting proteins | 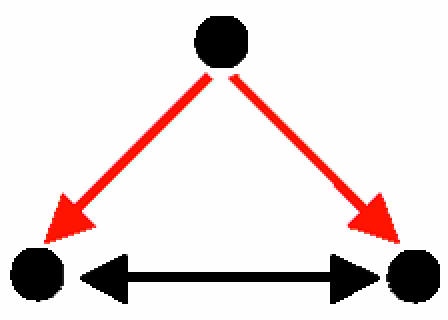 |
66 | 2 ± 1.4 | 46.5 | 285 | 107 ± 10.1 | 17.7 | 25 sets of coregulated interacting proteins, most of which act in concert or participate in a common complex |
| E. Mixed-feedback loop between transcription factors that coregulate a gene | 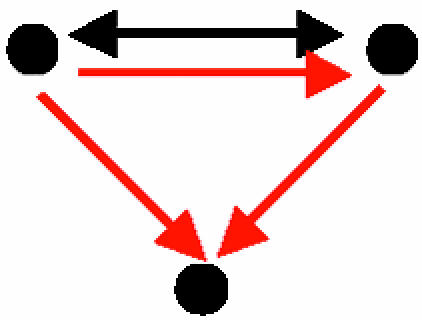 |
46 | 2.7 ± 1.6 | 26.3 | 118 | 8.2 ± 5.4 | 20.3 | Four distinct pairs of transcription factors that are also involved in a mixed-feedback loop |
These motifs were highly statistically significant in both the stringent and nonstringent networks; in all 1,000 randomized networks the number of their occurrences was lower than in the actual network.
A node represents a gene and its protein product; a red, directed edge represents a TRI; a black, bidirected edge represents a PPI.
A protein clique (Table 2, motif A): This is the most abundant motif and is composed entirely of PPIs. Of the occurrences of this motif, 92% correspond to known protein complexes [based on information from the Munich Information Center for Protein Sequences (21) and the Yeast Proteome Database (18)].
Interacting transcription factors that coregulate a third gene (Table 2, motif B): There were 243 occurrences of this motif corresponding to 21 distinct transcription factor pairs, each of which regulate a group of genes. For example, the pair of transcription factors Mbp1–Swi6, known as the MBF complex, regulate 34 different genes. In most pairs of interacting transcription factors that coregulate genes, the two pair mates are known to have the same function, either coactivating or corepressing genes. Two such examples include Pho2 and Pho4, which coactivate five genes, and Ssn6 and Tup1, which corepress nine genes. In the former, the interaction of Pho2 with Pho4 increases the accessibility of the activation domain of Pho4. In the latter, both proteins are part of a repressor complex that becomes an activator in a Hog1-dependent manner.
A feed-forward loop comprising solely TRIs (Table 2, motif C): This motif contains two transcription factors, one of which regulates the other, both jointly regulating a target gene. This motif has been detected within the transcription–regulation networks of E. coli (11) and S. cerevisiae (10, 12). There were 83 occurrences of this motif in the S. cerevisiae interaction network corresponding to 20 distinct pairs of transcription factors.
Coregulated interacting proteins (Table 2, motif D): This motif consists of a pair of genes that are regulated by a common transcription factor, and the protein products of which interact with each other. The 66 occurrences of this motif correspond to 25 sets of coregulated genes, where a set may consist of several proteins that are all coregulated by the same transcription factor and interact with each other. The proteins in most sets are known to work in concert or participate in a common complex. In many cases, the set of coregulated genes is regulated by more than one common transcription factor. This motif is found in a variety of cellular pathways. For example, the histones Hta1–Htb1, which are coregulated by four different transcription factors, play a role in chromatin structure; Fas1–Fas2, which are coregulated by three transcription factors, function in fatty acid biosynthesis; and Cdc10–Cdc11, which are coregulated by the transcription factor Swi4, function in cytokinesis. Although this motif is highly significant, it spans <1% of the pairs of coregulated genes in the current data set.
A mixed-feedback loop between transcription factors that coregulate a third gene (Table 2, motif E): This motif can be viewed as a combination of any pair of the following motifs: a mixed-feedback loop (Fig. 2D), two transcription factors that coregulate a third gene (Table 2, motif B), and the feed-forward loop (Table 2, motif C). The topology of this motif enables composite regulation schemes. A well known example for this mechanism is provided by Swi6–Swi4: Swi6 activates SWI4 transcription, and once Swi4 is synthesized, it interacts with Swi6 to form the SBF complex. This complex then regulates genes during the transition to S phase in the cell cycle.
Four-Protein Network Motifs. There are >3,000 possible four-protein patterns. The analysis of the stringent network revealed 201 distinct patterns, of which 63 were network motifs (statistically significant with P ≤ 0.05). Note that each motif appears at least five times in the network. A randomized network as a control shows virtually no motif with this significance and number of occurrences. Intriguingly, almost all of the four-protein network motifs contained one or more of the three-protein network motifs presented above: 36 motifs could be presented as a three-protein motif with a dangling fourth node, and 21 motifs could be presented as a combination of two or more three-protein motifs (Fig. 3a). Moreover, almost every pair of three-protein motifs could be combined, in at least one way, to produce a four-protein network motif (Fig. 3a). The exception is the combination of a feed-forward loop with a motif containing a pair of coregulated interacting proteins [Fig. 3a, entry (C,D)], which is present in the stringent network but is not statistically significant.
Fig. 3.

Four-protein network motifs discovered in the stringent network. (a) Motifs that can be represented as combinations of three-protein network motifs. When there is more than one possible way to generate a four-protein motif, the combination involving the more abundant three-protein motifs is presented. The three-protein motif of a mixed-feedback loop between coregulating proteins (Table 2, motif E) was not included here, because by itself it is a combination of two other three-protein motifs. Dangling motifs, where a fourth node is connected to only one of the nodes of the three-protein motif, are not presented. A three-protein motif may appear more than once in a combination that yields a four-protein motif [e.g., entry (A,D)]. (b) Motifs that cannot be constructed from three-protein motifs. i, the bi-fan motif; ii, a motif containing a feed-forward loop; iii–vi, motifs that appear as extensions of smaller network motifs, for which one of the PPIs in each smaller motif (Left) is extended to a series of PPIs by means of an intermediate protein (Right). A node represents a gene and its protein product; a red, directed edge represents a TRI; and a black, bidirected edge represents a PPI.
Six of the four-protein network motifs could not be constructed from a three-protein motif in combination with either another three-protein motif or a dangling node (Fig. 3b). Four of these motifs may be viewed as extensions of smaller network motifs, in which one of the PPIs in each smaller motif was extended to a series of PPIs (Fig. 3b, iii–vi). Of the two remaining motifs, one was built of two transcription factors that coregulate genes (Fig. 3b, i). This motif was termed “bi-fan” (12) and may lead to higher-order patterns of overlapping regulation similarly to the “dense-overlapping regulon” motif detected in E. coli (11). The other motif contained a transcriptional feed-forward loop (Fig. 3b, ii).
Most of the four-protein motifs contained protein triplets acting as higher-order hubs. Explicitly, each motif was associated with a limited set of recurring protein triplets, occurring in combination with various other proteins. At most there were 10 such protein triplets per motif, although the total number of motif occurrences was 10- to 100-fold greater.
Applying our analysis to the nonstringent network revealed 496 distinct patterns, of which 168 were statistically significant. Although some of the network motifs that were detected in the stringent network became insignificant in the nonstringent network, the description of four-protein network motifs as combinations of three-protein network motifs holds in both networks.
Discussion
We analyzed the local structure of an integrated cellular interaction network. This network has two types (colors) of edges, representing PPIs and TRIs. To analyze this network, we developed algorithms for detecting network motifs in networks with multiple types of edges. Our analysis revealed several highly significant network motifs of two, three, and four nodes. It would be intriguing to interpret the functionality of these motifs.
The Mixed-Feedback Loop, but Not Pure Transcriptional Feedback, Is a Motif. At the level of two-protein patterns, we found the mixed-feedback loop with one PPI edge and one TRI edge (Fig. 2D) to be a highly significant motif. In contrast, the feedback loop with two TRI edges (Fig. 2C) was not significantly more common than in randomized networks.
What could underlie the apparent preference for mixed feedback and the selection against pure TRI feedback? One possibility regards response time. Transcriptional regulation is generally slow: each transcription–regulation edge causes a delay of approximately one lifetime of the protein product, as was recently demonstrated theoretically and experimentally (27, 28). For negative-feedback loops, long delays can lead to instability and noisy oscillations (29–31), which may be undesirable in homeostatic systems. Thus, mixed feedback has an advantage over pure transcriptional feedback in that the slow transcriptional edge is closed by a fast PPI.
Selection in favor of mixed feedback over pure transcriptional feedback may be a design principle in other cell types as well. In mammalian cells, there are many examples of mixed feedback, such as p53 transcriptionally activating Mdm2, which in turn targets p53 for degradation by PPI (32), or in the control of nuclear factor κB expression (33). Heat-shock response in both eukaryotes (34) and bacteria (35) is controlled by mixed feedback. For example, in E. coli, σ32 activates the transcription of dnaKJ, which in turn targets σ32 for degradation (35).
Three-Protein Motifs: Cliques, Coregulation, and Regulatory Complexes. Five three-protein motifs were found (Table 2, motifs A–E). These motifs represent basic patterns of regulation and of organization of proteins into modules. Motif A, a protein clique, is a motif with three PPIs. It represents complexes of interacting proteins that work together as a multicomponent machine. Motifs B and D represent two proteins that interact at the protein level and that either regulate a common gene (motif B) or are regulated transcriptionally by the same transcription factor (motif D). Motif B represents a transcription regulator that is made of a complex of two proteins, a common scenario in eukaryotic cells. A well known example for motif B in higher eukaryotes is the complex formed by Jun and Fos transcriptional regulator proteins, which binds to promoters that bear the AP-1 site. Motif D, on the other hand, is found when interacting proteins are coregulated. It is widely accepted that coexpressed genes are coregulated (e.g., see ref. 36). In turn, several studies showed that genes with similar expression profiles are more likely to encode interacting proteins (37–39). Taken together, this suggests that some proteins that act in concert should be coregulated at the transcriptional level, as motif D demonstrates. Motif C is the feed-forward loop comprising purely transcription interactions. This motif was described previously in studies on transcription-regulatory networks. The function of the feed-forward loop depends on the signs of the regulations (activating/repressing). The most common configuration, with positive regulations, acts as a persistence detector (11, 40, 41). A second common feed-forward configuration (with two positive and one negative regulation) acts as a response accelerator or pulser (40). Motif E represents a more complex circuit, with a mixed-feedback loop between two regulators that jointly control a target gene. The mixed feedback can act as a control mechanism on the levels of the two proteins that interact to form a transcription-factor complex. Note that when considering only TRIs, this motif would be detected as a feed-forward loop. Only after taking PPIs into consideration does the specific structure of this motif reveal itself, highlighting the importance of the integration of the two types of interactions.
Four-Protein Motifs Are Largely Composed of Combinations of Three-Protein Motifs. At the level of four-protein patterns, we detected only 63 motifs out of the numerous possible patterns. Most of these motifs can be understood as combinations of three-node motifs. We represent this as a “multiplication table” in Fig. 3a. The operation of combining two motifs can be repeated, in principle, to produce even larger patterns; this may help in understanding the biological meaning of large motifs. For example, two cliques of three proteins can be combined to make a full four-protein clique, which can be a component of a large protein complex [Fig. 3a, entry (A,A)]. Another example is provided by the three-protein motifs A and D, which combine to make a three-protein clique, the members of which are regulated by the same transcription factor [Fig. 3a, entry (A,D)], representing a coordinately regulated interacting module.
In general, using small motifs as basic building blocks and combination operations to put them together can be viewed as an algebra that may allow the descriptions of composite motifs. Such motifs can be used, in turn, to explain more complicated patterns in terms of the basic ones. In forming such composite motifs, one can distinguish between repeated usages of the same hinge to combine patterns, as opposed to combinations that use different hinges in each step. Combinations of identical three-protein motifs (along the diagonal in Fig. 3a) are of special interest: Their repeated (composed) application forms a regular structure; for example, when applied to motif B, an interacting pair of regulators that coregulate many (rather than just two) different genes [Fig. 3a, entry (B,B)].
Of the 63 four-protein motifs, only five motifs do not contain any three-protein motif in their structures. Four of these motifs are extensions of smaller motifs, as illustrated in Fig. 3b, iii–vi, which suggests that each motif can be generalized, in principle, to a family of structures that share a common structural theme and potentially a common functionality, as exemplified for mixed-feedback loops (23). The remaining four-protein motif is the bi-fan made purely of TRIs, previously detected in the analysis of transcription networks (11). The bi-fan generalizes to higher-order arrays of transcription factors that combinatorially regulate arrays of genes known as dense-overlapping regulons (10, 11). Each gene receives multiple inputs, which are integrated in cis-regulatory input functions resembling logic AND- or OR-like gates (42–44). The bi-fan thus can be thought of as a hard-wired decision-making device. As such, it can be considered as another basic building block.
Interestingly, we found high-order analogues of network hubs within the three- and four-protein network motifs in the form of protein pairs and triplets that recur in motifs. It is conceivable that these higher-order hubs play a central role in the cellular network, similarly to their single-node counterparts (e.g., see ref. 45).
The present approach can be used to analyze any network with multiple types of interactions, both directed and undirected. A limitation of the current study is that the PPIs in the data set are undirected, which is natural for some PPIs such as those involved in dimers or higher-order complexes; however, it may mask the interpretation of naturally directed interactions such as phosphorylation or targeted degradation. As the amount of network data increases, future studies could aim at detecting network motifs in networks in which edge colors distinguish between different types of PPIs. Nevertheless, the present motifs provide insight into the structure of the cellular circuitry. It would be interesting to experimentally study the dynamic behavior of molecular systems bearing the present motifs (27, 41, 46) to determine whether they carry out defined functions in the network.
Acknowledgments
This study was supported by grants from the Israeli Ministry of Science and Israeli Science Foundation (administered by the Israeli Academy of Sciences and Humanities) and by European Union Grant QLRI-CT-2001-00015 (to H.M.). U.A. acknowledges support from the National Institutes of Health and The Natan and Ringel memorial funds. E.Y.-L. and R.M. are supported by the Horowitz Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PPI, protein–protein interaction; TRI, transcription–regulation interaction.
References
- 1.Thieffry, D., Huerta, A. M., Perez-Rueda, E. & Collado-Vides, J. (1998) BioEssays 20, 433–440. [DOI] [PubMed] [Google Scholar]
- 2.Ouzounis, C. A. & Karp, P. D. (2000) Genome Res. 10, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fell, D. A. & Wagner, A. (2000) Nat. Biotechnol. 18, 1121–1122. [DOI] [PubMed] [Google Scholar]
- 4.Guelzim, N., Bottani, S., Bourgine, P. & Kepes, F. (2002) Nat. Genet. 31, 60–63. [DOI] [PubMed] [Google Scholar]
- 5.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) Nature 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 6.Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M. & Sakaki, Y. (2001) Proc. Natl. Acad. Sci. USA 98, 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer, V. R., Horak, C. E., Scafe, C. S., Botstein, D., Snyder, M. & Brown, P. O. (2001) Nature 409, 533–538. [DOI] [PubMed] [Google Scholar]
- 8.Simon, I., Barnett, J., Hannett, N., Harbison, C. T., Rinaldi, N. J., Volkert, T. L., Wyrick, J. J., Zeitlinger, J., Gifford, D. K., Jaakkola, T. S. & Young, R. A. (2001) Cell 106, 697–708. [DOI] [PubMed] [Google Scholar]
- 9.Ren, B., Robert, F., Wyrick, J. J., Aparicio, O., Jennings, E. G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., et al. (2000) Science 290, 2306–2309. [DOI] [PubMed] [Google Scholar]
- 10.Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Science 298, 799–804. [DOI] [PubMed] [Google Scholar]
- 11.Shen-Orr, S. S., Milo, R., Mangan, S. & Alon, U. (2002) Nat. Genet. 31, 64–68. [DOI] [PubMed] [Google Scholar]
- 12.Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D. & Alon, U. (2002) Science 298, 824–827. [DOI] [PubMed] [Google Scholar]
- 13.Rives, A. W. & Galitski, T. (2003) Proc. Natl. Acad. Sci. USA 100, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bu, D., Zhao, Y., Cai, L., Xue, H., Zhu, X., Lu, H., Zhang, J., Sun, S., Ling, L., Zhang, N., Li, G. & Chen, R. (2003) Nucleic Acids Res. 31, 2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spirin, V. & Mirny, L. A. (2003) Proc. Natl. Acad. Sci. USA 100, 12123–12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wuchty, S., Oltvai, Z. N. & Barabasi, A. L. (2003) Nat. Genet. 35, 176–179. [DOI] [PubMed] [Google Scholar]
- 17.Zhu, J. & Zhang, M. Q. (1999) Bioinformatics 15, 607–611. [DOI] [PubMed] [Google Scholar]
- 18.Costanzo, M. C., Crawford, M. E., Hirschman, J. E., Kranz, J. E., Olsen, P., Robertson, L. S., Skrzypek, M. S., Braun, B. R., Hopkins, K. L., Kondu, P., et al. (2001) Nucleic Acids Res. 29, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xenarios, I., Fernandez, E., Salwinski, L., Duan, X. J., Thompson, M. J., Marcotte, E. M. & Eisenberg, D. (2001) Nucleic Acids Res. 29, 239–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bader, G. D., Donaldson, I., Wolting, C., Ouellette, B. F., Pawson, T. & Hogue, C. W. (2001) Nucleic Acids Res. 29, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mewes, H. W., Frishman, D., Guldener, U., Mannhaupt, G., Mayer, K., Mokrejs, M., Morgenstern, B., Munsterkotter, M., Rudd, S. & Weil, B. (2002) Nucleic Acids Res. 30, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sprinzak, E., Sattath, S. & Margalit, H. (2003) J. Mol. Biol. 327, 919–923. [DOI] [PubMed] [Google Scholar]
- 23.Yeger-Lotem, E. & Margalit, H. (2003) Nucleic Acids Res. 31, 6053–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman, M. E., Strogatz, S. H. & Watts, D. J. (2001) Phys. Rev. E Stat Nonlin. Soft Matter Phys. 64, 026118. [DOI] [PubMed] [Google Scholar]
- 25.Maslov, S. & Sneppen, K. (2002) Science 296, 910–913. [DOI] [PubMed] [Google Scholar]
- 26.Lohr, D., Venkov, P. & Zlatanova, J. (1995) FASEB J. 9, 777–787. [DOI] [PubMed] [Google Scholar]
- 27.Rosenfeld, N., Elowitz, M. B. & Alon, U. (2002) J. Mol. Biol. 323, 785–793. [DOI] [PubMed] [Google Scholar]
- 28.Rosenfeld, N. & Alon, U. (2003) J. Mol. Biol. 329, 645–654. [DOI] [PubMed] [Google Scholar]
- 29.Ferrell, J. E., Jr. (2002) Curr. Opin. Cell Biol. 14, 140–148. [DOI] [PubMed] [Google Scholar]
- 30.Elowitz, M. B. & Leibler, S. (2000) Nature 403, 335–338. [DOI] [PubMed] [Google Scholar]
- 31.Tyson, J. J., Chen, K. C. & Novak, B. (2003) Curr. Opin. Cell Biol. 15, 221–231. [DOI] [PubMed] [Google Scholar]
- 32.Vogelstein, B., Lane, D. & Levine, A. J. (2000) Nature 408, 307–310. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann, A., Levchenko, A., Scott, M. L. & Baltimore, D. (2002) Science 298, 1241–1245. [DOI] [PubMed] [Google Scholar]
- 34.Santoro, M. G. (2000) Biochem. Pharmacol. 59, 55–63. [DOI] [PubMed] [Google Scholar]
- 35.Straus, D., Walter, W. & Gross, C. A. (1990) Genes Dev. 4, 2202–2209. [DOI] [PubMed] [Google Scholar]
- 36.Ihmels, J., Friedlander, G., Bergmann, S., Sarig, O., Ziv, Y. & Barkai, N. (2002) Nat. Genet. 31, 370–377. [DOI] [PubMed] [Google Scholar]
- 37.Laub, M. T., McAdams, H. H., Feldblyum, T., Fraser, C. M. & Shapiro, L. (2000) Science 290, 2144–2148. [DOI] [PubMed] [Google Scholar]
- 38.Grigoriev, A. (2001) Nucleic Acids Res. 29, 3513–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kemmeren, P., van Berkum, N. L., Vilo, J., Bijma, T., Donders, R., Brazma, A. & Holstege, F. C. (2002) Mol. Cell 9, 1133–1143. [DOI] [PubMed] [Google Scholar]
- 40.Mangan, S. & Alon, U. (2003) Proc. Natl. Acad. Sci. USA 100, 11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mangan, S., Zaslaver, A. & Alon, U. (2003) J. Mol. Biol. 334, 197–204. [DOI] [PubMed] [Google Scholar]
- 42.Yuh, C. H., Bolouri, H. & Davidson, E. H. (1998) Science 279, 1896–1902. [DOI] [PubMed] [Google Scholar]
- 43.Buchler, N. E., Gerland, U. & Hwa, T. (2003) Proc. Natl. Acad. Sci. USA 100, 5136–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Setty, Y., Mayo, A. E., Surette, M. G. & Alon, U. (2003) Proc. Natl. Acad. Sci. USA 100, 7702–7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong, H., Mason, S. P., Barabasi, A. L. & Oltvai, Z. N. (2001) Nature 411, 41–42. [DOI] [PubMed] [Google Scholar]
- 46.Ronen, M., Rosenberg, R., Shraiman, B. I. & Alon, U. (2002) Proc. Natl. Acad. Sci. USA 99, 10555–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]