

Abstract
Filamentous hemagglutinin (FHA), the major 230-kDa adhesin of the whooping cough agent Bordetella pertussis, is one of the most efficiently secreted proteins in Gram-negative bacteria. FHA is secreted by means of the two-partner secretion (TPS) pathway. Several important human, animal, and plant pathogens also secrete adhesins and other virulence factors by using this mode of secretion. A TPS system is composed of two separate proteins, with TpsA the secreted protein and TpsB its associated specific outermembrane transporter. All TPS-secreted proteins contain a distinctive N-proximal module essential for secretion, the TPS domain. We report here the 1.7- Å structure of a functionally secreted 30-kDa N-terminal fragment of FHA. It reveals that the TPS domain folds into a β-helix, with three extrahelical motifs, a β-hairpin, a four-stranded β-sheet, and an N-terminal capping, mostly formed by the nonconserved regions of the TPS domain. The structure thus explains why the TPS domain is able to initiate folding of the β-helical motifs that form the central domain of the adhesin, because it is itself a β-helical scaffold. It also contains less conserved extrahelical regions most likely involved in specific properties, such as the recognition of the outer-membrane transporter. This structure is representative of the TPS domains found so far in >100 secreted proteins from pathogenic bacteria. It also provides a mechanistic insight into how protein folding may be linked to secretion in the TPS pathway.
The addressing of proteins to their proper compartment is an essential process in all organisms and involves specific signals and machineries. In Gram-negative bacteria, proteins destined to the extracellular milieu must be transported across two distinct membranes, the cytoplasmic membrane and the lipopolysaccharide-containing outer membrane. These organisms have thus developed specific systems for protein transport across that second membrane. Among them, the type V secretion stands out for its apparent simplicity. It comprises two distinct pathways, the autotransporter (AT) and the two-partner secretion (TPS) pathways (1–3). Both are devoted to the translocation of large proteins or protein domains, mostly adhesins, cytolysins, and enzymes, and have been identified in an increasing number of bacterial genera, including important human, animal, and plant pathogens, such as Neisseria meningitidis, Yersinia pestis, Pseudomonas aeruginosa, Haemophilus influenzae, and Erwinia chrysanthemi. TPS systems are composed of two separate proteins, with TpsA being the secreted protein and TpsB being its specific transporter (2). Little is known about the functionality of this secretion pathway, and structural data are needed to unravel the molecular aspects of TPS secretion.
A hallmark of the TPS pathway is the presence in the TpsA exoproteins of a conserved N-proximal module called the TPS domain, which is essential for secretion (2, 4). The TPS domain is thus hypothesized to bear secretion determinants mediating specific interactions of TpsA proteins with their TpsB transporters at the periplasmic side of the outer membrane (4, 5). These recognition events are thought to trigger the translocation of the TpsA proteins, starting from their N terminus, across that membrane. The TpsB transporters appear to form rather small β-barrel channels in the outer membrane, through which their TpsA partners pass most likely in an extended conformation (6–8). Folding of the TpsA proteins is thus hypothesized to take place at the cell surface in a progressive fashion.
Filamentous hemagglutinin (FHA), the major adhesin of the whooping cough agent Bordetella pertussis is one of the most efficiently secreted proteins in Gram-negative bacteria. The 230-kDa FHA is secreted by a TPS system involving a specific outer membrane transporter, FhaC. The FHA/FhaC system has served as a prototype for the definition of the TPS pathway (2). FHA is synthesized as a 370-kDa precursor and processed to yield a 230-kDa mature protein by the removal of a large C-terminal fragment of unknown function (9–11). Distinct binding determinants have been identified in mature FHA (12), including a heparin-binding domain, an arginine-glycineaspartate motif and a carbohydrate recognition domain.
The importance of FHA both in the pathogenesis of B. pertussis and as a prototype of the widespread bacterial TPS pathway provides an incentive for seeking information about its molecular structure. However, crystallographic studies of FHA have been hindered by the poor solubility and unwieldy dimensions of mature FHA, estimated to reach a length of 500 Å (13). We therefore focused on the characterization of the TPS domain of FHA. The shortest N-terminal fragment of FHA that could be secreted was identified by progressive truncations, to include all secretion determinants of the protein. We report here the structure at a 1.7- Å resolution of this secreted 30-kDa N-terminal FHA fragment. This protein reveals the first TPS domain architecture, the representative scaffold of the TpsA family, and several β-helical 19-residue repeat motifs that form the central right-handed β-helix domain of the full-length protein. Finally, this structure paves the way to model building of the complete 230-kDa FHA adhesin and other members of the TpsA family.
Materials and Methods
Construction of Strains, Protein Expression, and Purification. The TPS secretion system of FHA/FhaC has been reconstituted in Escherichia coli as described (14). Recombinant E. coli UT5600(pEC24,pEC97) was used for the production of Fha35. Plasmid pEC24 allows for the expression of fhaC in E. coli (6). pEC97 was obtained as follows. A 5′ segment of fhaB was amplified by PCR with the oligonucleotides 5′-ATGAACACGAACCTGTACAGG-3′ and 5′-GGTACCTCAGGATCCCTTGCCGCCGCCTTGCA-3′. The amplicon was cloned in pC-RII-Topo (Invitrogen) and sequenced by using the ABI 377 Prism sequencer, and the 1.1-kb SphI–KpnI fragment of the recombinant plasmid was exchanged for the 1.3-kb SphI–KpnI fragment of pBG12 (15), yielding pFJD95. The EcoRI–KpnI fragment of pFJD95, corresponding to the first 1.4 kb of fhaB, was then introduced into pUC19, generating pFJD117. The complementary oligonucleotides 5′-GATCTCACCATCACCACCATCACGGGCCCTAG-3′ and 5′-GATCCATGGGCCCGT-GATGGTGGTGATGGTGA-3′ were then annealed and introduced into the BamHI site of pFJD117, generating pFJD117-His. Finally, the 0.6-kb NotI–BamHI fragment of pFJD117-His was exchanged for the 2-kb NotI–BamHI fragment of pFJD12 (14), yielding pEC97. The C-terminal sequence of Fha35 coded by pEC97 is thus GlyGlyLysGlySerHisHisHisHisHisHisGlyPro, in which the last 10 residues do not belong to the authentic FHA sequence. For the production of Fha30, we used E. coli UT5600(pEC24, pEC102). pEC102 was constructed as follows. An internal fragment of fhaB was amplified by PCR with the primers 5′-GGGCGTCCCAGCGTCAACG-3′ and 5′-GTCGACTTAGGATCCCT-TCACCGCCCCGCCGCCCG-3′. The amplicon was cloned into PCRII-Topo, yielding pEC100, and sequenced. The 0.5-kb NotI–SalI fragment was then excised from pEC100 and exchanged for the 2-kb NotI–SalI fragment of pFJD12 (14), thus creating pEC102. This plasmid codes for the N-terminal portion of FHA up to residue 304. Fha30 terminates with the sequence GlyAlaValLysSerGly, in which the last two residues do not belong to the authentic FHA sequence.
Cultures of E. coli were performed under the following conditions. The bacteria were grown in liquid LB medium supplemented with 25 μg/ml kanamycin and 150 μg/ml ampicillin until the absorbance of the cultures reached 0.8 at 600 nm. The cultures were then treated by the addition of 1 mM isopropyl β-d-thiogalactoside to induce the expression of the recombinant fhaB genes, and the cultures were grown for three additional hours. Supernatants of isopropyl β-d-thiogalactoside-treated E. coli were collected, filtered on 0.45-μm membranes, and loaded onto a cation-exchange column Poros HS20 (Perkin–Elmer). The protein was eluted with a 0 to 1 M gradient of NaCl. Fractions containing pure Fha30 were pooled and dialyzed against 0.01 M imidazole (pH 6.5). The protein was concentrated to 4 mg/ml by ultrafiltration (Centricon, Millipore).
Crystallization and Preparation of Heavy-Atom Derivatives. Native crystals of Fha30 were obtained at 21°C by using the hanging-drop vapor diffusion method by mixing the protein and precipitant solutions in a 1:1 ratio. Crystals were grown at a protein concentration of 4 mg/ml in 24% PEG8000/15% glycerol/0.17 M sodium acetate/0.085 M Tris·HCl (pH 8.5). The platinum heavy-atom derivative was prepared by soaking crystals during 19 h at 21°C in a solution containing 24% PEG8000, 15% glycerol, 0.17 M sodium acetate, 0.085 M Tris·HCl (pH 8.5), and 0.01 M K2PtCl4. The mercuric derivative was obtained by cocrystallization. Crystals were grown in 22% PEG8000/15% glycerol/0.17 M sodium acetate/0.085 M Tris·HCl (pH 8.5)/0.02 M HgCl2.
Data Collection and Processing. The native data set and the mercuric and platinum derivative data sets were collected at 100 K on a MAR345 Image Plate Detector, with CuKα radiation produced by a Bruker–Nonius FR591 rotating-anode generator equipped with an Osmic's Confocal Max-Flux optical system and running at 100 mA, 50 kV. All the diffraction data were indexed, integrated, scaled, and merged with the xds package (Xray Detector Software) (16). The native high-resolution data set was collected at 100 K on beamline BM30A at the European Synchrotron Radiation Facility (Grenoble, France). These data were processed with xds. Statistics of data collection for the various data sets are presented in Table 1, which is published as supporting information on the PNAS web site. The volume of the unit cell suggested the presence of one molecule in the asymmetric unit.
Structure Determination and Refinement. The structure was solved by the multiple isomorphous replacement method with anomalous scattering by using the platinum and mercuric derivatives (Table 1). Heavy-atom positions were determined by difference Patterson maps and difference Fourier maps with the cns package (17). The initial multiple isomorphous replacement method with anomalous scattering phases were extended to 1.72 Å by density modification with cns. Model building was accomplished by the WarpNTrace procedure (18) implemented with the ccp4 package (19). The structure was refined in cns at 1.72 Å by using cross-validated maximum likelihood as the target function. The structure was inspected during the refinement by using turbo-frodo (20). Water molecules were added when peaks in the 2Fo - Fc density were >2σ and had a stereochemistry compatible with at least one hydrogen bond with a protein atom or another water molecule. In the final stage, the σ cutoff for picking water molecules was lowered to 1σ, and water molecules with a B factor > 50 Å2 were discarded. The final model refined at 1.72 Å has an Rwork of 14.4% and an Rfree of 19.0% and consists of 2,066 protein atoms and 295 water molecules. The stereochemistry of the final structure was evaluated by using the procheck program (21). Residues 13–18 displayed weak electron densities for many side chains. Residues 298–304 were not present in the electron density map and are not included in the final model.
The coordinates and structure factors have been deposited with the Protein Data Bank (PDB ID code 1RWR).
Results
Design of the Fha30 Fragment Used for Structure Determination. To determine the crystal structure of the TPS domain of FHA, N-terminal fragments of the adhesin were produced in an E. coli-reconstituted TPS secretion system including the membrane transporter FhaC (14). We started expression trials of an N-terminal FHA derivative of 32 kDa (designated as Fha35) engineered with a C-terminal His-6 tag. This FHA derivative was efficiently secreted, but its purification yielded a mixture of two protein species of 30 and 32 kDa, respectively, both shown to correspond to FHA fragments by peptide mass fingerprint. Crystals could be obtained by using this nonhomogeneous protein preparation, but they displayed diffraction patterns characteristic of fiber diffraction (not shown). An analysis by MS indicated that the 30-kDa protein included the 304 N-terminal residues of FHA. Therefore, a new variant called Fha30 was designed comprising only the first 304 residues of FHA, without C-terminal tag. This new FHA fragment was also efficiently secreted, and, after purification, it yielded only one protein species of 30 kDa. Crystallization conditions of Fha30 were determined that allowed us to obtain well diffracting crystals and to solve the structure of Fha30 at a resolution of 1.72 Å by the multiple isomorphous replacement method with anomalous scattering (Fig. 7, which is published as supporting information on the PNAS web site).
Overview of the Fha30 Structure. The core of Fha30 is folded into a right-handed parallel β-helix of nine coils (Fig. 1). A coil corresponds to a complete turn of the parallel β-helix (22). This fold was first observed in pectate lyase C of E. chrysanthemi and since then it has been reported in a dozen of proteins (23). The structural motifs of a right-handed parallel β-helix comprise three parallel β-sheets, named PB1, PB2, and PB3 (Fig. 1). The global topology of Fha30 is represented in Fig. 2. Starting from the N terminus, the coils become more regular to finally display, at the C-terminal part, an isosceles triangular-shaped section (Fig. 3). Concomitantly, the helix curvature progressively decreases from the N to the C terminus (Fig. 1). The helix interior of Fha30 is essentially filled with stacks of aliphatic residues (by decreasing numbers Val, Leu, Ile, Ala, and Gly), as often observed in right-handed parallel β-helices (22). The β-turns and loops that link the β-strands, in general, contain one glycine residue and are in four of nine coils stabilized by hydrogen bonds between the coils main chain and the side chain of a serine residue pointing toward the β-helix interior (Fig. 3).
Fig. 1.

Ribbon representation of the overall structure of Fha30. The helix β-sheets PB1, PB2, and PB3, the β-hairpin β7/β8, and the β-sheet β16/β25 are shown. The three β-strands, β1, β2, and β3, involved in the N-terminal β-helix capping are represented in red. N- and C-terminal extremities are also indicated.
Fig. 2.

Diagram illustrating the topology of Fha30. PB1, PB2, PB3, and extra β-helical motifs are represented.
Fig. 3.

Ball-and-stick view of the coil formed by β-strands β31, β32, and β33 that belong to PB1, PB2, and PB3, respectively. This coil corresponds to the first R1 repeat and displays an isosceles-triangular-shaped section. The hydrogen bonds between the coil's main chain and the side chain of Ser-257 are illustrated.
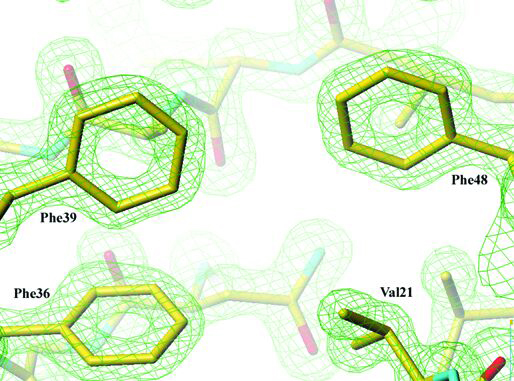
The first coil of the β-helix is strengthened by an aromatic cluster composed of three phenylalanine residues, Phe-36, Phe-39, and Phe-48 that belong to β4, β5, and β6, respectively. Almost all right-handed β-helices are N-terminally capped with an amphipatic α-helix that shields the hydrophobic core of the β-helix from the solvent. In contrast, the N terminus of Fha30 is capped by three β-strands, β1, β2, and β3, which cover the first coil of the β-helix (Fig. 4). β2 and β3 form a β-hairpin that extends the β-sheet PB3, whereas β1 forms hydrogen bonds with both β4 and β5, thus stabilizing the first coil of the β-helix. Among the 12 proline residues of Fha30, there is one cis-proline (Pro-44) localized in the turn between β5 and β6 in the first β-helix coil.
Fig. 4.

Ribbon representation of the N-terminal capping that shields the β-helix hydrophobic interior from the solvent. The helix β-sheets PB1, PB2, and PB3, the β-hairpin β7/β8, and the β-sheet β16/β25 are shown. The three β-strands β1, β2, and β3 involved in the capping are represented in red.
β7 and β8 form an extrahelical β-hairpin that interacts with β6 and β11 of PB3 (Fig. 2). The resulting β-sandwich is stabilized by hydrogen bonds between the main chain of the β7-β8 turn and the side chain of Asp-93 from β11. The β16/β25 β-sheet is made up of two antiparallel β-hairpins (Fig. 2). A salt bridge between Asp-139 (β17) and Arg-192 (β24) strengthens the cohesion between the two β-hairpins. β16/β25 and PB1 pack face to face to form a β-sandwich structure. Two salt bridges take part in the stabilization of the β16/β25–PB1 interactions. They involve Arg-198 (turn β24–β25) and Asp-161 (β20), and Arg-128 (β16) and Glu-79 (β9). β25 terminates the extra-β-helix sandwich and is followed by a loop of 12 residues containing seven alanines and three glycines.
The TPS Domain. To pinpoint the structural determinants of the TPS domain, the Fha30 structure was analyzed in the light of sequence similarities observed between Fha30 and the other TpsA proteins, including the hemolysins/cytolysins ShlA of Serratia marcescens, HpmA of Proteus mirabilis, EthA of Edwardsiella tarda, HhdA of Haemophilus ducreyi, the large supernatant proteins LspA1 and LspA2 of H. ducreyi, and the HecA adhesin of E. chrysanthemi (Fig. 5). This sequence comparison revealed that TpsA proteins display two conserved regions, C1 and C2, and two less-conserved regions, LC1 and LC2 (Figs. 5 and 6). C1 contains residues 20–127 of Fha30, encompassing the structural elements from β3 to β15. This region corresponds to the first three β-helix coils and the β7/β8 hairpin (Fig. 6). C2 extends from residue 216 to residue 245 and corresponds to a β-helix coil made up of β26 to β28 and β29. The first 19 N-terminal residues of Fha30 determine the less-conserved zone LC1 that participates in β-helix capping, including β1 and β2.
Fig. 5.

Sequence alignment of representative TpsA proteins. Secondary structure elements observed in the Fha30 structure are indicated. The conserved residues are boxed in black (cut off at 60% conservation), and the homologous residues are boxed in gray. The conserved regions C1 and C2 are delimited.
Fig. 6.

Ribbon view of Fha30 illustrating the LC1, C1, LC2, and C2 regions of the TPS domain. The R1 repeats that belong to the central right-handed β-helix domain of the full-length adhesin are also shown.
C1 and C2 are separated by LC2. The size of LC2 varies between 70 and 95 residues in the different TpsA proteins. In Fha30, this region corresponds to two β-helix coils (β18–β23) and the β16/β25 β-sheet. The two coils in LC2 complement the β-helical structure between C1 and C2 (Figs. 2 and 6), with a rather conserved sequence stretch corresponding in Fha30 to β20 (Fig. 5), whereas the β-sheet forms an extra-β-helix motif. Divergence in LC2 between TpsA proteins may reflect some of their functional specificities.
Residues 1–130 of Fha30 have been reported in the Pfam Protein Families Database under the codename PF05860 as a potential hemagglutination domain. However, we have no evidence that Fha30 expresses hemagglutination activity (24), and the present analyses indicate that the conservation of this TPS domain extends further. C-terminal deletions of ShlA (5), HpmA (25), and FHA (F.J.-D., unpublished data) have indicated that the minimal region needed for secretion includes at least LC1, C1, LC2, and C2. We thus propose to redefine the region composed of LC1, C1, LC2, and C2 as the secretion domain of FHA, which includes several coils of a right-handed β-helix structure with an N-terminal capping, a β-hairpin, and an extra-β-helix motif. Residues 1–245 of FHA thus define the representative scaffold for the TPS domain of TpsA proteins.
Discussion
We have determined the first structure of the TPS domain of a TpsA protein, that of FHA of B. pertussis. This domain folds into a β-helix with extrahelical motifs, a β-hairpin, a four-stranded β-sheet, and an N-terminal capping. The Fha30 structure also reveals several β-helical repeats that form the central right-handed β-helix domain of the full-length adhesin. The sequence of FHA contains two long 19-residue tandem amphipathic repeat regions called R1 and R2 (13), and each of the first R1 repeats found in Fha30 correspond to a β-helical coil. The most likely model for the mature adhesin is therefore that of an elongated β-helix (26), with the adherence determinants presented on loops or extrahelical motifs along the helix.
The TpsA exoproteins secreted by the TPS pathway in other organisms are also large, with masses >100 kDa and up to 400–500 kDa (27). So far, >100 TpsA proteins, including many adhesins and virulence factors, have been identified based on the presence of a conserved, N-proximal TPS domain, and the list will continue to grow with the ongoing efforts of microbial genome sequencing. Many of them present amphipatic repeated motifs with β-helical propensities (26). They are thus expected to share a similar architecture with Fha30 and to adopt right-handed β-helical structures, despite their limited overall sequence similarities, mainly confined to the TPS domain. FHA might thus serve as a structural model for a large number of virulence proteins.
Previous structural predictions suggested that the TPS domain might adopt an Ig-like fold (26). The Fha30 structure reveals that these predictions are incorrect and our results have important biological implications. Indeed, the structure demonstrates that TPS domain originates the β-helical architecture, with highly conserved sequence motifs representing the initial coils of the β-helical fold. The formation of a β-helix motif by the TPS domain is most likely an essential feature of the TPS mode of secretion. Our structural analysis thus provides a rationale of the folding of bacterial adhesins and virulence factors comprising a TPS domain. Several residues highly conserved between TpsA proteins have been shown to be important for secretion (5, 28, 29). The present structural analyses indicate that they are mainly devoted to the stabilization of the β-helix. In particular, the replacement of the first Asn of the conserved NPNL and NPNGI motifs in C1 (Asn-66 and Asn-105 in FHA) drastically affected the secretion of FHA and ShlA (4, 5). These motifs form type I β-turns, which might play important stabilizing roles. We have extended this analysis by replacing other conserved residues of the first β-helical coils. Most of these mutations abolish or strongly affect FHA secretion (H.H. and F.J.-D., unpublished observations). Therefore, the conserved residues of the TPS domain serve to drive the folding of the TPS domain into a β-helix and to stabilize the helix, which is essential for the efficient secretion and folding of the entire protein. This hypothesis is in good agreement with the current model of TPS secretion, according to which the TpsA proteins go through the outer membrane in an extended conformation and fold at the bacterial surface (6). The N-proximal position of the TPS domain is favorable to an early initiation of translocation across the outer membrane, which may be the best option to avoid periplasmic aggregation or degradation of such extremely large proteins. The transport of TpsA proteins to the cell surface is thus believed to proceed in a vectorial manner, from their N to their C terminus, and the exoproteins are expected to fold progressively as they emerge from the outer membrane. The frequent occurrence of repeated sequence motifs in TpsA proteins fits well with the sequential building of elongated β-helix structures. Therefore, the TPS domain most likely contributes to nucleate the folding of the rest of the molecule.
To reach the bacterial surface, the TpsA proteins must interact with their dedicated TpsB transporters at the periplasmic side of the outer membrane. It has been observed that neither the TPS domains nor the TpsB transporters are functionally interchangeable between TPS systems, arguing that the specific TpsA–TpsB interactions might involve variable rather than conserved regions (4). The structure of Fha30 nicely explains how the specific recognition between the secreted protein and its transporter might occur by using the β-helical scaffold of the TPS domain. This domain displays several non-β-helical motifs that essentially correspond to the variable regions between TpsA proteins. Extrahelical, antiparallel β-structures, such as the LC1-capping motif or the β-hairpin, are good candidates to confer specificity on the FHA–FhaC interaction. Indeed, LC1 is ideally positioned for secretion initiated from the N terminus. It is conceivable that it is involved in FhaC recognition in addition to its structural role.
Beside the TPS pathway, type V secretion comprises also the AT pathway, which is widely used by pathogenic bacteria to secrete adhesins and other large virulence proteins (1). Our results suggest that TPS and AT pathways are mechanistically related but function with inverse polarities. AT proteins contain their own transporter domain, covalently attached to the C-terminal extremity of the secreted “passenger” domain. The passenger domain is believed to be translocated vectorially across the outer membrane, from the C to the N terminus and to fold progressively in the same direction. Most AT proteins contain a well conserved junction region (PF03212 in the Pfam database) at the C terminus of the passenger domain, proposed to act as a scaffold that initiates the folding of the rest of the passenger (30, 31). The structure of P69 pertactin of B. pertussis, an AT passenger domain is also a right-handed β-helix (32). Whereas the N-terminal extremity of the pertactin β-helix is uncapped, its C-terminal end is capped by several antiparallel β-strands of the junction domain, similar to the N-terminal capping of Fha30. The C-terminal coils of pertactin are reinforced by an aromatic cluster, a structural characteristic also observed in the first N-terminal coil of Fha30. Fha30 and pertactin thus display comparable architectures with inverse polarities that parallel the direction of their secretion and folding. The fact that TpsA and most AT proteins contain domains acting as scaffolds indicates that these two pathways most likely represent convergent solutions to the secretion of large virulence proteins with certain folding characteristics, in the present case, right-handed β-helices.
Supplementary Material
Acknowledgments
We thank H. Drobecq for the MS measurements and the BM30 beamline staff for support during data collection. B.C., F.J.D., and V.V. are researchers of the Centre National de la Recherche Scientifique. H.H. is supported by a joint predoctoral fellowship from the Institut Pasteur de Lille and the Région Nord-Pas de Calais. This work was supported in part by the Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, the Institut Pasteur de Lille, and the Région Nord-Pas-de Calais. V.V. is a recipient of an Action Thématique et Incitative sur Programme Jeunes Chercheurs grant from the Centre National de la Recherche Scientifique.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FHA, filamentous hemagglutinin; TPS, two-partner secretion; AT, autotransporter.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1RWR).
References
- 1.Henderson, I. R., Navarro-Garcia, F. & Nataro, J. P. (1998) Trends Microbiol. 6, 370-378. [DOI] [PubMed] [Google Scholar]
- 2.Jacob-Dubuisson, F., Locht, C. & Antoine, R. (2001) Mol. Microbiol. 40, 306-313. [DOI] [PubMed] [Google Scholar]
- 3.Yen, M.-R., Peabody, C. R., Partovi, S. M., Zhai, Y., Tseng, Y.-H. & Saier, M. H. (2002) Biochim. Biophys. Acta 1562, 6-31. [DOI] [PubMed] [Google Scholar]
- 4.Jacob-Dubuisson, F., Buisine, C., Willery, E., Renauld-Mongenie, G. & Locht, C. (1997) J. Bacteriol. 179, 775-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schonherr, R., Tsolis, R., Focareta, T. & Braun, V. (1993) Mol. Microbiol. 9, 1229-1237. [DOI] [PubMed] [Google Scholar]
- 6.Guédin, S., Willery, E., Locht, C. & Jacob-Dubuisson, F. (1998) Mol. Microbiol. 29, 763-774. [DOI] [PubMed] [Google Scholar]
- 7.Jacob-Dubuisson, F., El-Hamel, C., Saint, N., Guedin, S., Willery, E., Molle, G. & Locht, C. (1999) J. Biol. Chem. 274, 37731-37735. [DOI] [PubMed] [Google Scholar]
- 8.K önninger, U. W., Hobbie, S., Benz, R. & Braun, V. (1999) Mol. Microbiol. 32, 1212-1225. [DOI] [PubMed] [Google Scholar]
- 9.Coutte, L., Antoine, R., Drobecq, H., Locht, C. & Jacob-Dubuisson, F. (2001) EMBO J. 20, 5040-5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domenighini, M., Relman, D., Capiau, C., Falkow, S., Prugnola, A., Scarlato, V. & Rappuoli, R. (1990) Mol. Microbiol. 4, 787-800. [DOI] [PubMed] [Google Scholar]
- 11.Lambert-Buisine, C., Willery, E., Locht, C. & Jacob-Dubuisson, F. (1998) Mol. Microbiol. 28, 1283-1293. [DOI] [PubMed] [Google Scholar]
- 12.Locht, C., Bertin, P., Menozzi, F. D. & Renauld, G. (1993) Mol. Microbiol. 9, 653-660. [DOI] [PubMed] [Google Scholar]
- 13.Makhov, A. M., Hannah, J. H., Brennan, M. J., Trus, B. L., Kocsis, E., Conway, J. F., Wingfield, P. T., Simon, M. N. & Steven, A. C. (1994) J. Mol. Biol. 241, 110-124. [DOI] [PubMed] [Google Scholar]
- 14.Jacob-Dubuisson, F., Buisine, C., Mielcarek, N., Clement, E., Menozzi, F. D. & Locht, C. (1996) Mol. Microbiol. 19, 65-78. [DOI] [PubMed] [Google Scholar]
- 15.Renauld-Mongénie, G., Cornette, J., Mielcarek, N., Menozzi, F. D. & Locht, C. (1996) J. Bacteriol. 178, 1053-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabsch, W. (1993) J. Appl. Crystallogr. 26, 795-800. [Google Scholar]
- 17.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 18.Perrakis, A., Morris, R. & Lamzin, V. S. (1999) Nat. Struct. Biol. 6, 458-463. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project No. 4. (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 20.Roussel, A. & Cambillau, C. (1992) Biographics (Architecture et Fonction des Macromolécules Biologiques, Marseille, France).
- 21.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 22.Jenkins, J. & Pickersgill, R. (2001) Prog. Biophys. Mol. Biol. 77, 111-175. [DOI] [PubMed] [Google Scholar]
- 23.Yoder, M. D., Keen, N. T. & Jurnak, F. (1993) Science 260, 1503-1507. [DOI] [PubMed] [Google Scholar]
- 24.Hannah, J. H., Menozzi, F. D., Renauld, G., Locht, C. & Brennan, M. J. (1994) Infect. Immun. 62, 5010-5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uphoff, T. S. & Welch, R. A. (1994) in Molecular Mechanisms of Bacterial Virulence, eds. Kado, C. I. & Crosa, J. H. (Kluwer, Dordrecht, The Netherlands), pp. 283-292.
- 26.Kajava, A. V., Cheng, N., Cleaver, R., Kessel, M., Simon, M. N., Willery, E., Jacob-Dubuisson, F., Locht, C. & Steven, A. C. (2001) Mol. Microbiol. 42, 279-292. [DOI] [PubMed] [Google Scholar]
- 27.Locht, C., Antoine, R. & Jacob-Dubuisson, F. (2001) Curr. Opin. Microbiol. 4, 82-89. [DOI] [PubMed] [Google Scholar]
- 28.Grass, S. & St. Geme, J. W., III (2000) Mol. Microbiol. 36, 55-67. [DOI] [PubMed] [Google Scholar]
- 29.St. Geme, J. W., III, & Grass, S. (1998) Mol. Microbiol. 27, 617-630. [DOI] [PubMed] [Google Scholar]
- 30.Ohnishi, Y., Nishiyama, M., Horinouchi, S. & Beppu, T. (1994) J. Biol. Chem. 269, 32800-32806. [PubMed] [Google Scholar]
- 31.Oliver, D. C., Huang, G., Nodel, E., Pleasance, S. & Fernandez, R. C. (2003) Mol. Microbiol. 47, 1367-1383. [DOI] [PubMed] [Google Scholar]
- 32.Emsley, P., Charles, I. G., Fairweather, N. F. & Isaacs, N. W. (1996) Nature 381, 90-92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}