

Abstract
As part of our program to explore the influence of small structural modifications of our drug candidate, 3β-(hydroxy)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (galeterone, 5) on the modulation of the androgen receptor (AR), we have prepared and evaluated a series of novel C-3, C-16 and C-17 analogs. Using structure activity analysis, we established that the benzimidazole moiety at C-17 is essential and optimal and also that hydrophilic and heteroaromatic groups at C-3 enhance both anti-proliferative (AP) and AR degrading (ARD) activities. The most potent anti-proliferative compounds were 3β-(1H-imidazole-1-carboxylate)- 17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (47), 3-((EZ)-hydroximino)-17-(1Hbenzimidazol- 1-yl)-androsta-4,16-diene (36), 3β-(pyridine-4-carboxylate)-17-(1H-benzimidazol- 1-yl)-androsta-5,16-diene (43), with GI50 values of 0.87, 1.91 and 2.57 μM, respectively. Compared to 5, compound 47 was 4- and 8-fold more potent with respect to AP and ARD activities, respectively. Importantly, we also discovered that our compounds, including 5, 36, 43 and 47 could degrade both full-length and truncated AR in CWR22rv1 human prostate cancer cells. With these activities, their potential for development as new drugs for the treatment of all forms of prostate cancer.
Introduction
Compelling laboratory and clinical evidences strongly indicates that incurable castrationresistant prostate cancer (CRPC) remains dependent on functional androgen receptor (AR), ARmediated processes,1 and the availability of intra-prostatic intracellular androgens.2 Unlike early stage prostate cancer (ESPC), CRCP is not responsive to classical AR antagonist, (hydroxyflutamide (1) or bicalutamide (2); Figure 1) or androgen deprivation therapy (luteinizing hormone-releasing hormone agonists/antagonists). Therefore, recent strategies have focused on the development of more potent androgen synthesis inhibitors2d or AR antagonists.3 These research efforts have led to ongoing clinical evaluations/approvals of three potent CYP17 inhibitors, abiraterone acetate (Zytiga, 3a),4 TAK-700 (Orteronel, 4)5 and VN/124-1 (TOK-001 or galeterone, 5),1e, 2d, 6 and two potent AR antagonists, MDV3100 (enzalutamide, 6)7 and ARN- 509 (7).3a The chemical structures of these clinical compounds are presented in Figure 1.
Figure 1.

Chemical Structures of: compounds 1 – 7.
Despite the substantial clinical efficacy with 3a in patients with post-docetaxel CRPC,8 resistance to this therapy has already been reported.9 Resistance to 6 treatment has also been reported.10 Reactivation of AR signaling following compounds 3a or 6 treatments might occur by several mechanisms, prominent of which is a switching of transcription program under the control of AR signaling.11 Indeed, it may not be possible to inhibit the new AR-regulated transcription program by currently available therapies and some of the promising agents in clinical development. If so, substantial degradation of AR (full length and truncated forms) expression would be a promising strategy for future studies.
Although our clinical agent, 5 was originally designed as a CYP17 inhibitor, we have unequivocally established through several in vitro and in vivo experiments that it also has other important desirable anti-prostate cancer activities, acting as a potent anti-androgen and an AR degrading agent.6, 12 Because of our desire to invent more efficacious anti-prostate cancer agents, we were eager to exploit compound 5’s scaffold as a strategy to novel potent/efficacious AR degrading agents (ARDAs) with improved drug-like properties. Specifically, we conducted a systematic structure modification of compound 5 to see if we could obtain more potent ARDAs. Herein, we report that lead optimization of 5 gave rise to several novel compounds which exhibit the abilities to induce AR (full length and truncated) ablation at low micromolar concentrations and with improved anti-proliferative (AP) activities. This study expands our current understanding of the optimal pharmacophore requirements for AR degradation/down-regulator (ARD) activity and their capabilities in regulating the activity of the AR (i.e., AR inactivation). A preliminary account of part of this work has recently been reported.13
Results and discussion
Design strategy
Modifications that allow for additional interactions between a small molecule and receptor appear to play key determinants for designing new AR down-regulators with potential clinical use.14 Synthetic modifications of 5 were considered because the resulting fundamental chemical and physical changes may affect molecular shapes, bond angles, and partition coefficients. Different substituents can have different hydrophobic interactions, size, and electrostatic effects that can influence interaction of a ligand with its target receptor.15 These rational considerations provided the impetus for the systematic modifications of moieties tethered to C-17, C-16 and C-3 as described below.
C-17 modifications
To explore the structure activity relationship (SAR) of the C-17 benzimidazole moiety of 5, we synthesized analogs with varied ring nitrogen atoms, with increased aliphatic/aromatic hydrophobicity and with aromatic substituents to generate compounds 16–22 as outlined in Scheme 1.
Scheme 1.

Synthesis of C-17 benzimidazole compounds (5, 16–22)a
aReagents and conditions: (i) POCl3-DMF, CHCl3, Ar, reflux; (ii) benzimidazole or indole-3- carbaldehyde or 5,6-dimethylbenzimidazole or 5(6)-cyanobenzimidazole or 5(6)- methoxybenzimidazole or naphtho43 imidazole, or 2-chlorobenzimidazole, K2CO3, DMF, Ar, 80° C (iii) 10% Pd on activated charcoal, PhCN, reflux; (iv) 10% methanolic KOH, Ar, rt (2 – 3 h). (v) 6-chloropurine, TBAF, THF, Ar, 50° C; (vi) chlorotri(triphenylphosphine)rhodium[I], PhCH3, Ar, reflux.
C-16 modifications
On the basis of previous studies by Roy et al.16 of novel C-16 steroids that exhibit strong AR binding affinity and anti-androgenic activity, we designed and synthesized several C-16 substituted analogs (compounds 25, 28 and 31) of 5, tethered with bulky aliphatic and aromatic groups (Scheme 2).
Scheme 2.
Synthesis of C-16 substituted compounds (25, 28 and 31)a
aReagents and conditions: (i) substituted amines, molecular sieves, EtOH, Ar, reflux (3 – 7 h); (ii) MeOH, NaBH4, ice cold (2 h), rt (3 h); (iii) MeOH, 10% methanolic-KOH, Ar, rt (2 – 3 h);
C-3 modifications
Based on studies of DHT/testosterone interactions with AR, it is well established that the interaction to Arg752 occurs via C3 ketone.17 Arginine is a polar hydrophilic amino acid which contains a positively charged guanidine group. On the basis of the hypothesis that any substitution at C3 which increases interaction with Arg752 may increase AR downregulating activity, we designed and synthesized various C-3 modified compounds (33–49, Scheme 3).
Scheme 3.

Synthesis of C-3 modified compound (33 – 49)a
aReagents and conditions: (i) Al(i-PrO)3, 1-methyl-4-piperidone, toluene, reflux; (ii) Dess- Martin periodinane, MDC, 0° C, rt (5 h); (iii) Mesyl chloride or Tosyl chloride, Pyridine, ice cold (5 h), rt (5 h ); (iv) substituted hydroxylamine HCl, sodium acetate, MeOH, EtOH, Ar, reflux (2 – 3 h); (v) MeLi, THF, Ar, −60° C (1 h) rt (2 h); (vi) pyridine-carboxylic acid, 2- methyl-6-nitobenzoic anhydride, DMAP, TEA, THF, rt (1 h); (vi*) acid anhydride, DMAP, pyridine, reflux; (vii) 1,1′-carbonylbisimidazole or 1,1′-carbonylbis(2-methylimidazole) or 1,1′-carbonyl-di-(1,2,4-triazole), CH3CN, Ar, rt/reflux.
Chemistry
In this study, twenty six novel compounds are described and are based on the structures of our clinical candidate, compound 5, as outlined in Scheme 1 (for C-17 modified series), Scheme 2 (C-16 modified series), and Scheme 3 (C-3 modified series). The preparation of new 17-hetereoaryl substituted compounds (16–22) from the key intermediate, 3β-acetoxy-17- chloro-16-formylandtrosta-5,16-diene (13) followed the sequence: 17-heteroaryl-16-formyl intermediate → 16-deformylated intermediate → 3-deacetylated final product (not shown in Scheme 1), similar to the synthetic route to compound 5 outlined in Scheme 1. The key intermediate in our synthesis of all the compounds, 13, was prepared following our established procedure for Vilsmeier-Haack reaction of the commercially available 3β-acetoxyandrost-5-en- 17-one (12) with phosphoryl chloride (POCl3) and dimethylformamide (DMF) as previously reported.12a, 18 For the synthesis of 3β-acetoxy-16-formyl-17-1H-heteroaryls (14, 17a, 18a, 19a, 20a, and 22a), the corresponding heteroaryls were each treated with 13 in the presence of K2CO3 in DMF at approximately 80 °C to give the desired intermediates (structures of intermediates not shown except 14) in near quantitative yields. However, because of weak basicity of indole, we used indole-3-carbaldehyde instead for the synthesis of 17-indole-3-carbaldehyde (16a) intermediate following the same procedure with excellent yield. Attempts to condense 6- chloropurine with 13 in the presence of K2CO3 in DMF resulted in inseparable N9/N7 isomers (~ 6/4 ratio as indicated by TLC) in very low yield. Therefore, we adopted a reported N9-purine alkylation procedure,19 in which 13 was reacted with 6-chloropurine in presence of tetrabutylammonium fluoride (TBAF) in THF at 50 °C to give the desired intermediate (21a) in excellent yield. TLC analysis indicated that N7-purine alkylation was almost negligible and the N9-purine was easily purified following recrystallization in ethanol. The positional isomers of the 16-formyl derivatives (6-methoxy-BzIm 19a1 and 5-methoxy-BzIm 19a2) were separated at this stage and their structures were confirmed on the basis of reported aromatic proton resonances for related 5- and 6-methoxy benzyl compounds. Various attempts to separate positional isomers of 5(6) nitrile-benzimidazole intermediates of compound 18 at all stages were unsuccessful. The 5(6)-nitrile-benzimidazole and 2,3-diaminonaphthalene required for synthesis of 18a and 20a were synthesized by following reported procedure starting from 3,4-diaminobenzonitrile and benzo[f]benzimidazole respectively by refluxing with formic acid. 20 The 16-formyl intermediates (14, 17a – 21a; only structure of 14 shown) were each smoothly deformylated with 10% palladium on activated charcoal (Pd/C) in refluxing benzonitrile to give the corresponding deformylated compounds 15, 17b, 18b, 19b, 20b and 21b, respectively (structures not shown except 15) in high yields.12a Similarly, the two formyl groups of 17-indole-3-carbaldehyde intermediate (16a) were deformylated with 10% Pd/C as described above with good yield to give 16b. Deformylation of 22a was achieved by refluxing with readily available chlorotris(triphenylphosphine) rhodium(I) in toluene to give 22b in low yield.12a Unexpectedly, the 5-methoxy-16-formyl derivative 19a2 did not undergo deformylation using both methods. Hydrolysis of 15, 16b-22b with 10% methanolic-KOH gave target compounds 5, 16, 17, 18, 19, 20, 21 and 22, respectively in high yields.
The C-16 substituted compounds were synthesized starting from 14 as illustrated in Scheme 2. The intermediate imines 23, 26 and 29 were synthesized by refluxing i-pentylamine, aniline and 3,4-dimethoxyaniline, respectively with 14 in ethanol in presence of molecular sieves. Subsequent reduction of these imines with sodium borohydride (NaBH4) in ice-cold methanol21 gave 3-acetoxy-16-alkylamine intermediates 24, 27 and 30, respectively. Following hydrolysis of the 3β-acetoxy groups in compounds 24, 27 and 30, we obtained the desired 16- substituted compounds, 25, 28, and 31, respectively in excellent yields.
The C-3 modified compounds were synthesized as depicted in Scheme 3. Δ4-3-Oxo compound (32) was synthesized as we previously described via modified Oppenauer oxidation of 5 by using N-methylpiperidone and aluminum isopropoxide.12a Oxidation of 5 with Dess-Martin periodinane in dichloromethane (DCM) 22 afforded the Δ5-3-oxo compound 33 in 70% yield. The mesyl (34) and tosyl (35) derivatives of 5 were readily synthesized by reacting with methanesulfonyl and toluenesulfonyl chloride, respectively. The C-3 oxime derivatives (hydroxime: 36, phenyloxime: 37, methyloxime: 38 and benzyloxime: 39) were obtained by refluxing ketone (32) with the respective substituted hydroxylamine hydrochloride, using ethanol/methanol solvent mixture in presence of sodium acetate.23 Of all oximes, only biologically active oxime (36) was further purified to separate E- and Z- geometrical isomers by combined purification methods (column chromatography, preparative TLC, and recrystallization). Addition of MeLi to the C-3-keto group of 32 afforded two distereomeric (3α- and 3β) alcohols (40) which we did not separate due to modest biological activity.
The ester derivatives (41 – 46) of 5 were synthesized from 5 by two different methods as described below. The pyridinecarboxylates (41, 42 and 43) and carboxylate of 1,3-phenyldiacetic acid (44) of 5 were prepared using the mixed anhydride method via condensations with the respective anhydrides (pyridinecarboxylic acid/1,3-phenyldiacetic acid and 2-methyl-6- nitrobenzoic) in the presence of 4-dimethylaminopyridine (DMAP) and triethylamine (TEA)24 with varying yields (39–90%). The ester 45 (72% yield) and 46 (28% yield) were synthesized by refluxing 1,2,3,6-tetrahydropthalic and diglycolic anhydrides respectively with 5 in the presence of DMAP in pyridine.25 Finally the carbamates (imidazole: 47, 2-mehtylimidazole: 48 and 1,2,4- triazole: 49) were synthesized in modest to high yield (67–80%) by reacting 5 with 1,1- carbonylbis(2-methylimidazole) (CDI) and carbonylditriazole (CDT), respectively in acetonitrile and DMC solvent mixture.26 Except for the 3-mesyl (34), 5 and 32, all the compounds described in this study are novel and were rigorously characterized by physical and spectroscopic (IR, 1H and 13C NMR, and HRMS) analysis. Most of our novel compounds were then subjected to in vitro biological activity studies as described in detail in the following sections.
Biological Studies: Effects of compounds on transcriptional activation of androgen receptor in LNCaP cells
After synthesizing the compounds, we used a luciferase reporter assay to determine whether the novel compounds also affect AR transcriptional activation (screening assay). Specifically, we performed a luciferase experiment utilizing LNCaP cells dual transfected with the probasin luciferase reporter construct ARR2-luc and the Renilla luciferase reporting vector pRL-null as we previously described and reported in the methods section.6, 12a, 12d Luciferase expression was increased by approximately 100-fold after 10 nM DHT treatment for 24 hours. The ability of the novel compounds (10 μM) to affect DHT mediated AR transcription was assessed. Figure 3 shows the effects of our most potent compounds. These compounds were able to substantially inhibit DHT mediated transcription, with inhibition ranging from ~65–100% and the order of potency was 32, 5, 47 > 36 > 38 > 28 > 25 > 39 > 34.
Figure 3.

Effects of compounds at 10 μM on dihydrotestosterone (DHT)-stimulated transcription of AR. LNCaP cells were transfected with the ARR2 reporter construct + the Renilla luciferase reporting vector pRL-null and treated with novel compounds for 24 h in the presence of 10 nM dihydrotestosterone (DHT). Control, baseline activity without androgen stimulation. Androgen-stimulated luciferase activity (luminescence) was measured in a Victor 1420 plate reader. The results are presented as the fold induction (i.e., the relative luciferase activity of the treated cells divided by that of the control) normalized to that of the Renilla.
Androgen Receptor binding assays
In addition to AR down-regulation, we have previously shown that compound 5 reduces androgen action through inhibition of androgen binding and subsequently reduces AR mediated transcriptional activity. We used whole cell competitive binding assays with the synthetic ligand methyltrienolone (R1881, 9) to assess the AR binding affinities of our novel compounds in comparison to 5, and the FDA approved anti-androgens 2 and 6, and CYP17 inhibitor 3b as shown in Figure 4A. The compounds with the greatest ability to displace [3H]9 were 5 and 6, with EC50 values of 670 nM and 915 nM, respectively. Compound 2 was slightly weaker with an EC50 of 1.4 μM. We did not calculate the EC50 value of 3b because of the shallow steepness of the AR binding curve, a phenomenon which indicates interaction of 3b with more than one receptor population.27 A recent study also noted unusual (shallow steepness of the AR binding curve) AR binding characteristics with 3b.28 Interestingly, AR-binding assays using MDA-MB-453 cell showed that 6 was not as potent as previously reported for assays using LNCaP cells transfected with wild type AR3a and was not significantly different from the binding affinity of 2. Specifically, the binding affinity data were as follows: 6 (EC50 = 49 nM) and 2 (EC50 = 31 nM).3a Our new compounds were not as potent as 5 at inhibiting androgen binding at the concentrations tested (Figure 4B). For example, compound 36 showed the strongest inhibition of [3H]9 binding of all the new compounds tested (~40%) at 10 μM. At 30 μM, 36 inhibited [3H]9 binding to by ~80%, while 43 inhibited by ~53%. Unexpectedly, our most effective AR antagonist, 47, did not strongly compete for the AR binding site, exhibiting only 20% displacement at a 30 μM concentration. It is relevant to state here that other investigators have recently reported the discovery of small-molecule androgen receptor down-regulators and anti-androgens that bind weakly to the AR.29
Figure 4.

Figure 4A. Competitive inhibition of [3H]R1881 binding of compounds 2, 3b, 5, 6 and 36 to AR in LNCaP cells. Error bars, SD; n = 3.
Figure 4B. Competitive inhibition of [3H]R1881 binding of compounds 5, 16, 36, 43 and 47 to AR in LNCaP cells. Error bars, SD; n = 3.
Effects of compounds on AR degradation, transactivation and anti-proliferative activity
To explore the effects of our compounds on AR degradation, LNCaP cells were treated with each of the compounds (5, 6, 16–20, 25, 28, 32, 34, 36, 38, 39, 42, 43, 47–49) of interest for 24 h followed by western blot analysis. As shown in Figures 5A-C most of the new compounds significantly caused AR degradation in LNCaP cells, with compound 47 being the most potent and proved to be greater than 8-fold more active than compound 5 at 15 μM. In contrast, we note that a few compounds (16, 17, 20 and 49) caused significant up-regulation of AR, a phenomenon that will be investigated in future studies. The ability of compounds 5 and 47 to suppress protein AR expression was further demonstrated by immunocytochemical analysis (Figure 5D). As shown, exposure of LNCaP cells to 5 μM of compounds 5 and 47 for 48 h led to significant decrease in AR levels in the nucleus, in a fashion that mimics the western blot analysis data (vide supra). These data are similar to those reported for analogs of ciglitazone, a novel class of ARablative agents.15c
Figure 5.

Figure 5 A–E. Differential effect of compounds on suppressing AR expression in LNCaP and CWR22Rv1 prostate cancer cells: (A – C) Western blot analysis of AR expression in LNCaP cells treated with various compounds. Cells were exposed to individual compounds (15 μM) for 24 h, and the protein lysates were subjected to Western blot analysis. (D) Immunocytochemical analysis of the effect of 5 μM each of compounds 5 and 47 on suppressing AR expression in LNCaP cells after 48 h of exposure. Cells were stained with an-AR x-terminal antibody (green). The nuclear counterstaining was achieved using 4′, 6-diamino-2-phenylindole (DAPI). (E) Effects of compounds on the expression of fAR and tAR-3 in CWR22Rv1 cells. Cells were exposed to individual compounds (15 μM) for 24 h, and the protein lysates were subjected to Western blot analysis. Western blots were done either in duplicate or triplicate. The numbers below the blots represent respective densitometry intensities.
Because of the reported implication of AR splice variants lacking the ligand-binding domain (truncated AR) in driving the progression of CRPC,30 we next determined the effects of our compounds on the down-regulation of AR-3 (also called AR-V7).30c, d As shown in Figure 5E, we observed that compound 5 and some of our new compounds, 31, 32, 36 and 47 caused significant down-regulation of both full-length and truncated AR in CWR22rv1 prostate cancer cell line. Interestingly, we found that AR-3 was more susceptible to our compounds than the full- length AR in this cell line. In contrast, compound 6 did not affect the expression levels of either full-length or splice variant forms of AR. It is important to state here that a number of natural products31 and related analogs32 have been shown to degrade both full-length and truncated AR in several human prostate cancer cell lines. However, except for the curcumin analog, ASC-J9 that possesses excellent drug-like properties,32 most of these compounds are poor drug candidates because of modest potencies and/or toxic nature. Irrespective of how our compounds and others cause degradation of both forms of AR, such unique AR depleting agents if adequately developed may be more effective against CRPC than agents that obligatorily bind to specific region(s) of AR to elicit inactivation of AR.33
To determine whether AR degradation or AR transcriptional deactivation (AR inactivation) was contributing to the anti-proliferative activity, we treated the LNCaP cells with 15μM of selected active compounds (5, 36, 32, 47 and 48,) for 24 hours and performed cell viability, AR transcriptional (luciferase) assay and AR western blot analysis. As shown in Figure 6 the degradation of AR and inhibition of AR mediated transcription occurs before cell growth inhibition which suggest that compound-induced AR inactivation contributes to their anti-proliferative activities. These compounds also induced significant PARP cleavage in LNCaP and CWR22rv1 cells which suggest their abilities to induce apoptosis (data not shown).
Figure 6.
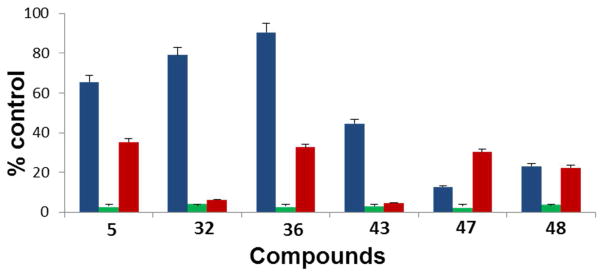
Effects of compounds 5, 32, 36, 43, 47 and 48 on: i) cell viability (blue); ii) DHTinduced AR transactivation (green); and iii) AR protein expression following treatment with 15 μM of each compound for 24 h using LNCaP cells. Error bars, SD; n = 3.
CYP17 (17α-hydroxylase activity) Inhibition Studies
A few compounds were evaluated for their ability to inhibit CYP17 enzyme. The assay was kindly performed by Dr. Emily Scott and colleagues according to their recently reported procedure in which a truncated version of human CYP17A1 (CYP171dH) was expressed in E. coli and then purified to homogeneity.34 IC50 values of the compounds were determined from dose-response curves and are listed in Table 1. The IC50 values of abiraterone alcohol (3b, a CYP17 inhibitor recently approved for prostate cancer therapy), galaterone and 3β-hydroxy-17-(1H-imidazole-1-yl)androsta-5,16-diene (VN/85-1, structure not shown, believed to be the most potent CYP17 inhibitor12a, 18) were also determined in the same assay system for comparison (used as positive controls). As expected, these new compounds (16, 36, 43, 47 and 48) with IC50 values in the high micromolar range (93.7 – 258 μM) were weak inhibitors of CYP17, reinforcing the previously established structural requirements for potent steroidal CYP17 inhibitors, including, no tolerance of bulky moieties at C-3 and appropriately positioned C-17 heterocyclic heteroatom.18, 35 As expected, the wellestablished CYP17 inhibitors exhibited exquisite inhibition of the enzyme with IC50 values in the nanomolar range (Table 1).18, 35b
Table 1.
IC50 values of select compounds for inhibition of CYP17 (17α-hydroxylase activity)
| Compounds | IC50 (μM)a |
|---|---|
| 16 | 130 |
| 36 | 258 |
| 43 | 132 |
| 47 | 122 |
| 48 | 93.7 |
| For comparison | |
| 3b | 0.206 |
| 5 | 0.752 |
| VN/85-1 | 0.125 |
IC50 value is the concentration of inhibitor to inhibit the CYP17 enzyme activity by 50%, each in duplicate. IC50 values were each determined from dose-response curves.
Anti-proliferative (anti-cancer) and androgen receptor down-regulating activities: Structure activity relationships (SAR)
Because of our hypothesis that the extent of AR degradation induced by compound 5 and possibly the new analogs would correlate with their ability to inhibit proliferation of prostate cancer cells (LNCaP), we assessed these two activities using Western blot analyses and MTT assays.
C-17 modification
Initially we synthesized and tested indole, 16 to assess the effect of decreased polarity at C17 position, due to absence of N-3 of BzIm ring. Unexpectedly, the compound caused up-regulation of AR (Figure 5A) and completely lost anticancer activity (GI50 >100 μM, Table 2) in comparison to lead compound 5 (GI50 = 3.35 μM). Increasing the number of nitrogen C-17 heterocycle by substituting with 6-chloropurine (21), caused a 4-fold reduction in antiproliferative activity (GI50 = 13.48 μM). Introducing cyano group (18) displayed potent antiproliferative activity (GI50 = 2.81 μM), but with diminished AR down-regulation (ARD) activity. Introduction of aliphatic hydrophobicity on BzIm ring by substituting methyl group on 5, 6 position (17) resulted into substantial loss of anti-proliferative (GI50 = 42.72 μM) and ARD activities, whereas substituting mono methoxy group (19) at 6th position of BzIm ring displayed no modulation of ARDA or anticancer activity (GI50 = 4.26 μM). Increasing aromatic hydrophobicity by replacing BzIm with naphtho[2,3-d]imidazole ring (20) caused significant loss of ARDA and anticancer activity (GI50 = 19.10 μM). Substituting 2-chloro BzIm (22) caused a 3-fold loss in anti-proliferative activity. None of the C17 modified molecules were superior to our lead compound 5, and this clearly indicates that the BzIm ring at C17 position of lead 5 is essential and optimal for ARDA and anti-proliferative activity.
Table 2.
GI50 values of C-17 modified compounds
| Compounds | GI50 (μM)a |
|---|---|
| 3b | 1.97 |
| 5 | 3.35 |
| 6 | 5.12 |
| 16 | >100 |
| 17 | 47.72 |
| 18 | 2.81 |
| 19 | 4.26 |
| 20 | 37.10 |
| 21 | 13.48 |
| 22 | 10.13 |
The GI50 were determined from dose-response curves (by nonlinear regression analysis using GraphPad Prism) compiled from at least three independent experiments, SEM < 10%, and represents the compound concentration required to inhibit cell growth by 50%.
C-16 modification
Our strategy to increase bulk at C16 position by tethering aliphatic hydrophobic groups (isopentyl: 25); aromatic (benzyl: 28; dimetoxybenzyl: 31) resulted in significant loss of ARD and anticancer activities (GI50s = 18.31, 22.13 and >100 μM, respectively; Table 3).
Table 3.
GI50 values of C-16 modified compounds
| Compounds | GI50 (μM)a |
|---|---|
| 25 | 18.31 |
| 28 | 22.13 |
| 31 | >100 |
The GI50 were determined from dose-response curves (by nonlinear regression analysis using GraphPad Prism) compiled from at least three independent experiments using LNCaP cells, SEM < 10%, and represents the compound concentration required to inhibit cell growth by 50%.
C-3 modification
In an attempt to better understand the role played by OH and O in the ARD/anti-proliferative activities of compounds 5 and 32, and to possibly achieve enhanced interaction with Arg in the AR ligand biding domain, we designed, synthesized and tested a number of C-3 modified analogs. First, oxidation of 5 or reductive alkylation of 32 to give 3- oxo-Δ5 compound, 33 and 3-hydroxy-3-methyl compound, 40, respectively, lead to significant loss (~5-fold) in anti-proliferative activity (Table 4). Conversion of compound 5 to the mesyl (34) and tosyl (35) derivatives also gave compounds with mediocre anti-proliferative activities, with GI50 values of 42.13 and 47.18 μM, respectively. On the contrary, introduction of oxime moieties at C-3 yielded compounds (E/Z oxime mixtures) with similar or better activities compared to compounds 5 and 32. Thus, the simple oxime (36), and the related methyl- (38) and benzyl- (39) analogs exhibited GI50 values of 1.91, 3.38 and 5.57 μM, respectively. We could not assess the biological activities of the phenyl oxime (37) because of its limited solubility in ethanol or DMSO. Considering the promising and superior activity of E/Z mixture of oximes 36, and the possibility that the pure E and Z had different anti-proliferative activities, we were surprised that 36E and 36Z isomers exhibited similar potencies, with GI50 values of 2.03 and 1.95 μM, respectively.
Table 4.
GI50 values of C-3 modified compounds
| Compounds | GI50 (μM)a |
|---|---|
| 32 | 2.64 |
| 33 | 15.96 |
| 34 | 42.13 |
| 35 | 47.18 |
| 36 | 1.91 |
| 36E | 2.03 |
| 36Z | 1.95 |
| 37 | NTb |
| 38 | 3.38 |
| 39 | 5. 57 |
| 40 | 13.34 |
| 41 | NTb |
| 42 | NTb |
| 43 | 2.57 |
| 44 | 7.78 |
| 45 | 8.22 |
| 46 | 9.13 |
| 47 | 0.87 |
| 48 | 5.34 |
| 49 | 6.67 |
The GI50 were determined from dose-response curves (by nonlinear regression analysis using GraphPad Prism) compiled from at least three independent experiments, SEM < 10%, and represents the compound concentration required to inhibit cell growth by 50%.
Not tested due to insolubility in ethanol.
On the basis of known ester based anticancer drugs, such as docetaxel, cabazitaxel36 and esters in clinical development such as bevirimat and related analogs,25 we first synthesized three pyridinecarboxylate derivatives of compound 5, including 41–43. Of these compounds, the isonicotinoyl derivative 43 exhibited similar anti-proliferative activity (GI50 = 2.57 μM) as 5. Here again, we could not assess the biological activities of compounds 41 and 42 because of their limited solubilities in ethanol or DMSO. The related analogs tethered to lipophilic ester side chain with a carboxylic acid terminus (44–46) exhibited potencies ~2.5-fold worse than compound 5. Finally, we considered evaluation of C-3 carbamates because of: 1) precedence of drugs with carbamate moieties such as the widely use anthielmintics albendazole, fenbendazole and mebendazole;37 2) the added feature of lowering the lipohilicity of compound 5, which should also increase solubilities and perhaps physiological relevance.38 Of the three heteroaryl carbamates tested, the imidazoly carbamate 47 with a GI50 value of 0.87 μM was shown to be the most active, being ~4-fold superior to compound 5. Introduction of 21-methyl as in carbamate 48 caused a 6-fold decrease in activity relative to 47, similar to ~8-fold decrease in activity
Concluding Remarks
Our study has shown that potent anti-proliferative (AP) and AR downregulating (ARD) activities can be retained in compound 5 by modification of the 3β-OH to appropriate carbamate (47). We also establish that the C-17 benzimidazole group is essential and optimal for both AP and ARD activities and substituents tethered to C-16 are not tolerated. A summary of the in vitro structure-activity relationship (SAR) of these steroidal compounds as androgen receptor degrading agents (ARDAs) is presented in Figure 7. Importantly; we show that binding affinity to the LBD of AR is not essential for potent AP/ARD activities and that some of our compounds also exhibit exquisite depletion of both full-length and splice variant ARs. The significance of these novel findings has important implications because our novel compounds, including galeterone currently in phase 2 clinical trials in prostate cancer patients can prevent androgen receptor activation by any known means. Although we are yet to conduct in vivo anti-prostate tumor efficacy studies with our carbamate compound 47, on the basis of the strong promising data presented in this study and also that the carbamate moiety plays an important role in medicinal chemistry, being found in many drugs as well as prodrugs,25, 36, 39 we strongly believe that 47 is a strong candidate for further development as a potential drug for the treatment of all forms of prostate cancer in humans. Consequently, in vivo anti-tumor efficacy evaluation of compound 47 and other promising compounds in castration resistant models of prostate cancer are in progress.
Figure 7.

In vitro SAR of 3,16/17-substituted steroid derivatives as androgen receptor degrading agents..
Experimental section
Chemistry
Melting points (mp) were determined with a Fischer-Johns melting point apparatus and are uncorrected. Proton magnetic resonance spectra (1H NMR) spectra were recorded in CDCl3 or DMSO-d6 at 500 or 400 MHz with Me4Si as an internal standard using a Varian Inova 500 or Bruker 400 MHz spectrometers. 13C NMR spectra were recorded in CDCl3 using Bruker 400 or 500 MHz spectrometers. High-resolution mass spectra (HRMS) were determined on a Bruker 12Tesla APEX-Qe FTICR-MS by positive ion ESI mode by Ms. Susan A. Hatcher, Facility Director, College of Sciences Major Instrumentation Cluster, Old Dominion University, Norfolk, VA. Epiandrosterone acetate, and all other chemicals, reagents were purchased from Sigma–Aldrich. Dihydrotestosterone (DHT) used in the biological experiments was synthesized following our recently reported procedure.40 Tritiated [3H]R1881 was purchased from Perkin Elmer LAS., while MDV3100 was purchased from Sequoiq Resrach Products Ltd., Pangbourne, UK. Compounds 3a and 3b were synthesized in our lab. All compounds were stored in the cold (0–8 °C). Silica gel plates (Merck F254) were used for thin-layer chromatography, while flash column chromatography (FCC) was performed on silica gel (230–400 mesh, 60 Å). The preparative TLC performed on Silica gel GF (Analtec 500 microns) plates. Pet ether refers to light petroleum, b.p. 40–60 °C. The purity of all final compounds was determined to be at least 95% pure by a combination of HPLC, NMR and HRMS.
3β-Acetoxy-17-chloro-16-formylandrosta-5,16-diene (13)
This compound prepared from 3β-acetoxyandrost-5-en-17-one (epinadrosterone acetate, 12) as previously described, provided spectral and analytical data as reported.18
General method A: Synthesis of 3β-Acetoxy-17-(1H-heteroaryl-1-yl)-16-formylandrosta- 5,16-diene (14, 16a-18a, 19a1, 19a2, 20a, and 22a)12a
A 25 mL RB flask equipped with a magnetic stir bar and condenser was charged with 3β-acetoxy-17-chloro-16-formylandrosta- 5,16-diene (13, 0.38 g, 1 mmol), corresponding heteroaryl (3 mmol) and K2CO3 (0.41 g, 3 mmol) in dry DMF (~7.5 mL) was stirred at 80 °C under Ar and monitored by TLC. After cooling to room temperature, the reaction mixture was poured onto ice-cold water (50 mL) and the resulting precipitate was filtered, washed with water and dried to give crude product. Purification by the FCC [petroleum ether/EtOAc/TEA (6:4:0.3)] gave the desired pure compounds. Above listed intermediate compounds were synthesized (using reactants, reagent and solvent ratio), isolated and purified by using this method unless otherwise stated.
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (14)
Compound 14 prepared by following general Method A, reacting 13 (2.5 g, 6.65 mmol) with benzimidazole (2.35 g, 19.9 mmol) in presence of K2CO3 (2.76 g, 19.9 mmol) in dry DMF at 80 °C for 1.5 h. Followed by FCC purification provided pure 14 with identical spectral and analytical data as we previously reported.12a
3β-Acetoxy-17-(3-formyl-1H-indol-1-yl)-16-formylandrosta-5,16-diene (16a)
Compound 16a prepared by following general method A, reacting 13 (1 g, 2.66 mmol) with indole-3- carbaldehyde (0.5 g, 3.44 mmol) in presence of K2CO3 (0.5 g, 3.62 mmol) in dry DMF (15 mL) at 80 °C for 8 h. Purification by FCC [petroleum ether/EtOAc (7:3)] gave 1.1 g (85%) of pure 16a: mp 206–208 °C; IR (Neat) 2935, 2852, 1729, 1665, 1635, 1453, 1374, 1239, 1032, 783 cm− 1; 1H NMR (500 MHz, CDCl3) δ 1.01 (s, 3 H, 18-CH3), 1.09 (s, 3 H, 19-CH3), 2.06 (s, 3 H, 3β-OCOCH3), 4.65 (dt, J = 12.2, 6.5 Hz, 1 H, 3 α-H), 5.46 (br, 1 H, 6-H), 7.29 (s, 1 H, 2′-H), 7.39 (m, 2 H, aromatic-Hs), 7.80 (d, J = 14.9 Hz, 1 H, aromatic-H), 8.36 (m, 1 H, aromatic-H), 9.58 (br, 1 H, 16-CHO) and 10.15 (s, 1 H, indole-CHO).
3β-Acetoxy-17-(5, 6-dimethyl-1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (17a)
Compound 17a prepared by following general method A, reacting 13 (0.5 g, 1.33 mmol) with 5,6-dimethylbenzimidazole (0.54 g, 4.0 mmol) in presence of K2CO3 (0.55 g, 4.0 mmol) in dry DMF (10 mL) at 80 °C for 5 h. Purification by FCC gave 0.46 g (70.7%) of pure 17a: mp 174–175 °C; IR (Neat) 2941, 2852, 1727, 1672, 1622, 1463, 1487, 1365, 1236, 1029, 897, 843, 717, 657 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.06 (s, 3 H, 18-CH3), 1.16 (br. s, 3 H, 19-CH3), 2.03 (s, 3 H, 3β-OCOCH3), 2.35 (s, 3 H, aromatic-CH3 ) 2.38 (s, 3 H, aromatic-CH3), 4.64 (m, 1 H, 3 α-H), 5.44 (br, 1 H, 6-H), 7.02 (br. s, 1 H, aromatic-Hs), 7.59 (s, 1 H, aromatic-H), 7.87 (s, 1 H, 2′-H) and 9.60 (s, 1 H, 16-CHO).
3β-Acetoxy-17-(5(6)-nitrile-1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (18a)
Compound 18a prepared by following general method A, reacting 13 (0.5 g, 1.33 mmol) with 5(6)-nitrilebenzimidazole20 (0.38 g, 2.65 mmol) in presence of K2CO3 (0.55 g, 4.0 mmol) in dry DMF (10 mL) at 80 °C for 5 h. Purification by short column [petroleum ether/EtOAc/TEA (6:4:0.1)] gave 0.28 g (43.5%) of pure 18a: mp 146–147 °C; IR (Neat) 2935, 2226, 1726, 1673, 1470, 1238 1032, 906, 728 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.07 (s, 3 H, 18-CH3), 1.19 (br. s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 4.62 (dt, J = 10.1, 5.3 Hz, 1 H, 3 α-H), 5.44 (br, 1 H, 6-H), 7.61 – 7.96 (m, 3 H, aromatic-H), 8.21 (s, 1 H, 2′-H) and 9.52 (s, 1 H, 16-CHO).
3β-Acetoxy-17-(6-methoxy-1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (19a1) and 3β-Acetoxy-17-(5-methoxy-1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (19a2)
Compound 19a1 and 19a2 prepared by following general method A, reacting 13 (0.5 g, 1.33 mmol) with 5(6)-methoxybenzimidazole (0.59 g, 4.0 mmol) in presence of K2CO3 (0.55 g, 4.0 mmol) in dry DMF (10 mL) at 80 °C for 3 h. Purification by FCC [petroleum ether/EtOAc/TEA (7.5:2:0.5)] gave first less polar 6-methoxy derivative (19a1) 0.15 g (24%): mp 242–245 °C; IR (Neat) 2935, 1721, 1673, 1502, 1440, 1249, 1220, 1032, 805, 759 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.07 (s, 3 H, 18-CH3), 1.18 (br. s, 3 H, 19-CH3), 2.03 (s, 3 H, 3β-OCOCH3), 3.82 (s, 3 H, -OCH3), 4.62 (dt, J = 11.2, 6.6 Hz, 1 H, 3 α-H), 5.44 (t, 1 H, J = 1.84 Hz, 6-H), 6.70 (m, 1 H, aromatic-H) 6.95 (m, 1 H, aromatic-H), 7.70 (m, 1 H, aromatic-H), 7.87 (s, 1 H, 2′-H) and 9.61 (s, 1 H, 16-CHO).
Subsequently more polar 5-methoxy derivative (19a2) 0.13 g (20%): mp 228–231 °C; IR (Neat) 2936, 2852, 1722, 1673, 1481, 1341, 1245, 1031, 897, 800, 739 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.06 (s, 3 H, 18-CH3), 1.16 (br. s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 3.88 (s, 3 H, -OCH3), 4.63 (m, 1 H, 3 α-H), 5.44 (d, J = 5.6 Hz, 1 H, 6-H), 6.98 (m, 1 H, aromatic-H) 7.29 (m, 1 H, aromatic-H), 7.30 (m, 1 H, aromatic-H), 7.92 (s, 1 H, 2′-H) and 9.61 (s, 1 H, 16-CHO). About 0.11 g of mixture of 19a1 and 19a2 also collected (overall yield is 61%)
3β-Acetoxy-17-(1H-benzo[f]benzimidazol-1-yl)-16-formylandrosta-5,16-diene (20a)
Compound 20a prepared by following general method A, reacting 13 (0.38 g, 1 mmol) with 1Hbenzo[ f]benzimidazole (0.2 g, 1.2 mmol) in presence of K2CO3 (0.207g, 1.5 mmol) in dry DMF (3 mL) at 80 °C for 2 h. Purification by FCC [petroleum ether/EtOAc/TEA (6:4:0.3)] gave 0.37 g (72%) of pure compound 20a: mp 158–160 °C; IR (CHCl3) 3691, 3024, 2951, 2359, 1725, 1670, 1604, 1491, 1452, 1375, 1253, 1032, 897, 852, 818, 700, 657, 618, 576, 565, 550, 529, 511, 476 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 6H, 18 and 19-CH3), 2.04 (s, 3 H, 3α-OCH3), 4.62 (m, 1 H, 3β-H), 5.44 (br, s, 6-H) 7.46 (br. s, 2 H, aromatic-H), 7.94 (s, 2 H, aromatic-H), 8.04 (m, 1 H, aromatic-H), 8.15 (s, 1 H, aromatic-H) 8.33 (s, 1 H, 2′-H) and 9.71 (s, 1 H, 16-CHO).
3β-Acetoxy-17-(6-Chloro-9H-purin-9-yl)-16-formylandrosta-5,16-diene (21a):19
A mixture of 13 (2.43 g, 6.46 mmol), 6-chloropurine (0.5 g, 3.23 mmol) and TBAF (1.69 g, 6.46) in dry THF (40 mL) was stirred at 50 °C under Ar for 48 h. After cooling to room temperature, the reaction mixture concentrated and poured onto ice-cold water (250 mL) and the resulting precipitate was filtered, washed with water and dried to give a crude product. Purification by FCC [DCM/Methanol (9.7:0.3)] and then recrystallized with hot ethanol to give 0.82 g (51.3%) of pure 21a: mp 140–142 °C; IR (Neat) 2943, 2853, 1729, 1672, 1584, 1556, 1435, 1236, 1032, 939, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.07 (s, 3 H, 18-CH3), 1.09 (s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 4.61 (m, 1 H, 3 α-H), 5.43 (br, 1 H, 6-H), 8.20 (s, 1 H, 2′-H), 8.79 (s, 1 H, aromatic-H), and 9.53 (s, 1 H, 16-CHO).
3β-Acetoxy-17-(2-chloro-1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (22a)
Compound 22a prepared by following general method A, reacting 13 (0.5 g, 1.33 mmol) with 2- chlorobenzimidazole (0.6 g, 4.0 mmol) in presence of K2CO3 (0.55 g, 4.0 mmol) in dry DMF (10 mL) at 80° C for 50 h. After cooling to room temperature, the reaction mixture was poured onto ice-cold water (250 mL) and the resulting emulsion was extracted with DCM, organic layer dried and evaporated. Purification by FCC [petroleum ether/EtOAc (8:2)] gave 0.27 g (41.1%) of pure 22a: mp 203 °C; IR (Neat) 2936, 1731, 1679, 1448, 1244, 1033, 734 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.06 (s, 3 H, 18-CH3), 1.16 (s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 4.62 (m, 1 H, 3 α-H), 5.43 (br, 1 H, 6-H), 7.17 (d, 1 H, J = 7.9 Hz, aromatic-H), 7.34 (m, 2 H, aromatic-Hs), 7.74 (d, 1 H, J = 7.4 Hz, aromatic-H) and 9.37 (s, 1 H, 16-CHO).
General method B: Synthesis of 3β-Acetoxy-17-(1H-heteroaryl-1-yl)-androsta-5,16-diene (15, 16b-21b)
A solution of 3β-Acetoxy-17-(1H-heteroaryl-1-yl)-16-formylandrosta-5,16-diene (14, 17a-21a) in dry benzonitrile (10 mL) was refluxed in the presence of 10% Pd/C (50% weight of reactant) under Ar and monitored by TLC. After cooling to room temperature, the catalyst was removed by filtration through a Celite pad. The filtrate was evaporated, and the residue was purified by FCC on silica gel, using petroleum ether/EtOAc/TEA (7.5:3:0.5) solvent system. Above listed intermediate compounds were synthesized (using reactants, reagent and solvent ratio), isolated and purified by using this method unless otherwise stated.
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (15)
Compound 15 prepared by refluxing 14 (2.04 g, 4.45 mmol), with 10% Pd/C (1.0 g) in dry benzonitrile (10 mL) for 5 h. Followed by FCC purification provided pure 15 with identical spectral and analytical data as we previously reported.12a
3β-Acetoxy-17-(1H-indol-1-yl)-androsta-5,16-diene (16b)
Compound 16b prepared by following general method B, refluxing 16a (0.17 g, 0.36 mmol), with 10% Pd/C (0.085 g) in dry benzonitrile (3 mL) for 24 h, then about 0.030 g of Pd/C and solvent (1 mL) added and further refluxed for 12 h. Purification by FCC gave 0.12 g (77.5%) of pure 16b: mp 182–185 °C; IR (Neat) 2936, 2854, 1727, 1631, 1455, 1368, 1249, 1030, 721, cm−1; 1H NMR (400 MHz, DMSOd6) δ 0.95 (s, 3 H, 18-CH3), 1.03 (s, 3 H, 19-CH3), 1.99 (s, 3 H, 3β-OCOCH3), 4.47 (m, 1 H, 3α-H), 5.42 (br, 1 H, 6-H), 5.88 (s, 1 H, 16-H), 6.57 (m, 1 H, 3′-H), 7.05 (m, 1 H, 2′-H), 7.15 (m, 1 H, aromatic-H), 7.37 (d, J = 3.2 Hz, 1 H, aromatic-H), 7.50 (d, J = 8.0 Hz, 1 H, aromatic-H), and 7.57 (d, J = 7.7 Hz, 1 H, aromatic-H).
3β-Acetoxy-17-(5,6-dimethyl-1H-benzimidazol-1-yl)-androsta-5,16-diene (17b)
Compound 17b prepared by following general method B, refluxing 17a (0.15 g, 0.308 mmol), with 10% Pd/C (0.075 g) in dry benzonitrile (2 mL) for 7 h. Purification by FCC gave 0.12 g (84.8%) of pure 17b: mp 159–162 °C; IR (Neat) 2926, 2852, 1729, 1626, 1491, 1462, 1369, 1236, 1030, 846, cm-−1; 1H NMR (400 MHz, CDCl3) δ1.03 (s, 3 H, 18-CH3), 1.09 (s, 3 H, 19-CH3), 2.06 (s, 3 H, 3β-OCOCH3), 2.40 (s, 6H, 2 X aromatic-CH3), 4.64 (m, 1 H, 3α-H), 5.45 (br, 1 H, 6-H), 5.96 (s, 1 H, 16-H), 7.26 (s, 1 H, aromatic-H), 7.58 (s, 1 H, aromatic-H), and 7.87 (s, 1 H, 2′-H).
3β-Acetoxy-17-(5(6)-nitrile-1H-benzimidazol-1-yl)-androsta-5,16-diene (18b)
Compound 18b prepared by following general method B, refluxing 18a (0.15 g, 0.31 mmol) 10% Pd/C (0.075 g) in dry benzonitrile (2 mL) for 24 h. Purification by FCC gave 0.09 g (63.5%) of pure 18b: mp 204–206 °C; IR (Neat) 2939, 2222, 1731, 1487, 1247, 1030, 822, cm−1; 1H NMR (400 MHz, CDCl3) δ1.07 (s, 3 H, 18-CH3), 1.18 (s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 4.62 (m, 1 H, 3α-H), 5.44 (m, 1 H, 6-H), 6.03 (m, 1 H, 16-H), 7.54 – 8.15 (m, 4 H, aromatic-H).
3β-Acetoxy-17-(6-methoxy-1H-benzimidazol-1-yl)-androsta-5,16-diene (19b)
Compound 19b prepared by following general method B, refluxing 19a1 (0.15 g, 0.307 mmol), with 10% Pd/C (0.075 g) in dry benzonitrile (2 mL) for 72 h, then about 0.030 g of Pd/C added and further refluxed for 12 h. Purification by FCC gave 0.05 g (35%) of pure sticky compound 19b: IR (Neat) 2940, 1713, 1496, 1363, 1237, 1216, 1030, 816, cm−1; 1H NMR (400 MHz, CDCl3) δ1.01 (s, 3 H, 18-CH3), 1.07 (s, 3 H, 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 3.88 (s, 3 H, -OCH3), 4.63 (m, 1 H, 3α-H), 5.44 (s, 1 H, 6-H), 5.96 (br, 1 H, 16-H), 6.92 (m, 2 H, aromatic-Hs), 7.69 (d, 1 H, J = 8.7 Hz, aromatic-H), and 7.85 (s, 1 H, 2′-H).
3β-Acetoxy-17-(1H-benzo[f]benzimidazol-1-yl)-androsta-5,16-diene (20b)
Compound 20b prepared by following general method B, refluxing 20a (0.2 g, 4.45 mmol), with 10% Pd/C (0.1 g) in dry benzonitrile (4 mL) for 5 h. Purification by FCC gave 0.14 g (73.8%) of pure 20b: mp 144–146 °C; IR (CHCl3) 3687, 2947, 2854, 2358, 2340, 1725, 1633, 1609, 1557, 1489, 1454, 1373, 1291,1253, 1195, 1136, 1031, 985, 910, 839, 735, 665, 590, 544, 533,513, 502, 488 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.08 (s, 3 H,18-CH3), 1.10 (s, 3 H, 19-CH3), 2.01 (s, 3 H, 3β- OCH3), 4.62 (m,1H, 3α-H), 5.45 (br,s,6-H), 6.11 (s, 1 H, 16-H), 7.42 (m, 2 H, aromatic-Hs), 7.92 (m, 2 H, aromatic-H), 8.04 (m, 1 H, aromatic-H), 8.15 (s, 1 H, aromatic-H) and 8.29 (s, 1 H, 2′-H).
3β-Acetoxy-17-(6-Chloro-9H-purin-9-yl)-androsta-5,16-diene (21b)
Compound 21b prepared by following general method B, refluxing 21a (0.4 g, 0.81 mmol), with 10% Pd/C (0.4 g, i.e., equal weight of 21a) in dry benzonitrile (7.5 mL) for 4 h. Cooled to room temperature, the catalyst was removed by filtration through a Celite pad. The filtrate was evaporated, and carried to next step without purification.
3β-Acetoxy-17-(2-chloro-1H-benzimidazol-1-yl)-androsta-5,16-diene (22b)
A solution of 3β- Acetoxy-17-(2-chlorobenzimidazol-1-yl)-16-formylandrosta-5,16-diene (22a), (0.15 g, 0.304 mmol) in dry toluene (3 mL) was refluxed in the presence of chlorotris (triphenylphosphine) rhodium (I) (0.29 g, 0.311 mmol) for 60 h. After cooling to room temperature, the catalyst was removed by filtration through a Celite pad. The filtrate was evaporated, and the residue was purified by FCC [petroleum ether/EtOAc (8:2)] to give 0.04 g (28%) of pure 22b: mp 161–165 °C; IR (Neat) 2926, 2853, 1629, 1403, 1462, 1369, 1233, 1035, 847 cm−1; 1H NMR (500 MHz, CDCl3) δ1.05 (d, 6H, 18 and 19-CH3), 2.04 (s, 3 H, 3β-OCOCH3), 4.62 (m, 1 H, 3α-H), 5.44 (m, 1 H, 6-H), 6.06 (s, 1 H, 16-H), 7.33 (m, 1 H, aromatic-H), 7.52 (m, 1 H, aromatic-H), and 7.68 (m, 2 H, aromatic-H).
General method C: Synthesis of 3β-Hydroxy-17-(1H-heteroaryl-1-yl)-androsta-5,16-diene (5, 16–22) and 3β-Hydoxy-17-(1H-benzimidazol-1-yl)-16-((alkyl/arylamino)methyl)- androsta-5,16-diene (25, 28 and 31)
The acetate (15, 16b-22b, 24, 27, 30) (1 g) was dissolved in methanol (15 mL) under an inert Ar atmosphere, and the resulting solution was treated with 10% methanolic KOH (5 mL). The mixture was stirred at room temperature, monitored by TLC. Reaction mixture concentrated under vacuum, ice water (100 mL) added, and the resulting white precipitate was filtered, washed with water and dried. FCC on a short silica gel column, eluting with petroleum ether/EtOAc (6:4) to obtain pure target compounds. Above listed final compounds were synthesized (using reactants, reagent and solvent ratio), isolated and purified by using this method unless otherwise stated.
3β-Hydroxy-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (5)12a
Compound 5 prepared by following general method C, treating acetate solution of 15 (1 g 3.02 mmol) in methanol (15 mL) with 10% methanolic KOH (5 mL) for 1.5 h. Purification by FCC over short column provided pure 5 with identical spectral and analytical data as we previously reported.
3β-Hydroxy-17-(1H-indol-1-yl)-androsta-5,16-diene (16)
Compound 16 prepared by slightly modifying general method C. The acetate solution of 16b (0.09 g 0.2 mmol) in methanol (1.5 mL) was refluxed with 10% methanolic KOH (1 mL) for 3 h. Purification by FCC over short column short column gave pure 16 (0.076 g, 98.7%), mp 142–145 °C; IR (Neat) 3305, 2931, 2836, 1625, 1455, 1327, 1225, 10598, 1042, 740 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.00 (s, 3 H, 18-CH3), 1.06 (s, 3 H, 19-CH3), 3.54 (m, 1 H, 3α-H), 5.41 (br, 1 H, 6-H), 5.85 (s, 1 H, 16-H), 6.55 (m, 1 H, 3′-H), 7.11 (m, 1 H, 2′-H), 7.19 (dd, J = 8.4, 5.7 Hz, 2 H, aromatic-Hs), 7.51 (d, 1 H, J = 8.3 Hz, aromatic-H), and 7.60 (d, 1 H, J = 7.8 Hz, aromatic-H); 13C NMR (500 MHz, CDCl3) δ 149.6, 141.2, 137.2, 128.4, 126.9, 122.0, 121.7, 120.6, 119.6, 111.3, 102.4, 71.7, 55.9, 50.6, 47.3, 42.0, 37.2, 36.8, 35.1, 31.6, 30.2, 20.8, 19.4, 16.0; HRMS calcd 410.2454 (C27H33ON.Na+), found 410.2460.
3β-Hydroxy-17-(5, 6-dimethyl-1H-benzimidazol-1-yl)-androsta-5,16-diene (17)
Compound 17 prepared by following general method C by treating acetate solution of 17b (0.1 g 0.22 mmol) in methanol (2 mL) with 10% methanolic KOH (1 mL) for 3 h. Purification by FCC over short column provided pure 17 (0.05 g, 55%), mp 194–196 °C; IR (Neat) 3262, 2925, 2896, 2848, 1628, 1493, 1481, 1371, 1058, 834, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.02 (s, 3 H, 18-CH3), 1.06 (s, 3 H, 19-CH3), 2.38 (s, 6H, 2 x aromatic-CH3), 3.55 (m, 1 H, 3α-H), 5.41 (m, 1 H, 6-H), 5.95 (t, J = 2.6 Hz, 16-H), 7.25 (s, 1 H, aromatic-H), 7.57 (s, 1 H, aromatic-H), and 7.87 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 147.3, 141.3, 132.7, 131.6, 123.4, 121.1, 119.9, 111.3, 71.6, 55.9, 50.5, 47.2, 42.3, 37.2, 34.9, 31.6, 30.37, 20.6, 19.3, 16.0; HRMS calcd 439.2719 (C28H36ON2.Na+), found 439.2726.
3β-Hydroxy-17-(5(6)-nitrile-1H-benzimidazol-1-yl)-androsta-5,16-diene (18)
Compound 18 prepared according to general method C by treating acetate solution of 18b (0.075 g 0.165 mmol) in methanol (1.5 mL) with 10% methanolic KOH (1 mL) for 2 h. Purification by FCC over short column provided pure 18 (0.055 g, 80.8%), mp 192–193 °C; IR (Neat) 3409, 3285, 2928, 2226, 1654, 1614, 1469, 1229, 1059, 801, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.01 (d, 3 H, 18-CH3), 1.06 (d, 3 H, 19-CH3), 3.55 (tdq, J = 9.0, 4.7, 2.6 Hz, 1 H, 3α-H), 5.40 (dp, J = 4.8, 1.7 Hz, 6-H), 6.02 (m, 1 H, 16-H), 7.52–8.15 (m, 4 H, aromatic-H); 13C NMR (500 MHz, CDCl3) δ 146.7, 144.8, 141.5, 127.0, 126.4, 125.5, 121.5, 119.8, 116.4, 112.4, 106.8, 106.1, 71.7, 56.1, 50.6, 47.5, 42.4, 37.3, 36.9, 34.9, 31.7, 30.6, 20.8, 19.5, 16.2, 15.0; HRMS calcd 414.2539 (C27H31ON3.H+), found 414.2532.
3β-Hydroxy-17-(6-methoxy-1H-benzimidazol-1-yl)-androsta-5,16-diene (19)
Compound 19 prepared according to general method C by treating acetate solution of 19b (0.05 g 0.11 mmol) in methanol (1 mL) with 10% methanolic KOH (1 mL) for 3 h. Purification by FCC over short column provided pure 19 (0.03 g, 55%), mp 169–179 °C; IR (Neat) 3339, 2933, 1614, 1501, 1450, 1283, 1068, 906, 813, 728 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.01 (s, 3 H, 18-CH3), 1.06 (s, 3 H, 19-CH3), 3.58 (m, 1 H, 3α-H), 3.86 (s, 3 H, -OCH3), 5.41 (t, 1 H, J = 2.42 Hz, 6-H), 5.95 (t, 1 H, J = 1.48 Hz,16-H), 6.92 (m, 2 H, aromatic-H), 7.67 (m, 1 H, aromatic-H), and 7.58 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 157.32, 147.6, 141.5, 137.9, 135.4, 124.0, 121.2, 120.7, 111.6, 95.2, 71.7, 56.2, 50.7, 47.5, 42.5, 37.4, 35.1, 31.8, 30.6, 20.9, 19.5, 16.2; HRMS calcd 441.2512 (C27H34O2N2.Na+), found 441.2507.
3β-Hydroxy-17-(1H-benzo[f]benzimidazol-1-yl)-androsta-5,16-diene (20)
Compound 20 prepared according to general method C by treating acetate solution of 20b (0.1 g, 0.32 mmol) in methanol (5 mL) with 10% methanolic KOH (1 mL) for 1.5 h. Purification by crystallization from EtOAc/Methanol gave 20 (0.075 g, 74%), mp 150–152 °C; IR (CHCl3) 2934, 2339, 1609, 1490, 1453, 1291, 1040, 837, 808, 705, 663, 608, 578, 550, 517 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.09 (s, 6H, 18 and 19-CH3), 3.57 (m, 1 H, 3α-H), 5.44 (br, s, 6-H), 6.13 (s,1 H, 16-H), 7.44 (m, 2 H, aromatic-Hs), 7.94 (m, 2 H, aromatic-H), 8.03 (m, 1 H, aromatic-H), 8.18 (s, 1 H, aromatic-H) and 8.31 (s, 1 H, 2′-H). HRMS calcd 461.2563 (C30H34N2O.Na+), found 461.2570.
3β-Hydroxy-17-(6-Chloro-9H-purin-9-yl)-androsta-5,16-diene (21)
Compound 21 prepared according to general method C by treating acetate solution of 21b (0.04 g 0.085 mmol) in methanol (1 mL) with 10% methanolic KOH (1 mL) for 3 h. Purification by FCC over short column [DCM/methanol/TEA (9.7:0.3:0.05)] to obtain pure 21 (0.03 g, 82.6%), mp 272–274 °C; IR (Neat) 3385, 2928, 2604, 2498, 1664, 1516, 1433, 1346, 1040, 805, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.12 (s, 3 H, 18-CH3), 1.23 (s, 3 H, 19-CH3), 3.50 (m, 1 H, 3α-H), 5.41 (br, 1 H, 6-H), 5.59 (s, 1 H, 16-H), 8.11 (s, 1 H, 2′-H), 8.40 (s, 1 H, aromatic-H); 13C NMR (500 MHz, CDCl3) δ 164.1, 153.4, 141.6, 139.4, 121.9, 120.8, 71.2, 56.3, 53.1, 50.1, 47.0, 46.0, 36.9, 31.2, 19.5, 15.0, 11.7, 8.9, 8.8; HRMS calcd 871.3952 (C24H29ClON4)2.Na+, found 871.3972
3β-Hydroxy-17-(2-chloro-1H-benzimidazol-1-yl)-androsta-5,16-diene (22)
Compound 22 prepared according to general method C by treating acetate solution of 22b (0.03 g 0.064 mmol) in methanol (0.75 mL) with 10% methanolic KOH (1 mL) for 3 h. Purification by FCC over short column [petroleum ether/EtOAc (7:3)] to obtain pure 22 (0.025 g, 91.6%), mp 83–86 °C; IR (Neat) 3346, 2929, 1449, 1267, 1121, 1071, 1040, 742 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.05 (br, 6H, 18 and 19-CH3), 3.54 (m, 1 H, 3α-H), 5.41 (br, 1 H, 6-H), 6.04 (m, 1 H, 16-H), 7.25 (m, 1 H, aromatic-H), 7.31 (m, 1 H, aromatic-H), and 7.68 (m, 2 H, aromatic-H); 13C NMR (500 MHz, CDCl3) δ 141.5, 133.2, 129.9, 123.3, 121.2, 111.5, 71.9, 55.9, 50.8, 42.5, 38.9, 37.3, 37.0, 34.0, 31.8, 30.6, 24.0, 23.2, 20.73, 19.5, 17.3, 16.4; HRMS calcd 445.2017 (C28H36ON2.Na+), found 445.2020.
General method D: Synthesis of 3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16- ((alkyl/arylimino)methyl)-androsta-5,16-diene (23, 26 and 29)
The title compounds were prepared by refluxing a solution of 3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-formylandrosta- 5,16-diene (14) (1 equivalent), corresponding primary amine (2 equivalent), molecular sieves (~25% weight of 14) and ethanol under Ar for 3–12 h. Reaction mixture was filtered, concentrated under vacuum, residue stirred with water and resulting crude product filtered. Purification by the FCC on silica gel column [petroleum ether/EtOAc (1:1)] gave the desired pure compounds. Above listed compounds were synthesized (using reactants, reagent and solvent ratio), isolated and purified by using this method unless otherwise stated.
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-((EZ)-(isopentylimino)methyl)-androsta-5,16- diene (23)
Compound 23 prepared by following general method D, refluxing 14 (0.4 g, 0.87 mmol), isopentylamine (0.15 g, 1.7 mmol), molecular sieves (0.2 g) in ethanol (5 mL) for 3 hours. Followed purification by FCC gave 0.41 g (89%) 23: mp sinters at 135 °C, melts at 145°C; IR (Neat) 2934, 2851, 1726, 1676, 1640, 1490, 1453, 1247, 1219, 1032, 744 cm−1; 1H NMR (400MHz, CDCl3) δ 0.87 (d, 6H, aliphatic-CH3), 1.07 (s, 3 H, 18-CH3), 1.16 (s, 3 H,19-CH3), 2.06 (s, 3 H, 3β-OCOCH3), 4.64 (m, 1 H, 3 α-H), 5.46 (br. s, 1 H, 6-H), 7.30 (s, 1 H, imine-CH), 7.34 (m, 2 H, aromatic-Hs), 7.72 (s, 1 H, aromatic-H), 7.87 (s, 1 H, aromatic-H), and 7.94 (s, 1 H, 2′-H).
General method E: Synthesis of 3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16- ((alkyl/arylamino)methyl)-androsta-5,16-diene (24, 27 and 30)
To ice cold solution of 16- enamines (23/ 26/30) (1 mole equivalent) in methanol added NaBH4 (0.5 mole equivalent) in three portions over 30 minutes. Reaction continued for 1.5–5 h then neutralized with acetic acid, evaporated, residue treated with water and filtered. Crude product carried to next step without purification.
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-((isopentylamino)methyl)-androsta-5,16-diene (24)
Compound 24 prepared by following general method E, reacting 23 (0.1 g, 0.2 mmol) in methanol (1.5 mL) with NaBH4 (0.0035 g, 0.09 mmol) at °C for 1.5 h. The crude product 24 (0.09 g, 89%) was carried to next step without purification.
3β-Hydoxy-17-(1H-benzimidazol-1-yl)-16-((isopentylamino)methyl)-androsta-5,16-diene (25)
Compound 25 prepared by following general method C, treating methanolic solution (1 mL) of crude acetate 24 (0.08 g 0.15 mmol) with 10% methanolic KOH (0.75 mL) for 3h. Followed purification by passing through short silica bed [DCM/ethanol (9.5:0.5)] to give 25 (0.065 g, 88%), mp 111–113 °C; IR (Neat) 3281, 2927, 2850, 1487, 1454, 1374, 1224, 1061, 1007, 765, cm−1; 1H NMR (500 MHz, CDCl3) δ 0.81 (d, 6H, alphatic-CH3), 1.04 (s, 6H, 18, 19- CH3), 3.55 (m, 1 H, 3α-H), 5.41 (br, 1 H, 6-H), 7.19–7.43 (m, 3 H, aromatic-Hs), 7.75–7.82 (m, 1 H, aromatic-H), and 8.1 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 142.8, 140.0, 134.8, 123.4, 122.4, 120.2, 110.8, 71.5, 55.9, 50.7, 48.9, 42.3, 38.9, 36.8, 34.6,, 32.4, 31.6, 30.3, 26.0, 22.6, 20.5, 19.3, 16.0, 15.8; HRMS calcd 510.3454 (C32H45ON3.Na+), found 510.34509
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-((EZ)-(phenylimino)methyl)-androsta-5,16-diene (26)
Compound 26 prepared by following synthetic method D, refluxing 14 (0.15 g, 0.33 mmol), aniline (0.06 g, 0.65 mmol), molecular sieves (0.04 g) in ethanol (2 mL) for 3 h. Purification by passing through a silica bed gave 0.15 g (85.9%) 26: mp sinters at 85–90 °C, melts at 125°C; IR (Neat) 2973, 2932, 2822, 1727, 1635, 1589, 1486, 1453, 1239, 1219, 1029, 764 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 3 H, 18-CH3) 1.23 (s, 3 H, 19-CH3), 2.06 (s, 3 H, 3β-OCOCH3), 4.65 (m, 1 H, 3α-H), 5.49 (br, 1 H, 6-H), 6.96 (m, 2 H, aromatic-Hs) 7.17 (m, 1 H, aromatic-H) 7.26 (s, 1 H, imine-CH), 7.35 (m, 4 H, aromatic-Hs), 7.87 (m, 1 H, aromatic-H), 7.94 (m, 1 H, aromatic-H) and 7.99 (s, 1 H, 2′-H).
3β-Acetoxy-17-(1H-benzimidazol-1-yl)-16-((phenylamino)methyl)-androsta-5,16-diene (27)
Compound 27 prepared by following General synthetic method E, reacting 26 (0.1 g, 0.19 mmol) in methanol (1.5 mL) with NaBH4 (0.0035 g, 0.09 mmol) at °C for 1.5 h. The crude 27 carried to next step without purification.
3β-Hydoxy-17-(1H-benzimidazol-1-yl)-16-((phenylamino)methyl)-androsta-5,16-diene (28)
Compound 28 prepared by following General method C, treating methanolic solution (1mL) of crude acetate 27 with 10% methanolic KOH (0.75 mL) for 3 h. Followed purification by passing through short silica bed [DCM/ethanol (9.5:0.5)] gave 28 (0.08 g, 86%), mp 130–132 °C; IR (Neat) 3329, 2928, 2852, 1602, 1418, 1375, 1217, 1058, 1007, 833, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.04 (s, 3 H, 19-CH3), 3.54 (m, 1 H, 3α-H), 3.65 (br. s, 2 H, - CH2), 5.38 (t, 1 H, J = 2.62 Hz, 6-H), 6.40 (t, 2 H, J = 8.8 Hz, aromatic-Hs), 6.69 (d, 1 H, J = 7.3 Hz, aromatic-H), 7.08 (m, 2 H, aromatic-Hs), 7.20–7.33 (m, 3 H, aromatic-Hs), 7.74–7.84 (m, 1 H, aromatic-H), and 7.79 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 147.6, 141.3, 138.7, 123.7, 122.5, 129.9, 120.4, 118.0, 113.0, 110.8, 71.6, 54.7, 50.6, 48.0, 42.2, 36.8, 34.4, 32.4, 31.1, 30.3, 20.5, 19.3, 15.8. HRMS calcd 516.2985 (C33H39ON3.Na+), found 516.2981
3β-Acetoxy-17-(1H-benzimidazol-1-yl)- 16-((EZ)-((3,4-dimethoxyphenyl)imino)methyl)- androsta-5,16-diene (29)
Compound 29 prepared by following general method D, refluxing 14 (0.3 g, 0.65 mmol), 3,4-dimethoxy aniline (0.2 g, 1.3 mmol), molecular sieves (0.075 g) in ethanol (2 mL) for overnight. Purification by FCC [petroleum ether/EtOAc (1:1)] gave 0.29 g (74.5%) 29: mp sinters at 115 °C, melts at 130°C; IR (Neat) 2937, 2904, 2852, 1729, 1586, 1509, 1451, 1372, 1233, 1125, 1026, 765 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 3 H, 18-CH3) 1.23 (s, 3 H, 19-CH3), 2.06 (s, 3 H, 3β-OCOCH3), 3.84 (m, 6H, 2 X OCH3), 4.64 (m, 1 H, 3α-H), 5.48 (br. s, 1 H, 6-H), 6.56 (m, 2 H, aromatic-Hs) 6.73 (m, 1 H, aromatic-H) 7.36 (m, 3 H, aromatic-2Hs and imine-CH), 7.88 (m, 1 H, aromatic-H), 7.95 (m, 1 H, aromatic-H), and 8.00 (s, 1 H, 21-H).
3β-Acetoxy-17-(1H-benzimidazol-1-yl)- 16-(((3,4-dimethoxyphenyl)amino)methyl)- androsta-5,16-diene (30)
Compound 30 prepared by following General synthetic method E, reacting 29 (0.15 g, 0.25 mmol) in methanol (2.5 mL) with NaBH4 (0.05 g, 0.126 mmol) at °C for 5 h. The crude 30 carried to next step without purification.
3β-Hydoxy-17-(1H-benzimidazol-1-yl)- 16-(((3,4-dimethoxyphenyl)amino)methyl)- androsta-5,16-diene (31)
Compound 31 prepared by following method C, treating methanolic solution of (2 mL) of crude acetate 30 with 10% methanolic KOH (0.75 mL). Subsequent purification by FCC [DCM/ethanol (9.7: 0.3)] to give 31 (0.11 g, 79.6%), mp sinters at 120 °C melts 135 °C; IR (Neat) 3351, 2929, 2852, 1612, 1514, 1454, 1229, 1136, 1025, 765, cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.09 (s, 3 H, 19-CH3), 3.53 (m, 1 H, 3α-H), 3.61 (br, 2 H, N-CH2), 3.74–3.77 (s, 6H, 2 X OCH3), 5.37 (br, 1 H, 6-H), 5.95 (br, 1 H, aromatic- 1″-H), 6.04 (d, J = 2.6 Hz, 1 H, aromatic-5″-H), 6.64 (br, 1 H, aromatic-6″-H), 7.21–7.31 (m, 3 H, aromatic-Hs), 7.74–7.83 (m, 1 H, aromatic-H), and 7.79 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 149.9, 142.2, 138.8, 123.7, 122.5, 120.9, 112.9, 110.3, 103.8, 99.4, 71.5, 56.6, 55.7, 50.6. 48.3, 42.8, 4.1, 34.7, 32.2, 31.1, 30.0, 20.5, 19.3, 15.8. HRMS calcd 576.3196 (C35H43O3N3.Na+), found 576.3188.
17-(1H-Benzimidazol-1-yl)-androsta-4,16-dien-3-one (32)
This compound prepared from 5 as previously described, provided spectral and analytical data as reported.12a,13C NMR (500 MHz, CDCl3) δ 199.4, 170.5, 147.2, 143.5, 141.1, 134.7, 124.3, 124.3, 123.5, 122.6, 122.5, 111.3, 54.3, 54.2, 47.4, 38.9, 35.9, 35.8, 34.1, 33.8, 32.8, 31.4, 30.4, 17.5, 17.3, 16.3.
17-(1H-Benzimidazol-1-yl)-androsta-5,16-dien-3-one (33)
To a ice cold solution of 5 (0.05 g, 0.13 mmol) in dry DCM (3 mL) was added Dess-Martin periodinane (0.11 g, 0.26 mmol) and the mixture was stirred at ice cold temperature for 5 h. Then it was diluted with ether and was quenched with a mixture of saturated aqueous NaHCO3/Na2S2O3 (1:3). The organic layer was washed with brine and dried over Na2SO4, then solvent was evaporated under vacuum and the crude product was purified by FCC [DCM/ethanol/TEA (30:1:0.05)] to give the title compound 33 (0.035 g, 70%): mp 170–172 °C; IR (Neat) 2941, 1711, 1491, 1451, 1226, 751 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 3 H, 18-CH3), 1.24 (s, 3 H, 19-CH3), 5.41 (t, 1 H, J = 2.5 Hz, 6-H), 5.99 (br, 1 H, 16-H), 7.30 (m, 2 H, aromatic-Hs), 7.49 (d, J = 6.9 Hz, 1 H, aromatic-H), 7.81 (m, 1 H, aromatic-H), and 7.96 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 209.9, 147.3, 143.5, 139.2, 134.8, 124.3, 123.5, 122.8, 122.6, 122.0, 120.5, 111.3, 56.0, 49.9, 49.7, 47.5, 37.74, 37.4, 37.0, 31.3, 31.1, 30.4, 19.3, 19.2, 16.8, 16.2. HRMS calcd 409.2250 (C26H30ON2.Na+), found 409.2258.
3β-Mesyloxy-17-(1H-benzimidazol-1-yl)-androsta-5,16-dien (34)
To ice cold solution of 5 (0.4 g, 1.03 mmol) in pyridine (5 ml), was added methanesulfonyl chloride (0.68 g, 6 mmol). Reaction mixture stirred at 0° C for 5 h, then room temperature for 8 h and quenched to 75 ml ice-water mixture. The resulting yellow solid was, filtered, washed, dried and the crude product was purified by FCC [DCM/ethanol (1.5%)] to give the title compound 34 (0.4 g, 83%), mp 177- 179 °C (lit.6 149–150 °C); IR (Neat) 2944, 1486, 1452, 1326, 1170, 938, 765 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.09 (s, 3 H, 19-CH3), 3.03 (s, 3 H, mesyl-Hs), 4.56 (m, 1 H, 3α-H), 5.49 (br, 1 H, 6-H), 6.0 (m, 1 H, 16-H), 7.30 (m, 2 H, aromatic-Hs), 7.49 (m, 1 H, aromatic-H), 7.82 (m, 1 H, aromatic-H), and 7.97 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 147.1, 143.3, 141.6, 139.1, 134.6, 123.4, 120.2, 81.6, 55.7, 50.3, 47.2, 39.2, 36.8, 34.8, 31.1, 28.9, 20.6, 19.1, 16.0. HRMS calcd 955.4472 (C26H30ON2)2Na+, found 955.4468.
3β-Tosyloxy-17-(1H-benzimidazol-1-yl)-androsta-5,16-dien (35)
To a cold (0° C) solution of 5 (0.1 g, 0.26 mmol) in pyridine (3 ml), was added tosyl chloride (0.06 g, 0.31 mmol). Reaction mixture stirred at 0° C for 5 h, then room temperature for 3 h and quenched to 30 ml ice-water mixture. The resulting yellow solid was filtered, washed, dried and the crude product was purified by FCC [DCM/Ethanol (1.0%)]. Resulting sticky solid was dissolved in 1.5 ml of EtOAc and about 10 ml of petroleum ether added slowly with stirring, the resulting turbid solution stirred at room temperature for 30 min, to give free flowing solid of title compound 35 (0.115 g, 84.5%), mp 139–141 °C; IR (Neat) 2948, 2850, 1490, 1451, 1329, 1171, 917, 740 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.99 (s, 3 H, 18-CH3), 1.01 (s, 3 H, 19-CH3), 2.44 (s, 3 H, 4″-CH3), 4.35 (m, 1 H, 3α-H), 5.37 (m, 1 H, 6-H), 5.97 (m, 1 H, 16-H), 7.25–7.34 (m, 3 H, aromatic- Hs), 7.35–7.37 (m, 2 H, 2″, 6″-Hs), 7.48 (m, 1 H, aromatic-H), 7.79 (m, 3 H, aromatic-H and 3″, 5″-H ), and 7.95 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 147.0, 144.5, 141.6, 139.3, 134.6, 129.8, 127.6, 123.5, 122.5, 120.6, 111.1, 82.1, 55.7, 50.3, 47.2, 38.9, 36.8, 34.8, 30.3, 28.5, 21.7, 20.57, 19.1. HRMS calcd 565.2495 (C33H38O3N2S.Na+), found 565.2506.
General method F: Synthesis of 3-(Substituted-oximino)-17-(1H-Benzimidazol-1-yl)- androsta-4,16-diene (36–39)
To a refluxing solution of ketone 32 (1 mole equivalent) in ethanol-methanol (2:1) solvent mixture, add a solution of sodium acetate (9.4 mole equivalent), corresponding substituted-oxamine hydrochloride (10.5 mole equivalent) in distilled water (10 mole equivalent). Reflux continued for 2–3 h, then concentrated, residue treated with water and crude product filtered. Purification FCC over silica using 5% ethanolic DCM gave pure oximes.
3-((EZ)-Hydroximino)-17-(1H-Benzimidazol-1-yl)-androsta-4,16-diene (36)
Compound 36 prepared by following general method F. To a refluxing solution of 32 (0.08 g, 0.194 mmol) in ethanol-methanol (2 mL) added a solution of sodium acetate (0.15 g, 1.83 mmol), hydroxylamine. HCl (0.07 g, 2.04 mmol) in 0.75 ml distilled water. The reflux continued for 2 h and subsequent purification by FCC gave compound (mixture of EZ isomers) 36 (0.06 g, 77%): mp sinters at 145 °C, melts 155–160 °C; IR (Neat) 3181, 2929, 2853, 1609, 1453, 1226, 847 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.02 (s, 3 H, 18-CH3), 1.11–1.15 (s, 3 H, 19-CH3), 5.81 and 6.52 (~57% and 33% for E and Z isomers respectively) of (s, 1 H, 4-H), 5.95 (br, 1 H, 16-H), 7.30 (m, 2 H, aromatic- Hs), 7.47 (m, 1 H, aromatic-H), 7.81 (m, 1 H, aromatic-H), and 7.95 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 158.64, 156.6, 154.5, 147.0, 142.9, 134.5, 124.3, 122.6, 117.8, 111.2, 55.3, 54.2, 47.3, 38.1, 34.6, 32.8, 30.3, 24.6, 20.9, 18.7, 17.9, 16.1. HRMS calcd 424.2359 (C26H31ON3.Na+), found 424.2363.
Separation of E and Z isomers of 36
Initially EZ mixture was purified by FCC using petroleum ether and EtOAc (1:1) mixture. This provided better purity of individual isomers with slight contamination of each in one another. The major product 36E was further purified by crystallization with hot EtOAc which resulted into pure single isomer 36E: mp 218–221 °C; 1H NMR (400 MHz, CDCl3) ™ppm 1.02 (s, 3 H, 18-CH3), 1.11 (s, 3 H, 19-CH3), 5.85 (s, 1 H, 4-H), 5.98 (s, 1 H, 16-H), 7.28 – 7.36 (m, 2 H, aromatic-Hs), 7.44 – 7.55 (m, 1 H, aromatic-H), 7.79 – 7.88 (m, 1 H, aromatic-H), 7.97 (s, 1 H, 2′-H), 9.04 (br. s., 1 H, -OH); 13C NMR (101 MHz, CDCl3) ™ ppm 156.7, 154.4, 147.1, 143.1, 141.6, 134.5, 124.1, 123.5, 122.5, 120.2, 117.9, 111.1, 55.3, 54.0, 47.3, 38.1, 34.8, 34.6, 34.2, 32.2, 31.5, 30.2, 21.1, 18.7, 17.6, 16.1. Where, 36Z was further purified by preparative TLC using petroleum-ether, EtOAc (1:1) as solvent system: mp 158–162 °C; 1H NMR (400 MHz, CDCl3) ™ ppm 1.02 (s, 3 H, 18-CH3), 1.15 (s, 3 H, 19-CH3), 5.97 (br s., 1 H, 16-H), 6.53 (s, 1 H, 4-H), 7.27 – 7.34 (m, 2 H, aromatic-Hs), 7.44 – 7.52 (m, 1 H, aromatic-H), 7.76 – 7.87 (m, 1 H, aromatic-H), 7.7 (s, 1 H, 2′-H), 8.87 (br. s., 1 H, -OH); 13C NMR (101 MHz, CDCl3) ™ ppm 158.5, 147.0, 143.1, 141.6, 134.5, 124.2, 123.5, 122.6, 120.2, 117.7, 111.1, 55.2, 54.2, 47.3, 39.0, 38.1, 36.1, 34.8, 34.2, 32.8, 31.8, 30.2, 24.7, 20.9, 17.9, 16.1.
3-((EZ)-O-Phenyloxime)-17-(1H-Benzimidazol-1-yl)-androsta-4,16-diene (37)
Compound 37 prepared by following general method F. To a refluxing solution of 32 (0.05g, 0.13 mmol) in ethanol-methanol (2ml) added a solution of sodium acetate (0.1 g, 1.22 mmol), phenoxamine. HCl (0.2 g, 1.35 mmol) in 0.5 ml distilled water. The reflux continued for 2 h and subsequent purification by FCC gave compound (mixture of EZ isomers) 37 (0.04 g, 64%): mp 96–98 °C; IR (Neat) 2935, 2854, 1627, 1590, 1487, 1216, 897 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 3 H, 18-CH3), 1.16–1.20 (s, 3 H, 19-CH3), 6.00 (s, 1 H, 4-H and 16-H), 6.00 and 6.67 (~55% and 45% for E and Z isomers respectively) (s, 1 H, 4-H), 7.01 (m, 1 H, aromatic-H), 7.22 (m, 2 H, aromatic-Hs), 7.32 (m, 4 H, aromatic-Hs), 7.52 (m, 1 H, Aromatic-H), 7.83 (m, 1 H, aromatic- Hs) and 7.97 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 160.6, 159.5, 158.0, 156.0, 147.1, 129.2, 124.2, 123.5, 121.7, 120.2, 117.4, 114.7, 111.2, 55.3, 55.0, 47.3, 38.2, 36.0, 34.1, 32.4, 30.2, 24.6, 21.0, 20.0, 17.6, 16.1. HRMS calcd 500.2672 (C32H35ON3.Na+), found 500.2677.
3-((EZ)-O-Methyloxime)-17-(1H-Benzimidazol-1-yl)-androsta-4,16-diene (38)
Compound 38 prepared by following general method F. To a refluxing solution of 32 (0.075g, 0.194 mmol) in ethanol-methanol (2 ml) added a solution of sodium acetate (0.15 g, 1.83 mmol), methoxyamine.HCl (0.17 g, 2.04 mmol) in 0.75 ml distilled water. The reflux continued for 3 h and subsequent purification by FCC gave compound (mixture of EZ isomers) 38 (0.072 g, 89%): mp 94–96 °C; IR (Neat) 2935, 2854, 1628, 1489, 1452, 1226, 1050, 743 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.04 (s, 3 H, 18-CH3), 1.11 (s, 3 H, 19-CH3), 3.89 (s, 3 H, OCH3), 5.83 and 6.44 (~69% and 31% for E and Z isomers respectively) (s, 1 H, 4-H), 6.03 (m, 1 H, 16-H), 7.35 (m, 2 H, aromatic-Hs), 7.53 (m, 1 H, aromatic-H), 7.87 (m, 1 H, aromatic-H), and 8.06 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 158.7, 156.0, 154.5, 153.1, 146.7, 125.2, 124.0, 123.3, 119.6, 117.7, 111.2, 61.6, 55.3, 54.2, 47.3, 38.0, 34.2, 32.2, 31.5, 30.3, 24.7, 21.0, 19.2, 17.6, 16.1. HRMS calcd 438.2515 (C27H33ON3.Na+), found 438.2520.
3-((EZ)-(O-Phenylmethyl)oxime)-17-(1H-Benzimidazol-1-yl)-androsta-4,16-diene (39)
Compound 39 prepared by following general method F. To a refluxing solution of 32 (0.075g, 0.194 mmol) in ethanol-methanol (2 ml) added a solution of sodium acetate (0.15 g, 1.83 mmol), benzyloxyamine.HCl (0.33 g, 2.04 mmol) in 0.75 ml distilled water. The reflux continued for 3 h and subsequent purification by FCC gave compound (mixture of EZ isomers) 39 (0.092 g, 96%) which solidifies on storage: mp sinters 66–68 °C, melts 77–79 °C; IR (Neat) 2935, 2854, 1627, 1609, 1489, 1452, 1225, 1015, 864 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.10 (s, 3 H, 19-CH3), 5.10 (s, 2 H, OCH2), 5.83 and 6.52 (~69% and 31% for E and Z isomers respectively) (s, 1 H, 4-H), 5.97 (s, 1 H, 16-H), 7.25 (br, 3 H, aromatic-Hs), 7.37 (m, 4 H, aromatic-Hs), 7.48 (m, 1 H, aromatic-H), 7.82 (m, 1 H, aromatic-H)and 7.95 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 156.4, 154.6, 153.5, 147.0, 138.1, 127.9, 122.8, 120.0, 117.8, 111.3, 55.4, 54.0, 47.3, 38.0, 34.6, 32.2, 30.3, 24.7, 21.0, 19.6, 17.9, 16.1. HRMS calcd 514.2828 (C33H37ON3.Na+), found 514.2834.
3-Methyl-3-hydroxy-17-(1H-benzimidazol-1-yl)-androsta-4,16-diene (40)
To a solution of ketone (32) (0.1 g, 0.26 mmol) in dry THF (3 mL) was added MeLi (1.6 M solution in ether, 0.41 mL, 0.60 mmol) at −60° C, and the resulting mixture was stirred at 0° C for 1 h then room temperature for 3 h. The reaction was quenched with saturated aqueous NH4Cl and was extracted with EtOAc. The organic layer was washed with brine and dried over Na2SO4, and the solvent was removed under vacuum. The residue was purified by short FCC [petroleum ether, EtOAc, TEA (60:40: 0.5)] to afford product 40 (0.05 g, 48%); mp 95–97 °C; IR (Neat) 3329, 2827, 2853, 1489, 1453, 1376, 1292, 1226, 1133, 918, 741 cm−1;1H NMR (500 MHz, CDCl3) δ 1.00 (s, 3 H, 18-CH3), 1.07 (s, 3 H, 19-CH3), 1.27 (s, 3 H, C3-CH3), 5.25 (t, J = 1.6 Hz, 1 H, 6-H), 5.96 (t, 1 H, J = 1.52 Hz, 16-H), 7.29 (m, 2 H, aromatic-Hs), 7.49 (m, 1 H, aromatic-H), 7.82 (dd, J = 7.0, 2.6 Hz, 1 H, aromatic- H), and 7.95 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 145.3, 127.6, 124.4, 123.6, 122.7, 120.4, 111.4, 70.1, 55.7, 54.8, 37.8, 35.6, 35.3, 34.7, 32.5, 30.4, 28.5, 21.1, 18.8, 16.3. HRMS calcd 425.2563 (C27H34ON2.Na+), found 425.2570.
General method G
Mixed anhydride method for the synthesis of aromatic/heteroaromatic esters (41–44): 2-Methyl-6-nitrobenzoic anhydride (0.39 mmol) was added to a solution of pyridinecaboxylic acid (0.386 mmol) and DMAP (0.29 mmol) in THF (1 ml), and the resulting mixture was allowed to stand at room temperature for 5min. A solution of 5 (0.193 mmol) in THF (1 ml) was mixed with the above reagent mixture and then with TEA (0.1 ml). This reaction mixture was allowed to stand at room temperature for 2 h. Reaction mixture absorbed on silica and purified by FCC using 2% ethanol in DCM in presence of traces of TEA (0.06%). The picolinoyl, nicotinoyl, isonoctinoyl and 1,3-phenyldiacetic acid esters derivatives were synthesized in a manner similar to the above. TLC and 1H NMR and HRMS analyses revealed that the presence of other esters derived from 2-methyl-6-nitrobenzoic anhydride is absent.
3β-(Pyridine-2-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (41)
Compound 41 prepared by following general method G, using 2-Methyl-6-nitrobenzoic anhydride (0.13 g, 0.39 mmol), picolinic acid (0.05 g, 0.39 mmol), 4-DMAP (0.04 g, 0.29 mmol), THF (1 ml), 5 (0.075 g, 0.19 mmol), THF (1 ml) and TEA (0.1 ml). FCC gave pure 41 (0.09 g, 90%): mp 243–44 °C; IR (Neat) 2942, 2852, 1729, 1496, 1286, 1227, 1139, 754 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.12 (s, 3 H, 19-CH3), 4.99 (m, 1 H, 3α-H), 5.49 (t, 1 H, J = 1.98 Hz, 6-H), 5.99 (t, 1 H, J = 1.42 Hz, 16-H), 7.32 (m, 2 H, aromatic-Hs), 7.46–7.50 (m, 2 H, picolinoyl-5-H and aromatic-H), 7.80–7.84 (m, 1 H, aromatic- H), and (1H, picolinoyl-4-H), 7.96 (s, 1 H, 2′-H), 8.15 (br, 1 H, picolinoyl-3-H), 8.79 (m, 1 H, picolinoyl-6- H); 13C NMR (500 MHz, CDCl3) δ 164.9, 150.1, 148.7, 143.4, 141.8, 140.2, 137.2, 134.7, 127.0, 125.4, 124.4, 123.6, 122.7, 120.3, 111.4, 75.6, 56.0, 50.6, 47.4, 38.2, 37.2, 35.0, 31.3, 30.5, 27.8, 20.82, 19.5, 17.0. HRMS calcd 516.2621 (C32H35O2N3.Na+), found 516.2614.
3β-(Pyridine-3-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (42)
Compound 42 prepared by following general method G, using 2-Methyl-6-nitrobenzoic anhydride (0.13 g, 0.39 mmol), nicotinic acid (0.05 g, 0.39 mmol), 4-DMAP (0.035 g, 0.29 mmol), THF (1 ml), 5 (0.075 g, 0.19 mmol), THF (1 ml) and TEA (0.1 ml). FCC gave pure 42 (0.85 g, 89%): mp 206–207 °C; IR (Neat) 3435, 2942, 2851, 1710, 1496, 1285, 1120, cm−1; 1H NMR (400 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.13 (s, 3 H, 19-CH3), 4.93 (m, 1 H, 3α-H), 5.49 (br, 1 H, 6-H), 5.99 (t, 1 H, J = 1.46 Hz, 16-H), 7.32 (m, 2 H, aromatic-Hs), 7.41 (m,1H, nicotinoyl-5-H), 7.50 (m, 1 H, aromatic-H), 7.83 (m, 1 H, aromatic- H), 7.98 (s, 1 H, 2′-H), 8.33 ( m, 1 H, nicotinoyl-4-H), 8.79 (m, 1 H, nicotinoyl-6-H), 9.23 (br. s, 1 H, nicotinoyl-2-H); 13C NMR (500 MHz, CDCl3) δ 164.9, 153.5, 151.1, 147.3, 141.8, 140.0, 137.3, 126.8, 124.4, 123.6, 122.7, 120.4, 111.4, 75.2, 55.0, 50.6, 47.4, 38.3, 37.1, 35.0, 31.3, 30.5, 20.8, 19.5, 16.2. HRMS calcd 516.2621 (C32H35O2N3.Na+), found 516.2617.
3β-(Pyridine-4-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (43)
Compound 43 prepared by following general method G, using 2-Methyl-6-nitrobenzoic anhydride (0.13 g, 0.39 mmol), isonicotinic acid (0.05 g, 0.39 mmol), 4-DMAP (0.035 g, 0.29 mmol), THF (1 ml), 5 (0.075 g, 0.19 mmol), THF (1 ml) and TEA (0.1 ml). FCC gave pure 43 (0.064 g, 67%): mp 184–85 °C; IR (Neat) 2944, 2953, 1719, 1489, 1282, 1124, 745 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.13 (s, 3 H, 19-CH3), 4.90 (m, 1 H, 3α-H), 5.49 (br, 1 H, 6-H), 5.99 (s, 1 H, 16-H), 7.30 (m, 2 H, aromatic-Hs), 7.49 (m, 1 H, aromatic-H), 7.81 (m, 1 H, aromatic- H), 7.85 (m, 2 H, isonicotinoyl-3, 5-Hs), 7.96 (s, 1 H, 2′-H), and 8.78 (m, 2 H, isonicotinoyl-2, 6-Hs); 13C NMR (500 MHz, CDCl3) δ 164.7, 150.8, 147.4, 143.5, 141.8, 139.9, 138.1, 134.8, 124.3, 123.6, 122.7, 120.4, 111.3, 75.6, 56.0, 50.6, 47.4, 38.2, 37.0, 35.0, 31.3, 30.5, 27.9, 19.5, 16.2. HRMS calcd 516.2621 (C32H35O2N3.Na+), found 516.2615.
3β-(3-(Oxycarbonyl)phenylacetic acid)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (44)
Compound 41 prepared by following general method G, using 2-Methyl-6-nitrobenzoic anhydride (0.18 g, 0.51 mmol) was added to a solution of 1, 3-phenyldiacetic acid (0.1 g, 0.51 mmol) and DMAP (0.05 g, 0.39 mmol) in THF (2 ml), 5 (0.1 g, 0.26 mmol), THF (1 ml) and TEA (0.15 ml). FCC gave pure 44 (0.055 g, 39.81%): mp 222–23 °C; IR (Neat) 2944, 1734, 1610, 1454, 1337, 1204, 1165, 1003 749 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.99 (s, 3 H, 18- CH3), 1.05 (s, 3 H, 19-CH3), 3.59 (s, 2 H, CH2-Hs), 3.64 (s, 2 H, CH2-Hs), 4.63 (m, 1 H, 3α-H), 5.40 (br, 1 H, 6-H), 5.98 (m, 1 H, 16-H), 7.18–7.23 (m, 3 H, aromatic-Hs), 7.27–7.31 (m, 3 H, aromatic-H), 7.47 (m, 1 H, aromatic-H), 7.81 (m, 1 H, aromatic-H) 8.01 (s, 1 H, 2′-H); 13C NMR (400 MHz, CDCl3) δ 171.2, 147.1, 141.8, 140.3, 135.0, 134.6, 130.5, 128.9, 128.0, 125.0, 123.9, 122.16, 120.0, 111.5, 74.4, 56.0, 50.5, 47.4, 45.6, 41.8, 38.2, 37.0, 37.0, 31.3, 30.5, 27.82, 20.8, 19.4, 16.1, 8.7. HRMS calcd 587.2880 (C36H40O4N2.Na+), found 587.2876
3β-(6-(Cyclohex-3-enecaboxylic acid)carboxylate)-17-(1H-benzimidazol-1-yl)-androsta- 5,16-diene (45)
A mixture of 5 (0.1 g, 0.26 mmol), DMAP (0.035 g, 0.28 mmol), 1,2,3,6- tetrahydrophthalic anhydride (0.13 g, 0.85 mmol) and pyridine (3 mL) was refluxed for 3 hrs. Cooled to room temperature and quenched to water. Precipitate was extracted with EtOAc, dried with Na2SO4, evaporated and the residue was purified by FCC [petroleum ether/EtOAc/TEA (9.5:0.3:0.2)] to give 0.1 g (71.9%) of pure compound 45: mp 178–179 °C; IR (Neat) 2931, 1724, 1453, 1225, 1195 and 743 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.99 – 1.04 (m, 6H, 18-CH3 and19-CH3), 4.64 (m, 1 H, 3α-H), 5.40 (br, 1 H, 6-H), 5.69 (m, 2 H, c-hexyl-4, c-hexyl-5, Hs), 5.96 (s, 1 H, 16-H), 7.30 (m, 2 H, aromatic-Hs), 7.50 (d, 1 H, aromatic-H), 7.84 (1 H, m, aromatic-H) 8.05 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 177.3, 173.5, 1401.0, 126.0, 125.3, 124.8, 123.8, 123.0, 121.9, 120.0, 111.4, 73.8, 55.9, 50.5, 47.4, 45.4, 40.7, 38.2, 37.1, 34.9, 31.3, 30.5, 27.7, 26.4, 19.4, 16.2, 8.8. HRMS calcd 563.2880 (C34H40N2O4.Na+), found 563.2879.
3β-(Oxycarbonyl-(methoxy) acetic acid)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (46)
A mixture of 5 (0.1 g, 0.26 mmol), DMAP (0.035 g, 0.28 mmol), diglycolic anhydride (0.1 g, 0.85 mmol) and pyridine (3 mL) was refluxed for 3 hrs. Cooled to room temperature and quenched to water. Precipitate was extracted with EtOAc, dried with Na2SO4, evaporated and the residue was purified by FCC [petroleum ether/EtOAc/TEA (9.5:0.3:0.2)] to give 0.05 g (28.6%) of pure compound 46: mp 214–215 °C; IR (Neat) 2934, 1722, 1456, 1225, 1147 and 745 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.01 (s, 3 H, 18-CH3), 1.07 (s, 3 H, 19-CH3), 4.25 (s, 2 H, CH2), 4.26 (s, 2 H, CH2), 4.74 (m, 1 H, 3α-H), 5.45 (br, 1 H, 6-H), 6.00 (m, 1 H, 16-H), 7.32 (m, 2 H, aromatic-Hs), 7.49 (m,1H, aromatic-H), 7.82 (m, 1 H, aromatic-H), 8.06 (s, 1 H, 2′ aromatic- H); 13C NMR (500 MHz, CDCl3); δ 172.9, 169.9, 147.0, 141.7, 140.0, 134.4, 125.4, 124.2, 123.4, 119.7, 111.6, 75.0, 69.1, 68.8, 56.0, 50.5, 47.4, 38.2, 37.0, 34.9, 31.3, 31.1, 30.5, 27.8, 20.8, 19.4, 16.2. HRMS calcd 527.2516 (C30H36N2O5.Na+), found 527.2516.
3β-(1H-Imidazole-1-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (47)
A solution of 5 (0.15 g, 0.38 mmol), CDI (0.125 g, 0.77 mmol) in anhydrous acetonitrile (2 mL) and DCM (1 mL) stirred at room temperature for 2 h. Then solvent evaporated, residue treated with water, and extracted with DCM. The crude white product obtained on evaporation of solvent was purified by FCC using 1.7% methanol in DCM in presence of traces of TEA (0.06%) to give 47 (0.135 g, 72%): mp 194–96 °C; IR (Neat) 2965, 2923, 2839, 1754, 1488, 1452, 1392, 1292, 834, 773 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.12 (s, 3 H, 19-CH3), 4.85 (m, 1 H, 3α-H), 5.51 (br, 1 H, 6-H), 5.99 (s, 1 H, 16-H), 7.07 (s, 1 H, 4″-H), 7.30 (m, 2 H, aromatic-Hs), 7.43 (s, 1 H, aromatic-H), 7.49 (m, 1 H, 5″-H) 7.81 (m, 1 H, aromatic- H), 7.96 (s, 1 H, 2′-H) and 8.13 (s, 1 H, 2″-H); 13C NMR (500 MHz, CDCl3) δ 148.1, 147.1, 143.3, 141.3, 139.1, 137.1, 134.6, 130.6, 124.1, 123.1, 120.2, 117.1, 111.1, 78.4, 55.7, 50.6, 47.2, 37.9, 36.8, 34.8, 31.1, 30.3, 27.6, 20.6, 19.3, 16.0. HRMS calcd 505.2573 (C30H34O2N4.Na+), found 505.2577.
3β-(2-Methyl-1H-imidazole-1-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (48)
A solution of 5 (0.075 g, 0.193 mmol), 1,1-carbonylbis(2-methylimidazole) (0.05 g, 0.214 mmol) in anhydrous acetonitrile (1.5 mL) and DCM (0.75 mL) was refluxed over-night. The solvent evaporated, residue treated with water, and extracted with DCM. The crude white product obtained on evaporation of solvent was purified by FCC using 4 % ethanol in DCM in presence of traces of TEA (0.06%). The product was triturated with petroleum ether to give 48 (0.065 g, 67% ): mp 186–187 °C; IR (Neat) 2935, 2855, 1749, 1452, 1394, 1291, 1146, 983 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.12 (s, 3 H, 19-CH3), 2.64 (s, 3 H, 2″-CH3), 4.80 (m, 1 H, 3α-H), 5.51 (m, 1 H, 6-H), 5.99 (m, 1 H, 16-H), 6.84 (s, 1 H, 5″-H), 7.29 (m, 2 H, aromatic-Hs), 7.35 (s, 1 H, aromatic-H), 7.48 (m, H, aromatic-H) 7.81 (m, 1 H, 4″-H), and 7.96 (s, 1 H, 2′-H); 13C NMR (500 MHz, CDCl3) δ 149.0, 147.9, 147.1, 143. 3, 141.6, 139.2, 134.6, 127.8, 123.4, 122.5, 120.2, 118.1, 111.1, 78.0, 55.7, 50.3, 47.2, 38.0, 36.8, 34.8, 31.1, 30.3, 27.7, 20.6, 19.3, 16.9, 16.0. HRMS calcd 519.2730 (C31H36O2N4.Na+), found 519.2730.
3β-(1H-1,2,4-Triazole-1-carboxylate)-17-(1H-benzimidazol-1-yl)-androsta-5,16-diene (49)
A solution of 5 (0.15 g, 0.386 mmol), CDT (0.19 g, 1.16 mmol) in anhydrous acetonitrile (3 mL) and DCM (1.5 mL) was refluxed for 3h. The solvent evaporated, residue treated with water, and extracted with DCM. The crude white product obtained on evaporation of solvent was purified by FCC using 4 % Ethanol in DCM in presence of traces of TEA (0.06%). The product was triturated with petroleum ether to give 49 (0.15 g, 80% ): mp 205–206 °C; IR (Neat) 2950, 2855, 1776, 1489, 1375, 1289, 978, 750 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.03 (s, 3 H, 18-CH3), 1.12 (s, 3 H, 19-CH3), 4.96 (m, 1 H, 3α-H), 5.52 (m, 1 H, 6-H), 5.99 (s, 1 H, 16-H), 7.30 (m, 2 H, aromatic-Hs), 7.50 (t, 1 H, J = 3.8 Hz, aromatic -H), 7.81 (m, H, aromatic-H), 7.96 (s, 1 H, 2′-H), 8.07 (s, 1 H, 5″-H), and 8.83 (s, 1 H, 3″-H); 13C NMR (500 MHz, CDCl3) δ 153.8, 147.3, 145.8, 143,5, 141.8, 139.2, 134.7, 124.3, 123.6, 122.7, 120.4, 111.3, 80.0, 55.9, 50.5, 47.4, 37.9, 37.0, 35.0, 31.3, 30.5, 27.6, 20.8, 19.4, 16.2. HRMS calcd 506.2526 (C29H33O2N5.Na+), found 506.2525.
Biology experiments
Cell Culture
LNCaP cells were purchased from American Type Culture Collection- ATCC (Rockville, MD, USA). Cells were maintained in ATCC recommended culture media with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA, USA) and 1% penicillin/streptomycin (invitrogen). Cells were grown as a monolayer in T75 or T150 tissue culture flasks in a humidified incubator (5% CO2, 95% air) at 37°C. CWR22rv1 cells are a gift from Dr. Marja Nevalainen of Thomas Jefferson University, Philadelphia.
Cell growth inhibition (MTT colorimetric assay)
The cells were seeded in 96-well plates (Corning Costar) at a density of 5 × 103 cells per well. Cells were allowed to adhere to the plate for 24 hours and then treated with various concentrations of compounds dissolved in 95% EtOH. Cells were treated for 7 days with renewal of test compound and media on day 4. On the 7th day, medium was renewed and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) (Sigma, St Louis, MO, USA) solution (0.5 mg MTT per ml of media) was added to the medium such that the ratio of MTT: medium was 1:10. The cells were incubated with MTT for 2 hours. The medium was then aspirated and DMSO was added to solubilize the violet MTTformazan product. The absorbance at 562 nm was measured by spectrophotometry (Biotek Inc.).
Transcriptional activation Luciferase assay
LNCaP cells were transferred to steroid-free medium 3 days before the start of the experiment and plated at 1 × 105 per well in steroid-free medium. The cells were dual transfected with ARR2-Luc and the Renilla luciferase reporter vector pRL-null. After a 24-h incubation period at 37 °C, the cells were incubated in fresh phenol red–free RPMI 1640 containing 5% charcoal-stripped fetal bovine serum and treated with 10 nmol/L dihydrotestosterone, ethanol vehicle, and/or the selected compounds in triplicate. After an 18-h treatment period, the cells were washed twice with ice-cold Dulbecco’s PBS and assayed using the Dual Luciferase kit (Promega) according to the manufacturer’s protocol.Cells were lysed with 100 CL of luciferase lysing buffer, collected in a microcentrifuge tube, and pelleted by centrifugation. Supernatants (20 CL aliquots) were transferred to corresponding wells of opaque 96-well multiwall plates. Luciferase Assay Reagent was added to each well, and the light produced during the luciferase reaction was measured in a Victor 1420 scanning multiwell spectrophotometer (Wallac, Inc.). After measurement, Stop and Glo reagent (Promega) was added to quench the firefly luciferase signal and initiate the Renilla luciferase luminescence. Renilla luciferase luminescence was also measured in the Victor 1420.The results are presented as the fold induction (i.e., the relative luciferase activity of the treated cells divided by that of the control) normalized to that of the Renilla.
Western blot analysis
For immunoblot detection of various proteins, LNCaP or CWR22v1 prostate cancer cells were cultured. Cells were treated with indicated compounds and whole cell lysates were prepared as described in41 using RIPA lysis buffer (Sigma Aldrich) and protease and phosphatase inhibitors (Sigma Aldrich). All of the antibodies were ordered from cell signaling technology. Protein content was determined using the Bradford Assay (Bio-Rad, Hercules, CA, USA). Protein was subjected to SDS–PAGE and transferred onto nitrocellulosemembrane. Membranes were then incubated with secondary antibody (cell signaling technology) at room temperature for 1 hour. Bands were visualized by chemiluminescence (Millipore). Protein expression was normalized to β-actin and densitometry was carried out using Image J or ImageQuant 5.0 (Molecular Dynamics, Sunnyvale, CA, USA). CWR22Rv1 cells were used for endogenous levels of splice variant AR-3. Protein levels were analyzed with respective antibodies; full length AR and β-actin antibodies were purchased from cell signaling, antibody specific for splice variant AR-3 was obtained from Dr. Yun Qiu, University of Maryland, School of Medicine, Baltimore.30c
Androgen receptor competitive binding assay
Competitive binding assays were performed with the synthetic androgen methyltrienolone [3H]R1881 essentially as described by Wong et al. and Yarbrough et al. 42. Wells in 24-well multiwell dishes were coated with poly-l-lysine (0.05 mg/ml) for 30 minutes, rinsed with sterilized, distilled water, and dried for 2 hours. To determine the kinetics of [3H]R1881 binding to the LNCaP AR cells were plated (2–3 × 105 cells/well) in 24 well multiwell dishes in steroid-free medium and allowed to attach. The following day the medium was replaced with serum-free, steroid free RPMI supplemented with 0.1 % BSA and containing [3H]R1881 (0.01–10 nM) in the presence or absence of a 200 fold excess of cold DHT, to determine nonspecific binding, and 1μM triamcinolone acetonide to saturate progesterone and glucocorticoid receptors. Following a 2 hour incubation period at 37°C, cells were washed twice with ice-cold DPBS and solubilized in DPBS containing 0.5 % SDS and 20 % glycerol. Extracts were removed and cell associated radioactivity counted in a scintillation counter. The data was analyzed, including Kd and Bmax determination, by nonlinear regression using Graphpad Prism software (GraphPad Software, Inc, San Diego, CA). When the concentration of [3H]R1881 required to almost saturate AR in both cell lines was established (5.0 nM), the ability of the test compounds (1 nM-10 μM) to displace [3H]R1881 (5.0 nM) from the receptors was determined as described above. The IC50 of each compound was determined by nonlinear regression with Graphpad Prism software (GraphPad Software, Inc, San Diego, CA).
Immunocytochemical analysis
LNCaP cells were plated in 8 chamber vessel tissue culture treated glass slide (0.025 X 106 cells/well), for 12h and then treated with 5uM of VN/124-1 or VNPT55 for 48h. Cells were washed twice with PBS and fixed in 3.7% formaldehyde for 10 mins and permeabilized with 0.25% triton in PBS for another 5mins after several washes. Cells blocked with 5% BSA with 0.5% NP40 in PBS and incubated with anti-AR (1:600 dilution; cell signaling) in 2.5% BSA in PBS overnight. Cells were incubated for 1h with secondary antibody Alexa Fluor 488-conjugate anti-rabbit IgG(H+L) at 1:1000(Cell Signaling) and nuclear counterstain for 5mins (DAPI at 1:5000). All images were taken using the Nikon TE2000 microscope.
CYP17 Inhibition assay
The assay was kindly performed by Dr. Emily Scott and colleagues according to their recently reported procedure in which a truncated version of human CYP17A1 (CYP171dH) was expressed in E. coli and then purified to homogeneity.34 IC50 values of the compounds were determined from dose-response curves. The IC50 values of abiraterone alcohol (3b, a CYP17 inhibitor recently approved for prostate cancer therapy), galaterone and 3β-hydroxy-17-(1H-imidazole-1-yl)androsta-5,16-diene (VN/85-1, structure not shown, believed to be the most potent CYP17 inhibitor12a, 18 ) were also determined in the same assay system for comparison (used as positive controls).
Supplementary Material
Figure 2.

Chemical structures of compounds 8 – 11.
Acknowledgments
This work was supported in part by grants from NIH and NCI (R21 CA11791-01, 1RO1CA129379-01 A2 and 5RO1CA129379-02) and from start-up funds from Jefferson School of Pharmacy, Thomas Jefferson University, Philadelphia, USA and University of Maryland School of Medicine and the Center for Biomolecular Therapeutics (CBT), Baltimore, USA to Professor Vincent C. O. Njar. We also thank Dr. Adejare Adeboye, Professor of Pharmaceutical Sciences, University of the Sciences, Philadelphia, PA, USA for help with some NMR acquisitions. Andrew K. Kwegyir-Afful was supported in part by University of Maryland School of Medicine Toxicology Program.
Abbreviations Used
- AR
Androgen receptor
- ARD
androgen receptor down-regulation
- AP
anti-proliferative
- BzIm
benzimidazole
- BSA
bovine serum albumin
- CRPC
castration resistant prostate cancer
- CYP
cytochrome P450
- CYP17A
17α-hydroxylase/17,20-lyase
- DAPI
6- diamino-2-phenylindole
- DHT
dihydrotestosterone
- DBPS
dulbecco’s phosphate-buffered saline
- ESPC
early stage prostate cancer
- FBS
fetal bovine serum
- FCC
flash column chromatography
- GI50
compound concentration required to inhibit cell growth by 50%
- HRMS
high-resolution mass spectra
- hAR
human androgen receptor
- IC50
compound concentration required to inhibit cell growth by 50%; compound concentration required to inhibit enzyme activity by 50%
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide)
- PDB
protein data bank
- SDS
sodium dodecyl sulfate
Footnotes
Disclosure of Potential Conflict of Interest: Vincent C. O. Njar is an inventor of compound 5 patents and technologies thereof owned by the University of Maryland, Baltimore, and licensed to Tokai Pharmaceuticals, Inc. The other authors declare no potential conflict of interest. A patent application to protect the novel compounds of this manuscript has been filed.
Supporting Information Available: HPLC chromatograms and high resolution mass spectral data for compounds 16 – 22, 25, 28, 31 and 33 – 49. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]; (b) Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10(10):981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. Short hairpin RNA knockdown of the androgen receptor attenuates ligandindependent activation and delays tumor progression. Cancer Res. 2006;66(21):10613–20. doi: 10.1158/0008-5472.CAN-06-0028. [DOI] [PubMed] [Google Scholar]; (d) Snoek R, Cheng H, Margiotti K, Wafa LA, Wong CA, Wong EC, Fazli L, Nelson CC, Gleave ME, Rennie PS. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castrationresistant prostate tumors. Clin Cancer Res. 2009;15(1):39–47. doi: 10.1158/1078-0432.CCR-08-1726. [DOI] [PubMed] [Google Scholar]; (e) Vasaitis TS, Njar VCO. Novel, potent anti-androgens of therapeutic potential: recent advances and promising developments. Future Medicinal Chemistry. 2010;2(4):667–680. doi: 10.4155/fmc.10.14. [DOI] [PubMed] [Google Scholar]
- 2.(a) Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68(11):4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mostaghel EA, Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab. 2008;22(2):243–58. doi: 10.1016/j.beem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]; (d) Vasaitis T, Bruno RD, Njar VCO. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol. 2010;2011:23–31. doi: 10.1016/j.jsbmb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cai L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, Yang G, Cao C, Sensintaffar J, Wasielewska T, Herbert MR, Bonnefous C, Darimont B, Scher HI, Smith-Jones P, Klang M, Smith ND, De Stanchina E, Wu N, Ouerfelli O, Rix PJ, Heyman RA, Jung ME, Sawyers CL, Hager JH. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72(6):1494–503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC) J Med Chem. 2010;53(7):2779–96. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryce A, Ryan CJ. Development and clinical utility of abiraterone acetate as an androgen synthesis inhibitor. Clin Pharmacol Ther. 2012;91(1):101–8. doi: 10.1038/clpt.2011.275. [DOI] [PubMed] [Google Scholar]
- 5.Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, Hara T, Yamaoka M, Kusaka M, Okuda T, Asahi S, Furuya S, Tasaka A. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20- lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem. 2011;19(21):6383–99. doi: 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- 6.Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AM, Njar VC. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76(12):1268–79. doi: 10.1016/j.steroids.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherji D, Pezaro CJ, De-Bono JS. MDV3100 for the treatment of prostate cancer. Expert Opin Investig Drugs. 2012;21(2):227–33. doi: 10.1517/13543784.2012.651125. [DOI] [PubMed] [Google Scholar]
- 8.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, Molife LR, Hunt J, Messiou C, Parker C, Dearnaley D, Swennenhuis JF, Terstappen LW, Lee G, Kheoh T, Molina A, Ryan CJ, Small E, Scher HI, de Bono JS. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–95. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR, Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72(14):3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17(18):5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009;16(6):458–62. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 11.(a) Godbole AMNVCO. New insights into the androgen-theargeted therapies and epigenetic therapies in prostate cancer. Prostate Cancer. 2011;2011:1–13. doi: 10.1155/2011/918707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Godbole AMN, VCO . Prostate Cancer: Current and Emerging Therapies. InTech; Rijika, Croatia: 2011. [Google Scholar]; (c) Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. Androgen receptor regulates a distinct transcription program in androgenindependent prostate cancer. Cell. 2009;138(2):245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, Newman D, Jr, Farquhar R, Guo Z, Qiu Y, Brodie AM. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48(8):2972–84. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]; (b) Schayowitz A, Sabnis G, Goloubeva O, Njar VC, Brodie AM. Prolonging hormone sensitivity in prostate cancer xenografts through dual inhibition of AR and mTOR. Br J Cancer. 2010;103(7):1001–7. doi: 10.1038/sj.bjc.6605882. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schayowitz A, Sabnis G, Njar VC, Brodie AM. Synergistic effect of a novel antiandrogen, VN/124-1, and signal transduction inhibitors in prostate cancer progression to hormone independence in vitro. Mol Cancer Ther. 2008;7(1):121–32. doi: 10.1158/1535-7163.MCT-07-0581. [DOI] [PubMed] [Google Scholar]; (d) Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, Guo Z, Fang HB, Njar VC, Brodie AM. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17- (1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol Cancer Ther. 2008;7(8):2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puranik PP, Godbole AM, Gediya LK, Ates-Alagoz Z, Njar VCO. Exploitation of multitarget prostate cancer clinical candidate VN/124-1 (TOK-001) to develop a novel class of androgen receptor down regulating agents for prostate cancer therapy. 242nd American Chemical Society (ACS) National Meeting; Denver, CO, USA. Aug 28 - Sept 1, 2011; p. Abstract #MEDI 35. [Google Scholar]
- 14.(a) Fan M, Rickert EL, Chen L, Aftab SA, Nephew KP, Weatherman RV. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res Treat. 2007;103(1):37–44. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]; (b) Hanson RN, McCaskill E, Tongcharoensirikul P, Dilis R, Labaree D, Hochberg RB. Synthesis and evaluation of 17alpha-(dimethylphenyl)vinyl estradiols as probes of the estrogen receptor-alpha ligand binding domain. Steroids. 2012;77(5):471–6. doi: 10.1016/j.steroids.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53(14):5061–84. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu W, Zhou J, Geng G, Shi Q, Sauriol F, Wu JH. Antiandrogenic, maspin induction, and antiprostate cancer activities of tanshinone IIA and its novel derivatives with modification in ring A. J Med Chem. 2012;55(2):971–5. doi: 10.1021/jm2015292. [DOI] [PubMed] [Google Scholar]; (c) Yang J, Wei S, Wang DS, Wang YC, Kulp SK, Chen CS. Pharmacological exploitation of the peroxisome proliferator-activated receptor gamma agonist ciglitazone to develop a novel class of androgen receptorablative agents. J Med Chem. 2008;51(7):2100–7. doi: 10.1021/jm701212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy J, Breton R, Martel C, Labrie F, Poirier D. Chemical synthesis and biological activities of 16alpha-derivatives of 5alpha-androstane-3alpha,17beta-diol as antiandrogens. Bioorg Med Chem. 2007;15(8):3003–18. doi: 10.1016/j.bmc.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Pereira de Jesus-Tran K, Cote PL, Cantin L, Blanchet J, Labrie F, Breton R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006;15(5):987–99. doi: 10.1110/ps.051905906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Njar VC, Kato K, Nnane IP, Grigoryev DN, Long BJ, Brodie AM. Novel 17-azolyl steroids, potent inhibitors of human cytochrome 17 alpha-hydroxylase-C17,20- lyase (P450(17) alpha): potential agents for the treatment of prostate cancer. J Med Chem. 1998;41(6):902–12. doi: 10.1021/jm970568r. [DOI] [PubMed] [Google Scholar]
- 19.Brik A, Wu CY, Best MD, Wong CH. Tetrabutylammonium fluoride-assisted rapid N9-alkylation on purine ring: application to combinatorial reactions in microtiter plates for the discovery of potent sulfotransferase inhibitors in situ. Bioorg Med Chem. 2005;13(15):4622–6. doi: 10.1016/j.bmc.2005.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devlin TA, Jebaratnam DJ. An Improved Procedure for the Synthesis of 1,3- Dideazaadenosine. Syn Comm. 1995;25(5):711–8. [Google Scholar]
- 21.Purushottamachar P, Khandelwal A, Vasaitis TS, Bruno RD, Gediya LK, Njar VC. Potent anti-prostate cancer agents derived from a novel androgen receptor downregulating agent. Bioorg Med Chem. 2008;16(7):3519–29. doi: 10.1016/j.bmc.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 22.Calogeropoulou T, Avlonitis N, Minas V, Alexi X, Pantzou A, Charalampopoulos I, Zervou M, Vergou V, Katsanou ES, Lazaridis I, Alexis MN, Gravanis A. Novel dehydroepiandrosterone derivatives with antiapoptotic, neuroprotective activity. J Med Chem. 2009;52(21):6569–87. doi: 10.1021/jm900468p. [DOI] [PubMed] [Google Scholar]
- 23.Holland HLKS, Tan L, Njar VCO. Synthesis of 6-hydroxyimino-3-oxo steroids, a new class of aromatase inhibitors. J Chem Soc Perkin Trans. 1992;1:585–587. [Google Scholar]
- 24.Yamashita K, Kobayashi S, Tsukamoto S, Numazawa M. Synthesis of pyridinecarboxylate derivatives of hydroxysteroids for liquid chromatography-electrospray ionization-mass spectrometry. Steroids. 2007;72(1):50–9. doi: 10.1016/j.steroids.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Qian K, Kim SY, Hung HY, Huang L, Chen CH, Lee KH. New betulinic acid derivatives as potent proteasome inhibitors. Bioorg Med Chem Lett. 2011;21(19):5944–7. doi: 10.1016/j.bmcl.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreira VM, Vasaitis TS, Guo Z, Njar VC, Salvador JA. Synthesis of novel C17 steroidal carbamates. Studies on CYP17 action, androgen receptor binding and function, and prostate cancer cell growth. Steroids. 2008;73(12):1217–27. doi: 10.1016/j.steroids.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Davenport APRFD. Radioligand Binding Assays: Theory and Practice. Kluwer Academic Publishers; Netherlands: 1996. [Google Scholar]
- 28.Soifer HS, Souleimanian N, Wu S, Voskresenskiy AM, Collak FK, Cinar B, Stein CA. Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. J Biol Chem. 2012;287(6):3777–87. doi: 10.1074/jbc.M111.261933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradbury RH, Hales NJ, Rabow AA, Walker GE, Acton DG, Andrews DM, Ballard P, Brooks NA, Colclough N, Girdwood A, Hancox UJ, Jones O, Jude D, Loddick SA, Mortlock AA. Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2011;21(18):5442–5. doi: 10.1016/j.bmcl.2011.06.122. [DOI] [PubMed] [Google Scholar]
- 30.(a) Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guo Z, Qiu Y. A new trick of an old molecule: androgen receptor splice variants taking the stage?! Int J Biol Sci. 2011;7(6):815–22. doi: 10.7150/ijbs.7.815. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107(39):16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Cao B, Liu X, Li J, Liu S, Qi Y, Xiong Z, Zhang A, Wiese T, Fu X, Gu J, Rennie PS, Sartor O, Lee BR, Ip C, Zhao L, Zhang H, Dong Y. 20(S)- protopanaxadiol-aglycone downregulation of the full-length and splice variants of androgen receptor. Int J Cancer. 2013 doi: 10.1002/ijc.27754. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li J, Cao B, Liu X, Fu X, Xiong Z, Chen L, Sartor O, Dong Y, Zhang H. Berberine suppresses androgen receptor signaling in prostate cancer. Mol Cancer Ther. 2011;10(8):1346–56. doi: 10.1158/1535-7163.MCT-10-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X, Liu Z, Xu X, Blair CA, Sun Z, Xie J, Lilly MB, Zi X. Kava components down-regulate expression of AR and AR splice variants and reduce growth in patient-derived prostate cancer xenografts in mice. PLoS One. 2012;7(2):e31213. doi: 10.1371/journal.pone.0031213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mashima T, Okabe S, Seimiya H. Pharmacological targeting of constitutively active truncated androgen receptor by nigericin and suppression of hormone-refractory prostate cancer cell growth. Mol Pharmacol. 2010;78(5):846–54. doi: 10.1124/mol.110.064790. [DOI] [PubMed] [Google Scholar]
- 32.Yamashita S, Lai KP, Chuang KL, Xu D, Miyamoto H, Tochigi T, Pang ST, Li L, Arai Y, Kung HJ, Yeh S, Chang C. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia. 2012;14(1):74–83. doi: 10.1593/neo.111436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Haile S, Sadar MD. Androgen receptor and its splice variants in prostate cancer. Cell Mol Life Sci. 2011;68(24):3971–81. doi: 10.1007/s00018-011-0766-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sadar MD. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011;71(4):1208–13. doi: 10.1158/0008-5472.CAN_10-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482(7383):116–9. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Njar VC, Klus GT, Brodie A. Nucleophilic Vinylic “addition-elimination” Substitution Reaction of 3 Beta-Acetoaxy-17-chloro-16-formylandrosta-5,16-diene: A Novel and General Route to 17-substituted-delta-steroids. Part 1. Synthesis of novel 17- azolyl Steroids; Inhibitors of 17 alpha-hydroxylase/17,20-lyase (P45017alpha) Bioorganic & Medicinal Chemistry Letters. 1996:2777–2782. [Google Scholar]; (b) Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38(13):2463–71. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 36.(a) Paller CJ, Antonarakis ES. Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther. 2011;5:117–24. doi: 10.2147/DDDT.S13029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vacondio F, Silva C, Mor M, Testa B. Qualitative structure-metabolism relationships in the hydrolysis of carbamates. Drug Metab Rev. 2010;42(4):551–89. doi: 10.3109/03602531003745960. [DOI] [PubMed] [Google Scholar]
- 37.(a) Chassaing C, Berger M, Heckeroth A, Ilg T, Jaeger M, Kern C, Schmid K, Uphoff M. Highly water-soluble prodrugs of anthelmintic benzimidazole carbamates: synthesis, pharmacodynamics, and pharmacokinetics. J Med Chem. 2008;51(5):1111–4. doi: 10.1021/jm701456r. [DOI] [PubMed] [Google Scholar]; (b) Geary TG, Conder GA, Bishop B. The changing landscape of antiparasitic drug discovery for veterinary medicine. Trends Parasitol. 2004;20(10):449–55. doi: 10.1016/j.pt.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 38.(a) Brandt GE, Schmidt MD, Prisinzano TE, Blagg BS. Gedunin, a novel hsp90 inhibitor: semisynthesis of derivatives and preliminary structure-activity relationships. J Med Chem. 2008;51(20):6495–502. doi: 10.1021/jm8007486. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chaudhaery SS, Roy KK, Shakya N, Saxena G, Sammi SR, Nazir A, Nath C, Saxena AK. Novel carbamates as orally active acetylcholinesterase inhibitors found to improve scopolamine-induced cognition impairment: pharmacophore-based virtual screening, synthesis, and pharmacology. J Med Chem. 2010;53(17):6490–505. doi: 10.1021/jm100573q. [DOI] [PubMed] [Google Scholar]
- 39.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 2009;280(2):211–21. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Purushottamachar P, Njar VC. A new simple and high-yield synthesis of 5alphadihydrotestosterone (DHT), a potent androgen receptor agonist. Steroids. 2012;77(14):1530–4. doi: 10.1016/j.steroids.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godbole AM, Purushottamachar P, Martin MS, Daskalakis C, Njar VC. Autophagy inhibition synergistically enhances anti-cancer efficacy of RAMBA, VN/12-1 in SKBR-3 cells and tumor xenografts. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Wong C, Kelce WR, Sar M, Wilson EM. Androgen receptor antagonist versus agonist activities of the fungicide vinclozolin relative to hydroxyflutamide. J Biol Chem. 1995;270(34):19998–20003. doi: 10.1074/jbc.270.34.19998. [DOI] [PubMed] [Google Scholar]; (b) Yarbrough WG, Quarmby VE, Simental JA, Joseph DR, Sar M, Lubahn DB, Olsen KL, French FS, Wilson EM. A single base mutation in the androgen receptor gene causes androgen insensitivity in the testicular feminized rat. J Biol Chem. 1990;265(15):8893–900. [PubMed] [Google Scholar]
- 43.Berges RR, Furuya Y, Remington L, English HF, Jacks T, Isaacs JT. Cell proliferation, DNA repair, and p53 function are not required for programmed death of prostatic glandular cells induced by androgen ablation. Proc Natl Acad Sci U S A. 1993;90(19):8910–4. doi: 10.1073/pnas.90.19.8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.