

Abstract
Anfinsen’s principle asserts that all information required to specify the structure of a protein is encoded in its amino acid sequence. However, during protein synthesis by the ribosome, the N-terminus of the nascent chain can begin to fold before the C-terminus is available. We tested whether this cotranslational folding can alter the folded structure of an encoded protein in vivo, versus the structure formed when refolded in vitro. We designed a fluorescent protein consisting of three half-domains, where the N- and C-terminal half-domains compete with each other to interact with the central half-domain. The outcome of this competition determines the fluorescence properties of the resulting folded structure. Upon refolding after chemical denaturation, this protein produced equimolar amounts of the N- and C-terminal folded structures, respectively. In contrast, translation in Escherichia coli resulted in a 2-fold enhancement in the formation of the N-terminal folded structure. Rare synonymous codon substitutions at the 5′ end of the C-terminal half-domain further increased selection for the N-terminal folded structure. These results demonstrate that the rate at which a nascent protein emerges from the ribosome can specify the folded structure of a protein.
Protein folding has been studied for decades in vitro using a carefully selected set of model proteins, but it is still unclear to what extent folding in the test tube mimics folding in vivo.1,2 Here we present results demonstrating that cotranslational folding during protein synthesis in vivo can alter the folded structure of a protein versus the structure formed in the test tube.
A key criterion for the selection of many model proteins used for in vitro folding studies is that they unfold and refold reversibly; i.e., their folding behavior is under thermodynamic control.2,4 Alternatively, some proteins fold under kinetic control (Figure 1), in which the conformations populated in the unfolded ensemble and early intermediates select a specific trajectory along the energy landscape that determines which of two (or more) final folded structures the protein will adopt.5 There are several well-characterized examples of proteins that fold under kinetic control (cf. refs (6−8)), although such proteins tend not to be selected as protein folding models because kinetic control complicates kinetic and thermodynamic characterization of folding mechanisms.
Figure 1.
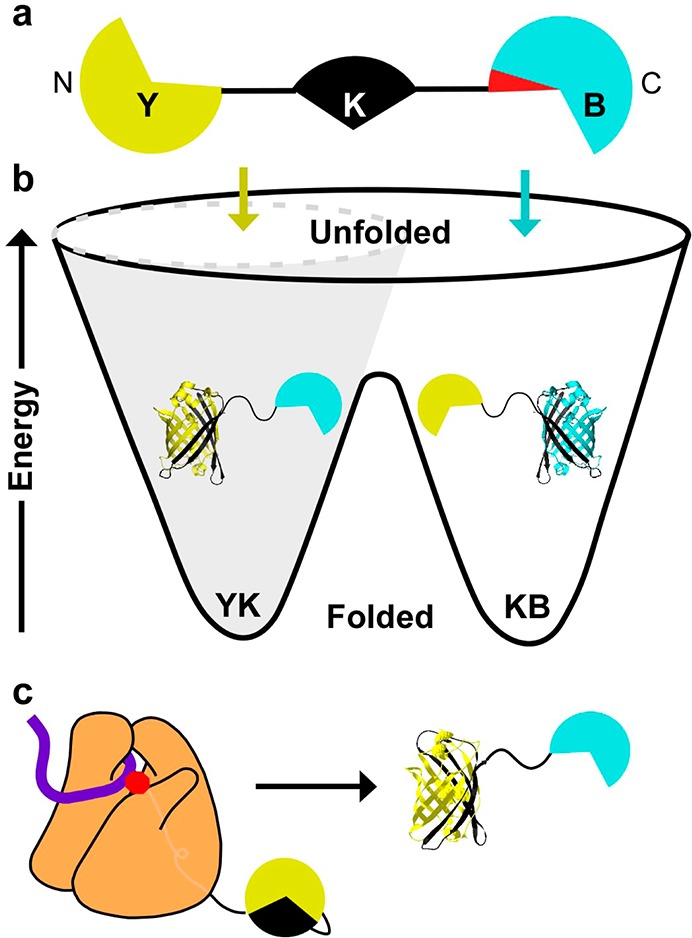
Experimental design for kinetically controlled folding. (a) Schematic of YKB, which consists of three half-domains connected by flexible (AGQ)5 linkers (black lines). The Y (yellow) and B (blue) half-domains compete to form a mutually exclusive kinetically trapped folded domain with the central K (black) half-domain. The red wedge indicates the location of synonymous codon substitutions (see text). (b) Energy landscapes for proteins that fold under kinetic control have multiple deep minima, representing alternative folded structures, separated by large barriers. The conformations of the unfolded protein and early folding intermediates (colored arrows) determine the final folded state of the protein. Forces that constrict the unfolded ensemble (gray cone) can bias folding toward one structure. (c) During translation of the nascent chain by the ribosome (orange), folding cannot be initiated from the untranslated C-terminus, which restricts the ensemble of unfolded states and leads to the preferential formation of one folded structure.
In the cell, proteins can begin folding cotranslationally, while the nascent chain is being synthesized.9−11 During translation, the nascent polypeptide chain emerges from the ribosome exit tunnel, whereupon N-terminal portions of the chain can start to form native-like interactions before C-terminal portions have been synthesized and/or are still confined within the tunnel.12,13 In contrast, protein refolding initiated by the dilution of full-length, unfolded polypeptides out of a chemical denaturant can begin via interactions formed anywhere along the polypeptide chain.14
We hypothesized that the proteins most likely to have native structures significantly affected by cotranslational folding would be (i) proteins that fold under kinetic control, i.e., can adopt two or more alternative native structures, depending on the conformations of the unfolded chain and early intermediates,4,5 and (ii) proteins whose native structures are kinetically stable and are therefore unlikely to unfold and refold over their lifetime in the cell. To test the hypothesis that cotranslational folding can globally alter a protein folded structure, we designed and constructed an Escherichia coli expression system encoding YKB (yellow-black-blue), a protein that can adopt two alternative folded structures. YKB consists of three half-domains derived from the BiFC split-fluorescent system15,16 connected by flexible (AGQ)5 linkers17 (Figure 1a). We designed YKB so that its folding represents a competition between the N- and C-terminal half-domains to fold with the central half-domain, with the result of this competition leading to either yellow (YK) or blue (KB) fluorescence, representing the formation of the mutually exclusive YK-B or Y-KB folded structures, respectively. The distinct fluorescent properties of the alternative structures enable the results of this structure-forming competition to be monitored in vivo using physiologically relevant translation rates. Moreover, the fluorescent protein folded structures are kinetically stable; once formed, they do not unfold and refold over a biologically relevant time scale.16,18,19
As expected, full-length YKB refolded via dilution from a chemical denaturant in vitro produced yellow and blue fluorescence in a ratio corresponding to equimolar formation of the YK and KB folded structures (Figure 2a,b). In contrast, YKB expressed in vivo produces more yellow fluorescence, indicating preferential formation of the YK folded structure and reflecting the preferential association of the N-terminal and central half-domains before the C-terminal half-domain is available for folding.
Figure 2.
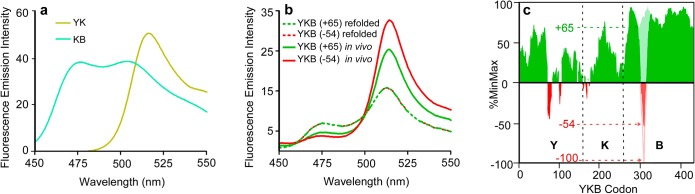
Translation alters YKB folded structure. (a) Fluorescence emission spectra of intact E. coli expressing the control fluorescent protein constructs YK (yellow) or KB (cyan). (b) Fluorescence emission spectra of intact E. coli expressing YKB constructs with common or rare codon usage (green versus red solid lines) versus the same YKB constructs folded in vitro upon dilution from a chemical denaturant (dashed lines). Numbers in parentheses correspond to synonymous codon usage; larger positive numbers correspond to more common codons. (c) E. coli MG1655 relative codon usage3 for codons encoding three representative YKB synonymous mutants: (+65) (light green), (−54) (red), and (−100) (pink line).
We hypothesized that altering the local rate of translation, and hence altering the rate of appearance of the YKB nascent protein chain, could be used to further modulate protein folding trajectories and select between its alternative folded structures. Changes to synonymous codon usage are known to alter local translation rate.3,20,21 All amino acids except methionine and tryptophan can be encoded by more than one mRNA codon. But these synonymous codons are not used with equal frequency, and rare codons are typically translated more slowly than common codons.20
To provide a translation rate-encoded switch to control folded structure formation, we used a simple algorithm3 to produce synonymous mRNA sequences encoding a short segment in the C-terminal half-domain of YKB, and selected sequences that had no significant effect on mRNA stability22 yet used synonymous codons with a wide variety of rarity (Figure 2c, Table S1, Supporting Information (SI); green, positive values represent codons more common than average, while red, negative values represent codons more rare than average). We hypothesized that increasing codon rarity would slow translation at this position, increasing the amount of time available for the N-terminal and central half-domains to interact during translation before the appearance of the competing C-terminal half-domain from the ribosome exit tunnel. We restricted codon substitutions to a short mRNA segment in order to alter YKB local translation rate while minimizing other, unwanted effects, including changes to mRNA stability, total cellular YKB levels or cellular tRNA availability, which could lead to premature translation termination or impaired cellular growth rate.
When this YKB switch region was encoded using synonymous rare codons, yellow fluorescence increased (Figure 2a), indicating that the translated polypeptide chains preferentially formed the N-terminal YK-B folded structure. Formation of YK versus KB was controllable by altering the relative rareness of the codons used to encode this region (Figure 3a), but not other more 5′ portions of the YKB mRNA sequence (Figure S1a,b, Table S1 (SI)). There were no significant differences in the intracellular accumulation of these codon-modified YKB variants, nor did we detect truncated products produced by premature translation termination (Figure S1c,d (SI)). LC–MS/MS analysis confirmed that translation of both rare and common YKB variants yields no detectable differences (<1%) in amino acid incorporation. The molar folding ratio ([YK]/[KB]) for these variants correlated more closely with changes in relative codon rareness (Pearson correlation coefficient = −0.85; P = 0.003) (Figure 3a) than tRNA concentration,23 relative wobble base translation velocity,21 mRNA stability22 or %GC content (Figure S2 (SI)), although the similarities between some of these correlations likely reflects the interdependence of these metrics on relative translation rate.
Figure 3.
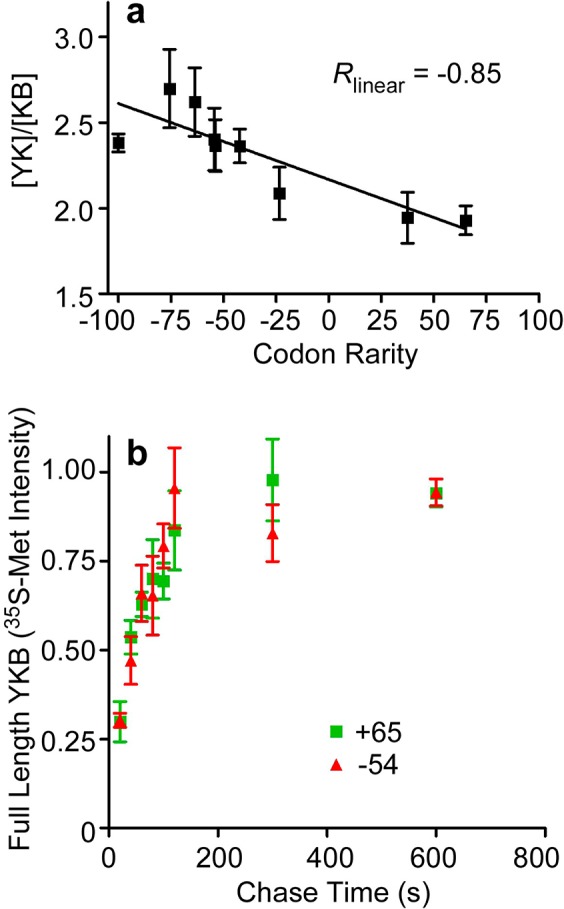
YKB synonymous codon substitutions predictably alter N-terminal versus C-terminal folding competition. (a) [YK]/[KB] molar ratios for synonymous mutants with altered codon usage, calculated as in Figure 2b. Error bars represent SEM of five measurements. (b) Pulse chase measurement of translation rate for YKB (+65) (green) versus (−54) (red). Error bars represent SEM of three measurements.
Each codon-modified YKB variant represents the substitution of only a few synonymous codons within an 18-codon window (Table S1 (SI)). These changes produced only a subtle effect on overall translation rate, as we were unable to detect a significant difference in the rate of synthesis of the codon-modified YKB variants using conventional pulse-chase labeling (Figure 3b). Yet this subtle alteration was sufficient to significantly alter the competition between the formation of the YK and KB alternative structures. These results demonstrate that significant changes in a protein folded structure can be triggered by very subtle differences in local translation rate, triggered by even small changes in codon usage.
Our results demonstrate that a protein native structure can be shaped by the vectorial appearance of the nascent chain during translation, a feature not present during in vitro refolding experiments. The coupling of folding to the process of translation is known to selectively stabilize specific folding intermediates,10,12,24,25 and altering translation rate has been shown to affect the folding efficiency of several proteins,9,26,27 suggesting that the formation of on-pathway folding intermediates during translation is partially dependent on translation rate. Our results demonstrate that, in addition to modulating folding yield (native versus aggregated), local translation rate can be adjusted in a predictable way to alter the selection between two alternative folded structures. We show for the first time that for a protein capable of forming alternative folded structures it is possible to predictably steer the protein folding mechanism to form one structure versus another by altering synonymous codon usage in specific portions of the mRNA sequence.
Synonymous mutations that affect protein structure, such as the ones described in this study, are likely to be particularly important for proteins that fold under kinetic control. More broadly, most proteins in the cell, when subject to chemical denaturation, cannot refold. Instead, these proteins misfold and aggregate. Some of these proteins have native and denatured states that are separated by an extremely high energy barrier28 and hence are expected to fold only once during their lifetime in the cell. In vivo, such proteins might be particularly dependent on the formation of cotranslational folding intermediates selected by local translation rate to most efficiently form the native protein structure.
Acknowledgments
We thank Jenna Leevy, Krastyu Ugrinov and Kay Finn for technical assistance and helpful discussions during the early stages of this project, and Alicia Specht and Jun Li for assistance with the statistical analyses. We thank Matthew Champion for performing the mass spectrometry analyses of translation fidelity and helpful discussions, and Chang-Deng Hu for providing the BiFC fragment plasmids. Supported by NIH grants to P.L.C. (R01 GM074807) and the Protein Translation Research Network (U54 GM105816), the Notre Dame CBBI Graduate Training Program GM075762 (I.M.S.), and a Clare Boothe Luce Graduate Fellowship (J.L.C.).
Supporting Information Available
Synonymous coding sequences, results from control experiments, and measured correlations between fluorescence ratios and other YKB sequence properties are detailed. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Anfinsen C. B. Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- Braselmann E.; Chaney J. L.; Clark P. L. Trends Biochem. Sci. 2013, 38, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T. F.; Clark P. L. PLoS One 2008, 3, e3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D.; Agard D. A. Biochemistry 1994, 33, 7505–7509. [DOI] [PubMed] [Google Scholar]
- Bryan P. N.; Orban J. Curr. Opin. Struct. Biol. 2010, 20, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl J. L.; Jaswal S. S.; Agard D. A. Nature 1998, 395, 817–819. [DOI] [PubMed] [Google Scholar]
- Luo X.; Tang Z.; Xia G.; Wassmann K.; Matsumoto T.; Rizo J.; Yu H. Nat. Struct. Mol. Biol. 2004, 11, 338–345. [DOI] [PubMed] [Google Scholar]
- Burmann B. M.; Knauer S. H.; Sevostyanova A.; Schweimer K.; Mooney R. A.; Landick R.; Artsimovitch I.; Rosch P. Cell 2012, 150, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar A. A.; Lesnik T.; Reiss C. FEBS Lett. 1999, 462, 387–391. [DOI] [PubMed] [Google Scholar]
- Frydman J.; Erdjument-Bromage H.; Tempst P.; Hartl F. U. Nat. Struct. Biol. 1999, 6, 697–705. [DOI] [PubMed] [Google Scholar]
- Fedorov A. N.; Baldwin T. O. J. Mol. Biol. 1999, 294, 579–586. [DOI] [PubMed] [Google Scholar]
- Evans M. S.; Sander I. M.; Clark P. L. J. Mol. Biol. 2008, 383, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola A. V.; Chen W.; Helenius A. Nat. Cell Biol. 1999, 1, 341–345. [DOI] [PubMed] [Google Scholar]
- Clark P. L. Trends Biochem. Sci. 2004, 29, 527–534. [DOI] [PubMed] [Google Scholar]
- Shyu Y.; Liu H.; Deng X.; Hu C.-D. BioTechniques 2006, 40, 61–66. [DOI] [PubMed] [Google Scholar]
- Robida A. M.; Kerppola T. K. J. Mol. Biol. 2009, 394, 391–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidus L. J.; Eaton W. A.; Hofrichter J. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7220–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. R.; Craggs T. D.; Christodoulou J.; Jackson S. E. J. Mol. Biol. 2007, 370, 356–371. [DOI] [PubMed] [Google Scholar]
- Do K.; Boxer S. G. J. Am. Chem. Soc. 2013, 135, 10226–10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen M. A.; Kurland C. G.; Pedersen S. J. Mol. Biol. 1989, 207, 365–377. [DOI] [PubMed] [Google Scholar]
- Spencer P. S.; Siller E.; Anderson J. F.; Barral J. M. J. Mol. Biol. 2012, 422, 328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Nucleic Acids Res. 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H.; Nilsson L.; Kurland C. G. J. Mol. Biol. 1996, 260, 649–663. [DOI] [PubMed] [Google Scholar]
- Clark P. L.; King J. J. Biol. Chem. 2001, 276, 25411–25420. [DOI] [PubMed] [Google Scholar]
- Ugrinov K. G.; Clark P. L. Biophys. J. 2010, 98, 1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller E.; DeZwaan D. C.; Anderson J. F.; Freeman B. C.; Barral J. M. J. Mol. Biol. 2010, 396, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Guo J.; Cha J.; Chae M.; Chen S.; Barral J. M.; Sachs M. S.; Liu Y. Nature 2013, 495, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K.; Manning M.; Hesham H.; Lin Q.; Bystroff C.; Colon W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17329–17334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.