

Abstract
Integrins are cell-surface adhesion receptors that bind to extracellular matrices (ECM) and mediate cell–ECM interactions. Some integrins are known to play critical roles in dental enamel formation. We recruited two Hispanic families with generalized hypoplastic amelogenesis imperfecta (AI). Analysis of whole-exome sequences identified three integrin beta 6 (ITGB6) mutations responsible for their enamel malformations. The female proband of Family 1 was a compound heterozygote with an ITGB6 transition mutation in Exon 4 (g.4545G > A c.427G > A p.Ala143Thr) and an ITGB6 transversion mutation in Exon 6 (g.27415T > A c.825T > A p.His275Gln). The male proband of Family 2 was homozygous for an ITGB6 transition mutation in Exon 11 (g.73664C > T c.1846C > T p.Arg616*) and hemizygous for a transition mutation in Exon 6 of Nance–Horan Syndrome (NHS Xp22.13; g.355444T > C c.1697T > C p.Met566Thr). These are the first disease-causing ITGB6 mutations to be reported. Immunohistochemistry of mouse mandibular incisors localized ITGB6 to the distal membrane of differentiating ameloblasts and pre-ameloblasts, and then ITGB6 appeared to be internalized by secretory stage ameloblasts. ITGB6 expression was strongest in the maturation stage and its localization was associated with ameloblast modulation. Our findings demonstrate that early and late amelogenesis depend upon cell–matrix interactions. Our approach (from knockout mouse phenotype to human disease) demonstrates the power of mouse reverse genetics in mutational analysis of human genetic disorders and attests to the need for a careful dental phenotyping in large-scale knockout mouse projects.
INTRODUCTION
Integrins are a large family of heterodimeric cell-surface receptors that are found in all metazoans. Composed of two non-covalently associated α and β subunits, they bind to extracellular matrices (ECM) and adjacent cells, mediating cell adhesion, migration and cell–environment communication. In mammals, 18 α subunits and 8 β subunits have been identified, which form at least 24 distinct integrin receptors. Many of them have been shown to play critical roles in various biological processes, including organogenesis, tissue maintenance and repair, immunity and hemostasis. Integrin mutations in humans have been reported to cause bleeding disorders, defective host defense, skin disorders and other diseases. Many corresponding mouse models also showed distinct disease phenotypes when specific integrins were ablated, which demonstrated their functional uniqueness and significance in biology (1).
Several integrins are expressed at different stages of murine tooth development (2). Also, specific integrin knockout mice showed distinct dental phenotypes that demonstrated critical roles for integrins in tooth development. For example, Itgb1 (integrin β1) was found to be expressed in both the epithelium and mesenchyme of early tooth development (3). Conditional knockout of Itgb1 in mouse dental epithelium led to altered tooth morphogenesis. Furthermore, the lower incisors of Itgb3 (integrin β3) null mice exhibited defective iron transportation and lacked pigmentation in dental enamel (4). Most recently, Itgb6 (integrin β6) null mice were reported to have enamel malformations that mimicked human amelogenesis imperfecta (AI) (5).
AI is a collection of inherited disorders that feature enamel malformations in the absence of non-dental phenotypes (6). The term is also used to describe the enamel phenotype in syndromes. Initial efforts to discover the genetic etiologies of AI focused on discovering ECM proteins and proteases in developing enamel and the genes encoding them. These approaches led to the discoveries that defects in AMELX, ENAM, MMP20 and KLK4 caused AI, but defects in these genes accounted for 25% or less of all isolated AI cases (7,8) and none of the AI in syndromes. Genome-wide searches and whole-exome sequencing helped identify other genes involved in the etiology of isolated AI, such as FAM83H (9), WDR72 (10), C4orf26 (11) and SLC24A4 (12), and AI in syndromes such as Nance–Horan syndrome (NHS) (13), cone-rod dystrophy and AI (CNNM4) (14), and enamel renal syndrome (FAM20A) (15,16).
Mutations in human ITGA6 and ITGB4 (integrin α6β4) cause junctional epidermolysis bullosa (JEB) with pyloric atresia, which includes enamel defects as a feature (17). JEB is a collection of recessive syndromic conditions featuring skin fragility and enamel malformations caused by defects in genes encoding hemidesmosome-anchoring filament complexes, such as laminin 332 (LAMA3, LAMB3, LAMC2), COL17A1, ITGA6 and ITGB4 (18). Isolated AI has been observed in heterozygous relatives of JEB patients with mutations in COL17A1 (19,20), LAMA3 (21) and LAMB3 (22,23). Despite the growing list of genes involved in the etiology of AI, the genetic causes of only about half of all isolated AI cases can be determined by the characterization of known candidate genes (7,8).
In this study, we establish ITGB6 as an AI candidate gene by identifying disease-causing ITGB6 mutations in two AI kindreds, which is supported by the recent finding that Itgb6 null mice display enamel defects (5).
RESULTS
The proband of Family 1 was a Hispanic girl, nearly 8 years old, who presented with enamel malformations in the absence of other symptoms (Fig. 1). The proband was the only affected person in the family, suggesting the disease was caused by a recessive or sporadic mutation. A pedigree was constructed based upon an interview with the mother. There was no history of consanguinity (Fig. 1A). The patient had a mixed dentition, with the first molars, mandibular incisors and the maxillary central incisors being the only erupted permanent teeth (Fig. 1B). Anterior open-bite and class-III malocclusion were also noticed. The enamel layer of the teeth was very thin (hypoplastic) with surface roughness. The erupted teeth had undergone noticeable attrition, so the permanent molars were covered with stainless steel crowns to maintain vertical dimension. The enamel layer of unerupted teeth, particularly the bicuspids, was thin but contrasted well with dentin on radiographs (Fig. 1C). Analysis of the proband's whole-exome sequence revealed no rare homozygous or potentially compound heterozygous variants, nor any de novo variants in the known candidate genes for AI, but two heterozygous missense mutations were observed in integrin beta-6 (ITGB6). The Exon 4 mutation (g.4545G > A c.427G > A p.Ala143Thr) was inherited from the father (II:2); the Exon 6 mutation (g.27415T > A c.825T > A p.His275Gln) was inherited from the mother (II:1). This distribution indicated that both ITGB6 alleles in the proband were altered and that the pattern of inheritance was autosomal recessive. Both of the potentially disease-causing ITGB6 mutations changed amino acids that are highly conserved among vertebrates (Fig. 2) and Polyphen-2 analysis gave a score of 1.0, indicating a high probability of adversely affecting protein structure and function.
Figure 1.
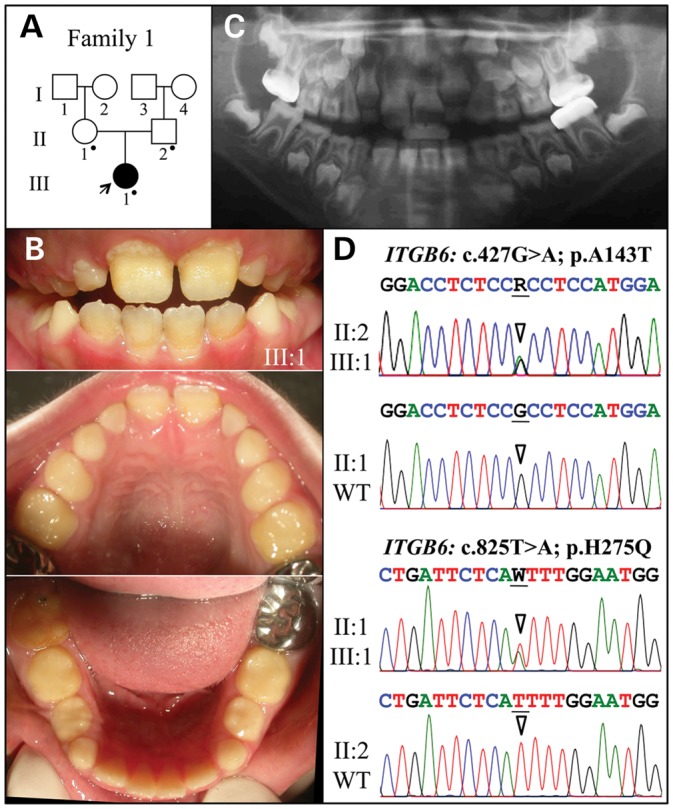
Family 1. (A) Pedigree. The proband (III:1) is the only person with enamel defects in the family. A dot marks the three study participants who donated samples for DNA sequencing. (B) The proband at almost 8 years is in the mixed dentition stage with the permanent maxillary central incisors and all mandibular incisors and first molars erupted. Oral photographs of the proband show very little enamel covering dentin and signs of rapid attrition. (C) Panoramic radiograph shows no contrasting enamel layer in erupted teeth and only a thin layer of enamel in unerupted teeth, which is characteristic of hypoplastic AI. (D) Sequencing chromatograms of heterozygous ITGB6 mutations in Exon 4 (g.4545G > A; c.427G > A; p.Ala143Thr) and Exon 6 (g.27415T > A; c.825T > A; p.His275Gln). The proband was the only compound heterozygote. The father (II:2) had only the Exon 4 mutation; the mother (II:1) had only the Exon 6 mutation. The sequence variations are named relative to the ITGB6 genomic (NC_000002.11) and cDNA (NM_000888.3) reference sequences. R = G or A; W = A or T.
Figure 2.

ITGB6 mutations causing AI. (A) ITGB6 gene structure diagram showing the positions of the three AI-causing mutations. The numbered boxes represent the 15 ITBG6 exons. The numbers below the exons correspond to the range of ITGB6 amino acids encoded by the exon. The bars are introns, which are not drawn to scale. The gene structure was determined by comparing the structure of the genomic reference sequence NC_000002.11 to the cDNA reference sequence NM_000888.3. (B) Multiple sequence alignments showing conservation of substitution sites in vertebrates.
The proband of Family 2 was a Hispanic 8-year-old boy with enamel malformations (Fig. 3). It was not clear if other members of the kindred were also affected as the mother reported her former husband and his brother had ‘yellow teeth’, but we were unable to examine these individuals (Fig. 3A). The proband was in the mixed dentition stage, with significant attrition on all posterior teeth. The maxillary incisors had a ‘Hutchisonian’ or screwdriver morphology (Fig. 3B). The secondary teeth were especially yellow-brown in color, rough surfaced and displayed very little enamel except for a thin collar around the cervical margin. A panorex radiograph revealed a thin enamel layer that contrasted with dentin (Fig. 3C), as well as agenesis of the mandibular left second molar (#18). Whole-exome sequencing identified a homozygous mutation in Exon 11 of ITGB6 (g.73664C > T c.1846C > T p.Arg616*) and a hemizygous mutation in Exon 6 of NHS (g.355444T > C c.1697T > C p.Met566Thr) in the proband. Sanger sequencing indicated that both parents were heterozygous for the ITGB6 sequence variation, whereas the mother was the carrier of the NHS variation.
Figure 3.
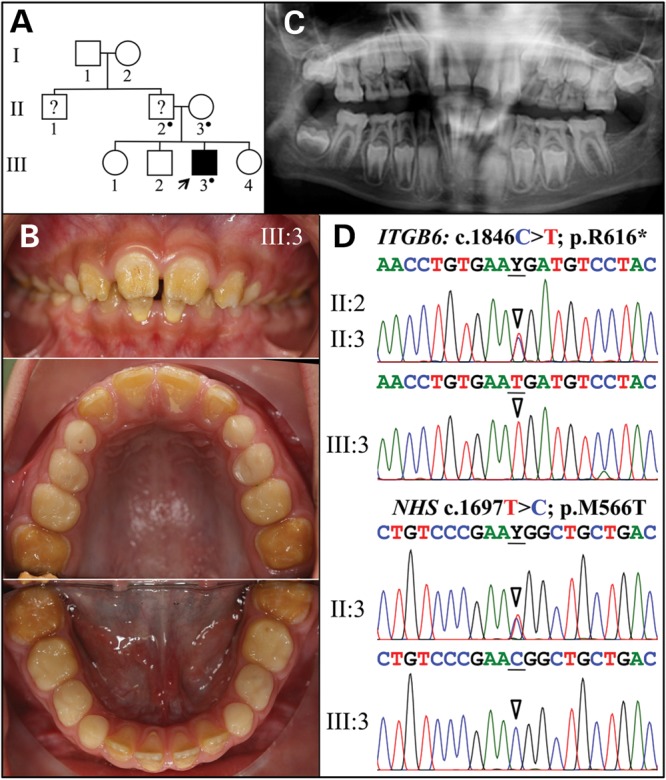
Family 2. (A) Pedigree. The proband (III:3) is the only person with known enamel defects in the family. The proband's father and uncle were reported by the mother to have ‘yellow teeth’ but their affection status is uncertain. A dot marks the three study participants who donated samples for DNA sequencing. (B) The proband at age 8 is in the mixed dentition stage with all of the permanent maxillary and mandibular incisors and first molars erupted. Oral photographs of the proband show very little enamel covering dentin and signs of rapid attrition. (C) Panoramic radiograph shows a thin, interrupted layer of contrasting enamel in some erupted teeth and a thin layer of continuous enamel in unerupted teeth. The mandibular left second molar is missing. (D) Sequencing chromatograms of ITGB6 exon 11 (top) and NHS Exon 6 (bottom). The mother (II:3) was heterozygous for both of these sequence variations. Y = T or C.
The proband was in good health, but exhibited anteverted pinnae and ptosis. He gave the impression of having average intelligence, but being undernourished (Supplementary Material, Fig. S1). The clinical and genetic findings supported a diagnosis of Nance–Horan syndrome (MIM #302350), a well-characterized X-linked syndrome that can explain all of the findings in the proband except the severity of the enamel defects. NHS is involved in actin remodeling (24) and serves key functions in the regulation of eye, tooth, brain and craniofacial development, and defects in NHS cause Nance–Horan syndrome (13). The NHS mutation in Family 2 has not been previously reported (Supplementary Material, Fig. S2), but the proband's condition is characteristic of a mild case of Nance–Horan syndrome. We examined all of the published color oral photographs from patients with NHS and found the enamel crowns to be misshapen, but white and glossy (Supplementary Material, Fig. S1) (13,25,26) In contrast, the enamel phenotype in the proband more closely resembles that of the proband in Family 1 and is attributed, in part, to the homozygous ITGB6 defect. In both families, the participating parents of the probands had dentitions that were within normal limits and showed little, if any, phenotype in the heterozygous condition (Supplementary Material, Fig. S3).
We performed immunohistochemistry on Day 14 mouse mandibular incisors using a commercial ITGB6 antibody (HPA023626; Sigma, St. Louis, MO, USA). ITGB6 signal localized to the distal membrane of differentiating ameloblasts and pre-ameloblasts, and within secretory stage ameloblasts (Fig. 4). ITGB6 was present on the ameloblast distal membrane at the onset of enamel formation and during formation of the initial enamel, and appeared to be internalized about the time ameloblasts developed their Tomes’ processes. This window of expression, along with the previous finding that Itgb6 null mice overexpressed the secreted enamel proteins amelogenin (21-fold) and enamelin (7.6-fold) but failed to make enamel rods, suggests that ITGB6 signaling regulates the onset of enamel formation by repressing the expression of enamel proteins and also regulates formation of the Tomes’ process (an ameloblast distal membrane specialization necessary for enamel rod/interrod organization). Enamel maturation is accomplished by the activities of maturation stage ameloblasts that modulate between ruffle-ended and smooth-ended forms (27,28). ITGB6 was most strongly expressed by maturation ameloblasts. ITGB6 localization along the distal membrane appeared to correlate with ameloblast modulations. An important role for ITGB6 during the maturation stage was suggested by the apparent enamel hypomineralization in Itgb6 null mice.
Figure 4.

ITGB6 immunohistochemistry of Day 14 mouse mandibular incisors. (A) The top left panel shows the cervical loop. Subsequent images move incisally. Single arrowheads mark ITGB6 signal along distal membrane of polarizing ameloblasts (note the varying levels of the nuclei and close proximity to the opposing sheet of odontoblasts). The early signal continues until the ameloblasts are fully polarized and the distance between the ameloblast and odontoblasts has increased. ITGB6 signal along the distal membrane diminishes and ends and ITGB6 appears to be internalized in secretory ameloblasts. Double arrowheads mark ITGB6 signal along the distal membrane in maturation stage ameloblasts. The signal along the distal membrane comes and goes, which presumably correlates with ameloblast modulations. (B) Higher magnification view of ITGB6 along the distal membrane of differentiating ameloblasts. (C) Higher magnification view of ITGB6 at the onset of enamel maturation. (D) Higher magnification view of ITGB6 late in maturation stage. Note: blue is DAPI staining of nuclei; red is ITGB6 immunofluorescence; Od, odontoblasts; Am, ameloblasts.
DISCUSSION
ITGB6 is one of the 8 members of mammalian integrin β family, which was first cloned from epithelial cells two decades ago. It was demonstrated to dimerize with the integrin αv (ITGAV) subunit and to bind fibronectin through RGD recognition. However, the physiological function of ITGB6 was unknown until Itgb6 knockout mice were generated and characterized (29,30). It was shown that Itgb6 null mice developed exaggerated inflammation and were protected from pulmonary fibrosis due to the lack of TGF-ß activation (31). Also, these mice developed age-related emphysema through alterations of macrophage MMP12 expression (32). Our probands did not show clinically detectable pulmonary problems as were demonstrated in Itgb6 null mice. This might be due to late onset of the lung phenotype or species differences. Despite the known expression of Itgb6 in many tissues, no other phenotypes were reported in the Itgb6 knockout mice until recently, when it was discovered that the null mice exhibited developmental enamel defects that phenocopied human AI. It had been reported that integrin αvβ6 is expressed in junctional epithelium, which links gingiva to tooth enamel and that this expression is down-regulated in human periodontal disease (33). Itgb6 null mice exhibited periodontal pockets and inflammation, suggesting that loss of integrin αvβ6 function might promote the initiation and progression of periodontal disease.
Integrins function as a heterodimer of α/β subunits, both of which are type-I transmembrane proteins with a large extracellular domain, a single-pass transmembrane domain, and, with the exception of integrin β4, a short cytoplasmic domain. The ‘head’ of the extracellular segment of some integrin β subunits contains a βI-domain (a.k.a. βA-domain, VWFA domain) that is crucial for its interaction with the α subunit and ligand (34,35). Many disease-causing human β integrin mutations are located within this domain. The ITGB6 nonsense mutation we identified in Family 2 would most likely lead to nonsense-mediated decay of the mutant transcripts and result in a complete null allele. For the ITGB6 missense mutations in Family 1, the Exon 6 mutation (p.His275Gln) substituted a histidine with a glutamine in the middle of the βI-domain. His275 is absolutely conserved among all the vertebral orthologs and human integrin β paralogs (ITGB1-8) and has been shown to reside at the α/β interface and mediate subunit interaction. Therefore, this p.His275Gln substitution in ITGB6 probably would attenuate its interaction with integrin αv and cause a loss of function. The Exon 4 mutation (p.Ala143Thr) replaced an evolutionarily conserved hydrophobic amino acid (Ala) with a polar amino acid (Thr). Ala143 is located within a specific metal ion-dependent adhesion site (MIDAS) at the beginning of βI-domain that coordinates a divalent cation (Mg2+) and is critical for ligand binding (34,35), so the p.Ala143Thr mutation would likely prevent ITGB6 from binding to ECM and result in a loss of function. While the p.Arg616* and p.His275Gln mutations of ITGB6 have not been reported in either the single nucleotide polymorphism database (dbSNP) (build 137) or 1000 Genome Project databases or NHLBI Exome Sequencing Project, the p.Ala143Thr mutation was identified in 2 out of 13 006 chromosomes in the NHLBI exome database and assigned a dbSNP ID of rs140015315 with very low (<0.001) minor allele frequency. Taken together, the genetic and structural biology evidence indicates that the ITGB6 mutations we identified are responsible for the defective enamel phenotype observed in our families.
Understanding how dental enamel forms is essential for appreciation of the fundamental principles underlying biomineralization. Historically, dental enamel formation has been understood as a process in which ameloblasts establish a well-delineated extracellular environment and control the mineralization process within it by secreting specialized proteins that initiate crystal formation through nucleation, selectively bind specific crystal faces to control crystal shape (36,37) and narrowly regulate extracellular ion concentrations, particularly calcium, to permit the growth of hydroxyapatite but exclude the deposition of competing mineral phases (38). Although there is still considerable interest in trying to grow enamel crystals ‘biomimetically’ with recombinant amelogenin, our findings here and many knockout mouse models with enamel defects strongly suggest that amelogenesis is a complicated biological process involving numerous critical components that depends upon extensive cell–matrix interactions and a mineralization front apparatus along the ameloblast distal membrane (39).
Early mutational analyses of AI kindreds employed a target gene approach that focused on candidate genes encoding enamel matrix proteins and proteases, which showed limited success. Afterwards, genome-wide approaches (linkage analysis, positional cloning and next-generation sequencing) identified more AI candidate genes. However, in this study, we included ITGB6 as an AI candidate gene based upon the phenotype of knockout mice, which demonstrated the power of reverse genetics (from genotype to phenotype) in unraveling the genetic etiology of human AI. Unfortunately, dental phenotypes are often ignored during the characterization of knockout mice. In the case of Itgb6, there was no mention of enamel defects in the original 1996 Itgb6 null mouse report and in all subsequent reports until finally they were described in 2013. Recently, the knockout mouse project (KOMP) and KOMP phenotyping plan were funded by National Human Genome Research Institute (NHGRI) and National Center for Research Resources. These projects include a concerted, centralized, high-throughput phenotyping effort to extend the scientific value of the knockout ES cell resources, which is also a precious resource for human genetics. If knockout mice could be routinely screened for apparent dental phenotypes, it could significantly advance our understanding of normal and pathological tooth development. Many tooth defects are apparent at a relatively young age and often precede the onset of potentially serious systemic conditions, as in the cases of oligodontia-colorectal cancer syndrome (MIM #608615), cone-rod dystrophy and AI (MIM #217080) and enamel renal syndrome (MIM 204690).
MATERIALS AND METHODS
Ethics statement
The human study protocol and subject consents were reviewed and approved by the Institutional Review Boards at the University of Michigan and the University of Texas Health Science Center at San Antonio. Study participants signed appropriate written consents after an explanation of their contents and after their questions about the study were answered. All procedures involving animals were reviewed and approved by the UACUC committee at the University of Michigan and all relevant guidelines were followed.
Mutational analyses
Genomic DNAs from the two probands were characterized by whole-exome sequencing (Edge BioSystems, Gaithersburg, MD, USA; or Yale Center for Genome Analysis, West Haven, CT, USA). The exome sequencing and subsequent analysis were modified from a previous report (40). Briefly, the genomic DNA was captured with NimblGen v2.0 exome capture reagent (Roche/NimblGen Inc, Madison, WI, USA) and sequenced with Illumina HiSeq 2000 for 75 base paired-end reads. Reads were aligned to human reference genome hg19 using ELAND v2. Single nucleotide variants and short insertions and deletions (indels) were called using SAMtools. The called variants were annotated using an in-house script. Potentially, disease-causing sequence variations were confirmed in the probands and all participating family members by Sanger sequencing.
Tissue preparation and immunohistochemistry
All procedures were carried out at 4°C, unless otherwise indicated. Day 14 mouse heads were dissected off skin, immersed in 4% paraformaldehyde fixative overnight, washed in PBS 4–5× (every 0.5–1 h) and decalcified by immersion in 1 liter of 4.13% disodium ethylenediaminetetraacetic acid (EDTA, pH 7.3) with agitation. The EDTA solution was changed every other day for 30 days. After decalcification, the tissues were embedded in paraffin. The blocks were sectioned at 5 μm thickness, and the sections were deparaffinized and heat treated (95°C) with antigen retrieval solution (ab973, Abcam, Cambridge, MA, USA) for 30 min. The slides were then rinsed with PBT buffer (0.1% Triton X-100 in PBS), blocked with 5% sheep serum (S-22, Chemicon, Billerica, MA, USA) in PBT for 30 min at room temperature, and serial sections were incubated overnight with anti-ITGB6 antibody (1:100, HPA023626, Sigma). The sections were washed with PBT for 15 min and incubated for 30 min at room temperature in solutions containing anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 594 (1:500, A11012, Invitrogen). Sections were rinsed in PBT for 15 min, mounted with ProLong® Gold antifade reagent with DAPI (P-36931, Invitrogen) and examined using an Olympus BX51 with fluorescence attachments and photographed using an Olympus DP71 camera with DP controller and manager software.
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by NIDCR/NIH research grant DE015846 and by grants from the Bio & Medical Technology Development Program (2011-0027790), the Science Research Center grant to Bone Metabolism Research Center (2012-0000487) by the Korea Research Foundation grant funded by the Korean Government (MEST).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the participants in this study for their cooperation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. doi:10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Salmivirta K., Gullberg D., Hirsch E., Altruda F., Ekblom P. Integrin subunit expression associated with epithelial-mesenchymal interactions during murine tooth development. Dev. Dyn. 1996;205:104–113. doi: 10.1002/(SICI)1097-0177(199602)205:2<104::AID-AJA2>3.0.CO;2-M. doi:10.1002/(SICI)1097-0177(199602)205:2<104::AID-AJA2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Chen B., Goodman E., Lu Z., Bandyopadhyay A., Magraw C., He T., Raghavan S. Function of beta1 integrin in oral epithelia and tooth bud morphogenesis. J. Dent. Res. 2009;88:539–544. doi: 10.1177/0022034509338008. doi:10.1177/0022034509338008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida T., Kumashiro Y., Iwata T., Ishihara J., Umemoto T., Shiratsuchi Y., Kawashima N., Sugiyama T., Yamato M., Okano T. Requirement of integrin beta3 for iron transportation during enamel formation. J. Dent. Res. 2012;91:1154–1159. doi: 10.1177/0022034512462722. doi:10.1177/0022034512462722. [DOI] [PubMed] [Google Scholar]
- 5.Mohazab L., Koivisto L., Jiang G., Kytomaki L., Haapasalo M., Owen G.R., Wiebe C., Xie Y., Heikinheimo K., Yoshida T., et al. Critical role for alphavbeta6 integrin in enamel biomineralization. J. Cell Sci. 2013;126:732–744. doi: 10.1242/jcs.112599. doi:10.1242/jcs.112599. [DOI] [PubMed] [Google Scholar]
- 6.Witkop C.J., Jr Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J. Oral Pathol. 1989;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. doi:10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 7.Chan H.C., Estrella N.M., Milkovich R.N., Kim J.W., Simmer J.P., Hu J.C. Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur. J. Oral Sci. 2011;119:311–323. doi: 10.1111/j.1600-0722.2011.00857.x. doi:10.1111/j.1600-0722.2011.00857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright J.T., Torain M., Long K., Seow K., Crawford P., Aldred M.J., Hart P.S., Hart T.C. Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells Tissues Organs. 2011;194:279–283. doi: 10.1159/000324339. doi:10.1159/000324339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J.W., Lee S.K., Lee Z.H., Park J.C., Lee K.E., Lee M.H., Park J.T., Seo B.M., Hu J.C., Simmer J.P. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am. J. Hum. Genet. 2008;82:489–494. doi: 10.1016/j.ajhg.2007.09.020. doi:10.1016/j.ajhg.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Sayed W., Parry D.A., Shore R.C., Ahmed M., Jafri H., Rashid Y., Al-Bahlani S., Al Harasi S., Kirkham J., Inglehearn C.F., et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am. J. Hum. Genet. 2009;85:699–705. doi: 10.1016/j.ajhg.2009.09.014. doi:10.1016/j.ajhg.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parry D.A., Brookes S.J., Logan C.V., Poulter J.A., El-Sayed W., Al-Bahlani S., Al Harasi S., Sayed J., Raif el M., Shore R.C., et al. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am. J. Hum Genet. 2012;91:565–571. doi: 10.1016/j.ajhg.2012.07.020. doi:10.1016/j.ajhg.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parry D.A., Poulter J.A., Logan C.V., Brookes S.J., Jafri H., Ferguson C.H., Anwari B.M., Rashid Y., Zhao H., Johnson C.A., et al. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am. J. Hum. Genet. 2013;92:307–312. doi: 10.1016/j.ajhg.2013.01.003. doi:10.1016/j.ajhg.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burdon K.P., McKay J.D., Sale M.M., Russell-Eggitt I.M., Mackey D.A., Wirth M.G., Elder J.E., Nicoll A., Clarke M.P., FitzGerald L.M., et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 2003;73:1120–1130. doi: 10.1086/379381. doi:10.1086/379381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parry D.A., Mighell A.J., El-Sayed W., Shore R.C., Jalili I.K., Dollfus H., Bloch-Zupan A., Carlos R., Carr I.M., Downey L.M., et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am. J. Hum. Genet. 2009;84:266–273. doi: 10.1016/j.ajhg.2009.01.009. doi:10.1016/j.ajhg.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Sullivan J., Bitu C.C., Daly S.B., Urquhart J.E., Barron M.J., Bhaskar S.S., Martelli-Junior H., Dos Santos Neto P.E., Mansilla M.A., Murray J.C., et al. Whole-exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am. J. Hum. Genet. 2011;88:616–620. doi: 10.1016/j.ajhg.2011.04.005. doi:10.1016/j.ajhg.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S.K., Aref P., Hu Y., Milkovich R.N., Simmer J.P., El-Khateeb M., Daggag H., Baqain Z.H., Hu J.C. FAM20A mutations can cause enamel-renal syndrome (ERS) PLoS Genet. 2013;9:e1003302. doi: 10.1371/journal.pgen.1003302. doi:10.1371/journal.pgen.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pulkkinen L., Rouan F., Bruckner-Tuderman L., Wallerstein R., Garzon M., Brown T., Smith L., Carter W., Uitto J. Novel ITGB4 mutations in lethal and nonlethal variants of epidermolysis bullosa with pyloric atresia: missense versus nonsense. Am. J. Hum. Genet. 1998;63:1376–1387. doi: 10.1086/302116. doi:10.1086/302116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Intong L.R., Murrell D.F. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin. Dermatol. 2012;30:70–77. doi: 10.1016/j.clindermatol.2011.03.012. doi:10.1016/j.clindermatol.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 19.McGrath J.A., Gatalica B., Li K., Dunnill M.G., McMillan J.R., Christiano A.M., Eady R.A., Uitto J. Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am. J. Pathol. 1996;148:1787–1796. [PMC free article] [PubMed] [Google Scholar]
- 20.Murrell D.F., Pasmooij A.M., Pas H.H., Marr P., Klingberg S., Pfendner E., Uitto J., Sadowski S., Collins F., Widmer R., et al. Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J. Invest. Dermatol. 2007;127:1772–1775. doi: 10.1038/sj.jid.5700766. [DOI] [PubMed] [Google Scholar]
- 21.Yuen W.Y., Pasmooij A.M., Stellingsma C., Jonkman M.F. Enamel defects in carriers of a novel LAMA3 mutation underlying epidermolysis bullosa. Acta Derm. Venereol. 2012;92:695–696. doi: 10.2340/00015555-1341. [DOI] [PubMed] [Google Scholar]
- 22.Poulter J.A., El-Sayed W., Shore R.C., Kirkham J., Inglehearn C.F., Mighell A.J. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur. J. Hum. Genet. 2013;1:76. doi: 10.1038/ejhg.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.-W., Seymen F., Lee K.-E., Ko J., Yildirim M., Tuna E.B., Gencay K., Shin T.J., Kyun H.-K., Simmer J.P., et al. LAMB3 mutations causing autosomal dominant amelogenesis imperfecta. J. Dent. Res. 2013;92:899–904. doi: 10.1177/0022034513502054. doi:10.1177/0022034513502054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks S.P., Coccia M., Tang H.R., Kanuga N., Machesky L.M., Bailly M., Cheetham M.E., Hardcastle A.J. The Nance-Horan syndrome protein encodes a functional WAVE homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology. Hum. Mol. Genet. 2010;19:2421–2432. doi: 10.1093/hmg/ddq125. doi:10.1093/hmg/ddq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coccia M., Brooks S.P., Webb T.R., Christodoulou K., Wozniak I.O., Murday V., Balicki M., Yee H.A., Wangensteen T., Riise R., et al. X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum. Mol. Genet. 2009;18:2643–2655. doi: 10.1093/hmg/ddp206. doi:10.1093/hmg/ddp206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tug E., Dilek N.F., Javadiyan S., Burdon K.P., Percin F.E. A Turkish family with Nance-Horan syndrome due to a novel mutation. Gene. 2013;6:00363–00366. doi: 10.1016/j.gene.2013.03.094. [DOI] [PubMed] [Google Scholar]
- 27.Warshawsky H., Smith C.E. Morphological classification of rat incisor ameloblasts. Anat. Rec. 1974;179:423–446. doi: 10.1002/ar.1091790403. doi:10.1002/ar.1091790403. [DOI] [PubMed] [Google Scholar]
- 28.Josephsen K., Fejerskov O. Ameloblast modulation in the maturation zone of the rat incisor enamel organ. A light and electron microscopic study. J. Anat. 1977;124:45–70. [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X.Z., Wu J.F., Cass D., Erle D.J., Corry D., Young S.G., Farese R.V., Jr, Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J. Cell Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. doi:10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X., Wu J., Zhu W., Pytela R., Sheppard D. Expression of the human integrin beta6 subunit in alveolar type II cells and bronchiolar epithelial cells reverses lung inflammation in beta6 knockout mice. Am. J. Respir. Cell Mol. Biol. 1998;19:636–642. doi: 10.1165/ajrcmb.19.4.3293. doi:10.1165/ajrcmb.19.4.3293. [DOI] [PubMed] [Google Scholar]
- 31.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. doi:10.1016/S0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 32.Morris D.G., Huang X., Kaminski N., Wang Y., Shapiro S.D., Dolganov G., Glick A., Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 2003;422:169–173. doi: 10.1038/nature01413. doi:10.1038/nature01413. [DOI] [PubMed] [Google Scholar]
- 33.Ghannad F., Nica D., Fulle M.I., Grenier D., Putnins E.E., Johnston S., Eslami A., Koivisto L., Jiang G., McKee M.D., et al. Absence of alphavbeta6 integrin is linked to initiation and progression of periodontal disease. Am. J. Pathol. 2008;172:1271–1286. doi: 10.2353/ajpath.2008.071068. doi:10.2353/ajpath.2008.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong J.P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D.L., Joachimiak A., Goodman S.L., Arnaout M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. doi:10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong J.P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S.L., Arnaout M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. doi:10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 36.Simmer J.P., Fincham A.G. Molecular mechanisms of dental enamel formation. Crit. Rev. Oral Biol. Med. 1995;6:84–108. doi: 10.1177/10454411950060020701. doi:10.1177/10454411950060020701. [DOI] [PubMed] [Google Scholar]
- 37.Aoba T. Recent observations on enamel crystal formation during mammalian amelogenesis. Anat. Rec. 1996;245:208–218. doi: 10.1002/(SICI)1097-0185(199606)245:2<208::AID-AR8>3.0.CO;2-S. doi:10.1002/(SICI)1097-0185(199606)245:2<208::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 38.Moreno E.C., Aoba T. Calcium binding in enamel fluid and driving force for enamel mineralization in the secretory stage of amelogenesis. Adv. Dent. Res. 1987;1:245–251. doi: 10.1177/08959374870010021301. [DOI] [PubMed] [Google Scholar]
- 39.Simmer J.P., Richardson A.S., Hu Y.Y., Smith C.E., Ching-Chun Hu J. A post-classical theory of enamel biomineralization... and why we need one. Int. J. Oral Sci. 2012;4:129–134. doi: 10.1038/ijos.2012.59. doi:10.1038/ijos.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloglu A., Ozen S., Sanjad S., et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. doi:10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.