

Abstract
The strategic importance of the genome sequence of the gray, short-tailed opossum, Monodelphis domestica, accrues from both the unique phylogenetic position of metatherian (marsupial) mammals and the fundamental biologic characteristics of metatherians that distinguish them from other mammalian species. Metatherian and eutherian (placental) mammals are more closely related to one another than to other vertebrate groups, and owing to this close relationship they share fundamentally similar genetic structures and molecular processes. However, during their long evolutionary separation these alternative mammals have developed distinctive anatomical, physiologic, and genetic features that hold tremendous potential for examining relationships between the molecular structures of mammalian genomes and the functional attributes of their components. Comparative analyses using the opossum genome have already provided a wealth of new evidence regarding the importance of noncoding elements in the evolution of mammalian genomes, the role of transposable elements in driving genomic innovation, and the relationships between recombination rate, nucleotide composition, and the genomic distributions of repetitive elements. The genome sequence is also beginning to enlarge our understanding of the evolution and function of the vertebrate immune system, and it provides an alternative model for investigating mechanisms of genomic imprinting. Equally important, availability of the genome sequence is fostering the development of new research tools for physical and functional genomic analyses of M. domestica that are expanding its versatility as an experimental system for a broad range of research applications in basic biology and biomedically oriented research.
Compilation and publication of the genome sequence of the gray short-tailed opossum, Monodelphis domestica, a metatherian (marsupial) mammal (Mikkelsen et al. 2007b), create unique opportunities for investigating the evolutionary processes that have shaped the structural and functional attributes of mammalian and other vertebrate genomes. These opportunities arise from both the unique phylogenetic position of metatherians in the vertebrate scheme and some fundamental characteristics of metatherians that distinguish them from other mammalian species.
Modern mammals comprise three major lineages (Fig. 1). Judged from multiple nuclear sequence and mtDNA alignments, the most closely related of these, the Metatheria and Eutheria (placental mammals), diverged from their most recent common ancestor ∼173–190 million years ago (Mya) (Kumar and Hedges 1998; Woodburne et al. 2003; van Rheede et al. 2006). The other major clade, the Prototheria, of which the egg-laying monotremes (platypus and four species of echidnas) are the only living representatives, branched off 20–35 million years (Myr) prior to the metatherian–eutherian divergence (Phillips and Penny 2003; Woodburne et al. 2003; van Rheede et al. 2006).2 By way of reference, the separation of mammals and birds occurred at least 310 Mya (discussed by Kumar and Hedges 1998; Hedges and Kumar 2004; Glazko et al. 2005; Donoghue and Benton 2007), while the most distantly related eutherian mammals appear to have shared a common ancestor no more than ∼95–112 Mya (Springer et al. 2003; Woodburne et al. 2003; Bininda-Emonds et al. 2007; Donoghue and Benton 2007; Murphy et al. 2007). The metatherian–eutherian relationship thus represents a midpoint in age relative to mammalian and nonmammalian vertebrates but is much older than the deepest division between living eutherian species (Fig. 1). This phylogenetic sister-group relationship makes metatherians the most appropriate outgroup for assessing the relative antiquity or novelty of the molecular and genetic changes that have occurred during the diversification of eutherian mammals.
Figure 1.
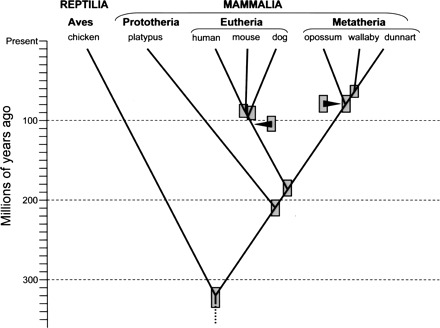
Phylogenetic splitting topology and approximate ages for mammalian divergences discussed in this article. Shaded boxes indicate approximate ranges for divergence date estimates extracted from multiple literature sources cited in the main text and footnote 2. Approximate dates for the earliest (deepest) divergences among extant species within the eutherian and metatherian lineages are indicated by arrowheads. As for the branching points, the shaded boxes associated with arrowheads indicate approximate range estimates for these basal divergence dates.
Consequent of their close common ancestry, metatherians and eutherians share basic genetic structures and molecular processes that reflect elemental and ancient mammalian characteristics; yet as a result of their long separation, each group has evolved its own distinctive morphologic, physiologic, and genetic variations on these elemental patterns. This combination of conserved and phylogenetically distinctive characteristics holds enormous potential for examining relationships between the molecular structures and functional attributes of mammalian genomes, especially for elucidating how variations in underlying genomic structural elements contribute to differences in gene regulation, expression, and function. In this light, the value of the opossum genome sequence for comparative analysis of structural evolution among vertebrate genomes is obvious. In addition, however, results from the genome project are also providing new tools for examining the biology of metatherian species, which are of interest both for their biomedical applications and in their own right as a fascinating group of mammals that has taken an alternative pathway to achieve a distinctive and highly successful variant on the fundamental mammalian way of life. This article summarizes key findings from the opossum genome project and discusses their general implications for furthering our ability to discover the elementary components of vertebrate genome structure and function, and for discerning how these components evolve. It also considers the use of genome data for better understanding of the basic biology of M. domestica and other metatherian species.
Metatherian mammals
Extant metatherians comprise ∼6% of the more than 5400 mammalian species currently recognized (Wilson and Reeder 2005). They are found on both American continents (∼96 species) and in Australasia (∼237 species), and range in size from tiny mouse-like predators of the family Dasyuridae to large grazing and browsing herbivores, such as kangaroos and other members of the Macropodidae. Metatherian species populate cool temperate, hot xeric, and tropical habitats, but there are no polar, alpine, or marine forms.
Of the many features that distinguish metatherian and eutherian mammals, the most salient are their reproductive characteristics. Both groups give birth to live young, but metatherian gestation is remarkably brief, and the young are born at an extremely early stage of development relative to eutherian infants (for reviews, see Behringer et al. 2006; Selwood and Johnson 2006). While the newborn has well-developed jaws and forelimbs, and an operational gut, its hind limbs are little more than buds (Fig. 2), it has no adaptive immune function, and its nervous system is rudimentary and shows remarkable powers of regeneration (references in Samollow 2006, and discussed beyond). Newborn metatherians complete the majority of their “fetal” development external to the mother, attached to a teat, and receiving nutrition and protective antibodies from their mother’s milk. In many but not all species this extended external phase of development takes place in a protective pouch, the marsupium, from which the designation “marsupial” derives. Placental attachments develop in all metatherians and involve the same general suite of extraembryonic membranes that form eutherian placentae (hence, the term “placental mammal” is a confusing descriptor when reserved exclusively for eutherians) but, as might be expected from the short gestation period, the magnitude and elaboration of placental structure is generally less extensive in metatherians than in eutherian species.
Figure 2.
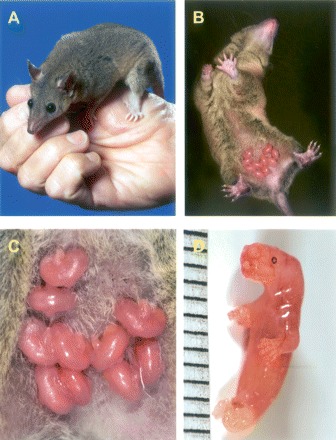
Monodelphis domestica. (A) Adult female. (B) Female with a litter of 10 pups. The newborns are ∼36 h postpartum age. Note that M. domestica does not possess a pouch. (C) Detail of litter seen in panel B. (D) Newborn, <12 h postpartum age. Scale is 1 mm between marks. (Photos: Larry Wadsworth, TAMU Media Resources.)
These distinctive reproductive characteristics have attracted considerable interest in the use of metatherian models for examining the earliest stages of embryonic development, sexual differentiation and development, hormonal effects on reproductive behavior, estrus cycling, lactation, and the ontogeny and evolution of placental structure and function. The underdeveloped state at birth has also elicited interest in the development of immune competence in newborns and has provided a model system for studying the evolution of developmental patterning and timing, growth, and maturation of neuromotor systems, and the capability of regeneration of damaged central nervous system structures.
The gray, short-tailed opossum
The rationale for targeting M. domestica for the first metatherian genome sequencing effort was based primarily on practical considerations of its status as a fully developed, laboratory research animal. M. domestica is a South American opossum (family Didelphidae) that is used as an experimental model in many basic, comparative, and biomedically oriented research applications (Samollow 2006). It is a small and docile animal (adult size ∼70–160 g), which grows rapidly (sexual maturity at 5–6 mo), is highly prolific (litter size range 1–13; average litter size ∼8; up to three litters per year), breeds year-round, and is easily maintained in standard rodent cages using commercial feed. M. domestica has been raised in pedigreed laboratory colonies for almost 30 yr (VandeBerg 1990, 1999; VandeBerg and Robinson 1997) and, owing to its favorable physical and reproductive characteristics, is the most widely used laboratory-bred research metatherian in the world. Although other metatherian species are used in various research applications to which they are better suited than M. domestica, especially in Australia, none combine the minimal space requirements, ease of husbandry, prolificacy, lineage continuity, availability, and transportability of M. domestica. Further details and references concerning opossum characteristics and husbandry, an international listing of colony locations, and an overview of recent research applications using this species can be found elsewhere (Samollow 2006). The potential impact of genome sequence data on several of these research areas is discussed in later sections of this article.
Characteristics of the opossum genome
The main opossum genome release (Mikkelsen et al. 2007b) and several focused companion articles have described the structure of the opossum genome in detail and have reported findings from comparisons of its components with those of eutherian mammals for which full-genome sequences are available. In addition to providing a window on the general characteristics of a metatherian genome, these initial analyses (discussed below) provide stunning evidence regarding the importance of noncoding elements in the evolution of mammalian genomes and the contributions of transposable elements in sculpting this structure. They also provide support for current theories regarding the relationship between meiotic recombination rate and characteristics of regional and whole-genome nucleotide composition, and the distributions of repetitive element families across the mammalian genomic landscape.
Genome assembly characteristics
Metatherian genomes are similar in size to those of eutherian mammals but are generally packaged into fewer chromosomes (see discussions by Samollow and Graves 1998; Graves and Westerman 2002; Ferreri et al. 2004). In this regard the opossum genome is typical, comprising ∼3.6–3.7 pg (3.52–3.62 Gb) of DNA per haploid genome (Mikkelsen et al. 2007b) distributed among a haploid complement of nine large autosomes and an X (or Y) sex chromosome (Pathak et al. 1993).
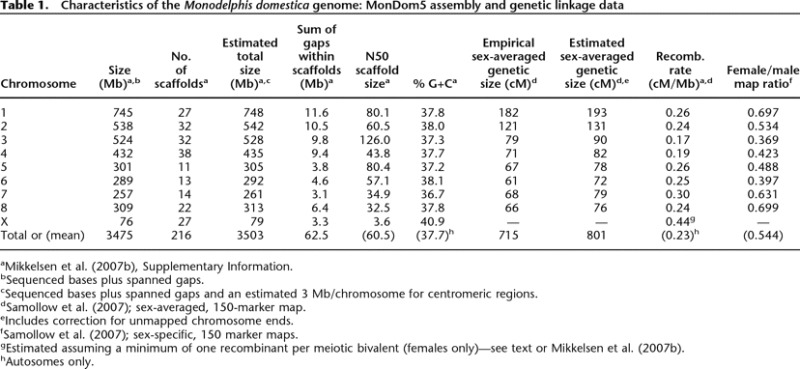
The most recent version of the opossum genome sequence assembly, MonDom5, is a high-quality, 6.76× depth (redundant) draft resulting from analysis of 38.8 million whole-genome-shotgun sequence reads derived from a partially inbred female M. domestica. Details regarding assembly statistics and quality are given in the main publication (Mikkelsen et al. 2007b) and its online supporting materials. In brief, the assembly is very good and compares favorably with those recently sequenced from other mammalian species. For example, the theoretical base error rate of one in 10,000 is on par with that of human finished sequence and equal to that of the recent draft of the dog genome (Lindblad-Toh et al. 2005). In addition, ∼98% of the opossum sequence is contained in regions with very low probability of structural errors, suggesting that very few genes should contain gaps or assembly errors. More than 50% of the genome is present in large, uninterrupted contigs of ∼108 kb or larger, which are anchored in scaffolds of ∼60 Mb or larger (Table 1). At least 2% of the assembly consists of small spanned sequence gaps that lie within the scaffolds, which is twice the corresponding proportion for the dog genome. However, the N50 scaffold size for the opossum genome (Table 1) is >30% larger than for dog, indicating substantially higher within-scaffold contiguity for the opossum assembly. Approximately 97% of the opossum assembly lies in 216 individual scaffolds that could be anchored to chromosomes (Duke et al. 2007) but could not be joined with one another without additional sequencing effort (Table 1). The average number of anchored scaffolds per chromosomes is 24. These scaffolds flank 205 unspanned (inter-scaffold) assembly gaps of relatively small but undetermined size. This large number of scaffolds per chromosome is due in part to the enormity of the eight autosomes, the smallest of which (chromosome 7, 261 Mb) is larger than any chromosome sequenced from any other vertebrate to date. Approximately 97% of the considerably smaller dog genome (2.45 Gb) also lies within anchored scaffolds, but with a haploid complement of 39 small chromosomes (mean = 61 Mb) and only 2.2 scaffolds per chromosome (Lindblad-Toh et al. 2005), the dog assembly has substantially higher overall contiguity than the opossum assembly. By comparison, then, the MonDom5 assembly has excellent accuracy, can be expected to be reasonably free of large-scale structural errors, and has very good, if not outstanding, contiguity.
Table 1.
Characteristics of the Monodelphis domestica genome: MonDom5 assembly and genetic linkage data
aMikkelsen et al. (2007b), Supplementary Information.
bSequenced bases plus spanned gaps.
cSequenced bases plus spanned gaps and an estimated 3 Mb/chromosome for centromeric regions.
dSamollow et al. (2007); sex-averaged, 150-marker map.
eIncludes correction for unmapped chromosome ends.
fSamollow et al. (2007); sex-specific, 150 marker maps.
gEstimated assuming a minimum of one recombinant per meiotic bivalent (females only)—see text or Mikkelsen et al. (2007b).
hAutosomes only.
Nucleotide composition and recombination
The base compositional features of the opossum genome provide support for recent concepts regarding the influence of meiotic recombination on overall genome sequence characteristics. The opossum genome exhibits unusually low overall proportion of G and C nucleotides (G+C). The autosomes average only 37.7% G+C, compared to values of 40.9%–41.8% for the autosomal portions among human, dog, mouse, and chicken genomes (Table 2 of Mikkelsen et al. 2007b), and 45% in platypus (http://www.ensembl.org/Ornithorhynchus_anatinus/index.html). This pattern is especially pronounced for CpG dinucleotide content, wherein opossum autosomes (0.9%) show half (or less) the average CpG content of other amniote autosomes (1.7%–2.2%). In contrast, G+C and CpG contents of the diminutive (79 Mb) opossum X chromosome (40.9% and 1.4%, respectively) are higher than its autosomes, which is the reverse situation found in eutherian species. In fact, G+C content of the opossum X chromosome exceeds levels for the X chromosomes of all other sequenced amniotes.
Table 2.
Imprinting status of metatherian homologs of eutherian imprinted genes
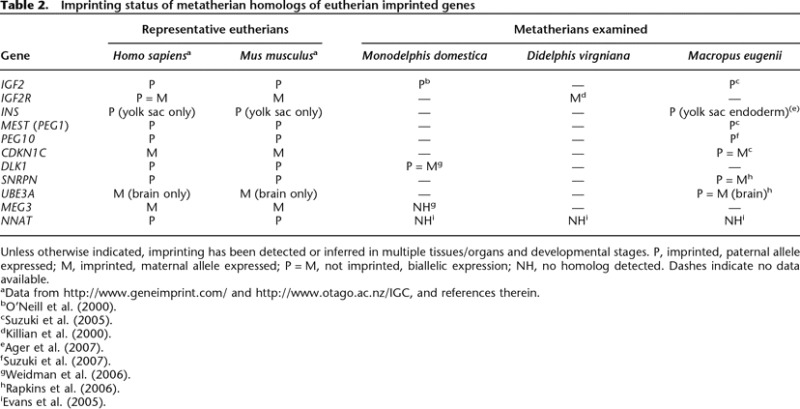
Unless otherwise indicated, imprinting has been detected or inferred in multiple tissues/organs and developmental stages. P, imprinted, paternal allele expressed; M, imprinted, maternal allele expressed; P = M, not imprinted, biallelic expression; NH, no homolog detected. Dashes indicate no data available.
aData from http://www.geneimprint.com/ and http://www.otago.ac.nz/IGC, and references therein.
Vigorous debate over the origin and dynamics of local base composition differences (for reviews, see Belle et al. 2004; Cohen et al. 2005; Duret et al. 2006b; Gu and Li 2006; Bernardi 2007) has culminated in a model proposing that the G+C content of a chromosomal region is determined largely by the interplay of two opposing mutational processes (Duret et al. 2006b). One is reduction of G+C content through a general GC to AT mutation bias, with methylated cytosines in CpG dinucleotides especially prone to mutation through deamination to thymine. In the absence of an opposing process, such as selection at specific sites, this mutational bias should lead to the inexorable decline of G+C content, and especially CpG dinucleotides, across the genome. Such an opposing process is postulated in the form of (G+C)-biased gene conversion, which increases G+C content through biased AT to GC mismatch repair during recombination-mediated gene conversion events (Hogstrand and Bohme 1999; Galtier et al. 2001). Under this model of opposing mutational pressures, there should be a positive correlation between recombination rate and G+C content, both on a regional basis, and when averaged across entire chromosomes or the genome as a whole.
Consistent with this model, M. domestica has one of the lowest recombination rates among vertebrates. The published M. domestica linkage map, comprising 150 loci in eight autosomal linkage groups and spanning 86%–89% of the physical genome (Samollow et al. 2007), yields a sex-averaged total map-length estimate of only 866 centiMorgans (cM), which is considerably shorter than the 1630 cM and 3763 cM estimates for mouse (Shifman et al. 2006) and human (Kong et al. 2004), respectively. This corresponds to an overall sex-averaged recombination rate of ∼0.23 cM/Mb (Table 1), a rate much lower than that for mouse (Shifman et al. 2006), which, at ∼0.63 cM/Mb, has the lowest rate known among non-metatherian vertebrates. Based on the principle that each chromosome pair must undergo at least one meiotic recombinational event to ensure proper disjunction (e.g., Paques and Haber 1999; Page and Hawley 2003; Petronczki et al. 2003), Mikkelsen et al. (2007b) estimated a minimum recombination rate of 0.44 cM/Mb for the opossum X chromosome, a rate substantially higher than that for the autosomes. This higher rate is consistent with the higher G+C and CpG contents of the X relative to the autosomes, although it alone cannot explain why the opossum X should have G+C levels equivalent to those of eutherian autosomes.
Another characteristic of opossum recombination that must be considered with regard to nucleotide composition is the huge disparity in sex-specific recombination rates. The length of the female linkage map is only about half that of the male map, suggesting that recombination is much less frequent in oogenesis than in spermatogenesis (Samollow et al. 2004, 2007). This sexual dimorphism in recombination rate is the largest observed to date for any mammalian species, and runs counter to that observed for all eutherian and nonmammalian vertebrates for which extensive linkage data exist, wherein recombination is either similar in both sexes or is biased toward higher female recombination rates (sheep also appear exceptional in this regard, but there is a slight possibility that the longer male sheep map results from an unusual pedigree structure in the mapping panel [J. Maddox, pers. comm.]). Lower female recombination was observed on every chromosome, resulting in an overall female to male (F/M) map ratio of 0.54 (Table 1). By comparison, F/M ratios for human and mouse are strongly female biased at 1.64 and 1.31, respectively (Kong et al. 2004; Shifman et al. 2006), whereas overall recombination rates in cattle (Bos taurus) are sex equal (Ihara et al. 2004). Evidence for reduced female recombination has also been detected in two Australian metatherians, the fat-tailed dunnart, Sminthopsis crassicaudata (Bennett et al. 1986), and tammar wallaby, Macropus eugenii (Zenger et al. 2002), suggesting that this might be a common metatherian attribute.
Cytologic studies of both M. domestica and S. crassicaudata meiotic cells (Bennett et al. 1986; Hayman et al. 1988) showed that chiasmata in female metaphase I nuclei are concentrated near the ends of chromosomes, whereas those of males were more evenly distributed. If this physical pattern mirrors the distribution of actual chromosomal exchange events, then we should expect female recombination, as assessed by map distance (cM), to outpace male recombination in subtelomeric regions. This is exactly the reverse of eutherian species, in which the F/M ratio drops near or below unity in subtelomeric regions, due largely to a dramatic rise in male recombination rates in these regions (e.g., Broman et al. 1998; Kong et al. 2002). We currently have insufficient linkage data in these regions to examine this hypothesis in detail, but the F/M ratio does appear to rise at the termini of some of the current M. domestica linkage-group maps, as is seen in the examples in Figure 3. Additional randomly selected markers have been genotyped for inclusion in the opossum linkage map, and recent analysis using 207 markers yielded an increased F/M ratio of 0.60, with only minimal increase in overall sex-averaged map length (P.B. Samollow, unpubl.). This is consistent with the expectation of increasing female recombination as new markers fall beyond previously mapped linkage-group ends. It must be stressed, however, that, regardless of increases in female recombination rate in subtelomeric regions, support for the proposed relationship between recombination rate and G+C content in these regions in M. domestica (Mikkelsen et al. 2007b) requires that the combined male and female recombination rate be higher in these regions than in interstitial ones. Similarly, it must be shown that the opossum X chromosome undergoes substantially more recombination than the autosomes. Based on a catalog of nearly 1.29 million potential single-nucleotide polymorphism (SNP) sites discovered in connection with the opossum genome project (http://www.broad.mit.edu/mammals/opossum/snps.html), developing a comprehensive panel of mapping markers across the X chromosome and all regions of the autosomes is now practical and will enable closer examination of the relationship between recombination rate, G+C composition, and neutral substitution rate.
Figure 3.
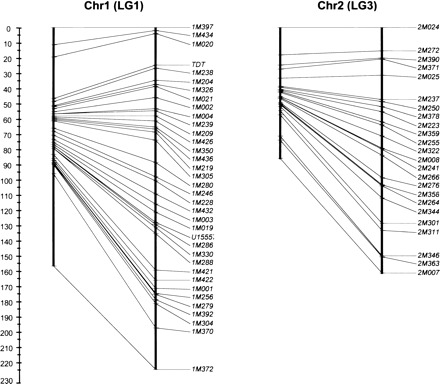
Sex-specific linkage maps for chromosomes 1 and 2 (linkage groups 1 and 3) of Monodelphis domestica. For each pair of maps, the female map is to the left. Scale is in centiMorgans (cM) with zero corresponding to the p terminus of the chromosome. Information for individual map markers may be found in Samollow et al. (2007).
In a broader context, M. domestica provides a unique model system with which to explore the roles of large-scale genome structure, local DNA sequence characteristics, variation in chromatin configuration, and sex in determining rates and patterns of meiotic recombination (for reviews, see Morelli and Cohen 2005; Hunt 2006; for more general reviews, see Petronczki et al. 2003; Kauppi et al. 2004; Ivanovska and Orr-Weaver 2006). For example, if sex-specific differences in chromatin structure, nucleosome distribution, cohesin complex localization, and other determinants of differential vulnerability to double-strand breaks and their ultimate resolution (reciprocal exchange vs. nonexchange) are generally important in shaping sexually disparate patterns and rates of recombination in vertebrates, then they can be expected to have predictably different (reversed) distributions in the meiotic (or premeiotic) cells of M. domestica as compared with those of a eutherian model species, such as mouse. M. domestica, then, embodies a natural experimental system for testing hypotheses about chromosomal characteristics and molecular processes that differentially promote or inhibit chiasma formation and distribution between the sexes.
Genome size, repetitive elements, and segmental duplications
The inflated size of the opossum genome is attributable largely to the proliferation of interspersed repeat family elements (Mikkelsen et al. 2007b). The proportion of the genome occupied by these elements (∼52%) substantially exceeds that in other amniote genomes (e.g., 38.6% in mouse and 44.8% human), and, when repetitive element contents are discounted, the total euchromatic size of the opossum genome differs little from those of eutherians. In a detailed companion article, Gentles et al. (2007) reported the abundances of nearly 500 repetitive element families, the majority of which were unknown before examination of the opossum genome. Members of the LINE1 family of non-long-terminal-repeat (non-LTR) retroposons are the most prominent class of interspersed elements, comprising ∼20% of the opossum genome, and all non-LTR retroposon family elements account for 29%, a far higher proportion than in mouse or human genomes. It was suggested (Mikkelsen et al. 2007b) that this accumulation might be related to low recombination rate because of a negative correlation between LINE1 density and recombination rate seen in model eutherian species (e.g., Jensen-Seaman et al. 2004; Myers et al. 2005; Shifman et al. 2006), but it remains to be determined if such a correlation exists for M. domestica. Other abundant repeat families included endogenous retrovirus (ERV)-derived elements (∼10.6% of the genome) and SINEs (∼10.4% of the genome) (Gentles et al. 2007). In a study of M. domestica from different geographic isolates, Gu et al. (2007) found evidence of very recent SINE1 family expansions, as well as genomic distributions of old and young SINE1 elements, that were consistent with a biased gene conversion model shaping local G+C content, especially in gene-rich regions of the genome.
In contrast with the huge fraction of interspersed repeat family elements, the contribution of segmental duplication to the mass and complexity of the opossum genome is relatively small. Only 1.7% of the sequence was identified as segmental duplicons, compared to 5.2%, 5.3%, and 10.4% in mouse, human, and chicken genomes, respectively (Table 2 of Mikkelsen et al. 2007b). Opossum segmental duplications also tended to be shorter, more closely spaced within chromosomes, and less likely to be dispersed among chromosomes than those of humans. This situation seems in line with the general structural conservatism exhibited by metatherian chromosomes in general (see following section).
Chromosome structure and evolution
Comparison of synteny relationships in the opossum genome with those of human, dog, mouse, rat, and chicken enabled construction of a five-way multispecies synteny map comprising 616 conserved blocks (allowing internal rearrangements) (Goodstadt et al. 2007; Mikkelsen et al. 2007b). Analysis of these blocks in a phylogenetic context yielded strong inferences regarding the number and kinds of rearrangements that occurred during the evolutionary divergence of these species from their shared common ancestor. These findings generally confirmed and refined the rearrangement trajectories inferred from human, rat, mouse, and dog genome data (Lindblad-Toh et al. 2005) and enabled reconstruction of 43 ancestral boreoeutherian synteny blocks (Mikkelsen et al. 2007b). These analyses also revealed the precise fusion point of the autosome-to-X-chromosome translocation that added what now comprises the larger portion of the short arm of the human X chromosome to the ancestral eutherian X subsequent to the metatherian–eutherian divergence (Lahn and Page 1999; Kohn et al. 2004; Sandstedt and Tucker 2004; Graves 2006).
The opossum genome sequence will also help to illuminate hidden aspects of chromosome evolution within the Metatheria. G-banding and chromosome-painting studies suggest that metatherian chromosomes have been remarkably conservative with regard to large-scale rearrangements. More than 90% of the 211 metatherian species examined have diploid numbers between 14 and 22 (Hayman 1990; updated by Tyndale-Biscoe 2005) and chromosome morphology manifests only minor deviations between most species, the majority of which are easily explained by a few simple internal rearrangements and/or a small number of centromeric fissions and fusions (Rofe and Hayman 1985; Hayman 1990; Rens et al. 2001, 2003; Ferreri et al. 2004; Metcalfe et al. 2007). Whether this strong karyotypic conservation is mirrored by parallel conservation in gene content and syntenic relationships has never been determined because of the sparseness of comparative gene mapping data from metatherian genomes. With the opossum genome assembly in hand, and low-coverage sequencing of the wallaby genome well underway (http://www.genome.gov/12512299), it will soon be possible to examine synteny relationships in fine detail in representatives of two very distinct and evolutionarily distant metatherian families. One reason this is so important is that chromosome painting studies have indicated that similar karyotypic structure in diverse metatherian families may reflect repeated reversals of chromosome fusions and fissions involving a small number of identical or very similar chromosomal breakpoints (Rens et al. 2003; also see Metcalfe et al. 2007). If so, the apparent structural conservation between the chromosomes of some groups of metatherians could be the result of character convergence rather than shared ancestry. The level of scrutiny afforded by whole-genome data will help delineate the precise boundaries of metatherian conserved syntenic blocks and possibly reveal the locations of past fission and fusion points that have gone undetected at lower levels of resolution.
Protein-coding genes
The protein-coding gene complement of the opossum genome is largely similar to that of eutherian mammals and other vertebrates, both in gene number and the kinds of genes and gene families present. Of the initial set of ∼18,650 predicted coding gene sequences detected in the opossum genome assembly (Goodstadt et al. 2007; Mikkelsen et al. 2007b), ∼96.7% had clear orthologs or candidate homologs in the human genome, and another ∼3.3% could be attributed to miscalled predictions arising from pseudogene sequences or assembly errors, leaving only eight predicted opossum genes without identifiable human counterparts (Supplemental Table S16 in Mikkelsen et al. 2007b). So few opossum-specific genes might indicate that the origin of truly novel genes was a very rare occurrence on the time scale of the eutherian–metatherian divergence. However, turning the comparison on its head by querying how many human genes have no opossum orthologs reveals another possible answer. Based on the same analytical strategy used for the opossum gene count, the human gene catalog comprises 20,806 predicted genes (Goodstadt and Ponting 2006), of which ∼1,100 had no obvious homologs in the initial opossum gene set. Revealingly, 57% of these genes could be aligned to unannotated sequences in the opossum assembly (Mikkelsen et al. 2007b), suggesting that a substantial fraction of the opossum gene set (including many from several rapidly evolving immune-related families; see “Immunogenetics” section) went undetected by the gene identification strategies employed in the annotation effort. If so, then it is possible that opossum-specific genes were overlooked as well. Why might this have happened?
In the absence of extensive expressed sequence data for the opossum, it was necessary to rely heavily on automated prediction strategies that identify putative genes by comparison of target genome sequence to documented protein sequences and general consensus characteristics known to be associated with transcriptionally active DNA in key model species, primarily human and mouse (e.g., Gross and Brent 2006; Hubbard et al. 2007; see Goodstadt and Ponting 2006; Goodstadt et al. 2007; Mikkelsen et al. 2007b for details). If truly novel genes encoding unknown proteins are encountered, there is a possibility that some of these characteristics will not be recognized by these algorithms, and the gene will be missed. The substantial evolutionary distance between the opossum and the eutherians upon which the prediction algorithms are based increases both the chance that novel, metatherian-specific genes exist in the opossum genome and that automated prediction strategies will overlook them. Some opossum genes clearly were missed by this approach, and the upper range of the estimate of 18,000–20,000 genes in the opossum genome (Mikkelsen et al. 2007b) is probably more realistic. Determining what these unknown genes are, and whether any of them encode novel, opossum (metatherian)-specific proteins will require the generation of an extensive expressed-sequence-tag (EST) catalog for M. domestica (see “Beyond structural analysis” section).
Despite nearly identical spectra of protein-coding gene families in eutherian and opossum genomes, certain gene families have diversified in the opossum lineage in ways that differ from eutherian lineages. About 15% of opossum gene families exhibit lineage-specific expansions in gene number and diversity, most of which (52%) have occurred in families with functions related to organismal/environmental interactions, and that commonly exhibit lineage-specific expansions or contractions in other mammals: e.g., genes related to species recognition and mating behavior (pheromone communication); other chemosensory functions; digestive processes; detoxification and nutrient scavenging pathways; and immune surveillance and host defense (Goodstadt et al. 2007; Mikkelsen et al. 2007b and references therein). Most of the remaining expansions are distributed among diverse functional categories (Table 5 in Goodstadt et al. 2007). Among these are a few duplications within families not known to be expanded in other mammalian genomes. Although small in number, these duplications could signal the existence of phylogenetically restricted de novo functionalizations that contribute to the metatherian mode of mammalian life, although such roles are not clear at this time.
Overall, then, the opossum genome yields only minimal evidence of innovation among the vast majority of protein-coding genes and gene families. If we are to identify genomic characteristics that underlie the metatherian and eutherian variants of the common mammalian pattern, apparently we must look elsewhere.
Conserved noncoding elements and evolution by transposition
Comparisons among eutherian genomes and between eutherian, avian, and other vertebrate genomes have confirmed the existence of substantial amounts of sequence that is identifiably similar, and thus conserved, between distant species, but which clearly does not encode proteins (e.g., Bejerano et al. 2004; Dermitzakis et al. 2005; Lindblad-Toh et al. 2005; Siepel et al. 2005; McEwen et al. 2006; Prabhakar et al. 2006; Muotri et al. 2007). It was therefore no surprise that such conserved, noncoding elements (CNEs) were found to be abundant in the opossum genome, accounting for ∼50% of all interspecifically alignable elements. What is remarkable is that the subset of all conserved elements shared between chicken and human, and that shared between chicken and opossum, is essentially identical, indicating that there exists a suite of “amniote conserved elements” (ACEs) (Mikkelsen et al. 2007b) that are of such fundamental importance that they cannot be lost. Specifically, considering genomic sequence without gaps, 99% of ∼68 Mb of sequence that could be aligned between chicken and opossum could also be aligned with the human genome, indicating a loss of only ∼1% of these ACEs in the human lineage. Similarly, <1% of ACEs were missing from the ungapped portion of the opossum sequence. Despite accounting for ∼50% of ACEs, protein-coding regions accounted for a disproportionately small fraction (∼4%) of these losses; the great majority (∼96%) occurred among CNEs. In light of the generally strong conservation of CNEs across sequenced amniotes, it is revealing that ∼20.5% of the ∼74 Mb of noncoding genomic elements in ungapped sequence that are conserved across eutherian species are missing from the opossum genome, and thus comprise a set of eutherian-specific CNEs. Such a large proportion of eutherian-specific CNEs indicates the introduction of many novel and strongly conserved genomic elements into the eutherian lineage subsequent to the metatherian–eutherian divergence, but not later than the common boreoeutherian ancestor. Importantly, the distributions and characteristics of these lineage-specific CNEs, relative to those of CNEs with known functions, suggest that a large fraction of them are likely to be functionally significant (Mikkelsen et al. 2007b). This surprising novelty in eutherian-specific CNEs, compared to the low level of innovation for protein-coding sequences, strongly supports the notion that mammalian evolution has been driven more by changes in families of noncoding elements that regulate protein-coding gene function than by tinkering with the structures or numbers of protein-coding genes themselves. It also suggests that species-specific and interindividual differences in CNEs may underlie a significant fraction of diversity in form and function between closely related species, and even contribute to important phenotypic and health-related variation within species, including our own.
Another surprise was that ∼16% of all eutherian-specific CNEs present in the human genome (and presumably those of other eutherians) display unambiguous characteristics of transposable elements (TEs) belonging to most major eutherian TE families (Gentles et al. 2007; Mikkelsen et al. 2007b). The retention of recognizable functional components from source TE family members suggests that in many cases portions of the TEs themselves may have been co-opted (exapted) and maintained as active regulatory structures by the host genome. In sharp contrast, only ∼1% of CNEs shared between the human and opossum genomes exhibited clear TE-like features. This low fraction makes sense because opossum–human shared CNEs must have originated prior to the eutherian–metatherian split, so would have experienced ∼170–190 Myr of sequence divergence, making it less likely that signatures of TE origin would be recognizable in them unless a particular TE structural feature was critical to an exapted regulatory functionality in the host genome. Nevertheless, maintenance of recognizable TE sequence features in CNEs has been observed across distant vertebrate classes (e.g., Siepel et al. 2005; Kamal et al. 2006; McEwen et al. 2006), and Gentles et al. (2007) detected traces of TE ancestry among 83 newly discovered families of repeat elements in the opossum genome whose origins may date to a time shortly before the divergence of birds and mammals. Such “transposon ghosts” in very old paralogs, together with other considerations (Mikkelsen et al. 2007b), suggest that a much larger fraction of eutherian-specific and other vertebrate CNEs probably originated via proliferation of endogenous transposable elements, and that many of these elements may have been integrated into the molecular machinery that regulates protein-coding gene expression.
Beyond structural analysis
Comparative structural analysis draws upon only one aspect of the opossum genome’s potential for biologic inquiry and discovery. Metatherian mammals are also versatile experimental models, and M. domestica in particular is used in a broad range of applications in basic biological research and in programs that are relevant to human development, physiology, and disease susceptibility (Samollow 2006). This diversity of research activities has created a need for tools that can be used for investigating variation in gene expression in normal and experimentally manipulated development and physiologic states, and in animals that exhibit variation in heritable, health-related phenotypes.
Resources for physical genomic analysis in M. domestica have proliferated rapidly over the past five years. In addition to the genome draft, SNP catalog, and FISH and linkage maps already mentioned, there are also fully arrayed, high-coverage BAC libraries for each sex (http://bacpac.chori.org/) and a fingerprinted BAC-based whole-genome physical map (http://www.bcgsc.ca/lab/mapping/data). However, tools for functional genomic studies are very limited. To begin remedying this shortfall, work is underway (in the author’s laboratory) to establish a comprehensive EST database via analysis of mRNAs from a broad range of opossum tissues and life stages that can be used to seek novel opossum transcripts, augment and refine the current genome annotation, and guide the design of basic gene-expression microarrays for genome-wide quantification of relative gene expression levels in tissue and cell samples.
In the remainder of this section, I discuss selected research areas in which the availability of physical and functional genomic resources for the opossum are likely to have significant impact for contributing to our understanding of basic metatherian biology and, by extension, the processes that impinge upon health-related physiologic characteristics, disease susceptibilities, and developmental anomalies in humans and other mammals.
Immunogenetics
Past efforts to identify structural components of the metatherian immune system resulted in the detection of various genes and processes (e.g., major histocompatibility complex [MHC] genes, T-cell receptors, immunoglobulins, generation of antigen-receptor diversity via somatic V–D-J recombination) that sufficiently resembled those of humans and mice to suggest that the metatherian immunologic toolkit was generally similar to that of eutherian species (in the absence of comprehensive reviews, see Stone et al. 1996, 1998; Miller and Belov 2000; Belov et al. 2002a, b, 2004, 2006; Browning et al. 2004; Miska et al. 2004; Baker et al. 2005; Gouin et al. 2006b; Samollow 2006). Nevertheless, the vast evolutionary distance between the metatherian and eutherian clades made it difficult to identify metatherian homologs of genes within the more rapidly evolving components of the immune system, so the overall complexity and diversity of gene families in major arms of the immune system remained largely unknown. Description of these components is particularly important in order to understand the molecular bases of certain immune responses (e.g., humoral response, isotype switching, allograft response, mixed lymphocyte response) of M. domestica and other metatherians that have been found to differ from those typically seen in eutherians (for review, see Stone et al. 1996; discussed by Samollow 2006). These functional differences generated speculation that the metatherian immune system might differ from that of eutherians in the genetic architecture and level of diversity of, for example, MHC Class I and Class II loci, or the ontogenetic characteristics and properties of T-cells and their receptors (Jurd 1994; Stone et al. 1996, 1998, 1999).
Recent studies facilitated by the opossum genome sequence reveal that the metatherian immune system is very similar to and every bit as complex as that of its eutherian counterpart, and indicate that all major components of the mammalian immune system were in place prior to divergence of the metatherian and eutherian lineages. For example, reconstruction and annotation of the opossum MHC region (Fig. 4) revealed the same basic components present in eutherian MHCs, but with key differences in spatial organization of MHC Class I, II, and III genes that resembled the MHC topologies of nonmammalian vertebrates (Belov et al. 2006; Gouin et al. 2006a, b). Studies of the broader immune system have revealed high levels of innovation through expansions of many other immune-related gene families in the opossum, indicating possible specializations to the metatherian variant of mammalian life. For example, Belov and colleagues (Wong et al. 2006; Belov et al. 2007) employed a combination of semiautomated and manual curation strategies to discover and describe the genomic organization of rapidly evolving immune-related families, many of whose members which went undetected in the initial genome annotation. These include: chemokines, cytokines, cytokine receptors, natural killer cell receptors, leukocyte receptors, natural killer-associated Ig-like receptors, and members of the wide-spectrum antimicrobial cathelicidin and defensin families. Expansions in some of these innate immune components, and especially the diversification of paralogs in the cathelicidin and defensin families, could be related to the unique need of metatherians to protect their tiny newborns during the postpartum period before adaptive immunity is acquired.
Figure 4.
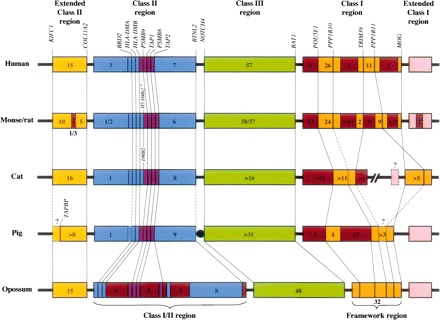
Comparison of MHC organization in representative mammals (figure and legend adapted from Belov et al. 2006, with permission). Of particular note in the opossum MHC is the arrangement of the Class I and II genes into a single interspersed cluster and the absence of Class I genes from the Framework region. These characteristics are reminiscent of arrangements seen in the MHCs of nonmammalian vertebrates. Lines between MHCs of different species indicate the positions of orthologous genes. The asterisk indicates the presence of a duplicated DMB locus (H2-DMb2) in mouse, but not in rat. Dashed lines and question marks represent missing or uncertain data for the particular gene or portion of the genome. Numbers within boxes indicate the number of predicted genes identified within that segment (pseudogenes not included). Unless otherwise indicated, the small boxes indicate individual genes. Map not drawn to scale. Additional details regarding this diagram and implications for the evolution of the mammalian MHC may be found in Belov et al. (2006).
Beyond revelation of immune system structural characteristics made possible by the opossum genome sequence itself, the distinctive nature of metatherian reproduction offers unique opportunities for using genome data for investigating, postnatally, events in immune development that take place in utero in eutherians. For example, lymphoid tissue development in the opossum occurs entirely postnatally (R.D. Miller, pers. comm.). The application of anticipated genome-wide gene-expression-profiling tools will enable identification of gene expression changes concomitant with the appearance of physiologic signals of immune system development and maturation. It can also be expected to promote exploration of the characteristics of maternal/fetal interactions, such as the composition and source(s) of maternally produced antimicrobials (as mentioned above) that may protect the newborn metatherian from a host of environmental exposures.
Genomic imprinting
Genomic imprinting is a special form of epigenetic modification that is established during gametogenesis and passed from parent to offspring, resulting in one of the two alleles at a locus being silenced in a parent-of-origin specific manner (Tycko and Morison 2002; Murphy and Jirtle 2003; Morgan et al. 2005; Swales and Spears 2005; Lewis and Reik 2006). Aberrant expression of imprinted genes can have serious outcomes, including embryonic failures, developmental anomalies, neurodevelopmental and neurobehavioral disorders, endocrine disorders, and neoplastic disease (e.g., Murphy and Jirtle 2003; Swales and Spears 2005; Dolinoy et al. 2007; Jirtle and Skinner 2007). Approximately 90 imprinted genes have been identified in one or another eutherian species (Morison et al. 2005; also http://www.geneimprint.com/ and http://www.otago.ac.nz/IGC), with estimated numbers as high as 600 in mice, and about half that many in humans (Luedi et al. 2005; Dolinoy et al. 2007).
Imprinting also occurs in metatherian mammals but has not been detected in prototherian mammals or other nonmammalian vertebrates. Of 11 eutherian imprinted genes that have been examined in metatherian species, five were found to be imprinted (Table 2). Despite the shared occurrence of imprinting in therian mammals, it is by no means clear whether the molecular basis of metatherian and eutherian imprinting arose from a single, common ancestral mechanism or instead reflects independent, convergent solutions driven by common evolutionary challenges. For example, among imprinted metatherian genes, only PEG10 possesses a clear differentially methylated region (DMR), a feature that is believed to be critical for the imprinting process in eutherian genes (Suzuki et al. 2007). Failure to identify a homolog of the eutherian DMR2 in the imprinted metatherian IGF2 gene (Weidman et al. 2004), or any DMRs at all in the imprinted IGF2R and MEST (PEG1) genes (Killian et al. 2000; Suzuki et al. 2005), hints that the mechanism of imprinting may differ between eutherian and metatherian orthologs, even though the functional outcome—uniparental expression—is the same.
Other differences are also illuminating in this regard. For example, the imprinted DLK1 gene of eutherian mammals lies within the imprinted CLPG (Callipyge) region, which also harbors the reciprocally imprinted MEG3 RNA gene. In M. domestica DLK1 is not imprinted, and no MEG3 ortholog has been identified in metatherians, leading to the proposal that LINE1 insertion of MEG3 subsequent to the eutherian–metatherian divergence was connected to attainment of imprinting capacity in this region (Weidman et al. 2006). A similar event might explain the origin of the imprinted eutherian NNAT gene, which lies within an intron of the nonimprinted BLCAP gene but has not been found outside of eutherians (Evans et al. 2005). Also, the eutherian CDKN1C and IGF2 loci both lie within the eutherian Beckwith-Wiedemann Syndrome imprinting cluster and both are imprinted. While IGF2 is imprinted in metatherians, CDKN1C is not (Suzuki et al. 2005). Finally, whereas the DMR associated with PEG10 imprinting in eutherians also influences parent-of-origin specific silencing of additional loci in the region, influence of the PEG10-associated DMR in metatherians appears restricted to the PEG10 locus (Suzuki et al. 2007).
Whether these differences in imprinting characteristics indicate distinct evolutionary pathways to uniparental expression for these genes in metatherians and eutherians or are the result interphyletic divergence from a common ancestral form of imprinting that has been elaborated or modified in one or the other mammalian lineages is uncertain; however, recent evidence regarding the origin of the eutherian Prader-Willi Syndrome/Angelman Syndrome (PWS/AS) region is quite instructive. In eutherians the imprinted SNRPN locus is expressed exclusively from the paternally derived homolog, and inactivation or deletion of this allele yields Prader-Willi syndrome (Nicholls and Knepper 2001). The nearby UBE3A locus is reciprocally imprinted, exhibiting exclusive maternal-allele expression, and Angelman syndrome in its absence. Homologs of these two loci are biallelically expressed in opossum and wallaby (Rapkins et al. 2006). Importantly, these loci are nonsyntenic in the genomes of metatherians, and a similar condition obtains for these genes or their precursors in the platypus, chicken, and zebrafish genomes (Rapkins et al. 2006; see also Chai et al. 2003). Together with the existence of several retroposed, eutherian-specific genes and snoRNA loci within or flanking the PWS/AS region, these findings indicate that the imprinted PWS/AS region arose through the fusion of two nonsyntenic, nonimprinted genomic regions and accumulation of transposable elements, which acquired regulatory functions subsequent to the separation of metatherian and eutherian mammals, but prior to the origin of the boreoeutherian ancestor (Rapkins et al. 2006).
Were such cobbling together of an imprinted region from nonimprinted precursor genes and transposed noncoding elements an isolated case, it might be dismissed as a curiosity; but, as summarized by Hore et al. (2007b), the universe of eutherian imprinted genes is populated by several such occurrences including the autosomal PWS/AS, CLPG, and PEG10 regions, and the X chromosomal X-inactivation center (XIC), which is at the heart of the random X-chromosome inactivation process of eutherian females. It seems likely that random assembly of the elements necessary for imprinting must have occurred at other genomic sites as well, some of which may have evolved imprinting capability, while others may have been actively prevented from doing so by natural selection against uniparental gene expression. In any case, it will be important to see if such additional examples are found among other imprinted genes. Finally, imprinting has been gained and lost among clades during eutherian evolution (e.g., Killian et al. 2001; Okamura and Ito 2006), and uniparental gene expression need not occur on a whole-animal basis but can be highly tissue-specific (e.g., Wu et al. 1997; Hu et al. 1998; Nicholls and Knepper 2001; Suzuki et al. 2005; Wagschal and Feil 2006; Ager et al. 2007). Both of these observations suggest that imprinted expression at a locus can be enhanced, diminished, or abolished rather easily on an evolutionary time scale. Viewed overall, the evidence suggests that imprinting may have evolved independently in mammals several times, both before and after the divergence of the metatherian and eutherian lineages.
Several theories have been advanced to explain the adaptive advantage of genomic imprinting in the face of the loss of protection that biparental gene expression provides against exposure of recessive mutations to the ravages of negative selection. The most durable and fully developed is the kinship model (often referred to as the “parental conflict model”), which proposes that parent-of-origin specific silencing of genes (imprinting) will be favored at loci that influence the extraction of resources from a parent by its offspring in such a way that the fitness benefits of provisioning the offspring differ for the two parents (Willson and Burley 1983; Haig and Westoby 1989; Moore and Haig 1991). For animals this hypothesis is most often couched in terms of parental conflict in the regulation of intrauterine fetal growth; and, while the theory is not restricted to animals or live-bearing species (Wilkins and Haig 2003; Haig 2004; Mills and Moore 2004; Wood and Oakey 2006), it is attractively consistent with the observed restriction of imprinting to live-bearing mammals, wherein the mother provides resources to the developing young via the placental connection (Iwasa 1998; Wilkins and Haig 2003; Haig 2004).
Nevertheless, neither the kinship model nor any of the others adequately explain the evolution of the majority of imprinted loci known in eutherian species (Wilkins and Haig 2003; Haig 2004). Indeed, there is no a priori reason to expect that a process that may have evolved many times through seemingly haphazard genetic events has a single adaptive explanation. It is possible that seeking enlightenment solely among genes that were initially discovered to be imprinted in eutherians is blinding us to alternative explanations for the evolution of imprinting that might be revealed if we were to discover, ab initio, genes that were imprinted in metatherians, prototherians, or even nonmammalian vertebrates. The fact that none were initially discovered (or found at all) in these groups may reflect our eutherian-centric search strategy; we simply have not looked for imprinted genes in noneutherian mammals except in cases where an imprinted eutherian ortholog was already known. Until recently, an efficient strategy for doing so has not been obvious, particularly in light of uncertainties about the role of differential methylation in the imprinted expression of metatherian genes (Peters and Beechey 2004). One exciting new approach that shows strong promise for choosing candidates for detailed parental-specific expression analysis is the recently developed ChIP-sequencing method for genome-wide analysis of promoter histone modification states (Mikkelsen et al. 2007a). This method appears highly efficient for detecting imprinted loci based on the presence of disparate histone modification states at the promoters of differentially expressed maternally and paternally derived alleles, and it should enable genome-wide strategies for seeking imprinted genes in sequenced noneutherian genomes, including those of opossum, wallaby, platypus, and nonmammalian species.
X-chromosome inactivation
X-chromosome inactivation (XCI) is a chromosome-wide phenomenon that results in the concerted silence of the majority of genes on one of the two X chromosomes in therian females. In eutherian females XCI occurs during early embryogenesis and is more or less random with regard to the parental source of the X chromosomes. As a result, somatic cells of the adult female are about equally divided between those bearing an inactive paternally derived X (Xp) and those in which the maternally derived X (Xm) is inactive. With some important exceptions (Disteche et al. 2002; Carrel and Willard 2005), most genes on the inactive eutherian X chromosome are severely transcriptionally repressed. In metatherians the process is decidedly nonrandom and less stringent. Invariably it is the Xp that is inactive, and genes on this chromosome may exhibit “leaky” or “partial” expression (for review, see Cooper et al. 1990, 1993; see also Samollow et al. 1995). A similar nonrandom XCI pattern also occurs in early differentiating extra-embryonic (trophoblast-derived) tissues of mouse (for review, see Latham 2005; Okamoto and Heard 2006) and bovine (Xue et al. 2002) embryos, and it has been suggested that this imprinted XCI system of trophoblast cells may be an evolutionary holdover from the metatherian pattern, upon which the random inactivation system of eutherian somatic cells has been elaborated (Migeon et al. 1989).
XCI in eutherian embryos results from interactions among noncoding transcriptional elements collectively known as the X-inactivation center (XIC). Through an incompletely understood process, these elements randomly target one of the X chromosomes to be inactivated and promote expression of the XIST gene from the targeted X only. This X is then decorated by its own XIST transcripts, and concomitantly inactivated and hypermethylated (Lee 2005; Heard and Disteche 2006; Wutz and Gribnau 2007). Sequencing of the opossum genome has revealed that the XIC is a novel construct of eutherian mammals. Specifically, XIST and several other components of the XIC region were elaborated through the reorganization and loss of protein-coding functions of existing genes in concert with transpositional recruitment of elements from various other locations in the ancestral eutherian genome (Duret et al. 2006a; Davidow et al. 2007; Hore et al. 2007a; Shevchenko et al. 2007). Lack of this critical control region is consistent with absence of the random XCI process in metatherians, but it does not illuminate the mechanism by which metatherian females achieve silence of the Xp, nor the extent to which this mechanism, whatever it might be, is similar to the one that leads to Xp inactivation in eutherian trophoblast derivatives. An obvious candidate mechanism for Xp inactivity in metatherian females is stable retention of the inactive state of the sperm X after fertilization and on into adult life (Cooper 1971; McCarrey 2001; Huynh and Lee 2005). In eutherian males the X and Y chromosomes are inactivated during spermatogenesis, through the XIST-independent process of meiotic sex chromosome inactivation (MSCI) (Turner 2007), and are reactivated at some yet undetermined point shortly after fertilization (Huynh and Lee 2003; Ferguson-Smith 2004; Heard and Disteche 2006). Two recent studies in the opossum have shown conclusively that the metatherian X also undergoes MSCI in a manner highly similar to that in mouse (Hornecker et al. 2007; Namekawa et al. 2007). The Xp is reactivated and the paternal imprint removed during metatherian oogenesis (Watson et al. 2000), but whether the sperm X is ever reactivated in somatic cells of the female metatherian embryo remains unknown. Availability of the full X-chromosome sequence can be expected to facilitate our understanding of the functional and evolutionary relationships between metatherian and eutherian forms of XCI by enabling more detailed study of the dynamics of multiple X-linked gene activity states in the earliest stages of metatherian embryonic development.
In addition, leaky expression of genes from the “inactive” paternally derived X chromosome in metatherian females (e.g., Samollow et al. 1987, 1995; VandeBerg et al. 1987; Cooper et al. 1993; Hornecker et al. 2007) indicates that repression of genes on the inactive metatherian X chromosome is less stringent and stable than in eutherians. This tissue- and age-specific variability in repression is consonant with the apparent absence of the differential methylation of paternally and maternally derived alleles at metatherian X-linked loci (Kaslow and Migeon 1987; Loebel and Johnston 1996; Hornecker et al. 2007) and defines a useful model for examining molecular changes that occur with dynamic loss of imprinting in a natural system. In particular, X-chromosome imprinting in metatherians might be based largely on histone-dependent chromatin modification (Wakefield et al. 1997; Keohane et al. 1998), which, when considering the dearth of DMRs among autosomal imprinted genes in metatherians, suggests the potential for mechanistic similarity between X chromosome and autosomal imprinting phenomena in metatherian species. Whole-genome-level analyses based on opossum genome sequence data will be invaluable for seeking such similarities.
Neurobiology
As in all metatherians, the newborn opossum exhibits a combination of relatively advanced and immature developmental features that has made it a favored subject for examining differentiation and growth in the developing brain, spinal cord, cranial sensory organs, and neuromotor systems. For example, the presence of relatively advanced skeletal and muscular features of the facial, jaw, and forelimb regions (necessary for crawling toward the mammary region and securing attachment to a teat), coincident with the immature state of the central nervous system, chemosensory systems, and hind limb structures (Fig. 1), provides an outstanding model system for investigating how relatively small changes in the timing and rates of early developmental events (heterochrony) can lead to large differences in adult form and function that distinguish species and higher taxa (for reviews, see Smith 2006; Karlen and Krubitzer 2007). The opossum also provides an extraordinary model for investigating the molecular properties of neurons that enable or prevent the capability of regeneration in the central and peripheral nervous systems. Unlike eutherian young, newborn opossums possess the ability to completely heal partial and complete transections of the spinal cord, a capacity that disappears in an anterior-to-posterior progression over the first two weeks of postnatal life (for reviews, see Nicholls and Saunders 1996; Nicholls et al. 1999; Ferretti et al. 2003; Wintzer et al. 2004). In the peripheral nervous system, stem cell transplantation experiments have revealed the potential of undifferentiated neuronal precursor cells to repair damage in the retina (Sakaguchi et al. 2003, 2004, 2005; Van Hoffelen et al. 2003; D.S. Sakaguchi, pers. comm.).
Describing parallel changes and differences in gene expression timing in components of the developing eutherian and metatherian nervous systems will highlight the importance of specific gene activities in heterochronic phenomena that lead to taxon-specific neurologic structures and functional capabilities (Smith 2003, 2006; Djavadian et al. 2006; Karlen and Krubitzer 2006; Karlen et al. 2006; Mihrshahi et al. 2006); and discovering what gene expression changes correlate with the loss of regeneration capacity of central and peripheral nervous system neurons (e.g., Wintzer et al. 2004; Mladinic et al. 2005, 2007; Lane et al. 2007; Mladinic 2007) could lead to the manipulation of gene expression to activate youthful growth abilities that are normally absent in mature neurons. Some research groups are already applying microarray-based gene-expression-profiling strategies in their investigations of neuroregeneration capacity in the opossum model, but they have had to rely on tools designed for eutherian species (e.g., Lane et al. 2007). The availability of the M. domestica genome sequence and ongoing development of the EST database will soon make possible the construction of M. domestica-specific expression microarrays for these and other studies of gene activity during neurologic development and in states associated with regeneration capability and its loss.
Reproduction and development
The immature state of the metatherian newborn has attracted considerable interest in their use as models for examining sexual differentiation, hormonal effects on reproductive behavior, estrus cycling, ontogeny and evolution of placental structure and function, and the earliest stages of embryonic development. For example, data from the wallaby (Renfree and Shaw 2001; Renfree et al. 2001a, b, 2002, 2006; Glickman et al. 2005; Renfree 2006) indicate that, as in eutherians, differentiation and development of male internal genitalia are directed by androgens; but, unlike the situation in eutherians, development of some external secondary sexual characteristics, such as scrotum, pouch, and mammary primordia, occurs prior to gonadal differentiation and is not influenced by circulating sex hormones. In light of these findings, the discoveries of steroidogenic enzymes in the gonads and androgen receptors in the scrotal anlage of prenatal M. domestica have created controversy over the involvement of sex hormones in the development of male and female secondary sex characteristics in metatherians (Sonea et al. 1997; Gilmore 2002; Shaw et al. 2006).
The role(s) of gonadal steroids in the development of sexual dimorphisms in metatherian brain and spinal cord structures is also unclear. These dimorphisms are established very early in postnatal development of M. domestica and other metatherians, at a time when there seem to be no clear differences in the levels of circulating androgens and estrogens (for review, see Gilmore 2002). Recent data from the wallaby suggest that androstanediol, rather than testosterone or dihydroxytestosterone, is the relevant circulating androgenic molecule (Shaw and Renfree 2006), so findings from M. domestica will need to be reevaluated; but in any case, the sexual behaviors of adult M. domestica can be modified by postnatal exposure to sex hormones (e.g., Fadem 2000, 2001; Fadem et al. 2000, and references therein), suggesting an early hormonal influence on brain sexual differentiation (see Gilmore 2002). There is also strong evidence that male pheromones are important effectors of female body growth, ovarian development and cycling, and sexual behavior in M. domestica (e.g., Jackson and Harder 2000; Harder and Jackson 2003; Zuri and Halpern 2005; Wang et al. 2007, and references therein), but the genetic pathways mediating these responses are virtually unexplored. The breadth of these observations suggest numerous avenues for the application of functional genomic analyses (e.g., temporal analysis of sets of specific candidate genes or, alternatively, genome-wide profiling during early development) for delineating genetic networks that underlie normal and experimentally altered (hormonally treated or deprived) patterns of sexual differentiation and adult sexual function in the opossum and wallaby models.
Concluding remarks
The power of comparative genomic analysis is strongly dependent on the breadth of taxonomic sampling (Pollock et al. 2000; O’Brien et al. 2001; Graves and Westerman 2002; Frazer et al. 2003; Margulies et al. 2005; King et al. 2007; Murphy et al. 2007). Rates of evolutionary change vary radically for different classes of genomic elements and, as a consequence, it is necessary to match the phylogenetic scope of any particular analysis to the scale of evolutionary divergence expected for the characters under scrutiny. The ability to do so requires the sequencing of genomes in key phylogenetic positions that can accommodate both the slowest and fastest evolving characters, and those that lie between (e.g., McEwen et al. 2006; Prabhakar et al. 2006). The opossum and platypus genomes (http://www.ensembl.org/Ornithorhynchus_anatinus/index.html) provide crucial intermediate comparators relative to those of the chicken and eutherian mammals, and they furnish alternate reference points for comparisons between mammalian, avian, and more distant vertebrate genomes. The revelations that have already emerged from comparative analysis of the opossum genome expand our awareness of the diversity of processes that determine the structural organization, complexity, and molecular functions of mammalian (and by extension, other vertebrate) genomes, and underscore the importance of understanding how these diverse elements operate and how variations in them lead to differences in gene regulation, expression, and action among and within species.
Nevertheless, a single metatherian reference genome provides inadequate resolution for some comparisons between the genomes of the eutherian and metatherian sister-groups, and is virtually useless for addressing questions about genomic diversity within the metatherian clade. Fortunately the genome of the tammar wallaby, M. eugenii, is also being sequenced, albeit at a low density (∼2× coverage) (http://www.genome.gov/12512299). This species was an excellent choice, not only because M. domestica and M. eugenii are the world’s most widely used metatherian research models, but also because they are among the most distantly related of metatherian species, being separated by ∼70–90 Myr of evolutionary divergence, a span only slightly less than the ∼80–95 Myr separating primates and rodents (Springer et al. 2003; Bininda-Emonds et al. 2007; Donoghue and Benton 2007; Murphy et al. 2007; Meredith et al. 2008). They also represent two of the largest metatherian families, the Didelphidae (∼87 species) and Macropodidae (∼65 species), which have evolved major anatomical, reproductive, and ecological distinctions. Data from these distantly related species will help to discriminate genomic characteristics that are idiosyncratic of individual species or clades from those that are fundamental to all metatherians, and are essential for the most basic reconstruction of the probable states of genic and chromosomal structures in the early metatherian ancestor. However, to accurately gauge the diversity of genomic structure among the major metatherian clades will require at least low coverage (2×) sequencing of additional strategically placed metatherian genomes. Moreover, given the position of the wallaby as the primary colony-bred metatherian research model in Australia, it seems that this species’ genome should be sequenced to greater depth to provide the best possible comparisons between the world’s two key metatherian models. In any case, the ability to perform broad sequence comparisons among a diversity of metatherian species would vastly increase the power of comparative genomic strategies for interpreting differences in the structures and functions of genes that influence reproductive patterns, early development, genomic imprinting, immunologic functions, and other processes that exhibit what appear to be phylogenetic distinctions between eutherian and metatherian mammals.
Note added in proof
While the current article was awaiting publication, Warren et al. (2008) reported the first full-genome sequence of a prototherian mammal, that of the platypus, Ornithorhynchus anatinus. The information described in that article, and in several companion articles which appeared simultaneously, greatly expands our understanding of the structural characteristics and evolution of vertebrate genomes, but does not materially impinge upon the conclusions or prospects for future research described in this paper.
Acknowledgments
The author thanks three anonymous reviewers for constructive comments on an earlier version of this article, and Dr. William J. Murphy for advice and guidance regarding mammalian phylogeny. The author’s work is supported in part by grant RR014214 from the National Center for Research Resources of the National Institutes of Health (USA).
Footnotes
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.065326.107.
The dating of the earliest mammalian divergence events is somewhat controversial. For example, a recent, widely cited supertree analysis by Bininda-Emonds et al. (2007), which incorporated 99% of all extant mammalian species, places the prototherian–therian divergence at only 166.2 Mya and, based partially on this fixed point, sets the origin of Metatheria and Eutheria at ∼147.7 Mya. Although that study involved extensive molecular data and was calibrated by 30 fossil-based time points, the date suggested for the base of the Mammalia (166.2 Myr) was constrained by an extinct vertebrate described from a single partial jawbone fragment posited by Bininda-Emonds et al. (2007) to represent the maximum age of ancestral mammals that gave rise to the prototherian and therian lineages. However, because a single fossil can only fix the minimum age for the existence of a lineage (e.g, Woodburne et al. 2003; Glazko et al. 2005; Donoghue and Benton 2007), the actual divergence of Prototheria and Theria could be considerably older. Moreover, the fossil itself, Ambondro mahabo, was placed with uncertain taxonomic affinity in its original description (Flynn et al. 1999), and later analysis suggests it is likely to be a eutherian (Woodburne et al. 2003) rather than ancestral to prototherians (sensu Luo et al. 2002). Considering the disputed and dubious phylogenetic position of A. mahabo, it seems prudent to abide by the older dates for the earliest mammalian divergences (see main text), which are supported by considerable molecular data from several different studies (cited in main text).
References
- Ager E., Suzuki S., Pask A., Shaw G., Ishino F., Renfree M.B. Insulin is imprinted in the placenta of the marsupial, Macropus eugenii. Dev. Biol. 2007;309:317–328. doi: 10.1016/j.ydbio.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Baker M.L., Belov K., Miller R.D. Unusually similar patterns of antibody V segment diversity in distantly related marsupials. J. Immunol. 2005;174:5665–5671. doi: 10.4049/jimmunol.174.9.5665. [DOI] [PubMed] [Google Scholar]
- Behringer R.R., Eakin G.S., Renfree M.B. Mammalian diversity: Gametes, embryos and reproduction. Reprod. Fertil. Dev. 2006;18:99–107. doi: 10.1071/rd05137. [DOI] [PubMed] [Google Scholar]
- Bejerano G., Haussler D., Blanchette M. Into the heart of darkness: Large-scale clustering of human non-coding DNA. Bioinformatics. 2004;(Suppl. 1) 20:i40–i48. doi: 10.1093/bioinformatics/bth946. [DOI] [PubMed] [Google Scholar]
- Belle E.M., Duret L., Galtier N., Eyre-Walker A. The decline of isochores in mammals: An assessment of the GC content variation along the mammalian phylogeny. J. Mol. Evol. 2004;58:653–660. doi: 10.1007/s00239-004-2587-x. [DOI] [PubMed] [Google Scholar]
- Belov K., Harrison G.A., Miller R.D., Cooper D.W. Molecular cloning of four lambda light chain cDNAs from the Australian brushtail possum Trichosurus vulpecula. Eur. J. Immunogenet. 2002a;29:95–99. doi: 10.1046/j.1365-2370.2002.00286.x. [DOI] [PubMed] [Google Scholar]
- Belov K., Nguyen M.A., Zenger K.R., Cooper D.W. Ontogeny of immunoglobulin expression in the brushtail possum (Trichosurus vulpecula) Dev. Comp. Immunol. 2002b;26:599–602. doi: 10.1016/s0145-305x(02)00015-0. [DOI] [PubMed] [Google Scholar]
- Belov K., Lam M.K., Colgan D.J. Marsupial MHC class II β genes are not orthologous to the eutherian β gene families. J. Hered. 2004;95:338–345. doi: 10.1093/jhered/esh049. [DOI] [PubMed] [Google Scholar]
- Belov K., Deakin J.E., Papenfuss A.T., Baker M.L., Melman S.D., Siddle H.V., Gouin N., Goode D.L., Sargeant T.J., Robinson M.D., et al. Reconstructing an ancestral mammalian immune supercomplex from a marsupial major histocompatibility complex. PLoS Biol. 2006;4:e46. doi: 10.1371/journal.pbio.0040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov K., Sanderson C.E., Deakin J.E., Wong E.S., Assange D., McColl K.A., Gout A., de Bono B., Barrow A.D., Speed T.P., et al. Characterization of the opossum immune genome provides insights into the evolution of the mammalian immune system. Genome Res. 2007;17:982–991. doi: 10.1101/gr.6121807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J.H., Hayman D.L., Hope R.M. Novel sex differences in linkage values and meiotic chromosome behaviour in a marsupial. Nature. 1986;323:59–60. doi: 10.1038/323059a0. [DOI] [PubMed] [Google Scholar]
- Bernardi G. The neoselectionist theory of genome evolution. Proc. Natl. Acad. Sci. 2007;104:8385–8390. doi: 10.1073/pnas.0701652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda-Emonds O.R., Cardillo M., Jones K.E., MacPhee R.D., Beck R.M., Grenyer R., Price S.A., Vos R.A., Gittleman J.L., Purvis A. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: Individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning T.L., Belov K., Miller R.D., Eldridge M.D. Molecular cloning and characterization of the polymorphic MHC class II DBB from the tammar wallaby (Macropus eugenii) Immunogenetics. 2004;55:791–795. doi: 10.1007/s00251-004-0644-7. [DOI] [PubMed] [Google Scholar]
- Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Chai J.H., Locke D.P., Greally J.M., Knoll J.H., Ohta T., Dunai J., Yavor A., Eichler E.E., Nicholls R.D. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet. 2003;73:898–925. doi: 10.1086/378816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N., Dagan T., Stone L., Graur D. GC composition of the human genome: In search of isochores. Mol. Biol. Evol. 2005;22:1260–1272. doi: 10.1093/molbev/msi115. [DOI] [PubMed] [Google Scholar]
- Cooper D.W. Directed genetic change model for X chromosome inactivation in eutherian mammals. Nature. 1971;230:292–294. doi: 10.1038/230292a0. [DOI] [PubMed] [Google Scholar]
- Cooper D.W., Johnston P.G., VandeBerg J.L., Robinson E.S. X-chromosome inactivation in marsupials. In: Graves J.A.M., et al., editors. Mammals from pouches and eggs: Genetics, breeding and evolution of marsupials and monotremes. CSIRO; Melbourne, Australia: 1990. pp. 269–275. [Google Scholar]
- Cooper D.W., Johnston P.G., Watson J.M., Graves J.A.M. X-inactivation in marsupials and monotremes. Semin. Dev. Biol. 1993;4:117–128. [Google Scholar]
- Davidow L.S., Breen M., Duke S.E., Samollow P.B., McCarrey J.R., Lee J.T. The search for a marsupial XIC reveals a break with vertebrate synteny. Chromosome Res. 2007;15:137–146. doi: 10.1007/s10577-007-1121-6. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond A., Antonarakis S.E. Conserved non-genic sequences—An unexpected feature of mammalian genomes. Nat. Rev. Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- Disteche C.M., Filippova G.N., Tsuchiya K.D. Escape from X inactivation. Cytogenet. Genome Res. 2002;99:36–43. doi: 10.1159/000071572. [DOI] [PubMed] [Google Scholar]
- Djavadian R., Bisti S., Maccarone R., Bartkowska K., Turlejski K. Development and plasticity of the retina in the opossum Monodelphis domestica. Acta Neurobiol. Exp. (Wars) 2006;66:179–188. doi: 10.55782/ane-2006-1605. [DOI] [PubMed] [Google Scholar]
- Dolinoy D.C., Weidman J.R., Jirtle R.L. Epigenetic gene regulation: Linking early developmental environment to adult disease. Reprod. Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Donoghue P.C., Benton M.J. Rocks and clocks: Calibrating the Tree of Life using fossils and molecules. Trends Ecol. Evol. 2007;22:424–431. doi: 10.1016/j.tree.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Duke S.E., Samollow P.B., Mauceli E., Lindblad-Toh K., Breen M. Integrated cytogenetic BAC map of the genome of the gray, short-tailed opossum, Monodelphis domestica. Chromosome Res. 2007;15:361–370. doi: 10.1007/s10577-007-1131-4. [DOI] [PubMed] [Google Scholar]
- Duret L., Chureau C., Samain S., Weissenbach J., Avner P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006a;312:1653–1655. doi: 10.1126/science.1126316. [DOI] [PubMed] [Google Scholar]
- Duret L., Eyre-Walker A., Galtier N. A new perspective on isochore evolution. Gene. 2006b;385:71–74. doi: 10.1016/j.gene.2006.04.030. [DOI] [PubMed] [Google Scholar]
- Evans H.K., Weidman J.R., Cowley D.O., Jirtle R.L. Comparative phylogenetic analysis of blcap/nnat reveals eutherian-specific imprinted gene. Mol. Biol. Evol. 2005;22:1740–1748. doi: 10.1093/molbev/msi165. [DOI] [PubMed] [Google Scholar]
- Fadem B.H. Perinatal exposure to estradiol masculinizes aspects of sexually dimorphic behavior and morphology in gray short-tailed opossums (Monodelphis domestica) Horm. Behav. 2000;37:79–85. doi: 10.1006/hbeh.1999.1561. [DOI] [PubMed] [Google Scholar]
- Fadem B.H. Evidence for extended action of gonadal hormones on the organization of sexually dimorphic behavior and morphology in gray short-tailed opossums (Monodelphis domestica) Horm. Behav. 2001;39:113–120. doi: 10.1006/hbeh.2000.1640. [DOI] [PubMed] [Google Scholar]
- Fadem B.H., Dziadosz D.R., Jackson L.M., Harder J.D. Partner preference of intact and ovariectomized female gray short-tailed opossums (Monodelphis domestica) Horm. Behav. 2000;38:187–192. doi: 10.1006/hbeh.2000.1618. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith A.C. X inactivation: Pre- or post-fertilisation turn-off? Curr. Biol. 2004;14:R323–R325. doi: 10.1016/j.cub.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Ferreri G.C., Marzelli M., Rens W., O'Neill R.J. A centromere-specific retroviral element associated with breaks of synteny in macropodine marsupials. Cytogenet. Genome Res. 2004;107:115–118. doi: 10.1159/000079580. [DOI] [PubMed] [Google Scholar]
- Ferretti P., Zhang F., O'Neill P. Changes in spinal cord regenerative ability through phylogenesis and development: Lessons to be learnt. Dev. Dyn. 2003;226:245–256. doi: 10.1002/dvdy.10226. [DOI] [PubMed] [Google Scholar]
- Flynn J.J., Parrish J.M., Rakotosamimanana B., Simpson W.F., Wyss A.R. A middle Jurassic mammal from Madigascar. Nature. 1999;401:57–60. [Google Scholar]
- Frazer K.A., Elnitski L., Church D.M., Dubchak I., Hardison R.C. Cross-species sequence comparisons: A review of methods and available resources. Genome Res. 2003;13:1–12. doi: 10.1101/gr.222003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N., Piganeau G., Mouchiroud D., Duret L. GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics. 2001;159:907–911. doi: 10.1093/genetics/159.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles A.J., Wakefield M.J., Kohany O., Gu W., Batzer M.A., Pollock D.D., Jurka J. Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res. 2007;17:992–1004. doi: 10.1101/gr.6070707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore D.P. Sexual dimorphism in the central nervous system of marsupials. Int. Rev. Cytol. 2002;214:193–224. doi: 10.1016/s0074-7696(02)14006-x. [DOI] [PubMed] [Google Scholar]
- Glazko G.V., Koonin E.V., Rogozin I.B. Molecular dating: Ape bones agree with chicken entrails. Trends Genet. 2005;21:89–92. doi: 10.1016/j.tig.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Glickman S.E., Short R.V., Renfree M.B. Sexual differentiation in three unconventional mammals: Spotted hyenas, elephants and tammar wallabies. Horm. Behav. 2005;48:403–417. doi: 10.1016/j.yhbeh.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Goodstadt L., Ponting C.P. Phylogenetic reconstruction of orthology, paralogy, and conserved synteny for dog and human. PLoS Comput. Biol. 2006;2:e133. doi: 10.1371/journal.pcbi.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstadt L., Heger A., Webber C., Ponting C.P. An analysis of the gene complement of a marsupial, Monodelphis domestica: Evolution of lineage-specific genes and giant chromosomes. Genome Res. 2007;17:969–981. doi: 10.1101/gr.6093907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin N., Deakin J.E., Miska K.B., Miller R.D., Kammerer C.M., Graves J.A., VandeBerg J.L., Samollow P.B. Linkage mapping and physical localization of the major histocompatibility complex region of the marsupial Monodelphis domestica. Cytogenet. Genome Res. 2006a;112:277–285. doi: 10.1159/000089882. [DOI] [PubMed] [Google Scholar]
- Gouin N., Wright A.M., Miska K.B., Parra Z.E., Samollow P.B., Baker M.L., Miller R.D. Modo-UG, a marsupial nonclassical MHC class I locus. Immunogenetics. 2006b;58:396–406. doi: 10.1007/s00251-006-0115-4. [DOI] [PubMed] [Google Scholar]
- Graves J.A. Sex chromosome specialization and degeneration in mammals. Cell. 2006;124:901–914. doi: 10.1016/j.cell.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Graves J.A., Westerman M. Marsupial genetics and genomics. Trends Genet. 2002;18:517–521. doi: 10.1016/s0168-9525(02)02772-5. [DOI] [PubMed] [Google Scholar]
- Gross S.S., Brent M.R. Using multiple alignments to improve gene prediction. J. Comput. Biol. 2006;13:379–393. doi: 10.1089/cmb.2006.13.379. [DOI] [PubMed] [Google Scholar]
- Gu J., Li W.H. Are GC-rich isochores vanishing in mammals? Gene. 2006;385:50–56. doi: 10.1016/j.gene.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Gu W., Ray D.A., Walker J.A., Barnes E.W., Gentles A.J., Samollow P.B., Jurka J., Batzer M.A., Pollock D.D. SINEs, evolution and genome structure in the opossum. Gene. 2007;396:46–58. doi: 10.1016/j.gene.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Haig D. Genomic imprinting and kinship: How good is the evidence? Annu. Rev. Genet. 2004;38:553–585. doi: 10.1146/annurev.genet.37.110801.142741. [DOI] [PubMed] [Google Scholar]
- Haig D., Westoby M. Parent-specific gene expression and the triploid endosperm. Am. Nat. 1989;134:147–155. [Google Scholar]
- Harder J.D., Jackson L.M. Male pheromone stimulates ovarian follicular development and body growth in juvenile female opossums (Monodelphis domestica) Reprod. Biol. Endocrinol. 2003;1:21. doi: 10.1186/1477-7827-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman D.L. Marsupial cytogenetics. In: Graves J.A.M., et al., editors. Mammals from pouches and eggs: Breeding and evolution of marsupials and monotremes. CSIRO; Melbourne, Australia: 1990. pp. 189–207. [Google Scholar]
- Hayman D., Moore H., Evans E. Further evidence of novel sex differences in chiasma distribution in marsupials. Heredity. 1988;61:455–458. [Google Scholar]
- Heard E., Disteche C.M. Dosage compensation in mammals: Fine-tuning the expression of the X chromosome. Genes & Dev. 2006;20:1848–1867. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- Hedges S.B., Kumar S. Precision of molecular time estimates. Trends Genet. 2004;20:242–247. doi: 10.1016/j.tig.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Hogstrand K., Bohme J. Gene conversion of major histocompatibility complex genes is associated with CpG-rich regions. Immunogenetics. 1999;49:446–455. doi: 10.1007/s002510050518. [DOI] [PubMed] [Google Scholar]
- Hore T.A., Koina E., Wakefield M.J., Graves J.A.M. The region homologous to the X-chromosome inactivation centre has been disrupted in marsupial and monotreme mammals. Chromosome Res. 2007a;15:147–161. doi: 10.1007/s10577-007-1119-0. [DOI] [PubMed] [Google Scholar]
- Hore T.A., Rapkins R.W., Graves J.A. Construction and evolution of imprinted loci in mammals. Trends Genet. 2007b;23:440–448. doi: 10.1016/j.tig.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hornecker J.L., Samollow P.B., Robinson E.S., VandeBerg J.L., McCarrey J.R. Meiotic sex chromosome inactivation in the marsupial Monodelphis domestica. Genesis. 2007;45:696–708. doi: 10.1002/dvg.20345. [DOI] [PubMed] [Google Scholar]
- Hu J.F., Oruganti H., Vu T.H., Hoffman A.R. Tissue-specific imprinting of the mouse insulin-like growth factor II receptor gene correlates with differential allele-specific DNA methylation. Mol. Endocrinol. 1998;12:220–232. doi: 10.1210/mend.12.2.0062. [DOI] [PubMed] [Google Scholar]
- Hubbard T.J., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt P.A. Meiosis in mammals: Recombination, non-disjunction and the environment. Biochem. Soc. Trans. 2006;34:574–577. doi: 10.1042/BST0340574. [DOI] [PubMed] [Google Scholar]
- Huynh K.D., Lee J.T. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature. 2003;426:857–862. doi: 10.1038/nature02222. [DOI] [PubMed] [Google Scholar]
- Huynh K.D., Lee J.T. X-chromosome inactivation: A hypothesis linking ontogeny and phylogeny. Nat. Rev. Genet. 2005;6:410–418. doi: 10.1038/nrg1604. [DOI] [PubMed] [Google Scholar]
- Ihara N., Takasuga A., Mizoshita K., Takeda H., Sugimoto M., Mizoguchi Y., Hirano T., Itoh T., Watanabe T., Reed K.M., et al. A comprehensive genetic map of the cattle genome based on 3802 microsatellites. Genome Res. 2004;14:1987–1998. doi: 10.1101/gr.2741704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovska I., Orr-Weaver T.L. Histone modifications and the chromatin scaffold for meiotic chromosome architecture. Cell Cycle. 2006;5:2064–2071. doi: 10.4161/cc.5.18.3253. [DOI] [PubMed] [Google Scholar]
- Iwasa Y. The conflict theory of genomic imprinting: How much can be explained? Curr. Top. Dev. Biol. 1998;40:255–293. doi: 10.1016/s0070-2153(08)60369-5. [DOI] [PubMed] [Google Scholar]
- Jackson L.M., Harder J.D. Evidence for spontaneous postlactational estrus in gray short-tailed opossums (Monodelphis domestica) Biol. Reprod. 2000;62:1823–1827. doi: 10.1095/biolreprod62.6.1823. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman M.I., Furey T.S., Payseur B.A., Lu Y., Roskin K.M., Chen C.F., Thomas M.A., Haussler D., Jacob H.J. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004;14:528–538. doi: 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle R.L., Skinner M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R.D. “Not proper mammals”: Immunity in monotremes and marsupials. Comp. Immunol. Microbiol. Infect. Dis. 1994;17:41–52. doi: 10.1016/0147-9571(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Kamal M., Xie X., Lander E.S. A large family of ancient repeat elements in the human genome is under strong selection. Proc. Natl. Acad. Sci. 2006;103:2740–2745. doi: 10.1073/pnas.0511238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlen S.J., Krubitzer L. The evolution of the neocortex in mammals: Intrinsic and extrinsic contributions to the cortical phenotype. In Percept, decision, action: Bridging the gaps. In: Chadwick D.J., et al., editors. Novartis Foundation Symposia 270. John Wiley; New York: 2006. pp. 146–159. [PubMed] [Google Scholar]
- Karlen S.J., Krubitzer L. The functional and anatomical organization of marsupial neocortex: Evidence for parallel evolution across mammals. Prog. Neurobiol. 2007;82:122–141. doi: 10.1016/j.pneurobio.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlen S.J., Kahn D.M., Krubitzer L. Early blindness results in abnormal corticocortical and thalamocortical connections. Neuroscience. 2006;142:843–858. doi: 10.1016/j.neuroscience.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Kaslow D.C., Migeon B.R. DNA methylation stabilizes X chromosome inactivation in eutherians but not in marsupials: Evidence for multistep maintenance of mammalian X dosage compensation. Proc. Natl. Acad. Sci. 1987;84:6210–6214. doi: 10.1073/pnas.84.17.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L., Jeffreys A.J., Keeney S. Where the crossovers are: Recombination distributions in mammals. Nat. Rev. Genet. 2004;5:413–424. doi: 10.1038/nrg1346. [DOI] [PubMed] [Google Scholar]
- Keohane A.M., Lavender J.S., O'Neill L.P., Turner B.M. Histone acetylation and X inactivation. Dev. Genet. 1998;22:65–73. doi: 10.1002/(SICI)1520-6408(1998)22:1<65::AID-DVG7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Killian J.K., Byrd J.C., Jirtle J.V., Munday B.L., Stoskopf M.K., MacDonald R.G., Jirtle R.L. M6P/IGF2R imprinting evolution in mammals. Mol. Cell. 2000;5:707–716. doi: 10.1016/s1097-2765(00)80249-x. [DOI] [PubMed] [Google Scholar]
- Killian J.K., Buckley T.R., Stewart N., Munday B.L., Jirtle R.L. Marsupials and Eutherians reunited: Genetic evidence for the Theria hypothesis of mammalian evolution. Mamm. Genome. 2001;12:513–517. doi: 10.1007/s003350020026. [DOI] [PubMed] [Google Scholar]
- King D.C., Taylor J., Zhang Y., Cheng Y., Lawson H.A., Martin J., Chiaromonte F., Miller W., Hardison R.C. Finding cis-regulatory elements using comparative genomics: Some lessons from ENCODE data. Genome Res. 2007;17:775–786. doi: 10.1101/gr.5592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn M., Kehrer-Sawatzki H., Vogel W., Graves J.A., Hameister H. Wide genome comparisons reveal the origins of the human X chromosome. Trends Genet. 2004;20:598–603. doi: 10.1016/j.tig.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G., et al. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Kong X., Murphy K., Raj T., He C., White P.S., Matise T.C. A combined linkage-physical map of the human genome. Am. J. Hum. Genet. 2004;75:1143–1148. doi: 10.1086/426405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Hedges S.B. A molecular timescale for vertebrate evolution. Nature. 1998;392:917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Lahn B.T., Page D.C. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lane M.A., Truettner J.S., Brunschwig J.P., Gomez A., Bunge M.B., Dietrich W.D., Dziegielewska K.M., Ek C.J., Vandeberg J.L., Saunders N.R. Age-related differences in the local cellular and molecular responses to injury in developing spinal cord of the opossum, Monodelphis domestica. Eur. J. Neurosci. 2007;25:1725–1742. doi: 10.1111/j.1460-9568.2007.05439.x. [DOI] [PubMed] [Google Scholar]
- Latham K.E. X chromosome imprinting and inactivation in preimplantation mammalian embryos. Trends Genet. 2005;21:120–127. doi: 10.1016/j.tig.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Lee J.T. Regulation of X-chromosome counting by Tsix and Xite sequences. Science. 2005;309:768–771. doi: 10.1126/science.1113673. [DOI] [PubMed] [Google Scholar]
- Lewis A., Reik W. How imprinting centres work. Cytogenet. Genome Res. 2006;113:81–89. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., Zody M.C., et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Loebel D.A., Johnston P.G. Methylation analysis of a marsupial X-linked CpG island by bisulfite genomic sequencing. Genome Res. 1996;6:114–123. doi: 10.1101/gr.6.2.114. [DOI] [PubMed] [Google Scholar]
- Luedi P.P., Hartemink A.J., Jirtle R.L. Genome-wide prediction of imprinted murine genes. Genome Res. 2005;15:875–884. doi: 10.1101/gr.3303505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.-X., Kielan-Jaworowska Z., Cifelli R.L. In quest for a phylogeny of Mesozoic mammals. Acta Palaeontol. Pol. 2002;47:1–78. [Google Scholar]
- Margulies E.H., Vinson J.P., Miller W., Jaffe D.B., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M. An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing. Proc. Natl. Acad. Sci. 2005;102:4795–4800. doi: 10.1073/pnas.0409882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarrey J.R. X-chromosome inactivation during spermatogenesis: The original dosage compensation mechanism in mammals. In: Xue G., et al., editors. Gene families: Studies of DNA, RNA, enzymes, and proteins. World Scientific; Teaneck, NJ: 2001. pp. 59–72. [Google Scholar]
- McEwen G.K., Woolfe A., Goode D., Vavouri T., Callaway H., Elgar G. Ancient duplicated conserved noncoding elements in vertebrates: A genomic and functional analysis. Genome Res. 2006;16:451–465. doi: 10.1101/gr.4143406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith R.W., Westerman M., Case J.A., Springer M.S. A phylogeny and timescale for marsupial evolution based on sequences for five nuclear genes. J. Mamm. Evol. 2008;15:1–36. [Google Scholar]
- Metcalfe C.J., Eldridge M.D., Johnston P.G. Mapping the distribution of the telomeric sequence (T2AG3)n in the Macropodoidea (Marsupialia) by fluorescence in situ hybridization. II. The ancestral 2n = 22 macropodid karyotype. Cytogenet. Genome Res. 2007;116:212–217. doi: 10.1159/000098189. [DOI] [PubMed] [Google Scholar]
- Migeon B.R., Jan de Beur S., Axelman J. Frequent derepression of G6PD and HPRT on the marsupial inactive X chromosome associated with cell proliferation in vitro. Exp. Cell Res. 1989;182:597–609. doi: 10.1016/0014-4827(89)90262-0. [DOI] [PubMed] [Google Scholar]
- Mihrshahi R., Lewis J.G., Ali S.O. Hormonal effects on the secretion and glycoform profile of corticosteroid-binding globulin. J. Steroid Biochem. Mol. Biol. 2006;101:275–285. doi: 10.1016/j.jsbmb.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007a;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T.S., Wakefield M.J., Aken B., Amemiya C.T., Chang J.L., Duke S., Garber M., Gentles A.J., Goodstadt L., Heger A., et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007b;447:167–177. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Miller R.D., Belov K. Immunoglobulin genetics of marsupials. Dev. Comp. Immunol. 2000;24:485–490. doi: 10.1016/s0145-305x(00)00011-2. [DOI] [PubMed] [Google Scholar]
- Mills W., Moore T. Polyandry, life-history trade-offs and the evolution of imprinting at Mendelian loci. Genetics. 2004;168:2317–2327. doi: 10.1534/genetics.104.030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska K.B., Wright A.M., Lundgren R., Sasaki-McClees R., Osterman A., Gale J.M., Miller R.D. Analysis of a marsupial MHC region containing two recently duplicated class I loci. Mamm. Genome. 2004;15:851–864. doi: 10.1007/s00335-004-2224-4. [DOI] [PubMed] [Google Scholar]
- Mladinic M. Changes in cyclic AMP levels in the developing opossum spinal cord at the time when regeneration stops being possible. Cell. Mol. Neurobiol. 2007;27:883–888. doi: 10.1007/s10571-007-9208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladinic M., Wintzer M., Del Bel E., Casseler C., Lazarevic D., Crovella S., Gustincich S., Cattaneo A., Nicholls J. Differential expression of genes at stages when regeneration can and cannot occur after injury to immature mammalian spinal cord. Cell. Mol. Neurobiol. 2005;25:407–426. doi: 10.1007/s10571-005-3150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladinic M., Del Bel E., Nicholls J. Increase of annexin 1 immunoreactivity in spinal cord of newborn opossum (Monodelphis domestica) at the time when regeneration after injury stops being possible. Histol. Histopathol. 2007;22:1205–1211. doi: 10.14670/HH-22.1205. [DOI] [PubMed] [Google Scholar]
- Moore T., Haig D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- Morelli M.A., Cohen P.E. Not all germ cells are created equal: Aspects of sexual dimorphism in mammalian meiosis. Reproduction. 2005;130:761–781. doi: 10.1530/rep.1.00865. [DOI] [PubMed] [Google Scholar]
- Morgan H.D., Santos F., Green K., Dean W., Reik W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005;14:R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- Morison I.M., Ramsay J.P., Spencer H.G. A census of mammalian imprinting. Trends Genet. 2005;21:457–465. doi: 10.1016/j.tig.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Muotri A.R., Marchetto M.C., Coufal N.G., Gage F.H. The necessary junk: New functions for transposable elements. Hum. Mol. Genet. 2007;16:R159–R167. doi: 10.1093/hmg/ddm196. [DOI] [PubMed] [Google Scholar]
- Murphy S.K., Jirtle R.L. Imprinting evolution and the price of silence. Bioessays. 2003;25:577–588. doi: 10.1002/bies.10277. [DOI] [PubMed] [Google Scholar]
- Murphy W.J., Pringle T.H., Crider T.A., Springer M.S., Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17:413–421. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S., Bottolo L., Freeman C., McVean G., Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Namekawa S.H., VandeBerg J.L., McCarrey J.R., Lee J.T. Sex chromosome silencing in the marsupial male germ line. Proc. Natl. Acad. Sci. 2007;104:9730–9735. doi: 10.1073/pnas.0700323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls R.D., Knepper J.L. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- Nicholls J., Saunders N. Regeneration of immature mammalian spinal cord after injury. Trends Neurosci. 1996;19:229–234. doi: 10.1016/0166-2236(96)10021-7. [DOI] [PubMed] [Google Scholar]
- Nicholls J.G., Adams W.B., Eugenin J., Geiser R., Lepre M., Luque J.M., Wintzer M. Why does the central nervous system not regenerate after injury? Surv. Ophthalmol. 1999;(Suppl. 1) 43:S136–S141. doi: 10.1016/s0039-6257(99)00008-9. [DOI] [PubMed] [Google Scholar]
- O’Brien S.J., Eizirik E., Murphy W.J. Genomics. On choosing mammalian genomes for sequencing. Science. 2001;292:2264–2266. doi: 10.1126/science.1059393. [DOI] [PubMed] [Google Scholar]
- Okamoto I., Heard E. The dynamics of imprinted X inactivation during preimplantation development in mice. Cytogenet. Genome Res. 2006;113:318–324. doi: 10.1159/000090848. [DOI] [PubMed] [Google Scholar]
- Okamura K., Ito T. Lessons from comparative analysis of species-specific imprinted genes. Cytogenet. Genome Res. 2006;113:159–164. doi: 10.1159/000090828. [DOI] [PubMed] [Google Scholar]
- O'Neill M.J., Ingram R.S., Vrana P.B., Tilghman S.M. Allelic expression of IGF2 in marsupials and birds. Dev. Genes Evol. 2000;210:18–20. doi: 10.1007/pl00008182. [DOI] [PubMed] [Google Scholar]
- Page S.L., Hawley R.S. Chromosome choreography: The meiotic ballet. Science. 2003;301:785–789. doi: 10.1126/science.1086605. [DOI] [PubMed] [Google Scholar]
- Paques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak S., Ronne M., Brown N.M., Furlong C.L., VandeBerg J.L. A high-resolution banding pattern idiogram of Monodelphis domestica chromosomes (Marsupialia, Mammalia) Cytogenet. Cell Genet. 1993;63:181–184. doi: 10.1159/000133529. [DOI] [PubMed] [Google Scholar]
- Peters J., Beechey C. Identification and characterisation of imprinted genes in the mouse. Brief. Funct. Genomic. Proteomic. 2004;2:320–333. doi: 10.1093/bfgp/2.4.320. [DOI] [PubMed] [Google Scholar]
- Petronczki M., Siomos M.F., Nasmyth K. Un menage a quatre: The molecular biology of chromosome segregation in meiosis. Cell. 2003;112:423–440. doi: 10.1016/s0092-8674(03)00083-7. [DOI] [PubMed] [Google Scholar]
- Phillips M.J., Penny D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol. 2003;28:171–185. doi: 10.1016/s1055-7903(03)00057-5. [DOI] [PubMed] [Google Scholar]
- Pollock D.D., Eisen J.A., Doggett N.A., Cummings M.P. A case for evolutionary genomics and the comprehensive examination of sequence biodiversity. Mol. Biol. Evol. 2000;17:1776–1788. doi: 10.1093/oxfordjournals.molbev.a026278. [DOI] [PubMed] [Google Scholar]
- Prabhakar S., Poulin F., Shoukry M., Afzal V., Rubin E.M., Couronne O., Pennacchio L.A. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapkins R.W., Hore T., Smithwick M., Ager E., Pask A.J., Renfree M.B., Kohn M., Hameister H., Nicholls R.D., Deakin J.E., et al. Recent assembly of an imprinted domain from non-imprinted components. PLoS Genet. 2006;2:e182. doi: 10.1371/journal.pgen.0020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfree M.B. Society for Reproductive Biology Founders' Lecture 2006—Life in the pouch: Womb with a view. Reprod. Fertil. Dev. 2006;18:721–734. doi: 10.1071/rd06072. [DOI] [PubMed] [Google Scholar]
- Renfree M.B., Shaw G. Germ cells, gonads and sex reversal in marsupials. Int. J. Dev. Biol. 2001;45:557–567. [PubMed] [Google Scholar]
- Renfree M.B., Coveney D., Shaw G. The influence of estrogen on the developing male marsupial. Reprod. Fertil. Dev. 2001a;13:231–240. doi: 10.1071/rd00123. [DOI] [PubMed] [Google Scholar]
- Renfree M.B., Pask A.J., Shaw G. Sex down under: The differentiation of sexual dimorphisms during marsupial development. Reprod. Fertil. Dev. 2001b;13:679–690. doi: 10.1071/rd01096. [DOI] [PubMed] [Google Scholar]
- Renfree M.B., Wilson J.D., Shaw G. The hormonal control of sexual development. In: Adinolfi M., editor. The genetics and biology of sex determination. John Wiley; New York: 2002. pp. 136–152. In. Novartis Foundation Symposium 244. [PubMed] [Google Scholar]
- Renfree M.B., Pask A.J., Shaw G. Sexual development of a model marsupial male. Aust. J. Zool. 2006;54:151–158. [Google Scholar]
- Rens W., O’Brien P.C., Yang F., Solanky N., Perelman P., Graphodatsky A.S., Ferguson M.W., Svartman M., De Leo A.A., Graves J.A., et al. Karyotype relationships between distantly related marsupials from South America and Australia. Chromosome Res. 2001;9:301–308. doi: 10.1023/a:1016646629889. [DOI] [PubMed] [Google Scholar]
- Rens W., O’Brien P.C., Fairclough H., Harman L., Graves J.A., Ferguson-Smith M.A. Reversal and convergence in marsupial chromosome evolution. Cytogenet. Genome Res. 2003;102:282–290. doi: 10.1159/000075764. [DOI] [PubMed] [Google Scholar]
- Rofe R., Hayman D. G-banding evidence for a conserved complement in the Marsupialia. Cytogenet. Cell Genet. 1985;39:40–50. doi: 10.1159/000132101. [DOI] [PubMed] [Google Scholar]
- Sakaguchi D.S., Van Hoffelen S.J., Young M.J. Differentiation and morphological integration of neural progenitor cells transplanted into the developing mammalian eye. Ann. N. Y. Acad. Sci. 2003;995:127–139. doi: 10.1111/j.1749-6632.2003.tb03216.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi D.S., Van Hoffelen S.J., Theusch E., Parker E., Orasky J., Harper M.M., Benediktsson A., Young M.J. Transplantation of neural progenitor cells into the developing retina of the Brazilian opossum: An in vivo system for studying stem/progenitor cell plasticity. Dev. Neurosci. 2004;26:336–345. doi: 10.1159/000082275. [DOI] [PubMed] [Google Scholar]
- Sakaguchi D.S., Van Hoffelen S.J., Grozdanic S.D., Kwon Y.H., Kardon R.H., Young M.J. Neural progenitor cell transplants into the developing and mature central nervous system. Ann. N. Y. Acad. Sci. 2005;1049:118–134. doi: 10.1196/annals.1334.012. [DOI] [PubMed] [Google Scholar]
- Samollow P.B. Status and applications of genomic resources for the gray, short-tailed opossum, Monodelphis domestica, an American marsupial model for comparative biology. Aust. J. Zool. 2006;54:173–196. [Google Scholar]
- Samollow P.B., Graves J.A.M. Gene maps of marsupials. ILAR J. 1998;39:203–224. doi: 10.1093/ilar.39.2-3.203. [DOI] [PubMed] [Google Scholar]
- Samollow P.B., Ford A.L., VandeBerg J.L. X-linked gene expression in the Virginia opossum: Differences between the paternally derived Gpd and Pgk-A loci. Genetics. 1987;115:185–195. doi: 10.1093/genetics/115.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samollow P.B., Robinson E.S., Ford A.L., VandeBerg J.L. Developmental progression of Gpd expression from the inactive X chromosome of the Virginia opossum. Dev. Genet. 1995;16:367–378. doi: 10.1002/dvg.1020160410. [DOI] [PubMed] [Google Scholar]
- Samollow P.B., Kammerer C.M., Mahaney S.M., Schneider J.L., Westenberger S.J., VandeBerg J.L., Robinson E.S. First generation linkage map of the gray, short-tailed opossum, Monodelphis domestica, reveals genome-wide reduction in female recombination rates. Genetics. 2004;166:307–329. doi: 10.1534/genetics.166.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samollow P.B., Gouin N., Miethke P., Mahaney S.M., Kenney M., VandeBerg J.L., Graves J.A., Kammerer C.M. A microsatellite-based, physically anchored linkage map for the gray, short-tailed opossum (Monodelphis domestica) Chromosome Res. 2007;15:269–281. doi: 10.1007/s10577-007-1123-4. [DOI] [PubMed] [Google Scholar]
- Sandstedt S.A., Tucker P.K. Evolutionary strata on the mouse X chromosome correspond to strata on the human X chromosome. Genome Res. 2004;14:267–272. doi: 10.1101/gr.1796204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwood L., Johnson M.H. Trophoblast and hypoblast in the monotreme, marsupial and eutherian mammal: Evolution and origins. Bioessays. 2006;28:128–145. doi: 10.1002/bies.20360. [DOI] [PubMed] [Google Scholar]
- Shaw G., Renfree M.B. Parturition and perfect prematurity: Birth in marsupials. Aust. J. Zool. 2006;54:139–149. [Google Scholar]
- Shaw G., Fenelon J., Sichlau M., Auchus R.J., Wilson J.D., Renfree M.B. Role of the alternate pathway of dihydrotestosterone formation in virilization of the Wolffian ducts of the tammar wallaby, Macropus eugenii. Endocrinology. 2006;147:2368–2373. doi: 10.1210/en.2005-1251. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.I., Zakharova I.S., Elisaphenko E.A., Kolesnikov N.N., Whitehead S., Bird C., Ross M., Weidman J.R., Jirtle R.L., Karamysheva T.V., et al. Genes flanking Xist in mouse and human are separated on the X chromosome in American marsupials. Chromosome Res. 2007;15:127–136. doi: 10.1007/s10577-006-1115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman S., Bell J.T., Copley R.R., Taylor M.S., Williams R.W., Mott R., Flint J. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.K. Time's arrow: Heterochrony and the evolution of development. Int. J. Dev. Biol. 2003;47:613–621. [PubMed] [Google Scholar]
- Smith K.K. Craniofacial development in marsupial mammals: Developmental origins of evolutionary change. Dev. Dyn. 2006;235:1181–1193. doi: 10.1002/dvdy.20676. [DOI] [PubMed] [Google Scholar]
- Sonea I.M., Iqbal J., Prins G.S., Jacobson C.D. Ontogeny of androgen receptor-like immunoreactivity in the reproductive tract of male Monodelphis domestica. Biol. Reprod. 1997;56:852–860. doi: 10.1095/biolreprod56.4.852. [DOI] [PubMed] [Google Scholar]
- Springer M.S., Murphy W.J., Eizirik E., O’Brien S.J. Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc. Natl. Acad. Sci. 2003;100:1056–1061. doi: 10.1073/pnas.0334222100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone W.H., Bruun D.A., Manis G.S., Holste S.B., Hoffman E.S., Spong K.D., Walunas T. The immunobiology of the marsupial, Monodelphis domestica. In: Stolen J.S., et al., editors. Modulators of immune responses; The evolutionary trail. SOS Publications; Fair Haven, NJ: 1996. pp. 149–165. [Google Scholar]
- Stone W.H., Bruun D.A., Foster E.B., Manis G.S., Hoffman E.S., Saphire D.G., VandeBerg J.L., Infante A.J. Absence of a significant mixed lymphocyte reaction in a marsupial (Monodelphis domestica) Lab. Anim. Sci. 1998;48:184–189. [PubMed] [Google Scholar]
- Stone W.H., Bruun D.A., Fuqua C., Glass L.C., Reeves A., Holste S., Figueroa F. Identification and sequence analysis of an Mhc class II B gene in a marsupial (Monodelphis domestica) Immunogenetics. 1999;49:461–463. doi: 10.1007/s002510050520. [DOI] [PubMed] [Google Scholar]
- Suzuki S., Renfree M.B., Pask A.J., Shaw G., Kobayashi S., Kohda T., Kaneko-Ishino T., Ishino F. Genomic imprinting of IGF2, p57(KIP2) and PEG1/MEST in a marsupial, the tammar wallaby. Mech. Dev. 2005;122:213–222. doi: 10.1016/j.mod.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Suzuki S., Ono R., Narita T., Pask A.J., Shaw G., Wang C., Kohda T., Alsop A.E., Marshall Graves J.A., Kohara Y., et al. Retrotransposon silencing by DNA methylation can drive mammalian genomic imprinting. PLoS Genet. 2007;3:e55. doi: 10.1371/journal.pgen.0030055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swales A.K., Spears N. Genomic imprinting and reproduction. Reproduction. 2005;130:389–399. doi: 10.1530/rep.1.00395. [DOI] [PubMed] [Google Scholar]
- Turner J.M. Meiotic sex chromosome inactivation. Development. 2007;134:1823–1831. doi: 10.1242/dev.000018. [DOI] [PubMed] [Google Scholar]
- Tycko B., Morison I.M. Physiological functions of imprinted genes. J. Cell. Physiol. 2002;192:245–258. doi: 10.1002/jcp.10129. [DOI] [PubMed] [Google Scholar]
- Tyndale-Biscoe H. Life of marsupials. CSIRO Publishing; Collingwood: 2005. [Google Scholar]
- VandeBerg J.L. The gray, short-tailed opossum (Monodelphis domestica) as a model didelphid for genetic research. Aust. J. Zool. 1990;37:235–247. [Google Scholar]
- VandeBerg J.L. The laboratory opossum (Monodelphis domestica) In: Poole T., English P., editors. UFAW handbook on the management of laboratory animals: Terrestrial vertebrates. 7th ed. Vol. 1. Blackwell Science; Oxford, U.K: 1999. pp. 193–209. [Google Scholar]
- VandeBerg J.L., Robinson E.S. The laboratory opossum (Monodelphis domestica) in laboratory research. ILAR J. 1997;38:4–12. doi: 10.1093/ilar.38.1.4. [DOI] [PubMed] [Google Scholar]
- VandeBerg J.L., Robinson E.S., Samollow P.B., Johnston P.G. X-linked gene expression and X-chromosome inactivation: Marsupials, mouse, and man compared. Isozymes Curr. Top. Biol. Med. Res. 1987;15:225–253. [PubMed] [Google Scholar]
- Van Hoffelen S.J., Young M.J., Shatos M.A., Sakaguchi D.S. Incorporation of murine brain progenitor cells into the developing mammalian retina. Invest. Ophthalmol. Vis. Sci. 2003;44:426–434. doi: 10.1167/iovs.02-0269. [DOI] [PubMed] [Google Scholar]
- van Rheede T., Bastiaans T., Boone D.N., Hedges S.B., de Jong W.W., Madsen O. The platypus is in its place: Nuclear genes and indels confirm the sister group relation of monotremes and Therians. Mol. Biol. Evol. 2006;23:587–597. doi: 10.1093/molbev/msj064. [DOI] [PubMed] [Google Scholar]
- Wagschal A., Feil R. Genomic imprinting in the placenta. Cytogenet. Genome Res. 2006;113:90–98. doi: 10.1159/000090819. [DOI] [PubMed] [Google Scholar]
- Wakefield M.J., Keohane A.M., Turner B.M., Graves J.A. Histone underacetylation is an ancient component of mammalian X chromosome inactivation. Proc. Natl. Acad. Sci. 1997;94:9665–9668. doi: 10.1073/pnas.94.18.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Chen P., Quan W., Halpern M. Suprasternal gland secretion of male short-tailed opossum induces IP3 generation in the vomeronasal organ. Biochim. Biophys. Acta. 2007;1770:725–732. doi: 10.1016/j.bbagen.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Warren W.C., Hillier L.W., Marshall Graves J.A., Birney E., Ponting C.P., Grützner F., Belov K., Miller W., Clarke L., Chinwalla A.T., et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson D., Jacombs A.S., Loebel D.A., Robinson E.S., Johnston P.G. Single nucleotide primer extension (SNuPE) analysis of the G6PD gene in somatic cells and oocytes of a kangaroo (Macropus robustus) Genet. Res. 2000;75:269–274. doi: 10.1017/s0016672300004523. [DOI] [PubMed] [Google Scholar]
- Weidman J.R., Murphy S.K., Nolan C.M., Dietrich F.S., Jirtle R.L. Phylogenetic footprint analysis of IGF2 in extant mammals. Genome Res. 2004;14:1726–1732. doi: 10.1101/gr.2774804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidman J.R., Maloney K.A., Jirtle R.L. Comparative phylogenetic analysis reveals multiple non-imprinted isoforms of opossum Dlk1. Mamm. Genome. 2006;17:157–167. doi: 10.1007/s00335-005-0116-x. [DOI] [PubMed] [Google Scholar]
- Wilkins J.F., Haig D. What good is genomic imprinting: The function of parent-specific gene expression. Nat. Rev. Genet. 2003;4:359–368. doi: 10.1038/nrg1062. [DOI] [PubMed] [Google Scholar]
- Willson M.F., Burley N. Mate choice in plants: Tactics, mechanisms, and consequences. Princeton University Press; Princeton: 1983. [Google Scholar]
- Wilson D.E., Reeder D.M. Mammal species of the world; A taxonomic and geographic reference. The Johns Hopkins University Press; Baltimore: 2005. [Google Scholar]
- Wintzer M., Mladinic M., Lazarevic D., Casseler C., Cattaneo A., Nicholls J. Strategies for identifying genes that play a role in spinal cord regeneration. J. Anat. 2004;204:3–11. doi: 10.1111/j.1469-7580.2004.00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E.S., Young L.J., Papenfuss A.T., Belov K. In silico identification of opossum cytokine genes suggests the complexity of the marsupial immune system rivals that of eutherian mammals. Immunome Res. 2006;2:4. doi: 10.1186/1745-7580-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.J., Oakey R.J. Genomic imprinting in mammals: Emerging themes and established theories. PLoS Genet. 2006;2:e147. doi: 10.1371/journal.pgen.0020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodburne M.O., Rich T.H., Springer M.S. The evolution of tribospheny and the antiquity of mammalian clades. Mol. Phylogenet. Evol. 2003;28:360–385. doi: 10.1016/s1055-7903(03)00113-1. [DOI] [PubMed] [Google Scholar]
- Wu H.K., Squire J.A., Song Q., Weksberg R. Promoter-dependent tissue-specific expressive nature of imprinting gene, insulin-like growth factor II, in human tissues. Biochem. Biophys. Res. Commun. 1997;233:221–226. doi: 10.1006/bbrc.1997.6431. [DOI] [PubMed] [Google Scholar]
- Wutz A., Gribnau J. X inactivation Xplained. Curr. Opin. Genet. Dev. 2007;17:387–393. doi: 10.1016/j.gde.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Xue F., Tian X.C., Du F., Kubota C., Taneja M., Dinnyes A., Dai Y., Levine H., Pereira L.V., Yang X. Aberrant patterns of X chromosome inactivation in bovine clones. Nat. Genet. 2002;31:216–220. doi: 10.1038/ng900. [DOI] [PubMed] [Google Scholar]
- Zenger K.R., McKenzie L.M., Cooper D.W. The first comprehensive genetic linkage map of a marsupial: The tammar wallaby (Macropus eugenii) Genetics. 2002;162:321–330. doi: 10.1093/genetics/162.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuri I., Halpern M. Modification of odor investigation and discrimination in female opossums (Monodelphis domestica) following the ablation of the accessory olfactory bulbs. Behav. Neurosci. 2005;119:612–621. doi: 10.1037/0735-7044.119.2.612. [DOI] [PubMed] [Google Scholar]