

Abstract
The ability to edit the yeast genome with relative ease has contributed to the organism being a model eukaryote for decades. Most methods for deleting, inserting, or altering genomic sequences require transformation with DNA that carries the desired change and a selectable marker. One-step genome editing methods retain the selectable marker. Seamless genome editing methods require more steps and a marker that can be used for both positive and negative selection, such as URA3. Here we describe the PCR-based Fifty-Fifty method for seamless genome editing that requires only two primers, one PCR with a URA3 cassette, and a single yeast transformation. Our method is based on pop-in/pop-out gene replacement and is amenable to the facile creation of genomic deletions and short insertions or substitutions. We used the Fifty-Fifty method to make two conservative loss-of-function mutations in MATALPHA1, with results that suggest the wildtype gene has a new function outside of that presently known.
Keywords: yeast, PCR, marker-free, seamless, mutation, MATALPHA1
INTRODUCTION
The budding yeast Saccharomyces cerevisiae has been a model eukaryote for decades in part because of the ease with which the genome can be precisely altered. For example, gene function can be studied in a number of ways by deleting the open reading frame (ORF), changing the coding sequence, inserting an epitope or fluorescent tag, or altering expression. Two properties that make yeast especially amenable to genome manipulation are the ease at which it can be transformed with exogenous DNA and the high fidelity of integrating the DNA into the genome by homologous recombination [Rothstein, 1991].
Although the methods used to alter the yeast genome have changed as new technologies have been developed, the underlying mechanism based on transformation and homologous recombination remains the same. Twenty years ago if one wanted to disrupt a gene, she would first clone the gene and surrounding DNA onto a plasmid. Using in vitro manipulations, all or part of the gene would be replaced with a selectable marker, such as the URA3 gene. The plasmid would then be digested with restriction enzymes to release a linear fragment used to transform yeast, with the result of replacing the wildtype gene with the disruption marked by URA3 [Rothstein, 1991]. The advent of PCR-based, one-step gene replacement made the method much simpler [Lorenz et al., 1995; Baudin et al., 1993]. The linear DNA used for yeast transformation is now created by PCR, using synthetic oligonucleotides (oligos) designed with yeast sequences up- and downstream of the deletion end points and sequences to amplify a selectable marker. This PCR product is used to transform yeast with the same result as the cloning-based method above.
Some experiments require seamless genome modification without a selectable marker or extraneous DNA remaining. For example, to study the effect of a single codon change on gene function, or to build a new strain requiring multiple changes that would make the accumulation of selectable markers impractical. Seamless genome modification requires more work than one-step gene replacement. The original method requires two steps and is referred to as “pop-in/pop-out” gene replacement [Scherer and Davis, 1979]. A plasmid is assembled in vitro that carries a segment of yeast DNA containing the desired alteration and the selectable/counterselectable URA3 marker. The first step is transformation of yeast with the plasmid that has been cut at a restriction site within the yeast DNA on one side or the other of the alteration to direct “pop-in” integration. Transformation with a gapped plasmid produces on the chromosome nearly a tandem duplication of the cloned DNA separated by the URA3 plasmid, with the altered copy on one side and wildtype on the other. The second step is initiated by growing the transformed cells in the absence of uracil selection. At a low frequency, cells arise in the population that have undergone recombination between the repeats that flank the URA3 plasmid. These rare “pop-out” recombinants can be selected for on 5-fluoro-orotic acid (FOA) medium, which is toxic to URA3 cells [Boeke et al., 1984]. Whether a pop-out strain retains altered or wildtype DNA depends on where the crossover takes place. If the crossover occurs in the yeast DNA between the alteration and the URA3 plasmid, then the alteration will remain while the wildtype is evicted. Likewise, if the crossover occurs outside the alteration relative to the URA3 plasmid, then wildtype DNA remains.
There are cloning-free methods for marker-free genome modification, but they are either not seamless, or they require multiple oligos, PCRs, and transformations. One method for marker-free gene deletion is easy to perform, but it leaves behind extraneous DNA, such as a fragment of bacterial hisG or a loxP site [Güldener et al., 1996; Schneider et al., 1996]. Strictly speaking, the products of these techniques are marker-free, but they are not seamless. The DNA remnants preclude use of the methods for applications such as in-frame deletions. Moreover, repeated use of the techniques in the same strain, so-called marker recycling, can lead to problems with subsequent transformation [Davidson and Schiestl, 2000] and genome rearrangements [Delneri et al., 2000]. Other PCR-based techniques are available that result in truly marker-free, seamless genome modification. Some require multiple PCR primers and PCR steps to assemble a DNA molecule for yeast transformation that carries sequences to target integration, the alteration, a repeat sequence for pop-out recombination, and a URA3 marker [Erdeniz et al., 1997; Akada et al., 2006]. Another method uses four long DNA oligos and two yeast transformations to integrate URA3 at the target locus and subsequently evict it with complementary oligos containing the alteration [Storici et al., 2001].
Here we describe the Fifty-Fifty method for marker-free, seamless genome editing that is almost as simple as PCR-based, one-step gene replacement. The technique requires only two primers, one PCR with a URA3 cassette, and a single transformation. It is a two-step method, with pop in of the URA3 cassette followed by selection with FOA for pop-out recombinants. The key is one of the two PCR primers: the 50/50 primer, a hybrid containing fifty percent pop-in sequences, the alteration, and fifty percent pop-out sequences. Together with a standard reverse primer and the URA3 cassette, the 50/50 primer provides all that is needed for PCR-based, seamless genome editing in yeast.
MATERIALS AND METHODS
Standard techniques were used for DNA manipulations (Ausubel, 1995). DNA oligos (Eurofins MWG Operon, Alabama) are listed in Table 1. Yeast media and culture conditions were as described by Amberg et al. [2005]. PCR cassettes for yeast transformation were amplified using Takara ExTaq polymerase, which gives a consistently high yield. Assays for α-factor and mating were as described by Sprague [1991]. A PCR-based, one-step gene replacement of MATALPHA1 (α1::kanMX4) was created using primers al1.52.1 and al1.52.2 with plasmid pFA6-kanMX4 (Wach et al., 1994).
Table 1.
PCR Primers
| Name | Purpose | Sequence (5′ to 3′) |
|---|---|---|
| U2 | Forward primer to amplify URA3 from pJH136 |
CGTACGCTGCAGGTCGAC |
| D2 | Reverse primer to amplify URA3 from pJH136 |
ATCGATGAATTCGAGCTCG |
| al1.46.1 | 50/50 primer for α1∆ |
TATGAAATGTATCAACCATATATAATAACTTAATAGAC GACATTCACAATAGTGTGGTCGTGGCGGAGGTTGTTTAT CTTTCGAGTACTGAATGTTGTCACGTACGCTGCAGGTCG ACa |
| al1.46.2 | 50/50 primer for α1-2x |
TATCAACCATATATAATAACTTAATAGACGACATTCAC AATATGTTTACTCGAGCCTGCTTTCAAAATTAAGAACAA AGCATCCAAATCATACAGAAACACACGTACGCTGCAGG TCGACb |
| al1.46.3 | Reverse primer for α1∆ and α1-2x |
TGTGTTTCTGTATGATTTGGATGCTTTGTTCTTAATTTTG AAAGCAGGCTATCGATGAATTCGAGCTCGc |
| al1.52.1 | Forward primer for α1::kanMX4 |
CTTCACTTTTTATGAAATGTATCAACCATATATAATAAC TTAATAGACGACATTCACAATCGTACGCTGCAGGTCGA Cd |
| al1.52.2 | Reverse primer for α1::kanMX4 |
GCGGAAAGCTGAAACTAAAAGAAAAACCCGACTATGCT ATTTTAATCATTGAAAACGAATATCGATGAATTCGAGCT CGe |
| URA3.54.1 | Amplify URA3 to make pJH136 |
CGTACGCTGCAGGTCGACTGTGGTTTCAGGGTCCATAA AGf |
| URA3.54.3 | Amplify URA3 to make pJH136 |
ATCGATGAATTCGAGCTCGGGTAATAACTGATATAATT AAATTGAAGCg |
| URA3.for | Forward primer for split-URA3 PCR |
CACAGTTAAGCCGCTAAAGGC |
| URA3.rev | Reverse primer for split-URA3 PCR |
AGTATATTCTCCAGTAGCTAGGGAGCC |
MATALPHA1 −50 to −1, double underline; 50 nts following stop codon, dotted underline; U2, single underline.
MATALPHA1 −41 to +9, double underline; 2 nt deletion and XhoI site, italics; +14 to +63, dotted underline; U2, single underline.
Reverse complement of MATALPHA1 +14 to +63, dotted underline; D2, single underline.
MATALPHA1 −60 to −1, double underline; U2, single underline.
Reverse complement of MATALPHA1 60 nts following stop codon, double underline; D2, single underline.
U2, single underline.
D2, single underline.
Plasmid pJH136 (Figure 1, Addgene 47554) carries URA3 flanked by PCR priming sites U2 and D2 [Chu and Davis, 2008]. It was constructed by using Phusion HSII (Thermo Scientific) to PCR amplify URA3 DNA from JHY222 (S288c background; [Lardenois et al., 2011]) with primers URA3.54.1 and URA3.54.3. The 1160 bp blunt-end PCR product was cloned into the SrfI site of pCR-Script Amp SK(+) (Agilent Technologies) and verified by DNA sequencing. The URA3 sequences in pJH136 extend from 243 bp upstream of the start codon to 79 bp downstream of the stop codon. Because U2 and D2 sequences flank the common heterologous markers [Goldstein and McCusker, 1999; Wach et al., 1994], pJH136 can also be used to make standard PCR-based, one-step gene replacements.
Figure 1.
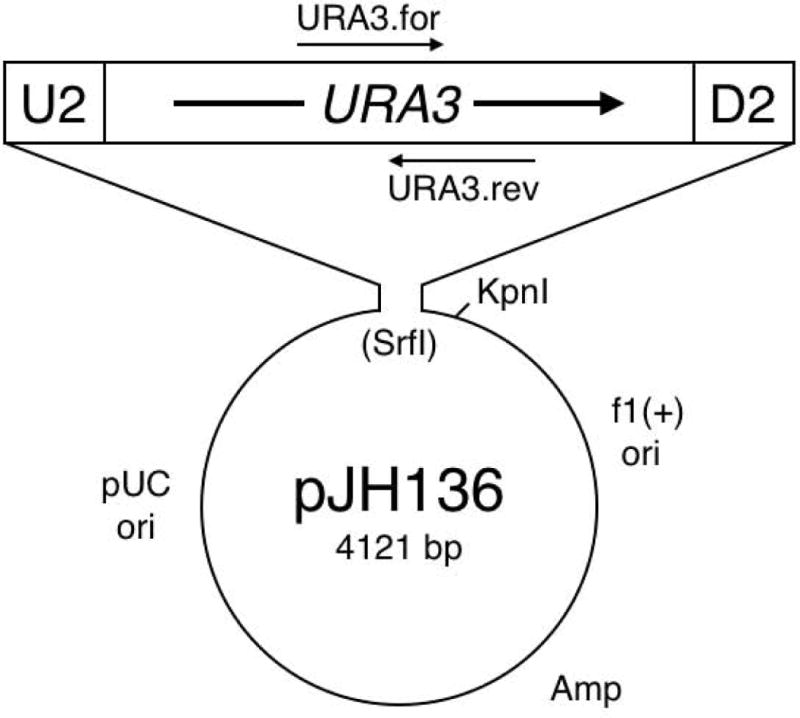
Plasmid pJH136, a template for URA3 cassette PCR. The U2 and D2 sequences provide robust PCR amplification of the 1160 bp cassette. URA3.for and URA3.rev primers can be used for split-URA3 PCR, if needed (see Materials and Methods).
The two PCR primers used to amplify URA3 for yeast transformation are the 50/50 primer and a standard reverse primer. Design of the 50/50 primer is straightforward: (5′ to 3′) 50 nts directly upstream of the alteration (pop-in sequence to target integration), the alteration, 50 nts directly downstream of the alteration (pop-out sequence), and a sequence for priming URA3 PCR (e.g., U2, Table 1). In our examples we use 50 nts for each of the pop-in and pop-out sequences, but they can be shorter or longer. The length of DNA inserted or changed is limited by oligo synthesis technology, which presently allows for ca. 125 nts total and therefore a maximum of 7 bp inserted or changed, although this can be increased if the 50/50 sequences are decreased. To create a deletion (Figures 2A and 3), the primer is 50 nts upstream of the deletion followed by 50 nts downstream of the deletion plus U2. For an insertion (Figure 2B), the primer is 50 nts upstream of the insertion, the insertion (e.g., GCA), followed by 50 nts downstream plus U2. For a single nucleotide change (Figure 2C), the primer is 50 nts upstream of the change, the single nucleotide change (e.g., C instead of G), followed by 50 nts downstream plus U2. Finally, in our example of deleting two non-consecutive basepairs (Figure 2D and see below), the primer is 50 nts upstream of the altered sequence, the alteration (CG instead of TCGA), followed by 50 nts downstream plus U2.
Figure 2.
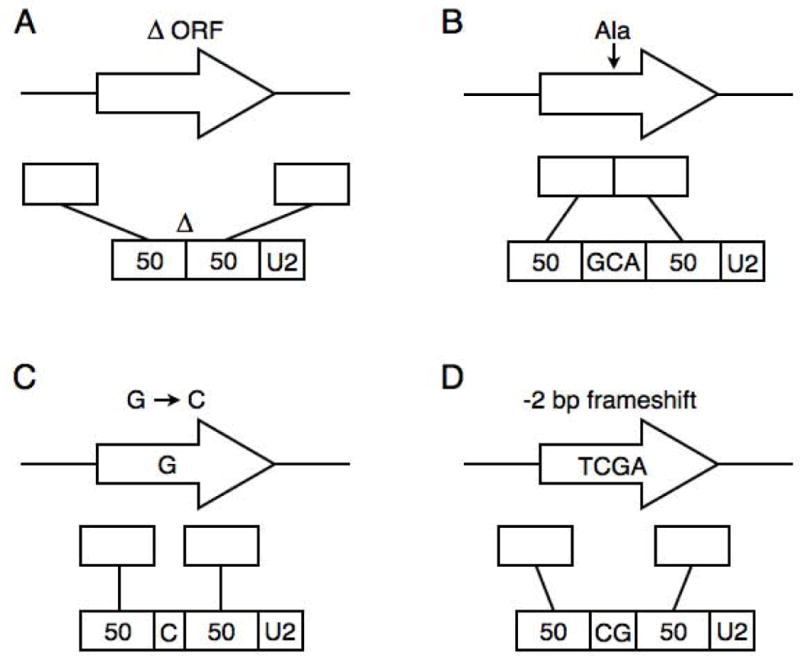
Examples of 50/50 primer design. For each alteration (A through D), the original genomic locus is shown on top. In the middle are the two 50 bp segments that will be incorporated into the 50/50 primer. On the bottom is the assembled 50/50 primer sequence, with U2 at the 3′ end for priming URA3 PCR. (A) Deletion of an ORF, (B) insertion of an alanine codon, (C) single nucleotide change, and (D) deletion of two non-consecutive basepairs.
Figure 3.
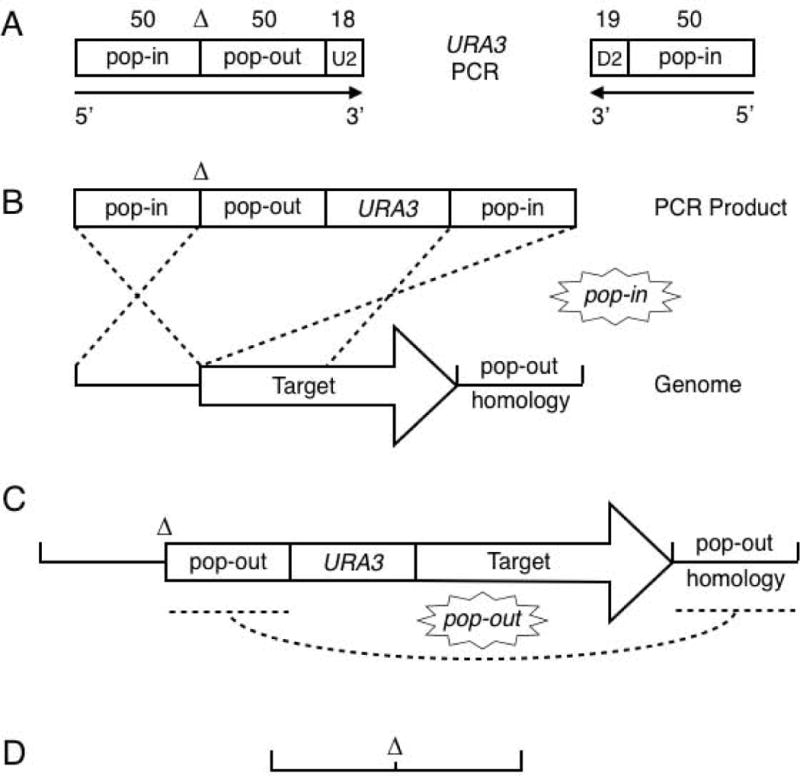
Outline of the Fifty-Fifty method. Seamless deletion of the target ORF is illustrated. Short insertions and base changes are similar (see text). (A) The two primers used for cassette PCR with pJH136. The 50/50 primer is composed of 50 nts upstream of the target ORF, the deletion (lack of ORF DNA, marked by ∆), 50 nts downstream of the ORF, and the 18 nt U2 priming sequence. The reverse primer is the reverse complement of 50 nts near the start of the ORF and the 19 nt D2 priming sequence. (B) Integration of the 50/50 URA3 cassette into the yeast genome. Dotted lines indicate DNA crossovers. (C) Structure of the 50/50 URA3 transformant. Recombination between the repeated pop-out segments leaves the seamless gene deletion, shown in (D).
The standard reverse primer is composed of 50 nts yeast sequence followed by a sequence for priming URA3 PCR (e.g., D2, Table 1). One function of the reverse primer’s yeast sequence is to target pop-in integration of the URA3 cassette. Depending on the alteration being created, the reverse primer’s yeast sequence can also serve as a direct repeat downstream of URA3 of the pop-out sequence present in the 50/50 primer. Thus for alterations that are not a deletion, the reverse primer should be the reverse complement of the 50/50 primer’s pop-out sequence plus D2. Beware that PCR with 50 bp direct repeats flanking URA3 can be problematic for some polymerases (see below). For deleting an ORF, the yeast sequences in the reverse primer can be the reverse complement of any 50 nts in the ORF or even the 50 nts immediately downstream of the ORF, which is equivalent to the 50/50 primer’s pop-out sequence.
We used the Fifty-Fifty method to create two MATALPHA1 mutations in JHY337 (MATα ura3∆0 leu2∆0 lys2∆0, a derivative of JHY222). Precise deletion of the ORF (α1∆) was accomplished with primers al1.46.1 and al1.46.3. A −2 bp frameshift mutation after the third codon (α1–2x) was accomplished with primers al1.46.2 and al1.46.3. For each mutation, primer pairs were used to amplify URA3 by PCR from pJH136. PCR products were either used directly to transform yeast [Gietz and Schiestl, 2007] or first concentrated by ethanol precipitation and resuspended in 0.1X TE buffer. We plated one-fifth of the transformation to SC-Ura plates and then replica plated to fresh plates after two days to eliminate the background lawn that is often present with PCR transformations. Several dozen colonies were obtained for each transformation. Correct transformants (18 of 20 scored) were identified by genomic PCR using primers specific to both sides of the integrated URA3 cassette. Four independent transformants for each mutation were streak purified on YPD plates and a single colony of each was used to inoculate 3 mL YPEG broth (2.5% ethanol, 2% glycerol; used to prevent growth of petites). Cultures were grown to saturation (~2 × 108 cells/mL) for 2 days at 30C. Fifty microlitres of the culture (~1×107 cells) was spread onto a plate of synthetic complete medium containing 0.8 mg/mL FOA (US Biological). An average of 74 colonies arose on each plate (range 32 to ~200, eight plates scored). Correct URA3 pop-out recombinants were identified by genomic PCR and confirmed by DNA sequencing.
PCR amplification of DNA segments that contain repeated sequences can be problematic, with results being dependent on length of the repeat and PCR conditions. PCR amplification of the URA3 cassette using primers al1.46.2 and al1.46.3 produces a product with a 50 bp direct repeat on each end. We found that Takara ExTaq polymerase can efficiently generate this product in a single reaction (Figure 4A). However, the same primers and template used with Phusion HSII polymerase failed (data not shown). A simple and robust solution to problematic PCRs with repeat-containing products is to perform split-URA3 PCRs, which separate the repeats into two tubes. Primers URA3.for and URA3.rev can be used with the long D2- and U2-tailed primers, respectively, to set up two PCRs that are later pooled and used for yeast transformation (Figures 1 and 4). The URA3 fragments share 280 bp overlap and recombine upon transformation to restore URA3. Single and split-URA3 PCRs using ExTaq polymerase are shown in Figure 4A. We have used both single and split-URA3 PCRs for transformation with similar results.
Figure 4.
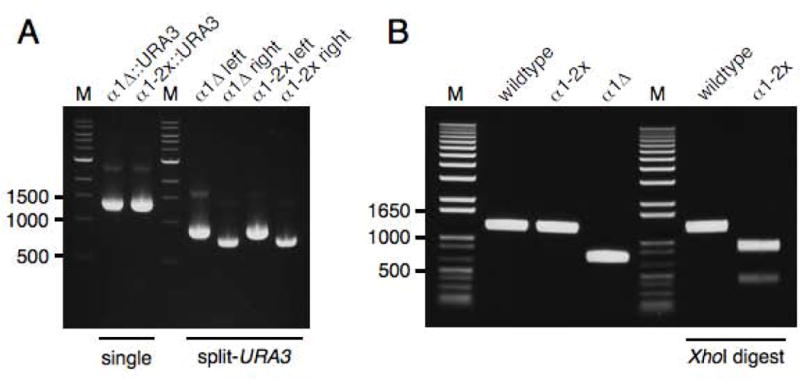
Gel analysis of 50/50 cassette PCR and genomic PCR of yeast strains created in this study. Marker (M) sizes are given in bp. (A) 50/50 PCR cassettes for yeast transformation. ExTaq was used for the 50 μL PCRs, of which 4 μL were loaded on the gel. On the left are single PCRs of the full 50/50 URA3 cassettes. On the right are split-URA3 PCRs performed with the same primers but in combination with URA3.for and URA3.rev. Either single or pooled split-URA3 PCRs can be used for yeast transformation. (B) Genomic PCR of MATALPHA1 alleles. Expected sizes (bp): wildtype (1278), α1-2x (1276), α1∆ (750), wildtype with XhoI (1278), α1-2x with XhoI (882, 394).
RESULTS
We wished to develop a simple method for seamless genome editing in yeast that satisfied the following criteria: cloning-free (no in vitro plasmid construction), based on synthetic DNA primers, requires only one PCR and one yeast transformation, and have no special strain requirements other than the ura3 genotype. The original and elegant two-step gene replacement technique [Scherer and Davis, 1979] (see Introduction) provided the framework for us to convert a plasmid-based method to a PCR-based one that satisfied our criteria.
What are the important features of the original two-step method, and can they be more-simply accomplished and even improved without cloning yeast DNA and introducing the alteration on a plasmid? One important feature is the selectable/counterselectable marker used to select for the pop-in and pop-out recombinants. URA3 is commonly used and has an advantage over another such marker, LYS2, because of its smaller ORF length (804 bp vs. 4179 bp). In a cloning-free method, only URA3 is required, plasmid sequences are not. For the pop-in step, linear DNA is used for transformation that has yeast sequences flanking URA3. In the original method, cutting within the yeast sequences cloned into a URA3 plasmid produces the linear DNA. The pop-in step can just as well be accomplished using linear DNA with yeast ends created by synthetic DNA primers and PCR amplification of a URA3 cassette. Indeed, the components of a cloning-free pop-in step are essentially PCR-based, one-step gene replacement using the URA3 marker.
This leaves two features that require adapting: the alteration and the duplicated yeast DNA that allows for pop-out recombination. Because of advances in DNA synthesis technology, we found that both can be incorporated into one of the two primers used to PCR amplify the URA3 cassette (Figures 2 and 3). This primer is a hybrid composed of two segments of yeast DNA flanking the alteration, followed by a sequence for priming URA3 PCR. The first segment serves as a pop-in sequence to target integration of URA3 into the genome. The second serves as a pop-out sequence, with homology to a segment located on the other side of the integrated URA3 marker. In between the pop-in and pop-out segments is the alteration. Because DNA primer length is limited by synthetic chemistry, the types of alterations are limited to deletions, short insertions or substitutions. In principle, the hybrid primer contains fifty percent pop-in sequence, the alteration, and fifty percent pop-out sequence. Thus, it is called the 50/50 primer, and it is used in the Fifty-Fifty method.
An additional advantage of Fifty-Fifty over the original two-step method is that none of the transformed sequences are duplicated upon integration, because the PCR cassette is linear DNA and not a gapped plasmid. The only sequences present in duplicate are the pop-out sequences designed into the 50/50 primer between the alteration and URA3, and the cognate sequences on the other side of URA3 (Figure 3C). Thus, unlike the original method where sequences up- and downstream of the alteration are duplicated and give rise to both wildtype and altered pop-out recombinants, the Fifty-Fifty method gives rise only to altered recombinants.
To demonstrate the technique, we used the Fifty-Fifty method to introduce two mutations in MATALPHA1. The α1 transcription factor is expressed only in MATα cells and functions in a complex with Mcm1 to activate α-specific genes, such as those encoding α mating pheromone and the a-factor receptor [Sprague, 2005]. Mutants lacking α1 are sterile and cannot mate to MATa cells. An α1::kanMX4 deletion strain yielded an unexpected and new result: although haploid α1::kanMX4 cells had the expected phenotypes of not producing α-factor and not mating to MATa cells, we found that, in contrast to wildtype MATα cells, α1::kanMX4 cells mated as MATa cells, albeit with low efficiency (Figure 5 and data not shown). One explanation for the ability of α1::kanMX4 cells to mate as MATa cells is that the kanMX4 cassette, with its strong A. gossypii TEF1 promoter, interferes with transcription of the adjacent MATALPHA2 gene, which encodes a repressor of a-specific genes [Sprague, 2005]. Reduced MATALPHA2 transcription would lead to a defect in a-specific gene repression, which in turn would allow mating as MATa cells. To test this hypothesis, we used the Fifty-Fifty method to create two MATALPHA1 mutations without the kanMX4 marker: one a precise ORF deletion and the other a deletion of two non-consecutive basepairs near the start of the ORF to introduce a frameshift and a diagnostic XhoI site.
Figure 5.
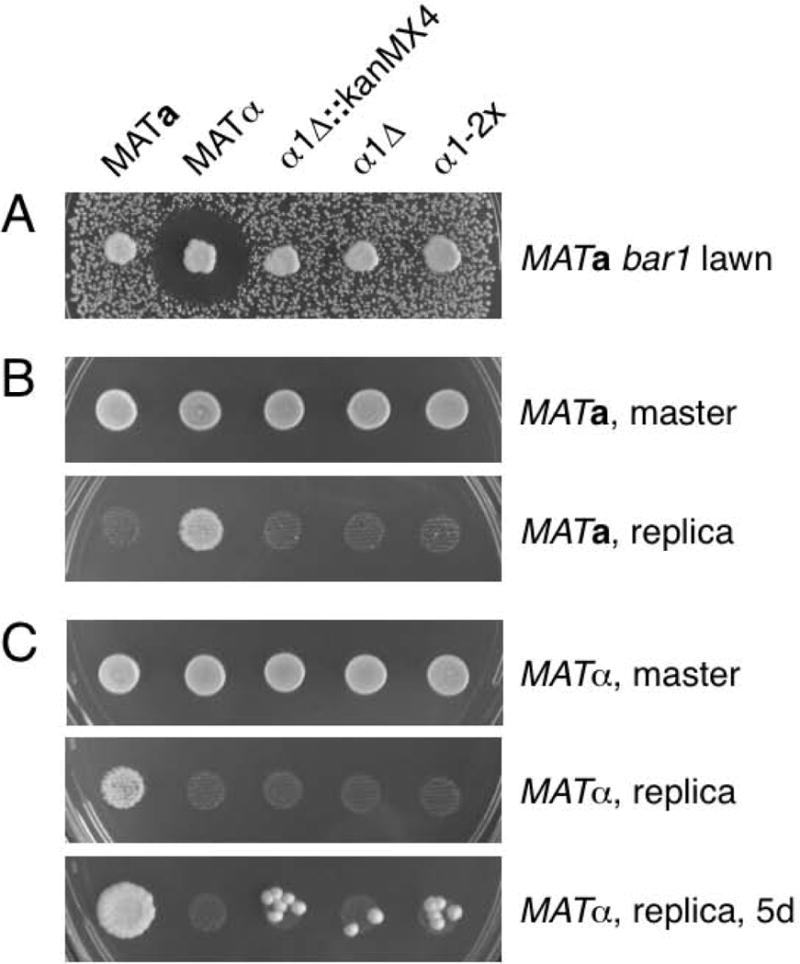
MATALPHA1 mutants are α-specific sterile but can mate as MATa cells with low efficiency. Plates were incubated at 30C. (A) Halo assay to measure α-factor production. Cells were patched onto a lawn of MATa bar1 cells on a YPD plate and incubated for one day. (B) Mating as MATα cells. Each of the five strains were mixed 1:1 with a MATa lys1 tester strain in YPD broth and spotted onto a YPD plate. The master plate was photographed after 1 day and then the cells were replica plated to selective medium, which was photographed after 1 day. (C) Mating as MATa cells. Similar to (B), except the five strains were mixed with a MATα lys1 tester strain. Mating was photographed after 1 and 5 days incubation.
To create the precise MATALPHA1 deletion (α1∆), we designed the 50/50 primer (al1.46.1) with 50 nts upstream of the MATALPHA1 start codon, the deletion (lack of the MATALPHA1 ORF), 50 nts downstream of the stop codon, and the 18 nt U2 sequence. The reverse primer (al1.46.3) for URA3 cassette PCR contained the reverse complement of the MATALPHA1 ORF from +14 to +63 followed by the 19 nt D2 sequence (Figure 3A). For deletions larger than 50 bp, such as the MATALPHA1 ORF, the URA3 cassette can be inserted at or upstream of the genomic pop-out homology. In this case, the Fifty-Fifty URA3 cassette was inserted in the first part of MATALPHA1, a site we chose so we could recycle the reverse primer when making the −2 bp deletion (see below). The transformed α1∆::URA3 yeast strain had the configuration 50 bp upstream of MATALPHA1 ATG, 50 bp downstream of MATALPHA1 stop codon, URA3, and then the MATALPHA1 ORF from +14 continuing into the wildtype genomic sequences (Figure 3C). Culture in the absence of uracil selection allowed for growth of FOA-resistant recombinants that had undergone a crossover between the 50 bp MATALPHA1 ORF pop-out sequence incorporated into the 50/50 primer and its cognate sequence downstream of URA3 on the chromosome (Figure 3C). The resulting strain carries a precise deletion of the MATALPHA1 ORF without URA3 or any other DNA (Figures 3D and 4B, DNA sequence data not shown). We tested the α1∆ strain and found it to be α-specific sterile, as expected (Figures 5A and 5B). We also found that α1∆ cells mated with low efficiency as MATa cells (Figure 5C). Thus, it is not the kanMX4 cassette per se that causes the MATa mating phenotype. It is either the lack of α1 activity or an off-target effect of deleting 528 bp DNA near MATALPHA2.
To discriminate between these two possibilities, we used the Fifty-Fifty method to introduce a frameshift mutation at the start of the MATALPHA1 ORF, a change that likely would not have an off-target effect MATALPHA2 transcription. The α1-2x mutation deletes two non-consecutive basepairs after the third codon and creates a diagnostic XhoI site (CTCGAG). Thus, the wildtype ORF begins ATGTTTACTTCGAAG, whereas α1-2x begins ATGTTTACTCGAG. We designed the 50/50 primer (al1.46.2) with sequences −41 to +9 relative to the wildtype ORF, the alteration (CG instead of TCGA following +9), sequences +14 to +63, and the 18 nt U2 sequence. The reverse primer (al1.46.3) for URA3 cassette PCR was the same as that used for α1∆. Note that the pop-out sequence in the second half of the α1-2x 50/50 primer is identical to the pop-in sequence in the reverse primer. Non-selective growth of α1-2x::URA3 cells followed by selection on FOA medium yielded pop-out recombinants that were verified as α1-2x based on a correct size genomic PCR product that could be digested with XhoI (Figure 4B) and DNA sequencing (data not shown). We found the α1-2x strain to be α-specific sterile (Figures 5A and 5B). As with α1::kanMX4 and α1∆, α1-2x mutants mated with low efficiency as MATa cells (Figure 5C). Given that α1-2x alters the MATα locus by deletion of only 2 bp at the start of the MATALPHA1 ORF, it is highly unlikely that the unexpected mating phenotype we observed is the result of an off-target effect. From these observations, we conclude that α1 has a function outside its known role as a positive regulator of α-specific gene expression. For example, α1 might positively regulate MATALPHA2 transcription. Another possibility is based on the fact that both α1 and α2 function by binding the Mcm1 transcription factor: absence of α1 might upset the balance of Mcm1 and α2 association, leading to a defect in a-specific gene repression. Whatever the mechanism, it is intriguing that a transcription factor that had only been assigned a role in activation of α-specific genes also seems to have a role in repression of a-specific genes.
DISCUSSION
Here we have described the Fifty-Fifty method for simplified marker-free, seamless genome editing in yeast. The utility of the method is that it requires only two primers, one PCR, and a single yeast transformation. The key component is the 50/50 primer, which provides the pop-in sequence, the alteration, and the pop-out sequence. Because the alteration is engineered upstream of the pop-out sequence relative to URA3, recombination between the 50/50 primer pop-out sequence and the cognate sequence downstream of URA3 only leaves the altered sequence on the chromosome. This is a significant advantage over the original, plasmid-based method that can produce both wildtype and altered recombinants. We used the S. cerevisiae URA3 marker, but if the host strain is not ura3∆0 the method can also be used with a heterologous URA3 marker, such as CaURA3MX4 [Goldstein et al., 1999]. One advantage of using S. cerevisiae URA3 is its smaller size (1160 vs. 1509 bp cassette). Another is that it does not contain the flanking A. gossypii TEF1 promoter and terminator sequences that are present in most heterologous markers. If a yeast strain already carries a heterologous marker, such as a kanMX4 gene replacement, the common sequences present in CaURA3MX4 will cause problems with obtaining correct integrants at the new locus and with obtaining correct pop-out recombinants that have not become FOA-resistant by way of gene conversion from kanMX4.
There are PCR-based methods that accomplish the same goal as the Fifty-Fifty method, although none are as efficient and economical. One method most similar to ours uses PCR primers that create a roughly 60 bp direct repeat flanking the Kl URA3 marker [Längle-Rouault and Jacobs, 1995]. The repeats contain 30 bp upstream of the desired alteration, the alteration itself, and 30 bp downstream. The targeting homology is short, leading to inefficient transformation even with the heterologous marker. Moreover, because the upstream-alteration-downstream sequences are present on both sides of the Kl URA3 cassette, there is the potential for undesired integration events at either the upstream sequence or the downstream sequence, popout of either of which would return wildtype. The Fifty-Fifty method reduces the possibility of incorrect integration because the upstream pop-in sequence is confined to one primer, which also makes the method more economical. Also, the stretches of pop-in homology on the 50/50 and reverse primers are longer. Together, these features contribute to nearly all Fifty-Fifty Ura+ transformants being correct (see Materials and Methods).
Other methods have been described that require only one yeast transformation, but they require multiple PCR primers and PCRs [Erdeniz et al., 1997; Akada et al., 2006]. The Akada method is limited to seamless gene deletion and requires four PCR primers and three PCRs divided into two steps [Akada et al., 2006]. The Erdeniz method adapted for de novo mutations likewise requires seven PCR primers and five PCRs divided into two steps [Erdeniz et al., 1997]. In both methods, one of the first-step PCRs uses yeast genomic DNA as a template to amplify several hundred basepairs of target locus DNA, which in a subsequent PCR is fused to either URA3 or Kl URA3. The long stretch of yeast DNA on one (Akada) or both (Erdeniz) sides of the marker can increase transformation efficiency. In the Erdeniz method, the long stretch of DNA is repeated on both sides of Kl URA3, which can also increase pop-out efficiency. Certainly one advantage of our method over these is that it requires only two PCR primers and one PCR. But another important advantage is that the length of chromosomal DNA affected by the Fifty-Fifty method is limited to about 50 bp up- and downstream of the alteration. When making a seamless genome alteration, we believe it is important to use DNA sequencing to confirm the change. In our experience, we have found a significant number of mutations that could be attributed to oligo synthesis or PCR. Genome editing methods that introduce longer than necessary stretches of synthetic DNA increase the chance of undesired mutations and the work required for DNA sequence confirmation. Regarding short flanking yeast homology and transformation efficiency, we have never encountered a problem obtaining transformants with the Fifty-Fifty method, or with any PCR-based gene deletion or modification method for that matter. Likewise, the 50 bp pop-out sequences in the 50/50 primer are sufficient to yield many FOA-resistant recombinants (see Materials and Methods).
Finally, another variation on cloning-free seamless genome editing is delitto perfetto, which requires two sets of long DNA oligos and two transformations [Storici et al., 2001]. Yeast is first transformed with a 3.2 kb kanMX4-Kl URA3 double heterologous marker (the CORE cassette) that is amplified by PCR with primers that target integration at the desired genomic locus. Correct integrants are then transformed a second time, typically with a pair of long, complementary oligos that have ends homologous to sequences up- and downstream of the integrated CORE cassette plus the desired change near the center. There is no direct selection with the second transformation, rather, cells are cultured non-selectively at first and then later plated to FOA medium with the goal of identifying ura3 cells that have replaced the CORE cassette with the altered oligo sequence. Recovering correct recombinants can be a challenge because transformation and homologous recombination of oligos is inefficient. To increase the frequency of obtaining recombinants in the second transformation, a 4.7 kb Kl URA3-kanMX4-GAL1-I-SceI endonuclease cassette has been introduced [Storici and Resnick, 2006]. In contrast to delitto perfetto, every cell with an integrated 50/50 URA3 cassette has the 50 bp pop-out sequences repeated on the chromosome up- and downstream of URA3. Besides not requiring a second transformation, having the pop-out repeats in every cell means that the efficiency of obtaining recombinants is not dependent on the efficiency of transformation with exogenous oligos.
In summary we have described the Fifty-Fifty method for PCR-based, seamless genome editing in yeast. There have been other methods described that achieve the same goal, but none are as simple or economical.
Acknowledgments
We thank Angela Chu for helpful discussions. Thanks to Angela, Rishi Rakhit, and Eric Foss for comments on the manuscript. This work was supported by National Institute of Health grant 5P01HG000205 to R.W.D.
References
- Akada R, et al. PCR-mediated seamless gene deletion and marker recycling in Saccharomyces cerevisiae. Yeast. 2006;23:399–405. doi: 10.1002/yea.1365. [DOI] [PubMed] [Google Scholar]
- Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2005. [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, LaCroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Chu AM, Davis RW. High-throughput creation of a whole-genome collection of yeast knockout strains. Methods Mol Biol. 2008;416:205–220. doi: 10.1007/978-1-59745-321-9_14. [DOI] [PubMed] [Google Scholar]
- Davidson JF, Schiestl RH. Mis-targeting of multiple gene disruption constructs containing hisG. Current Genetics. 2000;38:188–190. doi: 10.1007/s002940000154. [DOI] [PubMed] [Google Scholar]
- Delneri D, Tomlin GC, Wixon JL, Hutter A, Sefton M, Louis EJ, Oliver SG. Exploring redundancy in the yeast genome: an improved strategy for use of the cre-loxP system. Gene. 2000;252:127–135. doi: 10.1016/s0378-1119(00)00217-1. [DOI] [PubMed] [Google Scholar]
- Erdeniz N, Mortensen UH, Rothstein R. Cloning-free PCR-based allele replacement methods. Genome Res. 1997;7:1174–1183. doi: 10.1101/gr.7.12.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, Pan X, McCusker JH. Heterologous URA3MX cassettes for gene replacement in Saccharomyces cerevisiae. Yeast. 1999;15:507–511. doi: 10.1002/(SICI)1097-0061(199904)15:6<507::AID-YEA369>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardenois A, et al. Execution of the meiotic noncoding RNA expression program and the onset of gametogenesis in yeast require the conserved exosome subunit Rrp6. Proc Natl Acad Sci USA. 2011;108:1058–1063. doi: 10.1073/pnas.1016459108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Längle-Rouault F, Jacobs E. A method for performing precise alterations in the yeast genome using a recyclable selectable marker. Nucleic Acids Res. 1995;23:3079–3081. doi: 10.1093/nar/23.15.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, Heitman J. Gene disruption with PCR products in Saccharomyces cerevisiae. Gene. 1995;158:113–117. doi: 10.1016/0378-1119(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Roman H. The early days of yeast genetics: a personal narrative. Annu Rev Genet. 1986;20:1–12. doi: 10.1146/annurev.ge.20.120186.000245. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Meth Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BL, Steiner B, Seufert W, Futcher AB. pMPY-ZAP: a reusable polymerase chain reaction-directed gene disruption cassette for Saccharomyces cerevisiae. Yeast. 1996;12:129–134. doi: 10.1002/(sici)1097-0061(199602)12:2<129::aid-yea891>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Sprague GF. Assay of yeast mating reaction. Meth Enzymol. 1991;194:77–93. doi: 10.1016/0076-6879(91)94008-z. [DOI] [PubMed] [Google Scholar]
- Sprague GF. Three-pronged genomic analysis reveals yeast cell-type regulation circuitry. Proc Natl Acad Sci USA. 2005;102:959–960. doi: 10.1073/pnas.0409007102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Storici F, Resnick MA. The Delitto Perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Meth Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]