

Abstract
Autophagy, the major lysosomal pathway for recycling intracellular components including organelles, is emerging as a key process regulating tumorigenesis and cancer therapy. Most recently, we newly synthesized folate-appended methyl-β-cyclodextrin (FA-M-β-CyD), and demonstrated the potential of FA-M-β-CyD as a new antitumor drug. In this study, we investigated whether anticancer activity of FA-M-β-CyD in folate receptor-α (FR-α)-positive tumor cells is involved in autophagy. In contrast to methyl-β-cyclodextrin (M-β-CyD), FA-M-β-CyD entered KB cells (FR-α (+)) through CLIC/GEEC endocytosis. No significant depression in the DNA content was observed in KB cells after treatment with FA-M-β-CyD. Additionally, the transmembrane potential of mitochondria after treatment with FA-M-β-CyD was drastically elevated. Meanwhile, FA-M-β-CyD induced the formation of autophagic vacuoles, which were partially colocalized with mitochondria, in KB cells. Taken together, these results suggest that FR-α-expressing cell-selective cytotoxic activity of FA-M-β-CyD could be mediated by the regulation of autophagy, rather than the induction of apoptosis.
In cancer chemotherapy, to obtain the maximum treatment efficacy of anticancer agents, the drug delivery technique is extremely important. To confer an active targeting-ability, the chemical modification of tumor-specific ligands to a drug carrier is known. Of various tumor-specific ligands, folic acid (FA)1,2,3,4,5 has emerged as a remarkable targeting ligand capable of potent interaction with cancer cells expressing the folate receptor (FR) with high affinity (Kd: 10−9 ~ 10−10 M)6,7. FR is engaged the cell surface through a glycosylphosphatidylinositol-anchor, and is highly expressed in various tumor cells including malignancies of the brain, ovary, breast, kidney, and lung, and has negligible expression in normal tissues8. In addition, as a cancer progress, the expression level of FR increase remarkably9. Therefore, FR is one of the potent candidate for not only a promising marker but also a target protein for therapy of cancer.
Cyclodextrins (CyDs) are cyclic oligosaccharides forming inclusion complexes with a wide range of hydrophobic molecules, and are used widely in pharmaceutical region10,11. CyDs have been reported to interact with cell membrane components of cholesterol and phospholipids, resulting in the induction of hemolysis of red blood cells at high concentrations of CyDs12,13,14. In Addition, methyl-β-cyclodextrin (M-β-CyD) is often used to disrupt lipid rafts because of its ability to decrease cholesterol stores on cell membranes15. A number of studies have also demonstrated that the disruption of lipid rafts by M-β-CyD can harm cancer cells and cause cell-death. Notedly, Grosse et al. revealed that M-β-CyD significantly reduced tumor growth in tumor-bearing mice after intraperitoneal administration16. However, the cytotoxic reaction of M-β-CyD has a lack of a tumor cell-selectivity.
Most recently, to make an attempt to give a tumor-specific cytotoxic reaction to M-β-CyD, we previously prepared FA-conjugated M-β-CyD (FA-M-β-CyD)17, and evaluated its antitumor activity18. FA-M-β-CyD provided great antitumor activity, compared to M-β-CyD in KB cells, highly expressing folate receptor-α (FR-α). The single intravenous administration of FA-M-β-CyD significantly suppressed the tumor growth in Colon-26 cells (FR-α (+))-bearing mice. Additionally, the antitumor activity of FA-M-β-CyD was superior to that of doxorubicin after an intravenous administration, at the same dose. These results indicate that FA-M-β-CyD has the potential as a promising anticancer agent. However, the mechanism of antitumor activity of FA-M-β-CyD still remains unclear.
Autophagy, the major lysosomal pathway for recycling intracellular components including organelles, is emerging as a key process regulating tumorigenesis and cancer therapy19,20,21,22,23. The dynamic roles for autophagy in cancer is tumor suppressive effect in the early stage of cancer development, but is the growth effect of established tumors. Likewise, the stimulation of autophagy in response to therapeutics can contextually favor or weaken chemoresistance and antitumor immunity. Therefore, the understanding whether and how autophagy can be harnessed to kill cancer cells is essential for cancer chemotherapy. In this study, we investigated whether antitumor activity of FA-M-β-CyD in FR-α (+) cells is involved in autophagy. As a result, FA-M-β-CyD was found to induce autophagosome formation in FR-α (+) cells, indicating the involvement of autophagy in antitumor activity. Taking into the consideration of our previous results that FA-M-β-CyD drastically suppressed the tumor growth in mice inoculated FR-α (+) tumor cells18, FA-M-β-CyD can be applied as a novel anticancer drug through regulating autophagy for cancer chemotherapy against FR-α-overexpressing tumor.
Results
Antitumor effect of FA-M-β-CyD
To elucidate the antitumor effect of FA-M-β-CyD in FR-α (+) cells, we investigated antitumor effect of FA-M-β-CyD in FR-α (+) and FR-α (−) cells. FA-M-β-CyD had great antitumor effect in FR-α (+) cells such as KB and M213 cells, compared to control, after treatment for 2 h (Fig. 1A, B). However, there was no significant antitumor activity in FR-α-negative A549 cells (Fig. 1C). These results indicate that FA-M-β-CyD had FR-α (+) cell-selective antitumor effect.
Figure 1. Antitumor effect of FA-M-β-CyD.

(A) KB cells, (B) M213 cells, and (C) A549 cells. The concentration of FA-M-β-CyD was 10 mM. Results are represented as mean ± S.E.M. (n = 3–4 per group). *p < 0.05 vs. control.
Cellular association and intracellular distribution of FA-M-β-CyD
Previously, we reported that FA-M-β-CyD possesses FR-α (+) cell-specific antitumor effect18. To obtain the detail of the mechanism for the FR-α-mediated antitumor effect of FA-M-β-CyD, we studied whether TRITC-FA-M-β-CyD associates with KB cells (Fig. 2). Strikingly, TRITC-FA-M-β-CyD highly associated with KB cells (Fig. 2A), despite CyDs are known to be biomembrane-impermeable. Furthermore, the association of TRITC-FA-M-β-CyD was inhibited by the addition of FA as a competitor of FR (Fig. 2A). Similar results were observed in M213 cells (Fig. 2B). Additionally, cellular association of FA-M-β-CyD was significantly lowered in FR-α down-regulated KB cells, compared to KB cells (Fig. 2C). These data indicate that FA-M-β-CyD could associate with cells through FR-α.
Figure 2. Cellular association of TRITC-FA-M-β-CyD.

(A) KB cells, (B) M213 cells and (C) FR-α-knockdown cells. The fluorescence intensity derived from TRITC was determined 1 h after incubation at 37°C by a flow cytometer.
Next, we examined the intracellular distribution of TRITC-FA-M-β-CyD in KB cells after 1 h treatment (Fig. 3). It should be noted that cellular uptake of TRITC-FA-M-β-CyD in KB cells was observed. Furthermore, TRITC-FA-M-β-CyD mainly localized in cytoplasm rather than in nucleus after 1 h treatment. Collectively, these results indicate that FA-M-β-CyD distributed in cytoplasm after the cellular uptake into KB cells, and provided potent antitumor effects.
Figure 3. Intracellular distribution of TRITC-FA-M-β-CyD.
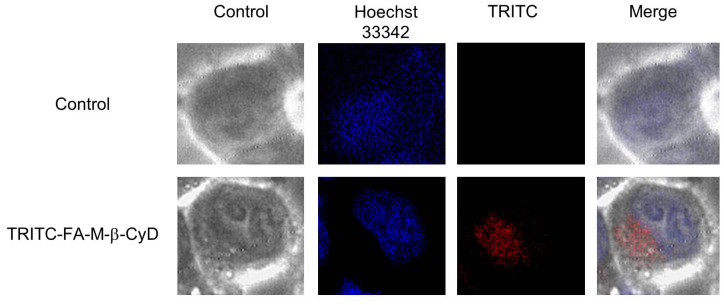
KB cells were treated with TRITC-FA-M-β-CyD (10 μM) for 1 h.
FA-M-β-CyD caused cytotoxic activity via apoptosis-independent pathway
To examine whether FA-M-β-CyD induces apoptosis or not, we investigated the DNA content in nucleus, transmembrane potential in mitochondria, TUNEL assay, and caspase 3 cleavage assay in KB cells. Here, we used 2, 6-di-O-methyl-β-CyD (DM-β-CyD) as a positive control of apoptosis inducer, because we previously demonstrated that DM-β-CyD elicited apoptosis through the suppression of the PI3K-Akt activity, through cholesterol extraction from plasma membranes in NR8383 cells24. The DNA content in KB cells after treatment with 10 mM DM-β-CyD for 2 h was significantly lowered, compared to that with control (Fig. 4A). Meanwhile, no significant decrease in the DNA content was observed in 10 mM FA-M-β-CyD in KB cells (Fig. 4A).
Figure 4.

DNA content (A) and mitochondrial transmembrane potential (B) after treatment with FA-M-β-CyD. KB cells were treated with FA-M-β-CyD (10 mM) for 2 h. Results are represented as mean ± S.E.M. (n = 3–4 per group). *p < 0.05 vs. control. †p < 0.05 vs. KB cells without treatment of FA. (C) TUNEL assay after treatment with FA-M-β-CyD in KB cells. (D) Cleaved caspase 3 assay after treatment with FA-M-β-CyD in KB cells. The cropped blots were indicated. (E) The band intensity of cleaved caspase 3/pro-caspase 3 ratio. Results are represented as mean ± S.E.M. (n = 3 per group). *p < 0.05 vs. control.
Next, we investigated the effects of CyDs on transmembrane potential in mitochondria using rhodamine 123 in KB cells (Fig. 4B). The transmembrane potential in mitochondria of KB cells treated with DM-β-CyD was drastically decreased, compared to control. In sharp contrast, the potential of KB cells treated with FA-M-β-CyD was drastically elevated. Furthermore, this increment of the potential by the addition of FA-M-β-CyD was decreased to control level in the presence of FA, a competitor of FR (Fig. 4B).
Next, we performed TUNEL assay. As shown in Fig. 4C, KB cells treated with DM-β-CyD were stained, compared to control, suggesting the induction of apoptosis. Meanwhile, the cells treated with FA-M-β-CyD were not stained.
Furthermore, in the cleaved caspase 3 assay (Fig. 4D, 4E), DM-β-CyD potently produced activated-caspase 3 through a cleavage of pro-caspase 3, indicating the induction of apoptosis. However, FA-M-β-CyD showed only slight cleavage activity for pro-caspase 3. Collectively, these data indicate that cell-death caused by FA-M-β-CyD was apoptosis-independent.
Involvement of autophagy in cell-death caused by FA-M-β-CyD
Autophagy is a normal physiological process in the body that deals with destruction of cells in the body, and can kill the cells under certain conditions. There are several reports on autophagy or autophagic cell-death activated in cancer cells after treatment with various anticancer drugs25. Next, we examined whether autophagosome formation in KB cells is elicited by FA-M-β-CyD, using Cyto-ID® Autophagy Detection Kit, which detects autophagic vacuoles in cells. As shown in Fig. 5A and 5B, the autophagic vacuoles in KB cells were observed after treatment with FA-M-β-CyD for 2 h. Additionally, the autophagic vacuoles elicited by the treatment with FA-M-β-CyD were overwhelmingly decreased by the pretreatment of LY294002, an autophagy inhibitor. These results suggest that FA-M-β-CyD induced the formation of autophagic vacuoles in KB cells.
Figure 5. Induction of autophagy by the treatment of FA-M-β-CyD.

(A, B) Effects of FA-M-β-CyD on the autophagosome formation in KB cells. (C, D) Effects of FA-M-β-CyD on the clearance of protein aggregation via autophagy. (E) Effects of chloroquine, bafilomycin A1, 3-MA, and LY294002 on antitumor activity of FA-M-β-CyD for KB cells. Results are represented as mean ± S.E.M. (n = 3–6 per group). *p < 0.05 vs. control. †p < 0.05 vs. FA-M-β-CyD without inhibitor.
Next, we performed autophagy assay using a kit of Premo™ Autophagy Sensors, which can monitor a clearance of protein aggregates via autophagy using GFP-labeled p62 in Fig. 5C and 5D. Here, the p62 protein is able to bind to both ubiquitin26 and LC327, thereby facilitating clearance of ubiquitinated proteins via autophagy. The fluorescence of GFP-p62 in control was drastically lowered by the addition of FA-M-β-CyD. Meanwhile, M-β-CyD did not show significant change in the fluorescence of GFP-p62, compared to control. These results suggest that the accumulated autophagosomes in KB cells were degraded by FA-M-β-CyD via autophagy.
Next, we examined the effects of autophagy inhibitors such as chloroquine, bafilomycin A1, 3-methyladenine (3-MA), and LY294002 on cell viability of KB cells after treatment with FA-M-β-CyD. Here, chloroquine and bafilomycin A1 prevent endosomal acidification, which leads to inhibition of both fusion of autophagosome with lysosome and lysosomal protein degradation. 3-MA and LY294002 were used as PI3K inhibitors. As shown in Fig. 5E, the cell viability of KB cells treated with FA-M-β-CyD in the presence of autophagy inhibitors was higher than that with FA-M-β-CyD alone. Taken together, these data indicate that FA-M-β-CyD is likely to cause autophagic cell-death.
The dysfunctional mitochondria are recognized and degraded within cells by both non-selective autophagy and mitophagy, a selective type of autophagy28,29. As shown in Fig. 4B, we found that FA-M-β-CyD significantly enhanced the mitochondrial membrane potential in KB cells, indicating the induction of mitochondrial stress. Therefore, we examined the involvement of mitophagy in cell-death caused by mitochondrial stress after treatment with FA-M-β-CyD (Fig. 6). The autophagic vacuoles and mitochondria, stained by Cyto-ID® Autophagy Detection Kit and rhodamine 123, respectively, were partially colocalized in KB cells after treatment with FA-M-β-CyD (Fig. 6A). Similar results were obtained in M213 cells (Fig. 6B). Therefore, these results suggest that the autophagic cell-death induced by FA-M-β-CyD could be associated with mitophagy elicited by a mitochondrial stress.
Figure 6. Colocalization of autophagosomes and mitochondria in KB cells and M213 cells after treatment with FA-M-β-CyD.

(A) KB cells and (B) M213 cells were treated with FA-M-β-CyD for 2 h, and then the cells were treated with Cyto-ID™ and rhodamine 123, to stain autophagosomes and mitochondria, respectively.
Discussion
Having a targeting ability of antitumor agents plays a key role to not only provide strong antitumor activity but also reduce a risk of side effects in cancer chemotherapy. Previously, we demonstrated that FA-M-β-CyD showed a FR-α (+) cell-selective antitumor effect17. Additionally, we revealed that the antitumor effect of FA-M-β-CyD was significantly suppressed in the presence of FA, indicating that FR-mediated endocytosis is crucial for the enhancement of antitumor effect by FA-M-β-CyD18. In generally, it is believed that the extent of cellular uptake of CyDs is negligible probably due to their hydrophilicity and high molecular weight, FA-M-β-CyD was actually internalized into KB cells (Fig. 3). Meanwhile, FR was thought to be endocytosed via clathrin-independent carrier/GPI-anchored proteins enriched early endosomal compartment (CLIC/GEEC)30. Therefore, FA-M-β-CyD could enter the cells via CLIC/GEEC after the recognition by FR-α. Actually, FA-M-β-CyD highly associated with KB cells rather than that with FR-α-knockdown KB cells (Fig. 2C), indicating the potential of FA-M-β-CyD as a FR-α (+) cell-selective anticancer drug.
Recently, an alternative process for controlling cell-death and novel drugs eliciting cancer cell demise have been discovered31. Among cell-death machinery, apoptosis plays crucial roles in cell survival and tumor growth. M-β-CyD is often utilized to impair lipid rafts due to the extraction of cholesterol from plasma membranes15,32. A number of studies have revealed that M-β-CyD can harm cancer cells through the impairment of lipid rafts. For instance, the decrease in cholesterol level by M-β-CyD provoked apoptosis in human epidermoid carcinoma cells33. In addition, we previously reported that DM-β-CyD elicited apoptosis by the impairment of the PI3K-Akt activity, through the depletion of cholesterol from lipid rafts in alveolar macrophages24. We also verified that DM-β-CyD provided apoptosis in KB cells through cholesterol depletion (Fig. 4). Additionally, M-β-CyD also elicited apoptosis in KB cells, due to the cholesterol extraction, leading to lowering DNA content in nucleus and transmembrane potential in mitochondria34. Interestingly, FA-M-β-CyD significantly released cholesterol from both KB cells (FR-α (+)) and A549 cells (FR-α (−)) to culture medium, compared to that of M-β-CyD and DM-β-CyD in our previous study18. However, FA-M-β-CyD showed potent cytotoxic activity without reducing the DNA content in nucleus and transmembrane potential in mitochondria (Fig. 4), suggesting that the cell-death mechanism in FA-M-β-CyD system could be different from apoptosis. Taken together, the modification of FA to M-β-CyD drastically changed the cell-death mechanism, probably due to entry into FR-α-overexpressing cells mediated by CLIC/GEEC endocytosis.
Most importantly, we revealed that involvement of autophagy in cell-death caused by FA-M-β-CyD in FR-α-expressing cells such as KB cells. That is, FA-M-β-CyD enhanced the expression of LC3-II, an autophagosome marker, in autophagic membranes in KB cells (Fig. 5A). Recently, autophagy is thought to be emerging as a key process regulating tumorigenesis and cancer therapy. At the early stage of tumor development, autophagy functions as a tumor suppressor. Meanwhile, at advanced stages of tumor development, autophagy promotes tumor progression. The tumor cells that are located in the central area of the tumor mass undergo autophagy to survive under low-oxygen and low-nutrient conditions. Autophagy protects some cancer cells against anticancer treatment by blocking the apoptotic pathway (‘protective autophagy'). In the present study, FA-M-β-CyD was found to induce autophagosome formation in FR-α-positive cells, suggesting the involvement of autophagy in antitumor activity. Taking into the consideration of our previous results that FA-M-β-CyD drastically suppressed the tumor growth in mice inoculated FR-α (+) tumor cells18, FA-M-β-CyD can be applied as a novel anticancer drug through regulating autophagy for cancer chemotherapy against FR-α-overexpressing tumor. However, it still remains unclear whether FA-M-β-CyD induces the dephosphorylation and inactivation of mTOR, which elicits autophagy, and activates class III PI3K, which involves in the autophagosome formation. Therefore, the further studies on not only the mechanism of autophagy caused by FA-M-β-CyD, but also the contribution of endocytosis pathway to cell-death mechanism are required.
Activation of mitochondrial permeability pore (mPTP) is associated with mitochondrial depolalization, uncoupling of oxidative dephosphorylation, swelling of mitochondria and release of death-promoting factors like cytochrome c35. Recently, Ziolkowski et al. demonstrated that M-β-CyD decreases the function of rat liver mitochondria, and suppresses the calcium chloride-induced swelling36. In the present study, FA-M-β-CyD internalized into KB cells may interact with mitochondrial raft-like microdomains, leading to the suppression of mPTP activity, resulting in the regulation of autophagosome formation. Further elaborate studies regarding the effects of FA-M-β-CyD on mitochondrial function are in progress.
In conclusion, in the present study, we revealed the involvement of autophagy in FR-α-expressing cell-selective antitumor effect of FA-M-β-CyD. These finding will give great information of FA-M-β-CyD as a novel autophagy inducer for cancer chemotherapy.
Methods
Materials
RPMI-1640 (FA-free) was obtained from GIBCO (Tokyo, Japan). Tetramethylrhodamine isothiocyanate (TRITC) and FR-α siRNA (sc-39969) were purchased from Funakoshi (Tokyo, Japan) and Santa Cruz Biotechnology (Delaware, CA), respectively. Lipofectamine™2000 reagent and Premo™ Autophagy Sensors were obtained from Invitrogen (Tokyo, Japan). Cyto-ID® Autophagy Detection Kit was purchased from Enzo Life Sciences (Farmingdale, NY).
Cell culture and In vitro antitumor activity
KB cells, a human oral squamous carcinoma cell line, A549 cells, a human lung carcinoma, and M213 cells, a human cholangiocarcinoma cell line, were cultured as reported previously17. The antitumor activity in vitro was performed by the WST-1 method, as reported previously18. Briefly, KB cells, M213 cells and A549 cells (5 × 104/96-well microplate) were treated with 150 μL of culture medium containing 10 mM FA-M-β-CyD for 2 h at 37°C. In the autophagy inhibitory study, KB cells were pretreated with RPMI-1640 culture medium containing 20 μM chloroquine, 1 nM bafilomycin A1, 5 mM 3-methyladenine (3-MA), and 50 μM LY294002 for 24 h. After washing with PBS (pH 7.4), 100 μL of fresh HBSS (pH 7.4) was supplemented. Then, the plates and incubated with WST-1 reagent for 30 min. The absorption wavelength and reference wavelength were 450 nm and 630 nm, respectively.
Cellular association of FA-M-β-CyD
KB cells and M213 cells (1 × 106/35 mm dish) were incubated with 1 mL of culture medium (FA-free) containing 10 μM tetramethylrhodamine isothiocyanate (TRITC)-labeled FA-M-β-CyD (TRITC-FA-M-β-CyD) at 37°C for 1 h. After washing with PBS (pH 7.4), the cells were scraped with 1 mL of PBS (pH 7.4). Data were obtained for 1 × 104 cells on a FACS Calibur flow cytometer using CellQuest software (Becton-Dickinson, Mountain View, CA).
Intracellular distribution of FA-M-β-CyD
KB cells (1 × 106/35 mm glass bottom dish) were treated with 10 μM TRITC-FA-M-β-CyD at 37°C for 1 h. Hoechst 33342 (10 μg/mL) was incubated at 37°C for 10 min. After washing with PBS (pH7.4), RPMI-1640 (FA-free) was added. KEYENCE Biozero BZ-8000, a fluorescence microscope, was used for the detection of TRITC and Hoechest33342.
DNA content and mitochondrial transmembrane potential
The DNA content and transmembrane potential in mitochondria in KB cells were determined as reported previously34. Briefly, KB cells (1 × 106/35 mm dish) were treated with RPMI-1640 (FA-free) containing 10 mM DM-β-CyD or FA-M-β-CyD for 2 h. Propidium iodide (PI, 20 μg/mL) and rhodamine 123, the indicators of DNA content and transmembrane potential in mitochondria, respectively, were quantified using a FACS Calibur flow cytometer with CellQuest software.
TUNEL assay
Detection of apoptosis was done by TUNEL assay. In short, KB cells (1 × 106/35 mm glass bottom dish) were incubated with 5 mM DM-β-CyD or FA-M-β-CyD at 37°C for 1 h. The cells were washed with PBS and fixed by incubation in 4% paraformaldehyde in PBS for 1 hr at room temperature. Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick and labeling (TUNEL) assays were performed by using the TACS® 2 TdT-DAB in situ Apoptosis Detection Kit (Trevigen Inc., Gaithersburg, MD) according to the manufacturer's instructions.
Cleaved caspase 3 assay
Caspase 3 cleavage assay was performed by western blotting. Briefly, KB cells (1 × 106/35 mm dish) were incubated with RPMI-1640 culture medium (FA-free) containing 10 mM DM-β-CyD or FA-M-β-CyD for 2 h. After washed with PBS, cells were lysed with 4× sample buffer (8% SDS, 40% glycerol, 24% β-mercaptoethanol in Tris-HCl buffer (pH6.8)) and boiled for 5 min. After determining protein concentrations using the bicinchoninic acid reagent from Pierce Chemical (Rockford, IL), samples (20 μg as proteins) were separated with 12% SDS-PAGE and transferred onto Immobilon P membranes (Nihon Millipore, Tokyo, Japan). The membranes were blocked with 5% skim milk in PBS containing 0.1% Tween 20 (PBS-T) and incubated with caspase 3 antibody (Santa Cruz, Delaware, CA) at 4°C for overnight. After washing with PBS-T, the membranes were incubated with secondary antibody of peroxidase-conjugated sheep (Amersham-pharmacia Biotech, Buckinghamshire, UK). Specific bands were detected using an ECL Western blotting analysis kit (Amersham Bioscience, Tokyo, Japan). The bands were detected using the Lumino-image analyzer LAS-1000 plus (Fujifilm, Tokyo, Japan). The band intensity ratio of cleaved caspase 3/pro-caspase 3 was analyzed by Image-J software.
Autophagosome formation
Briefly, KB cells (1 × 106/35 mm dish) were incubated with FA-M-β-CyD (5 mM) for 2 h, in the presences and absence of pretreatment with 1 μM LY294002, an autophagic inhibitor, for 4 h, and then the cells were treated with Cyto-ID® Autophagy Detection Kit. A fluorescence microscope of Biozero BZ-8000 (KEYENCE) was used for cell observation.
Clearance of protein aggregates via autophagy
The clearance of protein aggregates via autophagy was evaluated by the Premo™ Autophagy Sensors. Briefly, the cells (5 × 105/35 mm glass bottom dish) were incubated with RPMI-1640 culture medium (FA-free) for 24 h. After washing with PBS, 25 μL of GFP-p62 was added to the culture medium. After incubation for 16 h, the cells were washed with PBS and incubated with 50 μM chloroquine for 10 h. Then, the cells were incubated with 5 mM FA-M-β-CyD in the presence of 50 μM chloroquine for 2 h. After washing with RPMI-1640 culture medium (FA-free), a fluorescence microscope of Biozero BZ-8000 (KEYENCE) was used for cell observation.
Statistics
All experiments were performed in triplicate in each series of measurements, and each series was repeated more than three times. The experimental results are shown as means ± S.E.M. Significance levels for comparisons between samples were determined with Scheffe's test. The level of statistical significance was set at P < 0.05.
Author Contributions
R.O., K.M., N.T., A.O. and A.O. performed the experiments. R.O., K.M., T.H., R.K., S.O. and H.A. analysed the data. R.O., K.M. and H.A. designed the research. K.M., R.O. and H.A. wrote this manuscript. H.A. supervised this work.
Supplementary Material
Supplementary Figure 1
Acknowledgments
This work was funded by a Japan Society for the Promotion of Science (Grant-in-Aid for Young Scientists (B) (25870537)), a Ministry of Health Labour and Welfare (Grant-in-Aid for Third Term Comprehensive Control Research for Cancer program (24100701)), and The Japan Science Society (Sasakawa Scientific Research Grant).
References
- Chen H., Ahn R., Van den Bossche J., Thompson D. H. & O'Halloran T. V. Folate-mediated intracellular drug delivery increases the anticancer efficacy of nanoparticulate formulation of arsenic trioxide. Mol. Cancer Ther. 8, 1955–1963 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon A. et al. Improved therapeutic activity of folate-targeted liposomal doxorubicin in folate receptor-expressing tumor models. Cancer Chemother. Pharmacol. 66, 43–52 (2010). [DOI] [PubMed] [Google Scholar]
- Lu Y. & Low P. S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Deliv. Rev. 54, 675–693 (2002). [DOI] [PubMed] [Google Scholar]
- Mi Y., Liu Y. & Feng S. S. Formulation of Docetaxel by folic acid-conjugated d-α-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS(2k)) micelles for targeted and synergistic chemotherapy. Biomaterials 32, 4058–4066 (2011). [DOI] [PubMed] [Google Scholar]
- Nukolova N. V., Oberoi H. S., Cohen S. M., Kabanov A. V. & Bronich T. K. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials 32, 5417–5426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony A. C. The biological chemistry of folate receptors. Blood 79, 2807–2820 (1992). [PubMed] [Google Scholar]
- Low P. S. & Kularatne S. A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 13, 256–262 (2009). [DOI] [PubMed] [Google Scholar]
- Limmon G. V. et al. Scavenger receptor class-A is a novel cell surface receptor for double-stranded RNA. FASEB J. 22, 159–167 (2008). [DOI] [PubMed] [Google Scholar]
- Parker N. et al. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 338, 284–293 (2005). [DOI] [PubMed] [Google Scholar]
- Szente L. & Szejtli J. Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv. Drug Deliv. Rev. 36, 17–28 (1999). [DOI] [PubMed] [Google Scholar]
- Uekama K. & Otagiri M. Cyclodextrins in drug carrier systems. Crit. Rev. Ther. Drug Carrier Syst. 3, 1–40 (1987). [PubMed] [Google Scholar]
- Motoyama K. et al. Effect of 2,6-di-O-methyl-α-cyclodextrin on hemolysis and morphological change in rabbit's red blood cells. Eur. J. Pharm. Sci. 29, 111–119 (2006). [DOI] [PubMed] [Google Scholar]
- Motoyama K. et al. Involvement of lipid rafts of rabbit red blood cells in morphological changes induced by methylated β-cyclodextrins. Biol. Pharm. Bull. 32, 700–705 (2009). [DOI] [PubMed] [Google Scholar]
- Ohtani Y., Irie T., Uekama K., Fukunaga K. & Pitha J. Differential effects of α-, β- and γ-cyclodextrins on human erythrocytes. Eur. J. Biochem. 186, 17–22 (1989). [DOI] [PubMed] [Google Scholar]
- Galbiati F., Razani B. & Lisanti M. P. Emerging themes in lipid rafts and caveolae. Cell 106, 403–411 (2001). [DOI] [PubMed] [Google Scholar]
- Grosse P. Y., Bressolle F. & Pinguet F. Antiproliferative effect of methyl-β-cyclodextrin in vitro and in human tumour xenografted athymic nude mice. Br. J. Cancer 78, 1165–1169 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera R., Motoyama K. & Arima H. Design and evaluation of folate-appended methyl-β-cyclodextrin as a new antitumor agent. J. Incl. Phenom. Macrocycl. Chem. 70, 321–326 (2011). [Google Scholar]
- Onodera R., Motoyama K., Okamatsu A., Higashi T. & Arima H. Potential use of folate-appended methyl-β-cyclodextrin as an anticancer agent. Sci. Rep. 3(1109), 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C. et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699 (2006). [DOI] [PubMed] [Google Scholar]
- Liu E. Y. & Ryan K. M. Autophagy and cancer--issues we need to digest. J. Cell Sci. 125, 2349–2358 (2012). [DOI] [PubMed] [Google Scholar]
- Qu X. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, 1809–1820 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y. et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama K. et al. Involvement of PI3K-Akt-Bad pathway in apoptosis induced by 2,6-di-O-methyl-β-cyclodextrin, not 2,6-di-O-methyl-α-cyclodextrin, through cholesterol depletion from lipid rafts on plasma membranes in cells. Eur. J. Pharm. Sci. 38, 249–261 (2009). [DOI] [PubMed] [Google Scholar]
- Kondo Y., Kanzawa T., Sawaya R. & Kondo S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer. 5, 726–734 (2005). [DOI] [PubMed] [Google Scholar]
- Geetha T. & Wooten M. W. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 512, 19–24 (2002). [DOI] [PubMed] [Google Scholar]
- Pankiv S. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 (2007). [DOI] [PubMed] [Google Scholar]
- Kim I., Rodriguez-Enriquez S. & Lemasters J. J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Hernandez A. et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 5, 19–32 (2009). [DOI] [PubMed] [Google Scholar]
- Doherty G. J. & McMahon H. T. Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 (2009). [DOI] [PubMed] [Google Scholar]
- Long J. S. & Ryan K. M. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene 31, 5045–5060 (2012). [DOI] [PubMed] [Google Scholar]
- Simons K. & Ehehalt R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 110, 597–603 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E. K. et al. Cholesterol depletion induces anoikis-like apoptosis via FAK down-regulation and caveolae internalization. J. Pathol. 218, 337–349 (2009).19288501 [Google Scholar]
- Onodera R. et al. Involvement of cholesterol depletion from lipid rafts in apoptosis induced by methyl-β-cyclodextrin. Int. J. Pharm. 452, 116–123 (2013). [DOI] [PubMed] [Google Scholar]
- Bernardi P., Scorrano L., Colonna R., Petronilli V. & Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264, 687–701 (1999). [DOI] [PubMed] [Google Scholar]
- Ziolkowski W. et al. Methyl-β-cyclodextrin induces mitochondrial cholesterol depletion and alters the mitochondrial structure and bioenergetics. FEBS Lett. 584, 4606–4610 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1