

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disease in the elderly, and no cure or disease-modifying therapies exist. Several lines of evidence suggest that mitochondrial dysfunction and oxidative stress have a central role in the dopaminergic neurodegeneration of PD. In this context, mitochondria-targeted therapies that improve mitochondrial function may have great promise in the prevention and treatment of PD. In this review, we discuss the recent developments in mitochondria-targeted antioxidants and their potential beneficial effects as a therapy for ameliorating mitochondrial dysfunction in PD.
Keywords: mitochondrial dysfunction, mitochondria-targeted antioxidant, neuroprotection, oxidative stress, Parkinson’s disease
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects more than 1 million individuals over the age of 60 years in the United States [1]. According to a recent article, about 50,000 new cases are diagnosed annually, and this figure is expected to increase substantially as the median age of the population continues to rise in the coming decades [2]. Epidemiological studies suggest that sporadic late-onset PD accounts for 90% of cases, whereas the remaining 10% are early onset cases that mainly occur in familial clusters [3, 4]. Pathologically, PD is characterized by the loss of dopaminergic neurons within the substantia nigra pars compacta (SNc) and the ensuing depletion of dopamine in the striatum. This loss of dopaminergic neurons causes most of the motor symptoms of PD. By the time PD motor symptoms are clinically recognized, 60% of dopaminergic neurons and 80% of putamen dopamine have been lost [5]. PD is also associated with the presence of ubiquitin- and α-synuclein-positive cytoplasmic inclusions known as Lewy bodies within surviving dopaminergic neurons [6]. In addition to the nigrostriatal dopaminergic defects, emerging clinical evidence suggests that extranigral degeneration and non-motor symptoms are key features of early stages of PD pathogenesis [7-10].
Currently, the available therapies for PD only treat the symptoms; none slow or prevent progressive neuronal degeneration in the dopaminergic system [11, 12]. Dopamine replacement therapy, i.e., levodopa administered orally or stimulation of dopamine receptors, has been the most widely used treatment option for PD, but the beneficial effects of dopaminergic therapy wear off over time and its clinical efficacy gradually declines as the disease advances [13].
Despite extensive research, the precise cause of sporadic PD or non-familial PD remains unknown, but several pathogenic mechanisms have been proposed, including oxidative stress, mitochondrial dysfunction, impairment of the ubiquitin-proteasome system, and neuroinflammation. Convincing evidence from postmortem brain tissue, cell culture and animal models of PD and the analysis of human genetics support the involvement of oxidative stress and mitochondrial dysfunction in PD pathogenesis [14, 15]. Mitochondrial dysfunction due to oxidative stress, mitochondrial DNA deletions, altered mitochondrial morphology and the interaction of pathogenic proteins with mitochondria all result in dopaminergic neurodegeneration. Thus, therapeutic approaches targeting mitochondrial dysfunction and related oxidative stress may hold great promise of a cure for PD. One potential approach to ameliorating complications arising from PD is to suppress mitochondrial reactive oxygen species (ROS) generation with specific antioxidants. Several small antioxidant molecules, such as ubiquinol and creatine, have shown promising neuroprotective effects in different models of PD [16, 17]. However, a major limitation of using these compounds to treat PD is their failure to accumulate preferentially in the target organelle mitochondria. For this reason, several strategies to identify antioxidants with therapeutic potential that specifically target mitochondria have been developed. In this review, we will describe cellular changes in the progression of PD, and in our discussion of promising PD therapeutic strategies, we will focus on the mitochondrially targeted antioxidants as potential therapies for PD.
2. Production of mitochondrial ROS
Mitochondria play a central role in the life and death of cells. Physiologically, mitochondria perform a variety of fundamental regulatory processes in the cell, including ATP production [18], calcium homeostasis and modulation [19], amino acid and nitrogen metabolism [20], apoptotic cell death [21], ROS generation and detoxification [22, 23], and heme and iron-sulfur center biosynthesis [24]. They supply the vast majority of cellular energy in the form of ATP through oxidative phosphorylation. During oxidative phosphorylation, electrons from reduced cofactors are transferred through a series of respiratory chain complexes (Complexes I-IV) located in the mitochondrial inner membrane to oxygen, the ultimate electron acceptor. The flow of electrons simultaneously leads to the pumping of protons out of the mitochondrial matrix. This electrochemical reaction generates a transmembrane potential (ΔΨm) yielding the energy for ATP synthesis from ADP and inorganic phosphate.
ROS can be generated within mitochondria in several sites of mitochondrial electron transport chains (ETC), in particular on Complexes I and III, where electrons occasionally leak to oxygen and form a superoxide anion (O2•-), the predominant ROS in mitochondria (Fig. 1) [25, 26]. In fact, mitochondria are the major sites of cellular ROS production, with approximately 1-3% of mitochondrial oxygen consumption being converted to ROS [27]. In addition to formation from incomplete reduction of oxygen in ETC, a number of enzyme systems also generate superoxide, including the tricarboxylic acid cycle (TCA) enzymes α-ketoglutarate dehydrogenase [28] and aconitase [29], the non-TCA cycle enzymes pyruvate dehydrogenase, dihydroorotate dehydrogenase [30] and glycerol-3-phosphate dehydrogenase [31], and the mitochondrial outer membrane proteins such as methemoglobin reductase [32]. Because of its negative charge and poor membrane permeability, superoxide is relatively unreactive, but it can react rapidly with nitric oxide (NO) to form the potent oxidant and nitrating agent peroxynitrite (ONOO-) and subsequently other reactive nitrogen species (RNS). Moreover, it is able to damage some mitochondrial iron-sulfur cluster-containing proteins [33]. Most cellular superoxide is rapidly converted to hydrogen peroxide (H2O2) either through spontaneous dismutation or dismutation reactions catalyzed by superoxide dismutase (SOD) [34]. Hydrogen peroxide itself is a reactive free radical that is stable, membrane permeable and has a relatively long half-life enabling diffusion within the cell. As a redox active species, H2O2 can inactivate some enzymes by oxidizing their thiol groups [35], although it is unable to oxidize DNA or lipids directly [33]. Hydrogen peroxide can be decomposed by cytosolic and mitochondrial antioxidant systems such as glutathione peroxidase (GPx), catalase (CAT), and thioredoxin reductase (TPx). However, if not removed, it can further produce the highly reactive hydroxyl radical (OH•) in the presence of Fe2+ cations via the Fenton reaction [36]. The OH• has a strong oxidizing potential and can damage virtually every type of macromolecule close to their site of origin, making it an extremely dangerous compound to the organism. Furthermore, unlike superoxide and hydrogen peroxide, which can be detoxified by an enzymatic conversion, no enzymatic routes are known for eliminating hydroxyl radicals. Nonenzymatic mechanisms for scavenging peroxyl radicals include several antioxidants such as vitamin E and glutathione. Other radicals derived from oxygen include peroxyl radical (RO2•), hypochlorous acid (HOCl), alkoxyl radical (RO•), and hydroperoxyl radical (HO2•), which are high-energy species and exhibit a broad array of biological actions. Additional endogenous sources of cellular ROS are macrophages, neutrophils, and eosinophils. ROS generation can also occur through a host of exogenous sources including ultraviolet and high-energy irradiation, redox-cycling of quinones, xenobiotics, ions, metals, aging, and environmental toxins [37-41].
Figure 1.
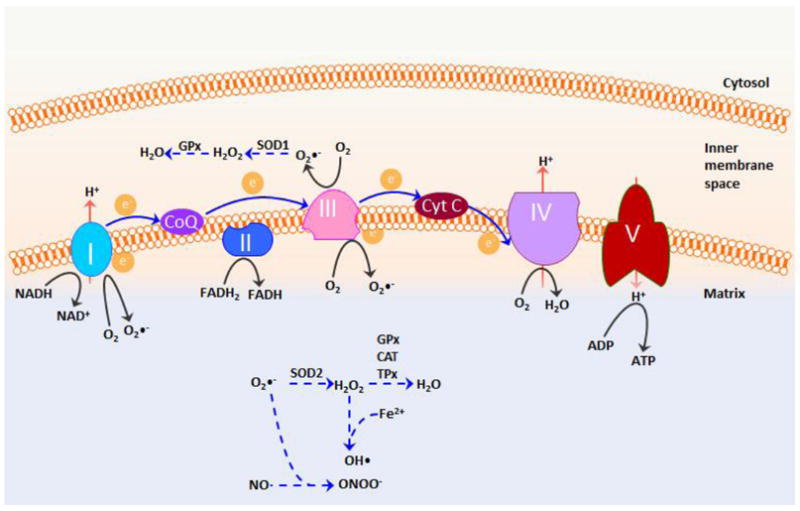
Schematic presentation of the generation of ROS in mitochondria. ROS are generated from the transfer of electrons (e-) to molecular oxygen to form superoxide (O2•-) at the mitochondrial electron transport chain complex I and III. Once generated, superoxide is decomposed enzymatically by superoxide dismutase 1 (SOD1) in the intermembrane space and by SOD2 (MnSOD) in the matrix to form hydrogen peroxide, which is further catabolized to water by the action of enzymes such as catalase (CAT), glutathione peroxidases (GPx), and thioredoxin reductase (TPx) to avoid possible buildup of oxidative stress. However, under mitochondrial stress, superoxide may react with nitric oxide to form the potent oxidant and nitrating agent peroxynitrite (ONOO-). Hydrogen peroxide can also form the highly reactive hydroxyl radical (OH•) in the presence of Fe2+ cations. These highly reactive radicals may cause damage to proteins, lipids, and nucleic acids. CoQ, coenzyme Q; Cyt C, cytochrome C.
It is worth pointing out that ROS production can be significant in both physiological and pathological situations depending on the environment. For example, it has been demonstrated that at lower physiological levels, mitochondrially generated H2O2 acts as an intracellular signaling molecule, affecting multiple cellular functions [42-44]. Thus, it is possible that an overuse of antioxidants may be detrimental. In contrast, at high concentrations, ROS may cause extensive damage to cells and the whole organism, consisting of the peroxidation of lipids, particularly phospholipids of biological membranes, the carbonylation of proteins, or oxidative damage to mtDNA [23, 26, 36, 45]. These noxious actions, often referred to as “oxidative stress”, either individually or collectively can disrupt mitochondrial function and cause ROS to flow to the cytosol, which in turn results in a further increase in ROS production, thus forming a vicious cycle [46]. In this sense, mitochondria are believed to have developed an excessive network of antioxidant defenses. Therefore, a delicate balance between ROS and antioxidants within mitochondria is essential for the functions of cells, tissues, and organs.
3. Scavenging of mitochondrial ROS
Mitochondria contain a series of well-defined and tightly controlled antioxidant defense systems, which work synergistically to intercept ROS, thereby minimizing oxidative damage. Disruption of these antioxidant defenses may result in extensive oxidative damage to mitochondria. There are two main types of mitochondrial antioxidant defense systems: enzymatic and nonenzymatic. Enzymatic antioxidants involve SOD, GPx, catalase, and TPx, all of which are encoded by the nuclear genome and are subsequently imported into the mitochondria. SOD is one of the most effective intracellular enzymatic antioxidants, and as mentioned in the previous section, SOD catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide. In mammals, three forms of SOD exist depending on the nature of the active metal center. SOD1 or Cu/Zn SOD, binds copper and zinc and is a cytoplasmic and mitochondrial inter-membrane space protein [47]. SOD2 (Mn-SOD) has manganese ion at its reactive center and is only expressed in the mitochondrial matrix, and SOD3 is secreted into the extracellular space after its protein translation [48]. Mn-SOD null mice die shortly after birth, confirming the importance of keeping superoxide in check in mammals [49]. GPx mainly detoxifies free H2O2 and lipid peroxides using reduced glutathione (GSH) as a cofactor. In this process, GSH is converted to its oxidized form, glutathione disulfide (GSSG). Once oxidized, GSH can be regenerated by the action of glutathione reductase using NADPH as its electron donor. In addition, GSH serves as a nonenzymatic antioxidant, scavenging hydroxyl radical and singlet oxygen directly and maintaining vitamin C and E in their reduced forms.
Other nonenzymatic antioxidants include α-tocopherol (vitamin E), ascorbic acid (vitamin C), coenzyme Q10 (CoQ10), β-carotene, flavonoids, and α-lipid acid [50]. Vitamin E is lipid-soluble, and therefore, is widely distributed in mammalian cell membranes. Of the different forms of vitamin E, α-tocopherol is the most biologically active form. As a lipid-soluble antioxidant, vitamin E acts as a scavenger of peroxyl radical, peroxynitrite, and hydroxyl radical by preventing the propagation of the radical chain, thus effectively inhibiting lipid peroxidation. During the antioxidant reaction, α-tocopherol is converted to α-tocopherol radical, which gets reduced to its original α-tocopherol form. Another vitamin antioxidant is vitamin C, which is water-soluble, making it able to work in the body’s aqueous environments. Vitamin C functions in association with vitamin E to regenerate α-tocopherol from α-tocopherol radical. CoQ10 is an essential component of the ETC, where it accepts electrons from Complexes I and II. It is also a coenzyme for Complex III and acts as a lipid-soluble antioxidant as well. The antioxidant activity of CoQ10 arises from its capacity to exchange two electrons in a redox cycle between its oxidized (ubiquinone) and its reduced form (ubiquinol). Ubiquinol is a very effective chain-breaking antioxidant that not only inhibits lipid peroxidation and protein and DNA oxidation, but also directly quenches oxidants such as superoxide and peroxyl radicals.
Despite the presence of elaborate defense systems to counteract oxidative damage, it seems that cellular ROS levels are not perfectly under control. Indeed, oxidative damage due to excessive ROS production may accumulate during both normal and stressful conditions. Oxidative stress accumulation and concomitant mitochondrial dysfunction have been implicated in PD pathogenesis.
4. Oxidative stress in PD
Oxidative stress has long been implicated in the process of neurodegeneration in PD pathogenesis. Oxidative stress, arising from excessive production of ROS and/or defective ROS removal, can potentially damage cellular lipids, proteins, and DNA. Postmortem studies have consistently shown high levels of oxidation of lipids, proteins, and nucleic acids in the SNc of sporadic PD brains [51-56]. Also, significant alterations of the antioxidant defense system, in particular reduced glutathione, were found in the SNc of PD patients [57]. As mentioned above, ETC is the major source of ROS, in particular the hydrogen peroxide and superoxide anions [58]. In the presence of ferrous iron, these ROS can be converted to even more potent ROS, such as the hydroxyl radical and hydroxyl anion [59, 60]. Not surprisingly, the level of iron was significantly increased in the SNc of PD brains [61-64].
Apart from being the main source of increased oxidative stress in PD brains, mitochondrial function itself can be affected by oxidative stress [26, 65, 66], which further contributes to the accumulation of ROS and mitochondrial damage in a vicious cycle. This feed-forward mechanism is what commonly underlies neuronal cell death in neurodegenerative diseases. In addition to mitochondria, the auto-oxidation of dopamine, a reaction known to generate superoxide and hydrogen peroxide, as well as reactive dopamine quinones, specifically contribute to cellular ROS in dopaminergic neurons [67, 68]. This dopamine-dependent oxidative stress is believed to partially explain the selective vulnerability of dopaminergic neurons in PD. Another important contributor to oxidative stress is NO, which is generated by nitric oxide synthase (NOS) [52]. Reaction of ROS with NO produces highly toxic RNS, such as the peroxynitrite and nitro-tyrosyl radicals [69]. Besides damaging cellular proteins, lipids, and DNA, oxidative stress also activates a variety of cell death pathways [70-79].
5. Mitochondrial dysfunction in PD
Considerable evidence exists suggesting a role for mitochondrial dysfunction in PD pathogenesis [38, 80]. Mutations in mitochondrial DNA (mtDNA) play a role in the demise of dopaminergic neurons. Indeed, high levels of somatic mtDNA point mutations in elderly PD patients have also been reported [81]. However, the most definitive evidence of mitochondrial dysfunction in PD has come from studies using MPTP, a Parkinsonian toxicant which causes Parkinsonian syndromes in humans, rodents and primates by inhibiting the mitochondrial complex-I of the electron transport chain [82]. Similar to MPTP, other complex-I inhibitors such as rotenone, maneb, paraquat, fenzaquin and trichloroethylene result in the loss of nigral dopaminergic neurons in the mouse model of PD, implicating mitochondrial dysfunction in its pathogenesis [83, 84]. Additionally, impairment of complex-I activity has been reported in the substantia nigra, platelets and skeletal muscles of PD patients [85, 86]. Moreover, reduced complex-I activity and an increased susceptibility to MPP+, the toxic metabolite of MPTP, were also observed in mitochondrial DNA from PD patients, clearly demonstrating mtDNA-encoded defects in PD [87, 88].
Recently, more convincing evidence of mitochondrial dysfunction in PD has been reported in conditional knockout ‘MitoPark’ mice, which have a disrupted mitochondrial transcription factor A (Tfam) gene in dopaminergic neurons [89]. These mice exhibit reduced mtDNA expression, attenuated expression of respiratory chain function in dopaminergic neurons in nigra along with behavioral impairments and striatal dopamine depletion, mimicking progressive PD phenotypes starting from 18 weeks of age [89].
Pathogenic mutations in several genes including α-synuclein, LRRK2, parkin, DJ-1 and PINK-1 also play an important role in mitochondrial dysfunction in PD patients [2, 90]. Impaired mitochondrial function in the MPTP mouse model of PD has been reported in cells overexpressing α-synuclein and in transgenic mice [91, 92]. Based on all these findings, it can be inferred that intervening in one or more of these processes could alleviate the harmful effects of mitochondrial dysfunction.
6. Antioxidant therapy
Since PD is a complex, multifactorial disease with oxidative stress and mitochondrial dysfunction playing central roles in nigrostriatal dopaminergic neurodegeneration, antioxidants targeting these factors have become attractive therapeutic agents in the treatment of PD. In this section, we will summarize the current evidence for preventing or slowing the development of PD by neuroprotective antioxidant agents.
6.1 Vitamin antioxidants
Various forms of vitamin antioxidants (Fig. 2A-B) have been tested for their neuroprotective potential. Vitamin E deficiency increased MPTP-induced dopaminergic neurotoxicity [93]. In another study, researchers found that administering vitamin E ameliorated oxidative stress induced by iron accumulation in the mouse brain [94], suggesting that vitamin E may be neuroprotective against PD. Additionally, vitamin E was neuroprotective in 6-OHDA-treated animals [95]. The observational data in humans suggest that the combined administration of high-dose vitamin E and vitamin C supplements was associated with a reduced progression of PD [96]. However, the results of double-blind, randomized controlled trials have been disappointing, where vitamin E showed no benefits in PD [97, 98]. More recently, a large cohort study demonstrated that high dietary vitamin E intake, but not vitamin C or carotenoids, reduced the risk of PD [99]. But in contrast, other studies contradict results regarding food intake of vitamin E and its efficacy to prevent PD progression [100-102]. The protective role of vitamin C in PD remains controversial since one epidemiological study found a decreased risk of PD in individuals consuming diets rich in vitamin C, whereas other studies showed no effects or even an increased risk of PD with consumption of vitamin C [99, 100, 103].
Figure 2.
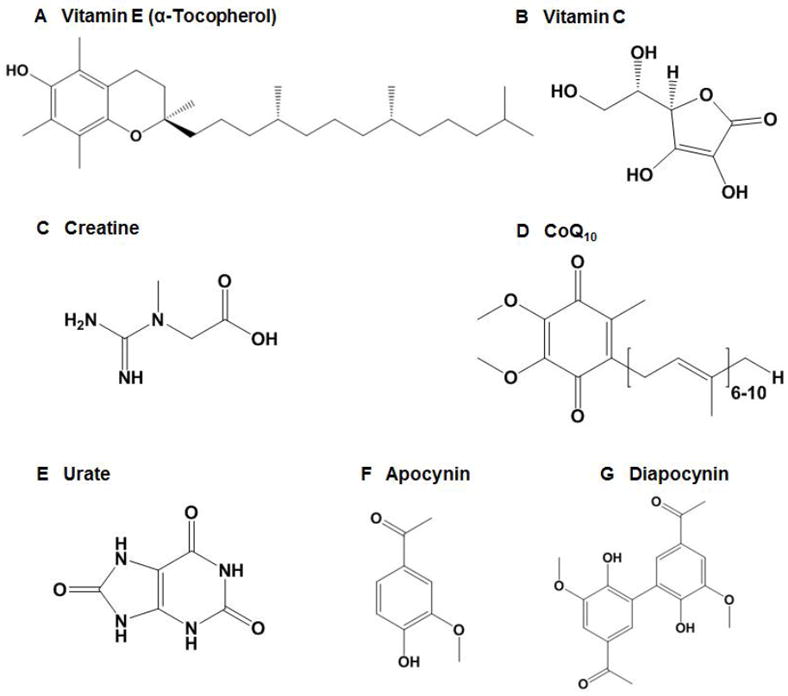
Structures of a series of small-molecule antioxidants showing protection in PD models, including (A) vitamin E, (B) vitamin C, (C) creatine, (D) CoQ10, (E) urate, (F) apocynin, and (G) diapocynin.
6.2 Creatine
Creatine (Fig. 2C) is a nitrogenous guanidine compound that forms high energy phosphate bonds, occurs naturally in vertebrates and supplies energy to muscle and nerve cells. Creatine also possesses antioxidant properties and can be an effective inhibitor of mitochondrial permeability transition pore opening and mitochondrial iron accumulation. Preclinical studies in various models have demonstrated its potential role as a neuroprotective agent [104]. Recently, a neuroprotective effect of creatine was demonstrated in MPP+ and 6-OHDA-treated dopaminergic neuronal cell cultures, where it protected tyrosine hydroxylase immunoreactive dopaminergic neurons and their fibers [105]. In the MPTP mouse model of PD, creatine restored the MPTP-induced loss of dopamine and protected dopaminergic neurons [106]. In early clinical studies, two grams daily creatine administration improved behavioral difficulties in a clinical trial of 200 subjects who were within 5 years of a PD diagnosis [16]. In an additional follow-up study, creatine continued to show neuroprotective efficacy 18 months after creatine administration [107]. Although a 2-year placebo-controlled study of 60 subjects demonstrated that creatine had no effect on PD scores or dopamine transporter imaging, improved mood behavior (a non-motor symptom of PD) was noticed in those patients [108]. Recently, a phase III clinical trial has been started by the NIH, where creatine is administered with a dose of 10 g in a large long-term study of PD targeting 1720 participants with the disease [109, 110]. This study involving 52 medical facilities is expected to be completed in 5-7 years.
6.3 CoQ10
CoQ10 also shows promise as a neuroprotective agent in PD (Fig. 2D). Postmortem studies have shown that CoQ10 levels in the plasma and platelets of PD patients were significantly lower compared to age-matched controls, and that the oxidized form of CoQ10 was elevated in PD patients, suggesting that taking CoQ10 supplements may be beneficial. Neuroprotective properties of CoQ10 have since been demonstrated in various in vivo and in vitro models of PD. More specifically, paraquat- and rotenone-induced mitochondrial dysfunction and neurodegeneration in rat mesencephalic primary neurons were inhibited by CoQ10 [111]. Also, a neuroprotective role for CoQ10 was shown in iron-induced apoptosis in dopaminergic neurons [112]. Pretreatment of neuronal cells with CoQ10 has maintained the mitochondrial membrane potential during oxidative stress and reduced the mitochondrial generation of ROS [113]. In the MPTP mouse model, CoQ10 protected against MPTP-induced dopamine depletion and the loss of dopaminergic neurons in aged mice [114]. Similarly, CoQ10 protected against MPTP toxicity in the chronic MPTP model, which mimics the progressive nature of PD [114].
A combination of creatine and CoQ10 exhibited a significant neuroprotective effect in chronic MPTP-treated mice [115]. Oral administration of CoQ10 also slowed the progression of PD in a primate model of PD [116]. Evidence from clinical trials suggests that high doses of CoQ10 are needed for beneficial results. A double-blind, placebo-controlled phase II study of CoQ10 with three different doses, such as 300, 600 and 1200 mg daily, in 80 early untreated patients for 16 months was conducted recently, resulting in a statistically improved PD rating only at the highest dose [117]. Next, this phase II trial was extended to test a higher dose (2400 mg/daily) of CoQ10 in early PD patients. Although a trend emerged, the improvement in clinical scores, unfortunately, was not statistically significant. One possible reason could be the lower number of patients [118]. Recently, an NINDS-funded phase III, multicenter, randomized, placebo-controlled, double-blind trial of CoQ10 at doses of 1200 and 2400 mg/daily was initiated on 600 early, non-medicated PD patients. Hopefully, some positive outcome will come from this clinical trial.
6.4 Urate
Urate (uric acid; Fig. 2E) plays a role in the regulation of oxidative stress by acting as a scavenger of superoxide, hydroxyl radical, and singlet oxygen and as an iron chelator [119-121]. Urate inhibits oxidative stress and prevents dopaminergic cell death in both cell culture and animal models of PD [122, 123]. In humans, several prospective cohort studies demonstrated that higher blood levels of urate were associated with a significantly reduced risk of developing PD [124-127]. In another prospective study, PD patients with high urate intake progressed slower toward the disability endpoint requiring dopamine treatment [128]. Although these findings indicate that urate intake could protect individuals from PD progression, the use of urate in PD treatment remains limited because elevating urate in the serum increases the risk of gout, a risk that is further compounded by the consumption of alcohol and fructose [129].
6.5 Apocynin and its derivative
Apocynin (4-hydroxy-3-methoxyacetophenone; Fig. 2F) is a plant-derived antioxidant widely used as an effective NADPH oxidase inhibitor, which interferes with the assembly of cytosolic NADPH oxidase components with the membrane components. NADPH is one of the major sources of cellular ROS during neurodegeneration. Apocynin protects dopaminergic neurons against MPP+-induced oxidative stress and cell death in cell cultures [130]. In contrast, no beneficial efficacy [131] or pro-oxidant property [132] of apocynin has been reported. Apocynin can be converted to diapocynin (Fig. 2G) in vivo, which is a more efficient inhibitor of NADPH oxidase than apocynin itself [133]. Additionally, diapocynin has higher lipophilicity than apocynin [134]. Interestingly, we recently demonstrated in a mouse MPTP model of PD, that the compound diapocynin protected against nigrostriatal damage and inhibited inflammatory and oxidative stress processes [135]. In LRRK2-R1441G transgenic mice, which are reported to develop PD-like symptoms at ~10 months of age, administration of diapocynin at an oral dose of 200 mg/kg three times per week protected against neurobehavioral dysfunction [136]. These findings suggest that diapocynin can be neuroprotective against PD, although additional animal and human studies are needed.
In summary, although a range of antioxidant moieties has been used with partial success in experimental cellular and animal models of PD, no human clinical studies to date have provided conclusive evidence of antioxidants benefitting PD patients. Evidence from these clinical trials, particularly with vitamin E, vitamin C, and creatine, is largely inconclusive. Furthermore, clinical trials involving CoQ10 have demonstrated that supplementation with very high doses of CoQ10 is necessary to show some benefit in PD subjects. This may be due to the fact that most small-molecule antioxidants get distributed throughout the body, with only a small fraction being taken up by mitochondria. Consequently, approaches selectively targeting mitochondria with antioxidants have been developed.
7. Mitochondria-targeted antioxidant therapy
During the past decade, considerable progress in developing mitochondrially targeted antioxidants has been made. A well-established approach is conjugation to a lipophilic cation, such as triphenylphosphonium (TPP) [137-139]. Phosphonium derivatives have been traditionally used to determine mitochondrial inner membrane potential. Triphenyl-phosphonium cations consist of a positively charged phosphorus atom surrounded by a large hydrophobic surface (Fig. 3A), thereby giving it the ability to directly and rapidly permeate lipid bilayers while retaining the positive charge. This positive charge is used to facilitate TPP cation accumulation within the mitochondrial matrix driven by the large mitochondrial membrane potential (ΔΨm) of 150-180 mV (negative inside), which was generated by the proton gradient during the transfer of an electron to oxygen. As shown in Fig. 3B, the plasma membrane potential (30-60 mV, negative inside) enables up to a 10-fold accumulation of TPP in the cytoplasm. Subsequently, the large, negative potential gradient across the mitochondrial inner membrane potentiates the redistribution of TPP from the intracellular space into the mitochondria, leading to a 100- to 500-fold higher concentration of TPP inside mitochondria [140, 141]. Based on this theory, Murphy and coworkers [139] discovered a series of orally bioavailable mitochondria-targeted antioxidants (MTAs), including MitoQ, MitoVitE, and MitoTEMPOL. These compounds are known to pass through all biological membranes and accumulate within mitochondria more easily than their non-targeted parent antioxidants, rendering them far more effective in protecting against mitochondrial oxidative damage. This section is dedicated to the most promising MTAs for treating neurodegenerative diseases, with emphasis on the evidence for managing PD.
Figure 3.
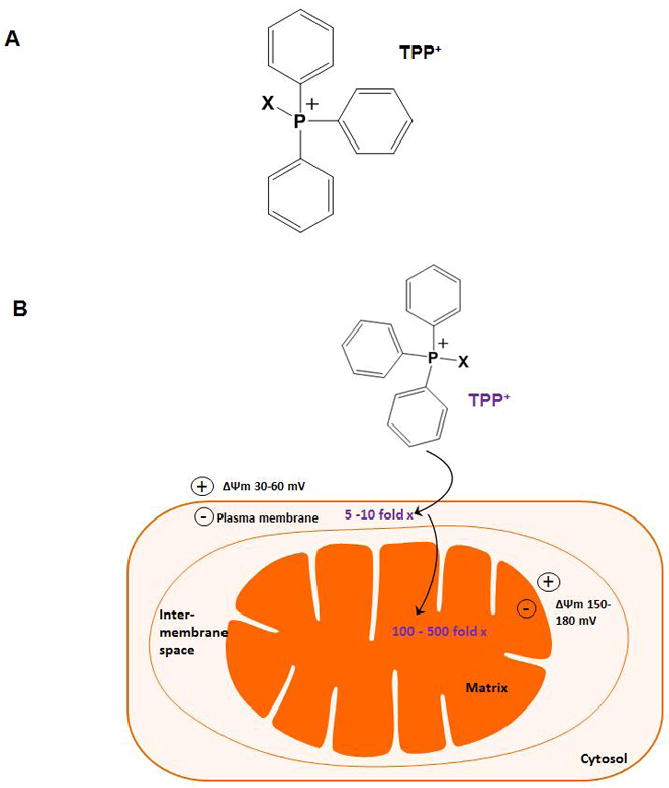
Mitochondrial accumulation of targeted lipophilic cationic antioxidants. A, Structure of TPP (triphenylphosphonium). B, Conjugation to a lipophilic cation such as TPP specifically targets the attached bioactive moiety (X) into the mitochondrial matrix in a ΔΨm-dependent fashion. This strategy leads to a 100-500 fold accumulation of the bioactive compound within mitochondria. TPP, triphenylphosphonium.
7.1 MitoQ
MitoQ (mitoquinone) is the most studied and widely used antioxidant targeted to mitochondria. It consists of TPP covalently attached to the ubiquinone moiety of the endogenous antioxidant CoQ10 through a ten-carbon aliphatic carbon chain (Fig. 4A). Multiple lines of evidence indicate that MitoQ predominantly accumulates within the mitochondria, where it is primarily absorbed to the matrix-facing surface of the inner mitochondrial membrane with the ubiquinone component penetrating deeply into the hydrophobic interior of the membrane [138]. MitoQ is a promising neuroprotective compound due to its direct antioxidant action. Like its parent antioxidant CoQ10, MitoQ continuously scavenges peroxyl, peroxynitrite, and superoxide, and thus can protect mitochondria against lipid peroxidation. After detoxifying oxidants, MitoQ is recycled back to the active ubiquinol antioxidant form by the respiratory chain Complex II [142]. MitoQ may also display concomitant anti-inflammatory and anti-hypoxic properties. Under certain conditions, however, it may become pro-oxidant and proapoptotic due to redox cycling of quinone and generation of superoxide [143, 144]. MitoQ was protective in a number of animal models of diseases involving oxidative stress, including neurodegenerative diseases [145], ischemia-reperfusion [146, 147], hypertension [148], sepsis [149, 150], fatty liver disease [151], alcoholic fatty liver disease [152], and kidney damage in type I diabetes [153].
Figure 4.
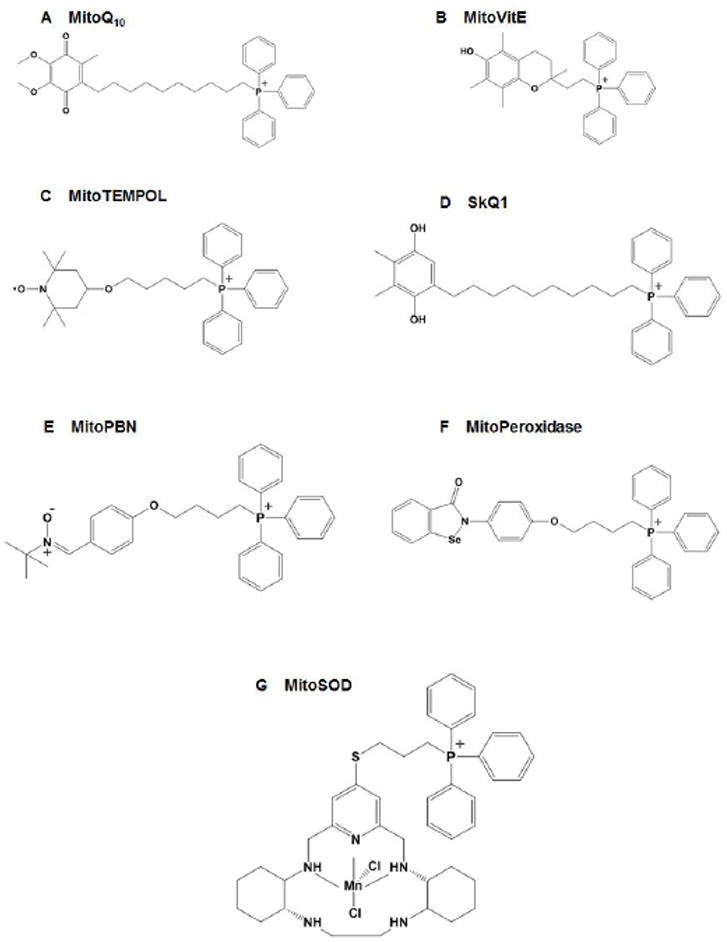
Structures of a range of TPP-based mitochondria-targeted antioxidants, including (A) MitoQ, (B) MitoVitE, (C) MitoTEMPOL, (D) SkQ1, (E) MitoPBN, (F) MitoPeroxidase, and (G) MitoSOD.
Although the therapeutic efficacy of MitoQ in PD needs to be further confirmed, several in vitro and in vivo studies have demonstrated beneficial effects (Table I). When used in SH-SY5Y cells, MitoQ markedly inhibited 6-OHDA-induced mitochondrial fragmentation [154]. In another study, we recently demonstrated the neuroprotective efficacy of MitoQ both in cell culture and in a pre-clinical animal model of PD [155]. MitoQ protected the MPP+-induced loss of neurons and neurites in a dopaminergic cell culture model of PD. At a dose of 4 mg/kg/day, MitoQ protected against the MPTP-induced loss of dopaminergic neurons and terminals in the nigrostriatum and it reversed both the MPTP-induced loss of dopamine and its metabolites as well as the MPTP-induced loss of behavioral activities [155]. Electron paramagnetic resonance analysis revealed that MitoQ inhibited the MPTP-mediated mitochondrial aconitase inactivation [155], suggesting that MitoQ indeed exerts its neuroprotective action at the intended target mitochondria. Administering MitoQ at a dose up to 500 μM through drinking water for 28 weeks showed no evidence of toxicity, indicating that MitoQ can be safely administered long-term in rodents [156]. MitoQ has now been developed as a pharmaceutical by Antipodean Pharmaceuticals Inc., and it has undergone Phase I and II clinical trials [157, 158]. In one clinical study, MitoQ at an oral dose of 40 - 80 mg/kg prevented liver damage in hepatitis C patients [157]. In contrast to this, another recent double-blind clinical trial failed to show any benefit from the oral intake of MitoQ in slowing the clinical progression of PD over the course of one year [158]. One explanation for the conflicting results, as put forward by the authors, is that MitoQ may not work for PD clinical remission, since nearly 50% of the dopaminergic neurons and 80% of striatal dopamine have been lost by the time PD is diagnosed. Hence, further studies are required to clarify the therapeutic effects of MitoQ in PD subjects.
Table I.
Mitochondria-targeted antioxidants in PD models
| In vitro Compounds | Cellular Models | Results | Referencesa |
|---|---|---|---|
| MitoQ | MPP+-induced cellular model | Reduced MPP+ toxicity in N27 cells and primary mesencephalic cultures | [1] |
| 6-OHDA-induced cellular model | Blocked 6-OHDA-induced mitochondrial fragmentation in SH-SY5Y cells | [2] | |
| SS-20 | MPP+-induced cellular model | Reduced MPP+ toxicity in SN4741 cells | [3] |
| SS-31 | MPP+-induced cellular model | Reduced MPP+ toxicity in SN4741 cells | [3] |
|
| |||
| In vivo Compounds | Model Systems | Results | |
|
| |||
| MitoQ | MPTP mouse model | Protected dopaminergic neurodegeneration, preserved striatal dopamine, and improved motor functions | [1] |
| MitoApocynin | MPTP mouse model | Showed neuroprotection in MPTP-treated mouse | [4] |
| SS-31 | MPTP mouse model | Decreased dopaminergic neuronal loss and restored dopamine levels | [3] |
| SS-20 | MPTP mouse model | Decreased dopaminergic neuronal loss and restored dopamine levels | [3] |
|
| |||
| Human studies Compounds | Mode Systems | Results | |
|
| |||
| MitoQ | Phase II clinical trial | Showed no neuroprotective benefit of taking MitoQ | [5] |
References:
A. Ghosh, K. Chandran, S.V. Kalivendi, J. Joseph, W.E. Antholine, C.J. Hillard, A. Kanthasamy, B. Kalyanaraman, Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model, Free radical biology & medicine, 49 (2010) 1674-1684.
M.E. Solesio, T.A. Prime, A. Logan, M.P. Murphy, M. Del Mar Arroyo-Jimenez, J. Jordan, M.F. Galindo, The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease, Biochimica et biophysica acta, 1832 (2013) 174-182.
L. Yang, K. Zhao, N.Y. Calingasan, G. Luo, H.H. Szeto, M.F. Beal, Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity, Antioxidants & redox signaling, 11 (2009) 2095-2104.
A. Ghosh, C. Hogan, A. Kanthasamy, V. Anantharam, B. Kalyanaraman, A.G. Kanthasamy, unpublished data.
B.J. Snow, F.L. Rolfe, M.M. Lockhart, C.M. Frampton, J.D. O’Sullivan, V. Fung, R.A. Smith, M.P. Murphy, K.M. Taylor, A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease, Movement disorders : official journal of the Movement Disorder Society, 25 (2010) 1670-1674.
7.2 MitoVitE
MitoVitE (mitotocopherol) was the first MTA to be discovered, and consists of TPP conjugated to the α-tocopherol moiety of vitamin E through a two-carbon chain (Fig. 4B). Internalized MitoVitE is immobilized by insertion in the lipid bilayer of the mitochondrial inner membrane. Like MitoQ, MitoVitE appears to protect mitochondria and cells from oxidative damage by inhibiting lipid peroxidation. MitoVitE is taken up rapidly by isolated mitochondria and cells in culture [140]. When administered to mice by intravenous injection, it rapidly accumulated in the tissues most affected by mitochondrial dysfunction and oxidative damage, including heart, brain, muscle, liver and kidney [141].
MitoVitE is protective in a number of cellular models of mitochondrial oxidative stress. MitoVitE was reported to reduce peroxide-mediated oxidative stress and to maintain proteasomal function in endothelial cells [159]. Likewise, another cell culture study demonstrated that MitoVitE protected against peroxide-induced caspase activation [160]. MitoVitE was protective against oxidative stress-induced cell death in fibroblasts from Friedrich ataxia (FRDA) patients [161]. However, it should be noted that higher levels of MitoVitE are cytotoxic in Jurkat cells [37]. There is also in vivo evidence for the beneficial effects of MitoVitE in several animal models of human diseases. Administering MitoVitE to rats 30 min post-induction of sepsis by pneumonia led to protection against cardiac damage [162]. MitoVitE was reported to decrease oxidative stress and reduce fat deposition in a mouse model of obesity [163]. In contrast, continuous striatal infusion of MitoVitE in rats was not protective against the hypoxia-ischemia-induced cell death of striatal medium-spiny neurons; moreover, MitoVitE at the higher dose of 435 μM was neurotoxic [164]. To date, neither the efficacy of MitoVitE in PD nor its therapeutic potential in humans has been investigated.
7.3 MitoTEMPOL
MitoTEMPOL is another TPP+ derivative, but one with the stable piperidine nitroxide radical TEMPOL (4-hydroxy-2,2,6,6,-tetramethyl-piperidine-1-oxyl; Fig. 4C), which accepts an electron from the potent radical scavenger hydroxylamine. MitoTEMPOL may also act as a cytosolic SOD mimetic, which converts superoxide molecules into water, and is able to detoxify ferrous iron by oxidizing it to ferric iron. The conjugated compound also accumulated inside energized, isolated mitochondria [165].
MitoTEMPOL has shown beneficial effects in several in vitro settings of mitochondrial oxidative stress. It has been demonstrated that MitoTEMPOL protected pancreatic β-cells against oxidative stress [166]. In a cellular model of ischemia-reperfusion, the related amide linked TPP, MitoTEMPO [167], partially inhibited ATP depletion-recovery mediated mitochondrial permeability transition pore opening and cell death [168]. In another cellular study, treatment with MitoTEMPOL significantly improved arteriolar endothelial function in vessels from type 2 diabetes mellitus patients [169]. Co-administering MitoTEMPO to rats simultaneously with angiotensin II (Ang II) infusion significantly protected them against Ang II-induced hypertension [170]. In animal models of diabetes, MitoTEMPOL prevented mitochondrial and cytosolic ROS production in the aorta and restored coronary collateral growth [171]. MitoTEMPOL has not yet been tested in animal models of PD.
7.4 “Sk” compounds
Skulachev et al. developed an alternative series of mitochondria-targeted antioxidants, termed “SkQs”, by using palstoquinone to replace the ubiquinone antioxidant moiety of MitoQ [172]. The compound SkQ1, which is a TPP+ derivative conjugated with palstoquinone itself (Fig. 4D), is the most studied “Sk” compound. SkQ1 performed as a potent antioxidant in isolated mitochondria [172]. Additional cell culture studies have confirmed that very low concentrations of SkQ1 and its analogs inhibited cell death induced by hydrogen peroxide [172, 173]. Extensive animal studies have demonstrated beneficial roles of SkQ1 and related compounds in a number of diseases associated with elevated oxidative stress [174-177], although little information regarding the effectiveness of “Sk” compounds against PD is currently available. Nevertheless, current evidence regarding “Sk” compounds is promising, and further exploration of these antioxidants in models of PD is warranted.
7.5 Other Mito compounds
Currently, other mitochondrially targeted antioxidants incorporating the TPP function have been developed. For instance, apocynin, a plant derived antioxidant and NADPH oxidase inhibitor, has been conjugated to TPP to form MitoApocynin. Recently, we observed significant beneficial effects of MitoApocynin in a preclinical MPTP mouse model of PD (unpublished data; Table I). MitoPBN is a similar TPP+ derivative with phenoxy-butyl-nitrone (Fig. 4E). The spin trap PBN was chosen based on PBN’s well-known reactivity with carbon-centered radicals [178]. MitoPBN was rapidly taken up by mitochondria and can block the oxygen-induced activation of uncoupled proteins [178]. MitoPeroxidase (Fig. 4F), a TPP-based mitochondria-targeted analog of ebselen, which has the peroxidase-like activity, was also prepared [179]. Growing evidence has indicated a class of SOD mimetics with neuroprotective effects in experimental models of PD [180-182]. Accordingly, more recently, Kelso et al. developed a mitochondrially targeted macrocyclic SOD mimetic, MitoSOD (Fig. 4G) [167]. In preliminary experiments, it rapidly accumulated within mitochondria and possessed SOD activity. The efficacy of MitoPeroxidase, MitoPBN and MitoSOD in vivo, particularly in the preclinical models of PD, however, remains to be determined.
7.6 SS tetrapeptides and alternative targeting approaches
To circumvent adverse effects of TPP+, alternate strategies have been explored for effective targeting of antioxidant to mitochondria. For example, small antioxidant molecules have been successfully targeted to mitochondria by incorporating mitochondria-targeted peptides. The Szeto-Schiller (SS) tetrapeptides contain an aromatic-cationic sequence motif that specifically enables them to be delivered to mitochondria, where they localize to the inner mitochondrial membrane with an approximate 1000-5000-fold accumulation [183, 184]. The mechanism behind the specific mitochondrial uptake of SS peptides is not fully clear, but it does not seem to depend on mitochondrial potential. Delivery of these peptides to tissues in vivo through intravenous, intraperitoneal or subcutaneous injection has been documented [142]. Currently, a number of SS tetrapeptides have been developed, of which SS-20 and SS-31 (Fig. 5A) are the most studied. Both of them comprise a dimethyltyrosine (Dmt) residue, which reacts with a variety of free radicals and inhibits lipid peroxidation [185].
Figure 5.
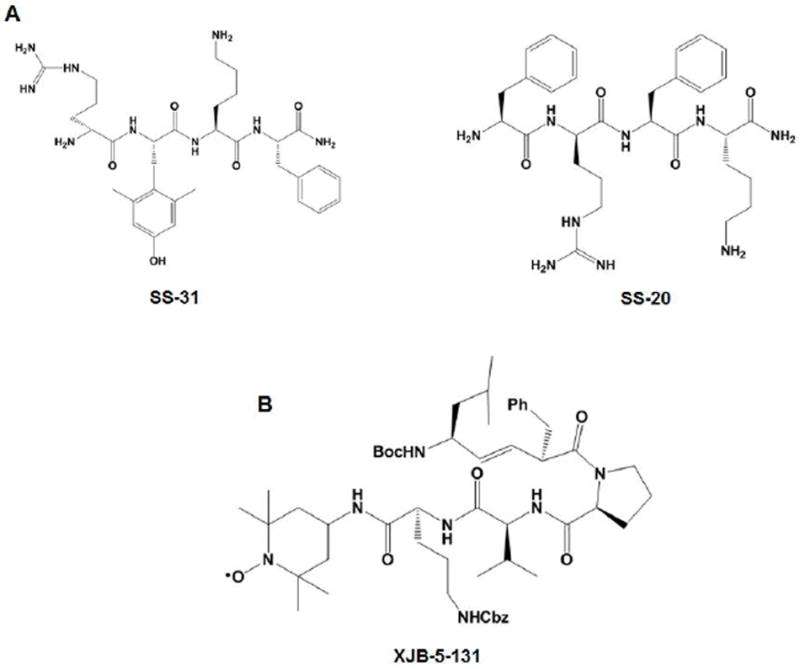
Structures of (A) SS peptides (SS-20 and SS-31) and (B) XJB peptide XJB-5-131.
In vitro studies have reported that the peptide antioxidants potently protected neurons against toxicity induced by amyloid beta (Aβ) [186, 187], herbicides [188], t-butyl hydroperoxide (tBHP) and 3-nitropropionic acid (3NP) [184, 189]. Interestingly, SS tetrapeptides have also shown promising benefits in animal models of PD (Table I). In the MPTP mouse model, administering SS-31 via intraperitoneal injection protected mice against nigrostriatal dopaminergic neurodegeneration and the loss of striatal dopamine and its metabolites [190]. Surprisingly, the SS-20 peptide, which lacks the Dmt residue and thus lacks antioxidant properties, also demonstrated significant protection in MPTP-treated mice [190], suggesting that the neuroprotective actions of SS peptides may not be attributable to the mechanism of scavenging ROS. Hence, additional preclinical studies on SS tetrapeptides are required to develop an effective drug capable of intervening in the progression of PD in humans.
In addition to SS peptides, several novel XJB peptides, including XJB-5-131 (Fig. 5B), have been invented. These peptides consist of an electron and ROS scavenger (4-NH2-TEMPO) conjugated to the Leu-D-Phe-Pro-Val-Orn fragment of gramicidin S. This pentapeptide fragment can specifically target the XJB peptides to mitochondria. XJB-5-131 improved mitochondrial function and enhanced the survival of neurons in a mouse model of Huntington’s disease [191].
Another approach to targeting mitochondria with small bioactive molecules is through a polymer based nano-carrier. This method involves biodegradable poly-lactide-co-gylcolide (PLGA) nanoparticles, which includes for example the PLGA-CoQ10 nanoparticles [192]. However, the biological efficacies of these CoQ10 nanoparticles remain to be investigated.
8. Conclusion and perspectives
PD is a complex, multifactorial disease triggered by both genetic and environmental factors. Substantial evidence has implicated mitochondrial dysfunction and oxidative damage as important components of PD pathogenesis. This has led to the enthusiastic use of mitochondria-targeted interventions as a tool for modulating mitochondrial function in the prevention and treatment of PD. Within the past 15 years, a class of compounds, referred to as mitochondria-targeted antioxidants, has been developed by conjugating the lipophilic triphenylphosphonium cation to an antioxidant moiety. These compounds have shown some promise in experimental models of neurodegenerative diseases against mitochondrial oxidative damage. Most notably, MitoQ and MitoApocynin have been efficacious in experimental models of PD, supporting the concept that mitochondria-targeted interventions would be effective in treating PD. However, a more recent, 1-year clinical study failed to show a link between MitoQ supplementation and prevention of PD progression. Although many explanations for the negative results may be postulated, one reason could be that the pharmacological effects of these Mito compounds are not completely known. For instance, MitoQ can act as a pro-oxidant and promote cell death due to redox cycling and generation of the superoxide anion. Therefore, an intriguing perspective would be to fine-tune the chemical biology of these compounds to remove the undesirable effects, as well as identify new compounds with bioactivity against mitochondrial oxidative damage. A second reason for the lack of efficacy may be that the preclinical assessment of these compounds in animal models of PD is woefully inadequate. Prior to entering the clinical development phase, a substantial preclinical data set should be developed to better define neuroprotective dose-response relationships, pharmacokinetic-pharmacodynamic correlations, therapeutic windows and optimum dosing regimens and treatment durations. Clearly, more preclinical studies in rodent and other mammalian models are necessary. Preclinical research in this area should not be limited to the traditional MPTP models of PD. Perhaps a more promising animal model for preclinical studies of mitochondria-targeted antioxidants is the MitoPark mouse, which exhibits spontaneous progressive PD-like pathology and behavior. Another potential explanation for the failure of MitoQ in the clinical trial could be due to the heterogeneous nature of PD, which will render any single therapeutic agent less beneficial than it appeared in simpler pre-clinical models. A more rational strategy would be to interfere at multiple points in the mitochondrial dysfunction by combining the mechanistic strategies offered by two or three antioxidants to achieve clinically demonstrable neuroprotection. Additionally, it should be noted that the tetraphenylphosphonium-based method changes mitochondrial potentials, which could be associated with toxic side effects. Thus, the future of mitochondria-targeted antioxidant therapies for PD will also depend on our developing better and more effective strategies targeting mitochondria with bioactive molecules. On the other hand, the mitochondria-targeted peptides (SS-31 and SS-20) have been effective in the MPTP models of PD, although their potential to treat or prevent PD needs further investigation. Finally, delivery of antioxidants using novel mitochondrially targeted nanomaterials may also be an attractive strategy for development of disease modifying drugs for treatment of neurodegenerative diseases such as PD.
Highlights.
MTAs are a novel class of antioxidant molecules targeted to the mitochondria
MTAs attenuate oxidative damage underlying mitochondrial dysfunction
MTAs show promise for treatment of many neurodegenerative diseases including PD
Recent advances and alternative strategies of MTA therapy in PD are discussed
Acknowledgments
The writing of this chapter was supported by the National Institutes of Health RO1 grants, NS039958 to BK and AGK, NS07443 to AGK and NS65167 to AK. The W. Eugene and Linda Lloyd Endowed Chair to AGK, and Harry R and Angeline E Quadracci Professor in Parkinson’s Research to BK are also acknowledged. We thank Gary Zenitsky for assistance in the preparation of this manuscript.
Abbreviations
- MTA
mitochondria-targeted antioxidant
- PD
Parkinson’s disease
- SNc
substantia nigra pars compacta
- ETC
electron transport chains
- O2•-
superoxide anion
- ROS
reactive oxygen species
- NO
nitric oxide
- TCA
tricarboxylic acid cycle
- ONOO-
peroxynitrite
- RNS
reactive nitrogen species
- H2O2
hydrogen peroxide
- SOD
superoxide dismutase
- GPx
glutathione peroxidase
- TPx
thioredoxin reductase
- OH•
hydroxyl radical
- RO2•
peroxyl radical
- HOCl
hypochlorous acid
- RO•
alkoxyl radical
- HO2•
hydroperoxyl radical
- GSH
glutathione
- GSSG
glutathione disulfide
- NOS
nitric oxide synthase
- CoQ10
coenzyme Q10
- mtDNA
mitochondrial DNA
- apocynin
4-hydroxy-3-methoxyacetophenone
- TPP
triphenylphosphonium
- MitoQ
mitoquinone
- MitoVitE
mitotocopherol
- TEMPOL
4-hydroxy-2,2,6,6,-tetramethyl-piperidine-1-oxyl
- Ang II
angiotensin II
- DMT
dimethyltyrosine
- PLGA
poly-lactide-co-gylcolide
- CAT
catalase
- tBHP
t-butyl hydroperoxide
- 3NP
3-nitropropionic acid
- Aβ
amyloid beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jankovic J, Stacy M. Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS drugs. 2007;21:677–692. doi: 10.2165/00023210-200721080-00005. [DOI] [PubMed] [Google Scholar]
- 2.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 3.Tanner CM. Is the cause of Parkinson’s disease environmental or hereditary? Evidence from twin studies. Advances in neurology. 2003;91:133–142. [PubMed] [Google Scholar]
- 4.Mizuno Y, Hattori N, Mori H, Suzuki T, Tanaka K. Parkin and Parkinson’s disease. Current opinion in neurology. 2001;14:477–482. doi: 10.1097/00019052-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Kirik D, Rosenblad C, Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Experimental neurology. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- 6.Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome science. 2008;6:8. doi: 10.1186/1477-5956-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knaryan VH, Samantaray S, Le Gal C, Ray SK, Banik NL. Tracking extranigral degeneration in animal models of Parkinson’s disease: quest for effective therapeutic strategies. Journal of neurochemistry. 2011;118:326–338. doi: 10.1111/j.1471-4159.2011.07320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolters E. Non-motor extranigral signs and symptoms in Parkinson’s disease. Parkinsonism & related disorders. 2009;15(Suppl 3):S6–12. doi: 10.1016/S1353-8020(09)70770-9. [DOI] [PubMed] [Google Scholar]
- 9.Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Parkinson disease: extranigral, multisystem, and {alpha}-synuclein “plus”. Archives of neurology. 2009;66:914–915. doi: 10.1001/archneurol.2009.140. [DOI] [PubMed] [Google Scholar]
- 10.Chou KL, Taylor JL, Patil PG. The MDS-UPDRS tracks motor and non-motor improvement due to subthalamic nucleus deep brain stimulation in Parkinson disease. Parkinsonism & related disorders. 2013 doi: 10.1016/j.parkreldis.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olanow CW. The scientific basis for the current treatment of Parkinson’s disease. Annual review of medicine. 2004;55:41–60. doi: 10.1146/annurev.med.55.091902.104422. [DOI] [PubMed] [Google Scholar]
- 12.Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. Missing pieces in the Parkinson’s disease puzzle. Nature medicine. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 13.Lewitt PA. Levodopa for the treatment of Parkinson’s disease. The New England journal of medicine. 2008;359:2468–2476. doi: 10.1056/NEJMct0800326. [DOI] [PubMed] [Google Scholar]
- 14.Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. Journal of cell science. 2007;120:1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- 15.Parker WD, Jr, Swerdlow RH. Mitochondrial dysfunction in idiopathic Parkinson disease. American journal of human genetics. 1998;62:758–762. doi: 10.1086/301812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.NINDS. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kalyanaraman B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free radical biology & medicine. 2010;49:1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 19.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell calcium. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 21.Green DR. Apoptotic pathways: the roads to ruin. Cell. 1998;94:695–698. doi: 10.1016/s0092-8674(00)81728-6. [DOI] [PubMed] [Google Scholar]
- 22.Akopova OV, Kolchinskaya LI, Nosar VI, Bouryi VA, Mankovska IN, Sagach VF. Cytochrome C as an amplifier of ROS release in mitochondria. Fiziol Zh. 2012;58:3–12. [PubMed] [Google Scholar]
- 23.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of signal transduction. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lill R, Muhlenhoff U. Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annual review of cell and developmental biology. 2006;22:457–486. doi: 10.1146/annurev.cellbio.22.010305.104538. [DOI] [PubMed] [Google Scholar]
- 25.Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of physiology. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free radical biology & medicine. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 27.Kirkinezos IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Seminars in cell & developmental biology. 2001;12:449–457. doi: 10.1006/scdb.2001.0282. [DOI] [PubMed] [Google Scholar]
- 28.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PloS one. 2009;4:e7095. doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forman HJ, Kennedy J. Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. The Journal of biological chemistry. 1975;250:4322–4326. [PubMed] [Google Scholar]
- 31.Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free radical biology & medicine. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 32.Galley HF. Bench-to-bedside review: Targeting antioxidants to mitochondria in sepsis. Crit Care. 2010;14:230. doi: 10.1186/cc9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiological reviews. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fridovich I. Superoxide radical and superoxide dismutases. Annual review of biochemistry. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 35.Vranova E, Inze D, Van Breusegem F. Signal transduction during oxidative stress. Journal of experimental botany. 2002;53:1227–1236. [PubMed] [Google Scholar]
- 36.Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods in enzymology. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 37.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochimica et biophysica acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Annals of neurology. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 39.Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free radical biology & medicine. 2001;31:1473–1485. doi: 10.1016/s0891-5849(01)00726-2. [DOI] [PubMed] [Google Scholar]
- 40.Kitazawa M, Wagner JR, Kirby ML, Anantharam V, Kanthasamy AG. Oxidative stress and mitochondrial-mediated apoptosis in dopaminergic cells exposed to methylcyclopentadienyl manganese tricarbonyl. The Journal of pharmacology and experimental therapeutics. 2002;302:26–35. doi: 10.1124/jpet.302.1.26. [DOI] [PubMed] [Google Scholar]
- 41.Carvour M, Song C, Kaul S, Anantharam V, Kanthasamy A. Chronic low-dose oxidative stress induces caspase-3-dependent PKCdelta proteolytic activation and apoptosis in a cell culture model of dopaminergic neurodegeneration. Annals of the New York Academy of Sciences. 2008;1139:197–205. doi: 10.1196/annals.1432.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Droge W. Free radicals in the physiological control of cell function. Physiological reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 43.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends in biochemical sciences. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell death & disease. 2011;2:e213. doi: 10.1038/cddis.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lenaz G, Bovina C, Formiggini G, Parenti Castelli G. Mitochondria, oxidative stress, and antioxidant defences. Acta biochimica Polonica. 1999;46:1–21. [PubMed] [Google Scholar]
- 46.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacological reviews. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 47.Milani P, Gagliardi S, Cova E, Cereda C. SOD1 Transcriptional and Posttranscriptional Regulation and Its Potential Implications in ALS. Neurology research international. 2011;2011:458427. doi: 10.1155/2011/458427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. The Journal of biological chemistry. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 50.Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. The Journal of the Association of Physicians of India. 2004;52:794–804. [PubMed] [Google Scholar]
- 51.Tsang AH, Chung KK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 52.Jenner P. Oxidative stress in Parkinson’s disease. Annals of neurology. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. discussion S36-28. [DOI] [PubMed] [Google Scholar]
- 53.Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 54.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 55.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 56.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 58.Migliore L, Coppede F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat Res. 2009;674:73–84. doi: 10.1016/j.mrgentox.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 59.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 60.Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. Biochim Biophys Acta. 2008;1780:1362–1367. doi: 10.1016/j.bbagen.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 62.Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology. 1996;47:S161–170. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
- 63.Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 64.Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, Youdim MB. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- 65.Cardoso SM, Pereira C, Oliveira R. Mitochondrial function is differentially affected upon oxidative stress. Free Radic Biol Med. 1999;26:3–13. doi: 10.1016/s0891-5849(98)00205-6. [DOI] [PubMed] [Google Scholar]
- 66.Cecarini V, Gee J, Fioretti E, Amici M, Angeletti M, Eleuteri AM, Keller JN. Protein oxidation and cellular homeostasis: Emphasis on metabolism. Biochim Biophys Acta. 2007;1773:93–104. doi: 10.1016/j.bbamcr.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 67.Hastings TG. The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr. 2009;41:469–472. doi: 10.1007/s10863-009-9257-z. [DOI] [PubMed] [Google Scholar]
- 68.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Dawson VL, Dawson TM. Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol Dis. 2000;7:240–250. doi: 10.1006/nbdi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 70.Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res. 2006;3:327–337. doi: 10.2174/156720506778249515. [DOI] [PubMed] [Google Scholar]
- 71.Mattson MP. Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1997–2006. doi: 10.1089/ars.2006.8.1997. [DOI] [PubMed] [Google Scholar]
- 72.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 73.Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. Role of proteolytic activation of protein kinase Cdelta in oxidative stress-induced apoptosis. Antioxidants & redox signaling. 2003;5:609–620. doi: 10.1089/152308603770310275. [DOI] [PubMed] [Google Scholar]
- 74.Perier C, Bove J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, Przedborski S, Vila M. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:8161–8166. doi: 10.1073/pnas.0609874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perier C, Tieu K, Guegan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 77.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci U S A. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–2255. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, Agid Y, Hirsch EC. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J Neurochem. 2001;76:1785–1793. doi: 10.1046/j.1471-4159.2001.00160.x. [DOI] [PubMed] [Google Scholar]
- 80.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 81.Simon DK, Lin MT, Zheng L, Liu GJ, Ahn CH, Kim LM, Mauck WM, Twu F, Beal MF, Johns DR. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiology of aging. 2004;25:71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 82.Sandy MS, Langston JW, Smith MT, Di Monte DA. PCR analysis of platelet mtDNA: lack of specific changes in Parkinson’s disease. Mov Disord. 1993;8:74–82. doi: 10.1002/mds.870080114. [DOI] [PubMed] [Google Scholar]
- 83.Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi DY, Hunter RL, Gerhardt GA, Smith CD, Slevin JT, Prince TS. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Annals of neurology. 2008;63:184–192. doi: 10.1002/ana.21288. [DOI] [PubMed] [Google Scholar]
- 84.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature neuroscience. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 85.Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7(Suppl):S136–145. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 86.Benecke R, Strumper P, Weiss H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain. 1993;116(Pt 6):1451–1463. doi: 10.1093/brain/116.6.1451. [DOI] [PubMed] [Google Scholar]
- 87.Gu M, Cooper JM, Taanman JW, Schapira AH. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Annals of neurology. 1998;44:177–186. doi: 10.1002/ana.410440207. [DOI] [PubMed] [Google Scholar]
- 88.Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD., Jr Origin and functional consequences of the complex I defect in Parkinson’s disease. Annals of neurology. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- 89.Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson’s disease. Journal of neurochemistry. 2007;100:503–519. doi: 10.1111/j.1471-4159.2006.04315.x. [DOI] [PubMed] [Google Scholar]
- 91.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. The American journal of pathology. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Experimental neurology. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- 93.Odunze IN, Klaidman LK, Adams JD., Jr MPTP toxicity in the mouse brain and vitamin E. Neuroscience letters. 1990;108:346–349. doi: 10.1016/0304-3940(90)90665-v. [DOI] [PubMed] [Google Scholar]
- 94.Lan J, Jiang DH. Desferrioxamine and vitamin E protect against iron and MPTP-induced neurodegeneration in mice. J Neural Transm. 1997;104:469–481. doi: 10.1007/BF01277665. [DOI] [PubMed] [Google Scholar]
- 95.Roghani M, Behzadi G. Neuroprotective effect of vitamin E on the early model of Parkinson’s disease in rat: behavioral and histochemical evidence. Brain research. 2001;892:211–217. doi: 10.1016/s0006-8993(00)03296-0. [DOI] [PubMed] [Google Scholar]
- 96.Fahn S. A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Annals of neurology. 1992;32(Suppl):S128–132. doi: 10.1002/ana.410320722. [DOI] [PubMed] [Google Scholar]
- 97.Mortality in DATATOP: a multicenter trial in early Parkinson’s disease. Parkinson Study Group. Annals of neurology. 1998;43:318–325. doi: 10.1002/ana.410430309. [DOI] [PubMed] [Google Scholar]
- 98.Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. The Parkinson Study Group. The New England journal of medicine. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 99.Zhang SM, Hernan MA, Chen H, Spiegelman D, Willett WC, Ascherio A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology. 2002;59:1161–1169. doi: 10.1212/01.wnl.0000028688.75881.12. [DOI] [PubMed] [Google Scholar]
- 100.Etminan M, Gill SS, Samii A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet neurology. 2005;4:362–365. doi: 10.1016/S1474-4422(05)70097-1. [DOI] [PubMed] [Google Scholar]
- 101.Weber CA, Ernst ME. Antioxidants, supplements, and Parkinson’s disease. The Annals of pharmacotherapy. 2006;40:935–938. doi: 10.1345/aph.1G551. [DOI] [PubMed] [Google Scholar]
- 102.Scheider WL, Hershey LA, Vena JE, Holmlund T, Marshall JR. Freudenheim, Dietary antioxidants and other dietary factors in the etiology of Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 1997;12:190–196. doi: 10.1002/mds.870120209. [DOI] [PubMed] [Google Scholar]
- 103.Seidl SE, Potashkin JA. The promise of neuroprotective agents in Parkinson’s disease. Frontiers in neurology. 2011;2:68. doi: 10.3389/fneur.2011.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gualano B, de Salles Painelli V, Roschel H, Lugaresi R, Dorea E, Artioli GG, Lima FR, da Silva ME, Cunha MR, Seguro AC, Shimizu MH, Otaduy MC, Sapienza MT, da Costa Leite C, Bonfa E, Lancha AH., Junior Creatine supplementation does not impair kidney function in type 2 diabetic patients: a randomized, double-blind, placebo-controlled, clinical trial. European journal of applied physiology. 2010;111:749–756. doi: 10.1007/s00421-010-1676-3. [DOI] [PubMed] [Google Scholar]
- 105.Andres RH, Huber AW, Schlattner U, Perez-Bouza A, Krebs SH, Seiler RW, Wallimann T, Widmer HR. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience. 2005;133:701–713. doi: 10.1016/j.neuroscience.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 106.Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Experimental neurology. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- 107.A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clinical neuropharmacology. 2008;31:141–150. doi: 10.1097/WNF.0b013e3181342f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bender A, Koch W, Elstner M, Schombacher Y, Bender J, Moeschl M, Gekeler F, Muller-Myhsok B, Gasser T, Tatsch K, Klopstock T. Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology. 2006;67:1262–1264. doi: 10.1212/01.wnl.0000238518.34389.12. [DOI] [PubMed] [Google Scholar]
- 109.Bloom MZ. NIH announces phase III clinical trial of creatine for Parkinson’s disease. Consult Pharm. 2007;22:378. [PubMed] [Google Scholar]
- 110.Couzin J. Clinical research. Testing a novel strategy against Parkinson’s disease. Science (New York, N Y. 2007;315:1778. doi: 10.1126/science.315.5820.1778. [DOI] [PubMed] [Google Scholar]
- 111.Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. Journal of neurochemistry. 2005;93:1199–1208. doi: 10.1111/j.1471-4159.2005.03112.x. [DOI] [PubMed] [Google Scholar]
- 112.Ono K, Hasegawa K, Naiki H, Yamada M. Preformed beta-amyloid fibrils are destabilized by coenzyme Q10 in vitro. Biochemical and biophysical research communications. 2005;330:111–116. doi: 10.1016/j.bbrc.2005.02.132. [DOI] [PubMed] [Google Scholar]
- 113.Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiology of disease. 2005;18:618–627. doi: 10.1016/j.nbd.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 114.Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain research. 1998;783:109–114. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- 115.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Annals of the New York Academy of Sciences. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Horvath TL, Diano S, Leranth C, Garcia-Segura LM, Cowley MA, Shanabrough M, Elsworth JD, Sotonyi P, Roth RH, Dietrich EH, Matthews RT, Barnstable CJ, Redmond DE., Jr Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson’s disease. Endocrinology. 2003;144:2757–2760. doi: 10.1210/en.2003-0163. [DOI] [PubMed] [Google Scholar]
- 117.Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Archives of neurology. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 118.NINDS. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68:20–28. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- 119.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cohen AM, Aberdroth RE, Hochstein P. Inhibition of free radical-induced DNA damage by uric acid. FEBS letters. 1984;174:147–150. doi: 10.1016/0014-5793(84)81094-7. [DOI] [PubMed] [Google Scholar]
- 121.Davies KJ, Sevanian A, Muakkassah-Kelly SF, Hochstein P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. The Biochemical journal. 1986;235:747–754. doi: 10.1042/bj2350747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Duan W, Ladenheim B, Cutler RG, Kruman, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. Journal of neurochemistry. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- 123.Guerreiro S, Ponceau A, Toulorge D, Martin E, Alvarez-Fischer D, Hirsch EC, Michel PP. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: potentiation by low-level depolarization. Journal of neurochemistry. 2009;109:1118–1128. doi: 10.1111/j.1471-4159.2009.06040.x. [DOI] [PubMed] [Google Scholar]
- 124.Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM. Observations on serum uric acid levels and the risk of idiopathic Parkinson’s disease. American journal of epidemiology. 1996;144:480–484. doi: 10.1093/oxfordjournals.aje.a008954. [DOI] [PubMed] [Google Scholar]
- 125.de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Annals of neurology. 2005;58:797–800. doi: 10.1002/ana.20663. [DOI] [PubMed] [Google Scholar]
- 126.Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson’s disease. American journal of epidemiology. 2007;166:561–567. doi: 10.1093/aje/kwm127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen H, Mosley TH, Alonso A, Huang X. Plasma urate and Parkinson’s disease in the Atherosclerosis Risk in Communities (ARIC) study. American journal of epidemiology. 2009;169:1064–1069. doi: 10.1093/aje/kwp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schwarzschild MA, Schwid SR, Marek K, Watts A, Lang AE, Oakes D, Shoulson I, Ascherio A, Hyson C, Gorbold E, Rudolph A, Kieburtz K, Fahn S, Gauger L, Goetz C, Seibyl J, Forrest M, Ondrasik J. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Archives of neurology. 2008;65:716–723. doi: 10.1001/archneur.2008.65.6.nct70003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363:1277–1281. doi: 10.1016/S0140-6736(04)16000-5. [DOI] [PubMed] [Google Scholar]
- 130.Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG. Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology. 2007;28:988–997. doi: 10.1016/j.neuro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003;23:6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vejrazka M, Micek R, Stipek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochimica et biophysica acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 133.Kanegae MP, Condino-Neto A, Pedroza LA, de Almeida AC, Rehder J, da Fonseca LM, Ximenes VF. Diapocynin versus apocynin as pretranscriptional inhibitors of NADPH oxidase and cytokine production by peripheral blood mononuclear cells. Biochemical and biophysical research communications. 2010;393:551–554. doi: 10.1016/j.bbrc.2010.02.073. [DOI] [PubMed] [Google Scholar]
- 134.Luchtefeld R, Luo R, Stine K, Alt ML, Chernovitz PA, Smith RE. Dose formulation and analysis of diapocynin. Journal of agricultural and food chemistry. 2008;56:301–306. doi: 10.1021/jf072792n. [DOI] [PubMed] [Google Scholar]
- 135.Ghosh A, Kanthasamy A, Joseph J, Anantharam V, Srivastava P, Dranka BP, Kalyanaraman B, Kanthasamy AG. Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson’s disease. Journal of neuroinflammation. 2012;9:241. doi: 10.1186/1742-2094-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dranka BP, Gifford A, Ghosh A, Zielonka J, Joseph J, Kanthasamy AG, Kalyanaraman B. Diapocynin prevents early Parkinson’s disease symptoms in the leucine-rich repeat kinase 2 (LRRK2) transgenic mouse. Neuroscience letters. 2013 doi: 10.1016/j.neulet.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liberman EA, Topaly VP, Tsofina LM, Jasaitis AA, Skulachev VP. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature. 1969;222:1076–1078. doi: 10.1038/2221076a0. [DOI] [PubMed] [Google Scholar]
- 138.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annual review of pharmacology and toxicology. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 139.Ross MF, Kelso GF, Blaikie FH, James AM, Cocheme HM, Filipovska A, Da Ros T, Hurd TR, Smith RA, Murphy MP. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry. Biokhimiia. 2005;70:222–230. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- 140.Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. European journal of biochemistry / FEBS. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 141.Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discovery medicine. 2011;11:106–114. [PubMed] [Google Scholar]
- 143.O’Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. The Journal of biological chemistry. 2006;281:39766–39775. doi: 10.1074/jbc.M608268200. [DOI] [PubMed] [Google Scholar]
- 144.Doughan AK, Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxidants & redox signaling. 2007;9:1825–1836. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]
- 145.McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 147.Neuzil J, Widen C, Gellert N, Swettenham E, Zobalova R, Dong LF, Wang XF, Lidebjer C, Dalen H, Headrick JP, Witting PK. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox report : communications in free radical research. 2007;12:148–162. doi: 10.1179/135100007X200227. [DOI] [PubMed] [Google Scholar]
- 148.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cocheme HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 149.Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction, American journal of physiology. Regulatory, integrative and comparative physiology. 2009;297:R1095–1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free radical biology & medicine. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 151.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free radical biology & medicine. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 152.Chacko BK, Srivastava A, Johnson MS, Benavides GA, Chang MJ, Ye Y, Jhala N, Murphy MP, Kalyanaraman B, Darley-Usmar VM. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology. 2011;54:153–163. doi: 10.1002/hep.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Annals of the New York Academy of Sciences. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 154.Solesio ME, Prime TA, Logan A, Murphy MP, Del Mar Arroyo-Jimenez M, Jordan J, Galindo MF. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochimica et biophysica acta. 2013;1832:174–182. doi: 10.1016/j.bbadis.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 155.Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kanthasamy A, Kalyanaraman B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free radical biology & medicine. 2010;49:1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rodriguez-Cuenca S, Cocheme HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM, Jones BA, Pope S, Heales SJ, Lam BY, Neogi SG, McFarlane I, James AM, Smith RA, Murphy MP. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free radical biology & medicine. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 157.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver international : official journal of the International Association for the Study of the Liver. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 158.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 159.Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. The Journal of biological chemistry. 2004;279:37575–37587. doi: 10.1074/jbc.M404003200. [DOI] [PubMed] [Google Scholar]
- 160.Hughes G, Murphy MP, Ledgerwood EC. Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappaB to modulate tumour necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. The Biochemical journal. 2005;389:83–89. doi: 10.1042/BJ20050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 162.Zang QS, Sadek H, Maass DL, Martinez B, Ma L, Kilgore JA, Williams NS, Frantz DE, Wigginton JG, Nwariaku FE, Wolf SE, Minei JP. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. American journal of physiology Heart and circulatory physiology. 2012;302:H1847–1859. doi: 10.1152/ajpheart.00203.2011. [DOI] [PubMed] [Google Scholar]
- 163.Mao G, Kraus GA, Kim I, Spurlock ME, Bailey TB, Zhang Q, Beitz DC. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. The Journal of nutrition. 2010;140:1425–1431. doi: 10.3945/jn.110.121715. [DOI] [PubMed] [Google Scholar]
- 164.Covey MV, Murphy MP, Hobbs CE, Smith RA, Oorschot DE. Effect of the mitochondrial antioxidant, Mito Vitamin E, on hypoxic-ischemic striatal injury in neonatal rats: a dose-response and stereological study. Experimental neurology. 2006;199:513–519. doi: 10.1016/j.expneurol.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 165.Trnka J, Blaikie FH, Smith RA, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free radical biology & medicine. 2008;44:1406–1419. doi: 10.1016/j.freeradbiomed.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 166.Lim S, Rashid MA, Jang M, Kim Y, Won H, Lee J, Woo JT, Kim YS, Murphy MP, Ali L, Ha J, Kim SS. Mitochondria-targeted antioxidants protect pancreatic beta-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2011;28:873–886. doi: 10.1159/000335802. [DOI] [PubMed] [Google Scholar]
- 167.Kelso GF, Maroz A, Cocheme HM, Logan A, Prime TA, Peskin AV, Winterbourn CC, James AM, Ross MF, Brooker S, Porteous CM, Anderson RF, Murphy MP, Smith RA. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chemistry & biology. 2012;19:1237–1246. doi: 10.1016/j.chembiol.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 168.Liang HL, Sedlic F, Bosnjak Z, Nilakantan V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free radical biology & medicine. 2010;49:1550–1560. doi: 10.1016/j.freeradbiomed.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, Widlansky ME. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2531–2539. doi: 10.1161/ATVBAHA.112.256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation research. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pung YF, Rocic P, Murphy MP, Smith RA, Hafemeister J, Ohanyan V, Guarini G, Yin L, Chilian WM. Resolution of mitochondrial oxidative stress rescues coronary collateral growth in Zucker obese fatty rats. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:325–334. doi: 10.1161/ATVBAHA.111.241802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Antonenko YN, Avetisyan AV, Bakeeva LE, Chernyak BV, Chertkov VA, Domnina LV, Ivanova OY, Izyumov DS, Khailova LS, Klishin SS, Korshunova GA, Lyamzaev KG, Muntyan MS, Nepryakhina OK, Pashkovskaya AA, Pletjushkina OY, Pustovidko AV, Roginsky VA, Rokitskaya TI, Ruuge EK, Saprunova VB, Severina, Simonyan RA, Skulachev IV, Skulachev MV, Sumbatyan NV, Sviryaeva IV, Tashlitsky VN, Vassiliev JM, Vyssokikh MY, Yaguzhinsky LS, Zamyatnin AA, Jr, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry. Biokhimiia. 2008;73:1273–1287. doi: 10.1134/s0006297908120018. [DOI] [PubMed] [Google Scholar]
- 173.Skulachev MV, Antonenko YN, Anisimov VN, Chernyak BV, Cherepanov DA, Chistyakov VA, Egorov MV, Kolosova NG, Korshunova GA, Lyamzaev KG, Plotnikov EY, Roginsky VA, Savchenko AY, Severia, Severin FF, Shkurat TP, Tashlitsky VN, Shidlovsky KM, Vyssokikh MY, Zamyatnin AA, Jr, Zorov DB, Skulachev VP. Mitochondrial-targeted plastoquinone derivatives. Effect on senescence and acute age-related pathologies. Current drug targets. 2011;12:800–826. doi: 10.2174/138945011795528859. [DOI] [PubMed] [Google Scholar]
- 174.Agapova LS, Chernyak BV, Domnina LV, Dugina VB, Efimenko AY, Fetisova EK, Ivanova OY, Kalinina NI, Khromova NV, Kopnin BP, Kopnin PB, Korotetskaya MV, Lichinitser MR, Lukashev AL, Pletjushkina OY, Popova EN, Skulachev MV, Shagieva GS, Stepanova EV, Titova EV, Tkachuk VA, Vasiliev JM, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 3. Inhibitory effect of SkQ1 on tumor development from p53-deficient cells. Biochemistry. Biokhimiia. 2008;73:1300–1316. doi: 10.1134/s0006297908120031. [DOI] [PubMed] [Google Scholar]
- 175.Anisimov VN, Bakeeva LE, Egormin PA, Filenko OF, Isakova EF, Manskikh VN, Mikhelson VM, Panteleeva AA, Pasyukova EG, Pilipenko DI, Piskunova TS, Popovich IG, Roshchina NV, Rybina OY, Saprunova VB, Samoylova TA, Semenchenko AV, Skulachev MV, Spivak IM, Tsybul’ko EA, Tyndyk ML, Vyssokikh MY, Yurova MN, Zabezhinsky MA, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 5. SkQ1 prolongs lifespan and prevents development of traits of senescence. Biochemistry. Biokhimiia. 2008;73:1329–1342. doi: 10.1134/s0006297908120055. [DOI] [PubMed] [Google Scholar]
- 176.Bakeeva LE, Barskov IV, Egorov MV, Isaev NK, Kapelko VI, Kazachenko AV, Kirpatovsky VI, Kozlovsky SV, Lakomkin VL, Levina SB, Pisarenko OI, Plotnikov EY, Saprunova VB, Serebryakova LI, Skulachev MV, Stelmashook EV, Studneva IM, Tskitishvili OV, Vasilyeva AK, Victorov IV, Zorov DB, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke) Biochemistry. Biokhimiia. 2008;73:1288–1299. doi: 10.1134/s000629790812002x. [DOI] [PubMed] [Google Scholar]
- 177.Neroev VV, Archipova MM, Bakeeva LE, Fursova A, Grigorian EN, Grishanova AY, Iomdina EN, Ivashchenko Zh N, Katargina LA, Khoroshilova-Maslova IP, Kilina OV, Kolosova NG, Kopenkin EP, Korshunov SS, Kovaleva NA, Novikova YP, Philippov PP, Pilipenko DI, Robustova OV, Saprunova VB, Senin, Skulachev MV, Sotnikova LF, Stefanova NA, Tikhomirova NK, Tsapenko IV, Shchipanova AI, Zinovkin RA, Skulachev VP. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 4. Age-related eye disease. SkQ1 returns vision to blind animals. Biochemistry. Biokhimiia. 2008;73:1317–1328. doi: 10.1134/s0006297908120043. [DOI] [PubMed] [Google Scholar]
- 178.Murphy MP, Echtay KS, Blaikie FH, Asin-Cayuela J, Cocheme HM, Green K, Buckingham JA, Taylor ER, Hurrell F, Hughes G, Miwa S, Cooper CE, Svistunenko DA, Smith RA, Brand MD. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. The Journal of biological chemistry. 2003;278:48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- 179.Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RA, Murphy MP. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. The Journal of biological chemistry. 2005;280:24113–24126. doi: 10.1074/jbc.M501148200. [DOI] [PubMed] [Google Scholar]
- 180.Peng J, Stevenson FF, Doctrow SR, Andersen JK. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. The Journal of biological chemistry. 2005;280:29194–29198. doi: 10.1074/jbc.M500984200. [DOI] [PubMed] [Google Scholar]
- 181.Liang LP, Huang J, Fulton R, Day BJ, Patel M. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:4326–4333. doi: 10.1523/JNEUROSCI.0019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Batinic-Haberle I, Reboucas JS, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxidants & redox signaling. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhao K, Luo G, Zhao GM, Schiller PW, Szeto HH. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. The Journal of pharmacology and experimental therapeutics. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- 184.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. The Journal of biological chemistry. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 185.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxidants & redox signaling. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 186.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. Journal of Alzheimer’s disease : JAD. 2010;20(Suppl 2):S609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Human molecular genetics. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Reddy TP, Manczak M, Calkins MJ, Mao P, Reddy AP, Shirendeb U, Park B, Reddy PH. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS31. International journal of environmental research and public health. 2011;8:203–221. doi: 10.3390/ijerph8010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochemical pharmacology. 2005;70:1796–1806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 190.Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxidants & redox signaling. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Xun Z, Rivera-Sanchez S, Ayala-Pena S, Lim J, Budworth H, Skoda EM, Robbins PD, Niedernhofer LJ, Wipf P, McMurray CT. Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell reports. 2012;2:1137–1142. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nehilla BJ, Bergkvist M, Popat KC, Desai TA. Purified and surfactant-free coenzyme Q10-loaded biodegradable nanoparticles. International journal of pharmaceutics. 2008;348:107–114. doi: 10.1016/j.ijpharm.2007.07.001. [DOI] [PubMed] [Google Scholar]