

Background: Filamins are actin cross-linking and signaling scaffolding proteins where C-terminal domains have inter-domain interactions but little is known about the N-terminal domains.
Results: Crystal structures of N-terminal domains 3–5 reveal novel domain packing and interaction details of domain 4.
Conclusion: Domain 4 is stabilized by interaction with domain 5.
Significance: Here, inter-domain interactions positively regulate domain 4 interactions with ligands.
Keywords: Cell Signaling, Filamin, Microfilaments, NMR, Protein-Protein Interactions, Structural Biology, X-ray Crystallography, X-ray Scattering, Immunogloblulin-like Domain, Platelet Glycoprotein Ib
Abstract
Immunoglobulin-like (Ig) domains are a widely expanded superfamily that act as interaction motifs or as structural spacers in multidomain proteins. Vertebrate filamins (FLNs), which are multifunctional actin-binding proteins, consist of 24 Ig domains. We have recently discovered that in the C-terminal rod 2 region of FLN, Ig domains interact with each other forming functional domain pairs, where the interaction with signaling and transmembrane proteins is mechanically regulated by weak actomyosin contraction forces. Here, we investigated if there are similar inter-domain interactions around domain 4 in the N-terminal rod 1 region of FLN. Protein crystal structures revealed a new type of domain organization between domains 3, 4, and 5. In this module, domains 4 and 5 interact rather tightly, whereas domain 3 has a partially flexible interface with domain 4. NMR peptide titration experiments showed that within the three-domain module, domain 4 is capable for interaction with a peptide derived from platelet glycoprotein Ib. Crystal structures of FLN domains 4 and 5 in complex with the peptide revealed a typical β sheet augmentation interaction observed for many FLN ligands. Domain 5 was found to stabilize domain 4, and this could provide a mechanism for the regulation of domain 4 interactions.
Introduction
Filamins (FLNs)3 are homodimeric actin cross-linking proteins that are required for multicellular tissue differentiation. All three FLN genes (FLNA, FLNB, and FLNC) are essential in mouse and truncation or substitution mutations cause developmental defects in humans (1, 2) (Fig. 1). The diversity of phenotypes caused by FLN mutations can be explained by at least 90 proteins that interact with FLNs (3). The interaction partners can be classified to at least three different categories: transmembrane proteins, cytoskeletal proteins, and intracellular signaling proteins. Thus, FLNs are involved in stabilization and regulation of plasma membrane, regulation of actin cytoskeleton, and intracellular signaling (3–8).
FIGURE 1.
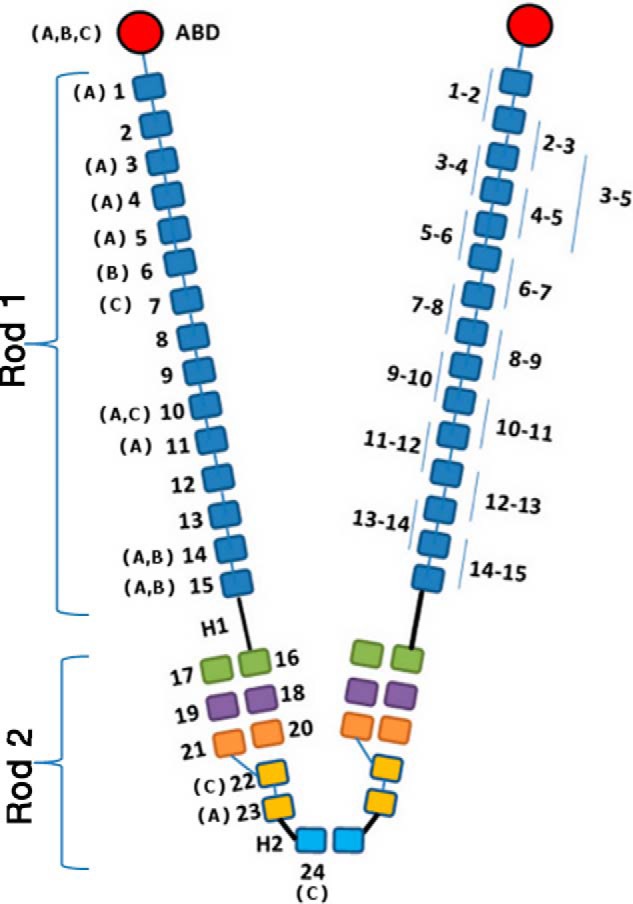
Schematic representation of filamin dimer structure. Actin-binding domains (ABDs) are red, followed by 24 Ig domains. Three known domain pairs in the rod 2 region (16–17, 18–19, and 20–21) are shown in green, purple, and orange, respectively. The fragments of the rod 1 region investigated in this study are labeled on the right subunit. Isoform-specific (FLNA, FLNB, and FLNC) substitution and local deletion mutations in human patients are shown on the left subunit.
Structurally, vertebrate FLNs consist of an N-terminal actin-binding domain and 24 immunoglobulin-like (Ig) domains (the dimer is shown in Fig. 1). Domains 1–15 are referred to as rod 1 and 16–24 as rod 2. The structure of the actin-binding domain is similar to that of α-actinin and is composed of two calponin homology domains (9, 10). The FLN Ig domains have a characteristic structure and they can be regarded as protein interaction modules. They often interact with other proteins by a β sheet augmentation mechanism. In this mechanism, the interaction partner forms an additional β-strand next to the strand C of the FLN domain and simultaneously interacts with the hydrophobic groove between the strands C and D, called the CD face (11–14). It is using this mode that ligands like transmembrane receptors of the integrin family (12, 15, 16), the platelet von Willebrand factor receptor subunit glycoprotein Ib (GPIb) (11), and the cystic fibrosis transmembrane conductance regulator (17, 18) interact with FLNa domains 17, 19, 21, and 23. Also, signaling protein FilGAP uses the same mechanism for interacting with FLNa domain 23 (4) and migfilin with FLNa domain 21 (13, 14). Biochemical studies have also suggested that FLNa domain 4 interacts in a similar way with many of the ligands (19). Interestingly, in the dimerization interface of FLNc and FLNa, domain 24 also utilizes the CD face, but the β sheet augmentation occurs through strand D (20, 21).
There are three structurally characterized, closely interacting domain pairs in the C-terminal rod 2 region of FLNa: domains 16–17, 18–19, and 20–21 (15, 22). Interestingly, in FLNa domain pairs 18–19 and 20–21, the function of the even numbered domain is to mask the CD face of the odd domain (15, 22) and this masking can be relieved by (a) low pico-Newton range mechanical forces that lead to a tighter interaction between FLN domain and ligand (23–25); or (b) displacement of the even numbered domain by the ligand itself, albeit with a lower affinity (26, 27). To find out if similar structural mechanisms could regulate the interactions of FLN domain 4, we solved the crystal structures of FLNa domains 3–5, FLNc domains 4–5, and FLNc domains 4–5 in complex with GPIb peptide. These structures disclosed a completely new type of interaction between three Ig domains.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Fragments of the human FLNC and FLNA cDNA (GenBankTM AJ012737 and AB593010.1) were PCR amplified according to the predicted domain boundaries (7) and cloned to the GST fusion protein vector pGTVL1 (Structural Genomics Consortium, University of Oxford) according to the ligation-independent cloning method (28). The final products were verified by DNA sequencing. The proteins were expressed in Escherichia coli BL21 Gold cells (Agilent Technologies) at 37 °C for 4 h with 0.4 mm isopropyl β-d-1-thiogalactopyranoside. The bacterial pellets were lysed using French press. The proteins were captured with glutathione-agarose column (Protino glutathione-agarose 4B, Macherey-Nagel), released with Tobacco Etch virus protease (Invitrogen), and further purified by size exclusion chromatography with a HiLoad 26/60 Superdex 75 column (GE Healthcare) in 100 mm NaCl, 1 mm dithiothreitol (DTT), 20 mm Tris, pH 7.5 (FLNc fragments), and 100 mm NaCl, 1 mm DTT, 20 mm Tris, pH 8.0 (FLNa fragments). Finally, the proteins were concentrated using Centriprep YM-10000 (Millipore). Mutants were generated using the QuikChange Multisite-directed mutagenesis kit (Agilent Technologies). 13C/15N-Labeled FLNc4–5, FLNc5, and FLNa3–5 were expressed in E. coli in standard d-glucose/M9 minimal medium. These proteins were purified in 50 mm sodium phosphate, 100 mm NaCl, 1 mm DTT using the same protocol as described above for the unlabeled fragments.
Protein Crystallography
Crystallization trials with hanging drop vapor diffusion method were set up for the purified recombinant proteins at room temperature. First crystals for FLNc4–5 were obtained in 1.4 m sodium potassium phosphate. The condition was optimized using a gradient of salt concentration and the final crystals were mounted from 1.6 m sodium potassium phosphate. These were first cryo-protected by adding 30% glycerol (final concentration) to the mother liquor and then frozen in liquid nitrogen. The crystals for FLNa3–5 were obtained in 0.1 m sodium malonate, pH 4, 12% (w/v) polyethylene glycol (PEG) 3,350, and frozen in liquid nitrogen using 0.1 m sodium malonate, pH 4, 35% (w/v) polyethylene glycol 3,350. Equimolar mixture (1 mm each) of FLNc4–5 with GPIb peptide (residues 573–596, RGSLPTFRSSLFLWVRPNGRVGPL, numbering according to Uniprot ID P07359) was used to obtain co-crystals in 0.1 m HEPES, pH 7.5, 20% (w/v) PEG 8,000 using a 1:2 ratio of protein to mother liquor. The crystals were cryo-protected with 6.6% ethylene glycol and 16.6% glycerol.
The diffraction data were collected at 100 K at the ESRF beamline ID23-1 (wavelength = 1.07227 Å) (FLNc4–5) and ID29 (wavelength = 0.976250 Å) (FLNa3–5 and FLNc4–5/GPIb). The data were processed with XDS (29) and the structures were solved using molecular replacement with the program Phaser (30) using FLNc23 (PDB code 2NQC) chain A as the search model for FLNc4–5, FLNc4–5 (PDB code 3V8O), and FLNc23 (2D7Q) as models for FLNa3–5 structure, and FLNc4–5 (PDB code 3V8O) for the FLNc4–5/GPIb structure. Refinement and model building were performed by programs Refmac5 (31, 32) and Coot (33) for FLNc4–5 and FLNc4–5/GPIb. The model for FLNa3–5 was built using ARP/wARP 7.3 (34) and Coot and refined using Refmac5. The structure of FLNc4–5 was refined using medium noncrystallographic symmetry restraints between chains A and B. Structure factors and coordinates were deposited in the PDB with codes 3V8O (FLNc4–5), 4M9P (FLNa3–5), and 4MGX (FLNc4–5/GPIb). For FLNc4–5, 89.2% of amino acids were in the most favored region, 9.8% in additionally allowed regions, and 1% in generously allowed region of the Ramachandran plot. The values for FLNa3–5 were 92.1, 7.5, and 0.4%, respectively, and for FLNc4–5/GPIb, 77.6, 19.0, and 3.4%, respectively. Crystal structure figures were generated using PyMOL (Schrödinger LLC, Portland, OR). The domain-domain interface was analyzed using the PISA server (35). Rendering was done in Chimera (36) according to the sequence alignment made with Clustal Omega (37) using FLNa/b/c3–5, FLNa3–5 of Mus musculus, Gallus gallus, Danio rerio, and Drosophila melanogaster.
Small Angle X-ray Scattering Measurements
Small angle x-ray scattering (SAXS) data were collected on the EMBL (European Molecular Biology Laboratory) X33 beamline at the DESY, Hamburg (38) (MAR345 image plate, sample-detector distance of 2.7 m and wavelength λ = 0.15 nm, covering the momentum transfer range 0.08 < s < 4.9 nm−1 (where s = 4π sin(θ)/λ, 2θ = scattering angle)); MAX IV Laboratory, beamline Cassiopeia I911-SAXS at the MAX II storage ring (Lund, Sweden) (39) (PILATUS 1M image plate, sample-detector distance of 2.2 m and wavelength 0.091 nm, covering the momentum transfer range 0.1 < s < 3 nm−1) and ESRF (European Synchrotron Radiation Facility), beamline BM29 (40) (PILATUS 1M image plate, sample-detector distance of 2.9 m and wavelength 0.10 nm, covering the momentum transfer range 0.01 < s < 5 nm−1). The protein concentrations were in the range of 1–10 mg/ml in purification buffer supplemented with 10 mm DTT. Buffer subtractions were conducted with either BioXTAS RAW software (41) or ATSAS package program PRIMUS (42) and the scattering intensity (I) was extrapolated to zero solute concentration. The forward scattering I(0) and the radius of gyration (Rg) were calculated using the program GUINIER (43), where at very small angles (s × Rg < 1.3), the scattering intensity is the following.
The distance distribution functions p(r) and the maximum particle dimensions Dmax were calculated for all fragments using the program GNOM (44). The molecular mass of the constructs were evaluated by comparing the forward scattering with that from reference solution of bovine serum albumin (BSA) with molecular mass 66 kDa using Equation 2.
Assuming the samples are monodisperse, Porod's law was applied to find out the excluded volume of the hydrated particle as,
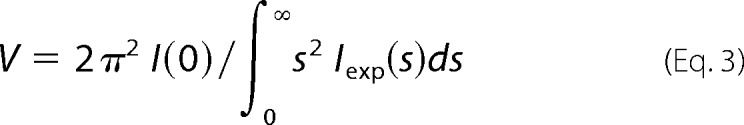 |
and to check the S−4 decay in scattering intensity at higher angles (45). Kratky plot (46) (I(s) × s2 versus s) was evaluated to check for the flexibility of the protein at higher scattering angles. The scattering patterns were further used to generate low resolution ab initio models of FLNc4–5 and FLNa3–5 by the programs DAMMIF (47) or GASBOR (48). Ten rounds of DAMMIF or GASBOR were done to generate models that were averaged using the program DAMAVER (49) to find the best model with common structural features. The scattering intensities of the crystal structures of FLNc4–5 (Protein Data Bank code 3V8O) and FLNa3–5 (PDB code 4M9P) were calculated using CRYSOL (50) and the superposition of the DAMAVER generated envelope with the respective crystal structures are done with the SUPCOMB program of the ATSAS package and the figures are made using PyMOL. To assess the flexibility of domain 3 in the FLNa and FLNc3–5 fragments, the ensemble optimization method (EOM) (51) was used. Similar analysis was performed for FLNc4–5 as an internal control. First, 10,000 randomized models (pool) were generated for each fragment using the native chain option in the RanCH program of the EOM package. The scattering profiles of each of these models were compared with the experimental scattering of the respective fragments (FLNa3–5, FLNc3–5, and FLNc4–5). A genetic algorithm was used to select a set of representative models (FLNa3–5 = 24; FLNc3–5 = 18, and FLNc4–5 = 12) from the pool such that the average scattering from the selected models fits the experimental scattering. The results were represented as Rg and Dmax distribution profiles using GraphPad Prism version 4 for Windows (GraphPad Software Inc., La Jolla, CA).
Protein Interaction Assay
Pull-down assays were performed using GST-tagged FLNc4 bound to glutathione-agarose resin. 1–20 μm FLNc5 and FLNc5R755D was allowed to interact for 1 h at 23 °C after which the resin was washed three times with 500 μl of PBS, 1 mm DTT and eluted with 10 μl of the SDS-PAGE sample buffer. Proteins were fractionated by SDS-PAGE and visualized with Coomassie stain. Intensities of the protein bands were quantified by ImageJ (52). Data were plotted using GraphPad Prism version 5 for Windows to produce a non-linear regression curve for one site total and nonspecific binding (specific = Bmax × X/(X + Kd); nonspecific = NS × X + background, where X = ligand concentration).
ThermoFluor Assay
Thermal stability of FLNc4, FLNc5, and FLNc4–5 was determined using Bio-Rad C1000 thermal cycler, CFx96 Real-Time system. Unfolding of the proteins was monitored using the fluorescent dye SYPRO Orange (Invitrogen), which binds to the hydrophobic core of the protein as it unfolds. A temperature gradient was set up from 20 to 95 °C with 0.5 °C/30-s increments. Each sample contained 100 μm protein, except FLNc4 (150 μm), and 5× dye in 25 μl total volume.
NMR Experiments
NMR samples were prepared in 50 mm NaH2PO4, 100 mm NaCl, 1 mm DTT buffer at pH 7.0. D2O was added to obtain ∼8% solutions. Protein concentrations were 0.4–1.2 mm. Measurement temperatures were 28 °C for FLNc5 and FLNc4–5, and 35 °C for FLNa3–5. For the chemical shift assignment HNCACB and CBCA(CO)NH spectra were recorded with a Varian INOVA 500 MHz spectrometer equipped with a 15N,13C,1H triple-resonance z-PFG probehead for FLNc5 and a Varian INOVA 800 MHz spectrometer equipped with a cryogenically cooled, 15N,13C,1H triple-resonance z-PFG probehead for FLNc4–5 and FLNa3–5. For FLNc4–5, the backbone amide assignment percentage was 95, whereas for FLNc5 all backbone amides were assigned. Peptide binding assays were performed by stepwise addition of the model peptide resulting in approximate concentration ratios of 1 (protein):0 (peptide), 1:0.5, 1:1, 1:2, 1:3, and 1:5. At each step a 1H,15N-heteronuclear single quantum coherence (HSQC) spectrum for the detection of backbone amide group chemical shift changes, and a 1H,13C-HSQC spectrum for the detection of methyl group chemical shift changes in the case of FLNa3–5 was recorded. Chemical shift changes were calculated as Δδ = ((ΔδH)2 + (0.15 × ΔδN)2)1/2. All spectra were processed with Vnmr 6.1C (Varian Inc.) and analyzed with Sparky 3.110 (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
Molecular Dynamics Calculations
The x-ray structure of FLNa3–5 (PDB code 4M9P) was used as the starting structure for MD simulations. Before energy minimizations and MD simulations, TIP3P water molecules extending 13 Å in all dimensions around the solute and counter ions to obtain neutrality were added with LEAP (53). All energy minimizations and MD simulations were performed with NAMD 2.9 (54) using ff03 force field parameters (55). Before MD simulations, the system was relaxed and equilibrated. First the water molecules and counter ions were minimized using the steepest descent algorithm (3000 steps) keeping the rest of the system fixed. This was followed by similar minimization but only Cα-atoms were restrained to their crystallographic positions with a harmonic force of 5 kcal mol−2 Å−1, whereas the rest of the system moved freely. Before starting the production of MD simulations, the equilibrium MD simulations for the whole system with Cα-atoms restrained were performed first at constant volume (for 300 ps) and then at constant pressure (for further 300 ps). Finally the whole system was equilibrated by simulating it at constant pressure without any restrains for 450 ps. After the system was fully equilibrated, the 50-ns production simulation was performed without constraints. Five parallel simulations were performed. During the simulations, the system was kept at constant temperature (300 K) using Langevin damping coefficient of 5 ps−1 and constant pressure (1 atm) using a Nosé-Hoover Langevin piston (56) with an oscillation timescale of 200 fs and damping time scale of 100 fs. 2-fs integration time steps were used under a multiple time stepping scheme (57), where bonded and short-range interactions were computed every time step and long-range electrostatic interactions every third time step. A cutoff of 12 Å for van der Waals and short-range electrostatic interactions was used. To ensure a smooth cutoff for van der Waals interactions, a switching function was started at 10 Å. Long-range electrostatic interactions were calculated using the particle-mesh Ewald method (58). Periodic boundary conditions were used in all simulations. The SHAKE algorithm was applied to constrain the bonds containing hydrogen atoms (59). Snapshots from trajectories obtained from MD simulation were extracted with PTRAJ in ANTECHAMBER (53).
RESULTS
Crystal Structures of FLNa Domains 3–5 and FLNc Domains 4–5 Show a Novel Domain-Domain Organization
The crystal structures of FLNa domains 3–5 (Fig. 2A and Table 1) and FLNc domains 4–5 (Fig. 2B and Table 1) disclosed a completely new type of interaction between three Ig domains (Fig. 2A) where the interfaces are highly conserved (Fig. 3 and supplemental Fig. S1). The domains interact side by side with each other along their β sheets. This leads to a sandwich structure where the β sheets of each domain stack roughly parallel on top of each other (Fig. 2A, left side), with domain 3 slightly shifted (Fig. 2A, right side). In this arrangement, the interaction between domains 3 and 4 is mediated via the edges of the β sheets, whereas domains 4 and 5 interact along three β-strands from each side. The buried interface area is about 500 and 830 Å2 for domains 3–4 and 4–5, respectively, suggesting that the interaction between 4 and 5 is tighter than the other. There is a large hydrophobic interaction surface between domains 4 and 5 around a Trp residue in the A strand of domain 4 (Trp-582 in FLNa and Trp-577 in FLNc) (Fig. 2C), whereas the interaction between domains 3 and 4 has mostly polar characteristics. The main chain structure of FLNc domains 4–5 is largely identical to FLNa domains (root mean square deviation of 0.66 Å between 165 C-α atoms) (Fig. 2B).
FIGURE 2.
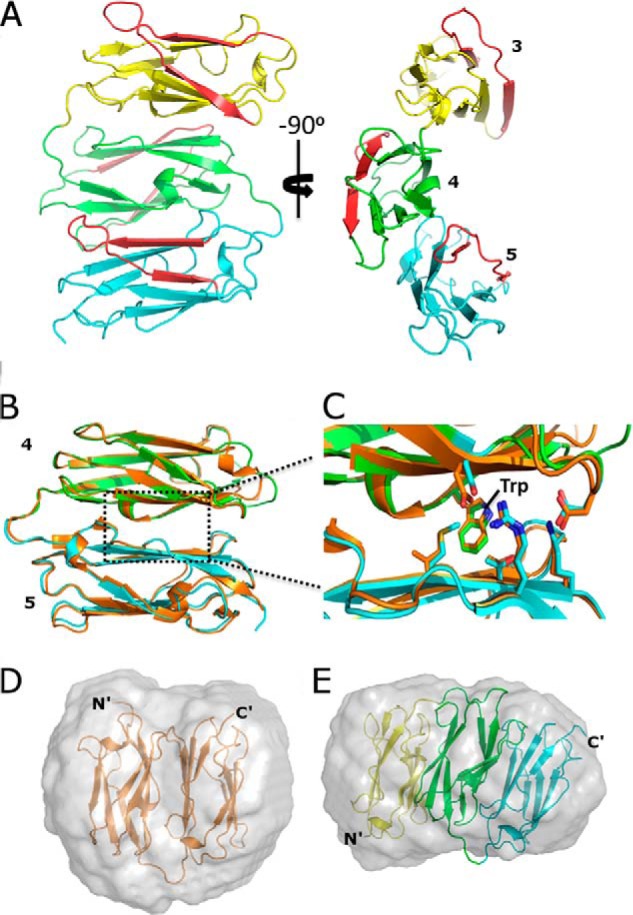
New domain packing in FLNs. A, two views of the crystal structure of FLNa3–5, where domains 3, 4, and 5 are shown in yellow, green, and cyan, respectively. β-Strands C and D are shown in red. B, superposition of FLNc4–5 (orange) with FLNa4–5 (domain coloring as in panel A). C, zoom-in view of the interface showing key residues. D and E, superposition of the FLNc4–5 and FLNa3–5 structures with their respective ab initio SAXS envelopes. Normalized spatial discrepancy of superposition for FLNc4–5 is 2.41 and FLNa3–5 is 1.63.
TABLE 1.
Crystallography data collection and refinement statistics
| FLNc4–5 (PDB code 3V8O) | FLNa3–5 (PDB code 4M9P) | FLNc4–5/GPIb (PDB code 4MGX) | |
|---|---|---|---|
| Data collection | |||
| Space group | P 212121 | P65 | P4322 |
| Cell dimensions | |||
| a, b, c, Å | 63.62, 91.35, 103.72 | 63.04, 63.04, 136.60 | 111.80, 111.80, 59.88 |
| α, β, γ, degree | 90, 90, 90 | 90, 90, 120 | 90, 90, 90 |
| Resolution range, Å | 46.63–2.8 (2.87–2.8)a | 42.65–1.72 (1.76–1.72) | 47.73–3.16 (3.24–3.16) |
| Rsymb (%) | 15.8 (76.3) | 9.7 (67.9) | 20.5 (242.7) |
| I/σI | 9.92 (2.7) | 18.24 (3.3) | 9.53 (0.7) |
| Completeness, % | 100 (98) | 99.7 (96.5) | 98.2 (100) |
| Redundancy | 4.2 (4.2) | 10.9 (8.0) | 10.42 (7.8) |
| Refinement | |||
| Resolution (Å) | 46.63–2.8 (2.87–2.8) | 42.65–1.72 (1.76–1.72) | 47.73–3.16 (3.24–3.16) |
| No. of reflections | |||
| Refinement | 13602 (1012) | 30839 (1623) | 6556 (456) |
| Test set | 1512 (113) | 2204 (115) | 345 (24) |
| Rwork/Rfree (%) | 20/25.5 (33.4/40.1) | 19.9/24.3 (24.5/30.6) | 22.6/27.7 (35.9/45.6) |
| No. of atoms | |||
| Protein | 2854 | 2173 | 1439 |
| Heterogen | 1 | Null | Null |
| Solvent | 47 | 219 | 2 |
| Root mean square differences | |||
| Bond lengths (Å) | 0.022 | 0.019 | 0.012 |
| Bond angles, degree | 1.8 | 1.98 | 1.80 |
| Average B-factor, Å2 | 29.3 | 22.18 | 108.48 |
| Protein | 29.28 | 21.95 | 108.48 |
| Solvent | 23.41 | 27.04 | 58.79 |
a Values of the last resolution shell in parentheses.
b Diederichs and Karplus (1997).
FIGURE 3.

Surface conservation of the domain-domain interfaces in FLN3–5. Corresponding schematic models made using PyMOL are shown below the surface visualization. Sequence alignment is shown in supplemental Fig. S1.
The domain-domain interactions observed in these crystal structures are, to our knowledge, unique among the entire Ig superfamily. Therefore, we used several complementary techniques to confirm that the domain arrangement and interactions exist also in solution and are not induced by crystal packing. First, we compared the crystal structures with the SAXS scattering data obtained in solution. The χ values for comparison of calculated scattering from the structures and observed solution scattering profile were 1.13 and 1.80 for FLNa domains 3–5 and FLNc domains 4–5, respectively. The structures fitted well in the SAXS-generated ab initio envelopes (Fig. 2, D and E). Mutations of the contact residues seen in the crystal structure made the two-domain fragment FLNc4–5 more extended in SAXS (Table 2) and the same mutations also diminished the interaction between individual FLNc domains 4 and 5 in pulldown assays (Fig. 4, A and B). Comparison of NMR chemical shifts between FLNc domain 5 and the ins 4–5 pair also verified the interaction surface (Fig. 4, C and D, and supplemental Fig. S2).
TABLE 2.
Parameters derived from SAXS measurements of FLN domains 4–5, mutants of FLNc4–5 and FLN 3–5
| Protein | Rga | ∼Dmaxb |
|---|---|---|
| Nm | ||
| FLN domains 4–5 constructs | ||
| FLNc4–5 | 1.71 ± 0.01 | 4.8 |
| FLNa4–5 | 1.90 ± 0.01 | 5.3 |
| Mutation in FLNc4–5 | ||
| FLNc4 W577A | 1.88 ± 0.01 | 6.5 |
| FLNc4 D627R | 1.85 ± 0.01 | 7.2 |
| FLNc4 R755D | 1.86 ± 0.01 | 6.7 |
| FLNc4 W577A/D627R | 1.85 ± 0.01 | 7.0 |
| FLNc4 W577A/FLNc5R755D | 2.04 ± 0.01 | 7.5 |
| FLNc4D627R/FLNc5 R755D | 1.85 ± 0.01 | 6.2 |
| FLN domains 3–5 constructs | ||
| FLNa 3–5 | 2.32c | 7.7c |
| FLNc 3–5 | 2.41c | 7.9c |
a Rg, radius of gyration (mean square of the distances from the center of mass of the particle weighted by electron densities) from Equation 1.
b ∼Dmax, maximum dimension inside the particle estimated from distance distribution probability function. The values written here are with an approximate error of 0.5 nm.
c Average values reported using EOM.
FIGURE 4.

Confirmation of the FLNc4–5 interface. A, quantification of the binding observed from the pulldown assay of FLNc4 and FLNc5. B, plot obtained from the pulldown assay of FLNc4 and FLNc5R755D showing a decline in the interaction. Bars show S.E. between three independent experiments. C, structural representation of the NMR measurements showing residues in blue that have chemical shift differences of more than 0.25 ppm in the 1H,15N-HSQC spectra of FLNc5 and FLNc4–5 (spectra shown in supplemental Fig. S2). D, plot of the chemical shift differences between FLNc5 and FLNc4–5.
In line with the relatively small interaction area between FLNa domains 3 and 4, closer analysis of SAXS data using EOM suggested that in both, FLNa and FLNc, three domain fragments of domains 3–5 can be flexible in solution. This is shown by the broader Dmax range for three domain fragments as compared with FLNc4–5 (Fig. 5, A and B). To further test flexibility of the FLNa3–5 structure, we performed MD simulations on FLNa3–5 without any restraints. Analysis of the snapshot structures obtained from 50-ns MD simulations showed that domain 3 changes its orientations with respect to domains 4 and 5 whose interactions stay intact (Fig. 5C). This flexibility of the three-domain fragment observed in the MD simulations is in accordance with the observed solution scattering profile of FLNa3–5 (χ values for snapshots of two extreme orientations in the range of 1.15–1.23) and the snapshots fit well in the SAXS-generated ab initio envelopes (Fig. 5D).
FIGURE 5.
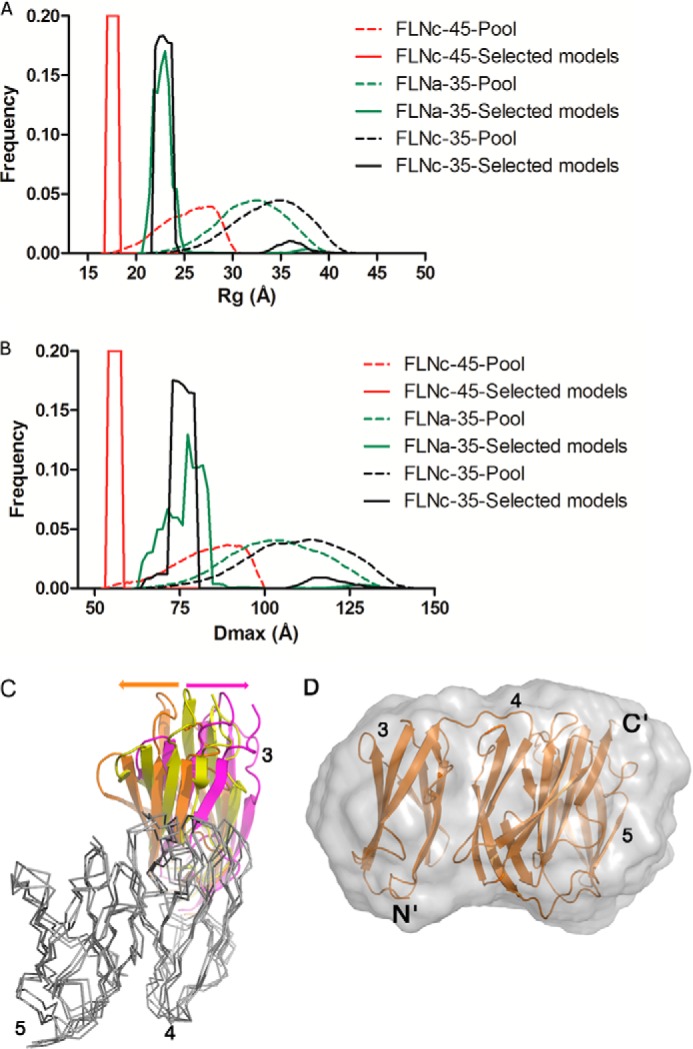
EOM and MD data showing the flexibility of the three-domain fragment. A, Rg and B, Dmax of the pools (dashed lines) and the selected models (solid lines) for FLNa3–5 (green), FLNc3–5 (black), and FLNc4–5 (red) as internal control. Note that the selected models of FLNc4–5 have a narrow Rg and Dmax distribution, whereas the selected models of FLNc3–5 and FLNa3–5 show a broader distribution. This suggests that there is more flexibility between domains 3–4 than between domains 4–5. C, the superposition of FLNa3–5 crystal structure (domain 3, in yellow) with two snapshots from MD simulations (one snapshot with domain 3 in orange and the second snapshot with domain 3 in magenta). Domains 4 and 5 are shown in gray for the crystal structure and MD snapshots and are superimposed using VERTAA module in Bodil (71). The snapshot structures shown represent the most extreme orientations between which domain 3 oscillates during MD simulations. D, superposition of one snapshot structure (orange) from MD simulations of FLNa3–5 with the SAXS generated ab initio envelope (normalized spatial discrepancy of superposition is 2.05).
Taken together, the crystal structures of FLNa domains 3–5 and FLNc domains 4–5 reveal a new type of Ig-domain packing that fits well with analyses performed in solution stage. The domains 4 and 5 interact relatively tightly, whereas the interface between domains 3 and 4 may be partly flexible.
Ligand Peptide Binds to Domain 4 in the Three Domain Module
Earlier, FLNa domain 4, but not 3 and 5, has been shown to interact with β-strand forming peptides (19). In FLNa3–5 and FLNc4–5 structures, the CD surfaces of all three domains are exposed. However, in domains 3 and 5, the CD loop partially occludes the groove between the β-strands (Fig. 6, A and C), which is not seen in domain 4. This suggests that only domain 4 may interact via its CD face. However, in FLNc domain 4 but not FLNa, the β-strand C is preceded by a 310 helix (Fig. 6B) that might interfere with β-strand augmentation interactions. To test if FLNc domain 4 can still bind a ligand, we co-crystallized the FLNc4–5 fragment with the model peptide derived from GPIb (residues 573–596, RGSLPTFRSSLFLWVRPNGRVGPL), which commonly has the highest interaction affinity among test peptides (19). The structure (Fig. 6D and Table 1) revealed that the peptide (residues 577–585) interacts with the typical β sheet augmentation mechanism originally discovered for FLNa domain 17 (11) (Fig. 6, E and F). Interestingly, the electron density for the 310 helix and the BC loop is not visible in this structure, suggesting local flexibility. We confirmed these interactions in solution by recording 15N- and 13C-HSQC spectra of FLNa3–5 and 15N-HSQC spectra of FLNc4–5 in the presence and absence of the GPIb peptide. Chemical shift perturbations mapped at or close to the CD face of domain 4, indicating that it is available for interaction (Fig. 6, G and H, and supplemental Fig. S3). This shows that the novel domain-domain interactions between domains 3–5 do not prevent the binding of small β-strand forming peptides to the CD face of domain 4.
FIGURE 6.

CD face conformation and interactions. A–C, conformation of the CD face of individual domains. In each case, the CD face is shown in red. A, FLNa3 is shown in yellow. B, FLNa4 is green and FLNc4 is gray. Note the 310 helix in FLNc4. C, FLNa5 is shown in cyan color and FLNc5 is gray; closed conformation of the CD loop is marked with an arrow. D, co-crystal structure of FLNc4–5 with the GPIb peptide (blue). E, superposition of FLNc4 and GPIb peptide with FLNa17 (gray) and GPIb peptide (lemon) (root mean square deviation = 0.75 Å between 89 C-α atoms). F, detailed view of the peptide conformation with FLNc4 (peptide in blue) and FLNa17 (peptide in lemon). N and C termini of the CD face of FLNc4 and FLNa17 are labeled. G, NMR titration of GPIb with FLNa3–5. Residues undergoing changes in peak intensity and chemical shifts are shown as blue (amide group changes) and pink side chains (methyl group changes). H, residues mapped for the GPIb interaction with FLNc4–5: peaks vanish (blue), chemical shift difference more than 0.025 ppm (red). 1H,15N-HSQC spectra for titration of GPIb with FLNa3–5 and FLNc4–5 are shown in supplemental Fig. S3.
Domains 5 Stabilizes the Structure of Domain 4
Isolated domain 4 has been shown to be partially unstable in previous studies (19). To investigate if neighboring domains have an influence on domain 4 stability, we probed the thermal stability of the domains using a hydrophobic fluoroprobe (Fig. 7) (60). Comparison of the temperature-dependent dye binding curves shows that isolated domain 4 starts binding the dye in the range of 35–45 °C, whereas domain 5 binds only in the 70–80 °C range. This indicates that the structure of domain 4 is less stable than domain 5. Furthermore, the dye binding curve of FLNc4–5 shows two transition temperatures. The lower temperature at around 55 °C is considerably higher than that observed with domain 4 alone, whereas the higher transition temperature is close to that of isolated domain 5. This suggests that domain 5 stabilizes domain 4 in the two domain fragment. A similar curve is seen for FLNc3–5 indicating that domain 3 does not have an additional effect on the stability of domain 4.
FIGURE 7.
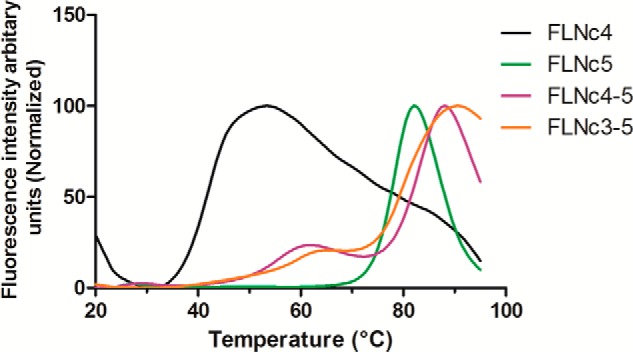
Thermal stability assay showing domain 5 stabilizes domain 4. Temperature-dependent fluoroprobe binding curves of FLNc4 (black), FLNc5 (green), FLNc4–5 (magenta), and FLNc3–5 (orange) are shown.
DISCUSSION
Traditionally, long multidomain proteins are thought to work as beads on a string that allow great flexibility, but where individual domains perform their functions largely independent of each other. This seems not to be the case in FLNs. Although FLNs are flexible molecules composed of 24 similar Ig domains, we have earlier reported three functional domain-domain interactions in the C-terminal rod 2 region of FLNa. In this paper, we tested if there are similar inter-domain interactions in the rod 1 region around domain 4. Our major findings are: 1) domains 3 and 5 interact with domain 4 and this module of FLN domains 3–5 shows a new type of domain-domain arrangement; 2) we show the details of ligand binding to the CD face of FLN domain 4; and 3) provide evidence of domain 4 stabilization by interactions with neighboring domains. We believe that each of these findings has wide implications to our understanding of the FLN function. The findings and their implications are discussed below.
Based on sequence similarity with the pair-forming domains in rod 2 (19, 61), we hypothesized that domain 4 might be involved in domain-domain interactions. To study this, we crystallized FLNa domains 3–5 and FLNc domains 4–5. The structures revealed a novel type of domain packing that has not been observed in Ig domains before. Typically, FLN-type Ig domains interact with each other via the edges of the β sheets and via loop regions (10, 15, 22). On the other hand, many other Ig domains form dimers by interacting perpendicularly face-to-face along their β sheets (62–64). In the current structures, the domains stack on each other and interact face-to-back so that their β sheets are approximately parallel. Because β sheets are rather flat in the FLN-type Ig domains, this kind of arrangement allows maximal interface surface area. This seems to be true particularly between domains 4 and 5, where the interface is governed by several hydrophobic interactions. These features suggest that the interaction between domains 4 and 5 is rather stable. The interface between domains 3 and 4 is slightly shifted, has a smaller area than between 4 and 5, and is mainly polar. These observations suggest that the 3–4 interface may be more flexible than that of 4–5. This was also supported by analysis of SAXS results using EOM and MD simulations. Residues on both interfaces are conserved throughout the animal kingdom and thus it is likely that the arrangement of domains 3–5 observed here is a general feature in FLNs.
To study how domain 4 interacts with the GPIb peptide derived from GPIb, which has been previously shown to interact with FLNa domain 4 and other similar domains (19), we co-crystallized the peptide with FLNc domains 4–5 and used NMR methods to verify the interaction site in solution. The crystal structure showed that interaction of the peptide with FLNc4 is almost identical as with FLNa17 (11): the peptide forms an extra β-strand next to the strand C of domain 4 and interacts hydrophobically with the groove between strands C and D. Using NMR chemical shift perturbation mapping, we were able to show that also in solution the peptide interacts with FLNc4–5 and FLNa3–5 via the CD face of domain 4. This fits well with the observation that in both domains 3 and 5 (FLNa3, FLNa5, and FLNc5), the CD face seems not to be accessible for similar β sheet augmentation interaction because of the CD loop conformation. It is noteworthy that in the unligated FLNc domain 4 there is a 310 helical turn just before strand C that partially occludes the CD groove, but the peptide can induce a conformation change and displace this turn. In the absence of the peptide, NMR spectra revealed two sets of cross-peaks that could be assigned to the residues belonging to the 310 helix, suggesting that this structure has conformational flexibility in a relatively slow (kex < 103 s−1) time scale.4 This 310 helix is not seen in the structure of FLNa domain 4 and the corresponding residues show only a single set of peaks in 15N-HSQC spectrum. Taken together, these results demonstrate that the CD face of domain 4 is the preferred binding site for β-strand forming peptides within the module of FLN domains 3–5 and that this site is accessible for interactions in this module. Functionally, our findings are in consensus with the hypothesis that there are multiple domains in FLNs to which the same ligand binds (18, 19, 61). In the case of FLNa, this finding is relevant as the interaction between FLNa and GPIb is essential for platelet activation (11, 65). Although mutations in FLNa17 disrupt GPIb interaction in a cell culture model, the highest affinity of GPIb is for FLNa21, and evidence has shown that GPIb binds to multiple sites on FLN (66). It is possible that FLNa17 is the initial binding site for GPIb. This interaction is then complemented by other FLNa domains, including FLNa4. Under force, FLNa21 may then be unmasked and become the strongest binder to facilitate the tight interaction required for platelet adhesion in high shear (67). It is also of interest that the spleen tyrosine kinase, Syk, has been reported to interact with FLNa domain 5 (68). In our current structures the CD faces of FLNa and FLNc domain 5 are occluded by the closed conformation of the CD loop. Thus, the current structures suggest that this interaction of Syk with FLNa domain 5 may use another kind of structural mechanism than β sheet augmentation next to strand C.
In the rod 2 region of FLN the function of domain-domain interactions seems to mask the binding sites on domains 19 and 21. This masking can be relieved by mechanical force and thus these domain-domain interactions provide the basis for the mechanosensor function of FLN (5, 23, 25, 69). In the module of domain 3–5, reported here, we do not see this kind of masking of the known interaction site in domain 4. Thus, CD face masking cannot explain the function of this conserved module. An alternative possibility might be similar to the interaction of FILGAP with domain 23 where mechanical forces on FLN may destabilize interactions (24). It has been observed before that FLNa domain 4 is partially unstructured when studied by NMR (19). Here we show by a hydrophobic fluoroprobe binding assay that domain 5 is required for stabilization of domain 4. Structurally, this can be explained by the hydrophobic surface in domain 4 (particularly the conserved Trp residue at the domain-domain interface), which may cause problems in domain folding when not allowed to interact with domain 5.
In context of diseases, substitution mutations causing familial cardiac valvular dystrophy have been reported in FLNa domain 4 (P637E) and domain 5 (V711D) (70). As this is the first study showing the high resolution structures of these domains, it was of interest to map the location of these residues on our crystal structure of FLNa3–5. We noticed that even though these amino acids were not at the domain-domain interface, the side chains of these residues faced the hydrophobic core made by the β sheets (Fig. 8). Hence, we speculate that mutating the proline or valine to glutamine or aspartate, respectively, will lead to a substantial change in the hydrophobic core and most likely destabilize the individual β sheet domain-fold.
FIGURE 8.
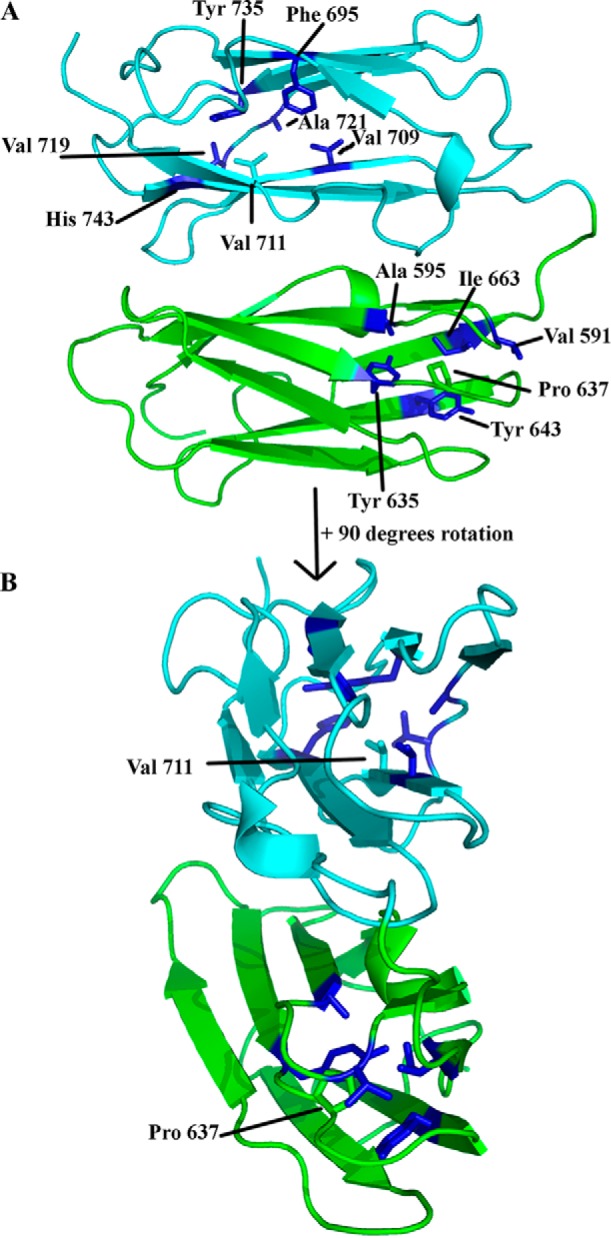
Residues that are mutated in FLNa domains 4 (P637E) and 5 (V711D) causing familial cardiac valvular dystrophy. A, FLNa4 is shown in green and FLNa5 in cyan. Pro-637 in domain 4 is shown in green. The neighboring residues forming the hydrophobic core are shown in blue. Val-711 in domain 5 is shown in cyan. Neighboring residues forming the hydrophobic core are shown in blue. B, view rotated by 90 degrees toward the reader.
In conclusion, our results highlight that multiple mechanisms have evolved for the inter-domain interactions in FLNs. We report here the presence of a novel three-domain module between domains 3–5 that is conserved in FLNs and partially flexible. We suggest that one function of the three-domain module is to stabilize domain 4 and allow its interactions via the previously characterized β-strand augmentation mechanism.
Supplementary Material
Acknowledgments
We thank Arja Mansikkaviita for excellent technical assistance. Ulla-Maria Jokipii and Susanna Sinisaari-Kaislo helped in the expression and purification of the proteins. We acknowledge European Molecular Biology Lab (EMBL), European Synchrotron Radiation Facility (ESRF), and MAX-LAB for providing synchrotron access for SAXS and crystal data collection. We express gratitude to Michal J. Gajda (EMBL Hamburg), Matthew Bowler, Adam Round, Petra Pernot (ESRF, Grenoble), Sylvio Haas and Tomás Plivelic (MAX-LAB, Lund) for assistance during data collection at the beamlines. Biocentrum Helsinki and Biocenter Finland are acknowledged for the NMR infrastructure support.
This work was supported by grants from the Marie Curie Actions FP 7 grant to Initial Training Network MUZIC (Muscle Z-disk Protein Complexes: from atomic structure to physiological function) (to R. S. and J. Y.), the National Doctoral Programme in Informational and Structural Biology (to M. Y.), and Sigrid Juselius Foundation and Academy of Finland Grants 114713 and 138327 (to J. Y.) and 263794 and 259447 (to P. P.), and CSC-Finnish IT Center for Science computational Grants jyy2516 and jyy2586 (to O. T. P.).

This article contains supplemental Figs. S1–S3.
The atomic coordinates and structure factors (codes 3V8O, 4M9P, and 4MGX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shifts for NMR assignments have been deposited to the BioMagResBank database under accession numbers 19496 (FLNa3–5) and 19495 (FLNc4–5).
Tossavainen, H., Seppälä, J., Sethi, R., Pihlajamaa, T., and Permi, P. (2014) NH, HN, Cα, Cβ, and methyl group assignments of filamin multidomain fragments IgFLNc4–5 and IgFLNa3–5. Biomol. NMR Assign. 10.1007/s12104-014-9542-6.
- FLN
- filamin
- GPIb
- glycoprotein Ib
- EOM
- ensemble optimization method
- MD
- molecular dynamics
- SAXS
- small angle x-ray scattering
- HSQC
- heteronuclear single quantum coherence
- PDB
- Protein Data Bank.
REFERENCES
- 1. Fürst D. O., Goldfarb L. G., Kley R. A., Vorgerd M., Olivé M., van der Ven P. F. (2013) Filamin C-related myopathies. Pathology and mechanisms. Acta Neuropathol. 125, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kley R. A., Serdaroglu-Oflazer P., Leber Y., Odgerel Z., van der Ven P. F., Olivé M., Ferrer I., Onipe A., Mihaylov M., Bilbao J. M., Lee H. S., Höhfeld J., Djinović-Carugo K., Kong K., Tegenthoff M., Peters S. A., Stenzel W., Vorgerd M., Goldfarb L. G., Fürst D. O. (2012) Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain 135, 2642–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakamura F., Stossel T. P., Hartwig J. H. (2011) The filamins. Organizers of cell structure and function. Cell Adh. Migr. 5, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakamura F., Heikkinen O., Pentikäinen O. T., Osborn T. M., Kasza K. E., Weitz D. A., Kupiainen O., Permi P., Kilpeläinen I., Ylänne J., Hartwig J. H., Stossel T. P. (2009) Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PloS One 4, e4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Razinia Z., Mäkelä T., Ylänne J., Calderwood D. A. (2012) Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 41, 227–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou A.-X., Hartwig J. H., Akyürek L. M. (2010) Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 20, 113–123 [DOI] [PubMed] [Google Scholar]
- 7. van der Flier A., Sonnenberg A. (2001) Structural and functional aspects of filamins. Biochim. Biophys. Acta 1538, 99–117 [DOI] [PubMed] [Google Scholar]
- 8. Stossel T. P., Condeelis J., Cooley L., Hartwig J. H., Noegel A., Schleicher M., Shapiro S. S. (2001) Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2, 138–145 [DOI] [PubMed] [Google Scholar]
- 9. Gorlin J. B., Yamin R., Egan S., Stewart M., Stossel T. P., Kwiatkowski D. J., Hartwig J. H. (1990) Human endothelial actin-binding protein (ABP-280, nonmuscle filamin). A molecular leaf spring. J. Cell Biol. 111, 1089–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruskamo S., Ylänne J. (2009) Structure of the human filamin A actin-binding domain. Acta Crystallogr. D Biol. Crystallogr. 65, 1217–1221 [DOI] [PubMed] [Google Scholar]
- 11. Nakamura F., Pudas R., Heikkinen O., Permi P., Kilpeläinen I., Munday A. D., Hartwig J. H., Stossel T. P., Ylänne J. (2006) The structure of the GPIb-filamin A complex. Blood 107, 1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kiema T., Lad Y., Jiang P., Oxley C. L., Baldassarre M., Wegener K. L., Campbell I. D., Ylänne J., Calderwood D. A. (2006) The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell 21, 337–347 [DOI] [PubMed] [Google Scholar]
- 13. Lad Y., Jiang P., Ruskamo S., Harburger D. S., Ylänne J., Campbell I. D., Calderwood D. A. (2008) Structural basis of the migfilin-filamin interaction and competition with integrin tails. J. Biol. Chem. 283, 35154–35163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ithychanda S. S., Das M., Ma Y.-Q., Ding K., Wang X., Gupta S., Wu C., Plow E. F., Qin J. (2009) Migfilin, a molecular switch in regulation of integrin activation. J. Biol. Chem. 284, 4713–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heikkinen O. K., Ruskamo S., Konarev P. V., Svergun D. I., Iivanainen T., Heikkinen S. M., Permi P., Koskela H., Kilpeläinen I., Ylänne J. (2009) Atomic structures of two novel immunoglobulin-like domain pairs in the actin cross-linking protein filamin. J. Biol. Chem. 284, 25450–25458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takala H., Nurminen E., Nurmi S. M., Aatonen M., Strandin T., Takatalo M., Kiema T., Gahmberg C. G., Ylänne J., Fagerholm S. C. (2008) β2 Integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood 112, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 17. Smith L., Page R. C., Xu Z., Kohli E., Litman P., Nix J. C., Ithychanda S. S., Liu J., Qin J., Misra S., Liedtke C. M. (2010) Biochemical basis of the interaction between cystic fibrosis transmembrane conductance regulator and immunoglobulin-like repeats of filamin. J. Biol. Chem. 285, 17166–17176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Playford M. P., Nurminen E., Pentikäinen O. T., Milgram S. L., Hartwig J. H., Stossel T. P., Nakamura F. (2010) Cystic fibrosis transmembrane conductance regulator interacts with multiple immunoglobulin domains of filamin A. J. Biol. Chem. 285, 17156–17165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ithychanda S. S., Hsu D., Li H., Yan L., Liu D. D., Liu D., Das M., Plow E. F., Qin J. (2009) Identification and characterization of multiple similar ligand-binding repeats in filamin. Implication on filamin-mediated receptor clustering and cross-talk. J. Biol. Chem. 284, 35113–35121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pudas R., Kiema T.-R., Butler P. J., Stewart M., Ylänne J. (2005) Structural basis for vertebrate filamin dimerization. Structure 13, 111–119 [DOI] [PubMed] [Google Scholar]
- 21. Seo M.-D., Seok S.-H., Im H., Kwon A.-R., Lee S. J., Kim H.-R., Cho Y., Park D., Lee B.-J. (2009) Crystal structure of the dimerization domain of human filamin A. Proteins 75, 258–263 [DOI] [PubMed] [Google Scholar]
- 22. Lad Y., Kiema T., Jiang P., Pentikäinen O. T., Coles C. H., Campbell I. D., Calderwood D. A., Ylänne J. (2007) Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 26, 3993–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pentikäinen U., Ylänne J. (2009) The regulation mechanism for the auto-inhibition of binding of human filamin A to integrin. J. Mol. Biol. 393, 644–657 [DOI] [PubMed] [Google Scholar]
- 24. Ehrlicher A. J., Nakamura F., Hartwig J. H., Weitz D. A., Stossel T. P. (2011) Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature 478, 260–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rognoni L., Stigler J., Pelz B., Ylänne J., Rief M. (2012) Dynamic force sensing of filamin revealed in single-molecule experiments. Proc. Natl. Acad. Sci. U.S.A. 109, 19679–19684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ithychanda S. S., Qin J. (2011) Evidence for multisite ligand binding and stretching of filamin by integrin and migfilin. Biochemistry 50, 4229–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruskamo S., Gilbert R., Hofmann G., Jiang P., Campbell I. D., Ylänne J., Pentikäinen U. (2012) The C-terminal rod 2 fragment of filamin A forms a compact structure that can be extended. Biochem. J. 446, 261–269 [DOI] [PubMed] [Google Scholar]
- 28. Gileadi O., Burgess-Brown N. A., Colebrook S. M., Berridge G., Savitsky P., Smee C. E., Loppnau P., Johansson C., Salah E., Pantic N. H. (2008) High throughput production of recombinant human proteins for crystallography. Methods Mol. Biol. 426, 221–246 [DOI] [PubMed] [Google Scholar]
- 29. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 36. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 37. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40, s190–s194 [Google Scholar]
- 39. Labrador A., Cerenius Y., Svensson C., Theodor K., Plivelic T. (2013) The yellow mini-hutch for SAXS experiments at MAX IV Laboratory. J. Phys. Conf. Ser. 425, 072019 [Google Scholar]
- 40. Pernot P., Round A., Barrett R., De Maria Antolinos A., Gobbo A., Gordon E., Huet J., Kieffer J., Lentini M., Mattenet M., Morawe C., Mueller-Dieckmann C., Ohlsson S., Schmid W., Surr J., Theveneau P., Zerrad L., McSweeney S. (2013) Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nielsen S. S., Noergaard Toft K., Snakenborg D., Jeppesen M. G., Jacobsen J. K., Vestergaard B., Kutter. J. P., Arleth L. (2009). BioXTAS RAW, a software program for highthroughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 42, 959–964 [Google Scholar]
- 42. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003). PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 43. Guinier A. (1939) La diffraction des rayons X aux très petits angles. Application à l'étude de phénomènes ultramicroscopiques. Ann. Phys. 12, 161–237 [Google Scholar]
- 44. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 45. Rambo R. P., Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kratky O., Glatter O. (1982) Small angle x-ray scattering, Academic Press, London [Google Scholar]
- 47. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 51. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 52. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J., Wang W., Kollman P. A., Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 54. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 56. Feller S. E., Zhang Y. H., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation. The Langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 57. Schlick T., Skeel R., Brunger A. T. D., Kale L. V., Board J. A., Jr., Hermans J., Schulte K. (1999) Algorithmic challenges in computational molecular biophysics. J. Comput. Phys. 151, 9–48 [Google Scholar]
- 58. Darden T., York D., Pedersen L. (1993) Partiicle mesh Ewald-an N Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 59. Ryckaert J. P., Ciccotti G., Berendsen H. J. (1977) Numerical integration of the Cartesian equations of motion of a system with constraints. Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 60. Pantoliano M. W., Petrella E. C., Kwasnoski J. D., Lobanov V. S., Myslik J., Graf E., Carver T., Asel E., Springer B. A., Lane P., Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 [DOI] [PubMed] [Google Scholar]
- 61. Light S., Sagit R., Ithychanda S. S., Qin J., Elofsson A. (2012) The evolution of filamin. A protein domain repeat perspective. J. Struct. Biol. 179, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wurzburg B. A., Jardetzky T. S. (2009) Conformational flexibility in immunoglobulin E-Fc 3–4 revealed in multiple crystal forms. J. Mol. Biol. 393, 176–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Müller R., Gräwert M. A., Kern T., Madl T., Peschek J., Sattler M., Groll M., Buchner J. (2013) High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl. Acad. Sci. U.S.A. 110, 10183–10188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Biersmith B. H., Hammel M., Geisbrecht E. R., Bouyain S. (2011) The immunoglobulin-like domains 1 and 2 of the protein-tyrosine phosphatase LAR adopt an unusual horseshoe-like conformation. J. Mol. Biol. 408, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Feng S., Reséndiz J. C., Lu X., Kroll M. H. (2003) Filamin A binding to the cytoplasmic tail of glycoprotein Ibα regulates von Willebrand factor-induced platelet activation. Blood 102, 2122–2129 [DOI] [PubMed] [Google Scholar]
- 66. Meyer S. C., Zuerbig S., Cunningham C. C., Hartwig J. H., Bissell T., Gardner K., Fox J. E. (1997) Identification of the region in actin-binding protein that binds to the cytoplasmic domain of glycoprotein IBα. J. Biol. Chem. 272, 2914–2919 [DOI] [PubMed] [Google Scholar]
- 67. Williamson D., Pikovski I., Cranmer S. L., Mangin P., Mistry N., Domagala T., Chehab S., Lanza F., Salem, H. H., Jackson S. P. (2002) Interaction between platelet glycoprotein Ibα and filamin-1 is essential for glycoprotein Ib/IX receptor anchorage at high shear. J. Biol. Chem. 277, 2151–2159 [DOI] [PubMed] [Google Scholar]
- 68. Falet H., Pollitt A. Y., Begonja A. J., Weber S. E., Duerschmied D., Wagner D. D., Watson S. P., Hartwig J. H. (2010) A novel interaction between FlnA and Syk regulates platelet ITAM-mediated receptor signaling and function. J. Exp. Med. 207, 1967–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen H., Chandrasekar S., Sheetz M. P., Stossel T. P., Nakamura F., Yan J. (2013) Mechanical perturbation of filamin A immunoglobulin repeats 20–21 reveals potential non-equilibrium mechanochemical partner binding function. Sci. Rep. 3, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kyndt F., Gueffet J.-P., Probst V., Jaafar P., Legendre A., Le Bouffant F., Toquet C., Roy E., McGregor L., Lynch S. A., Newbury-Ecob R., Tran V., Young I., Trochu J. N., Le Marec H., Schott J. J. (2007) Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 115, 40–49 [DOI] [PubMed] [Google Scholar]
- 71. Lehtonen J. V., Still D.-J., Rantanen V.-V., Ekholm J., Björklund D., Iftikhar Z., Huhtala M., Repo S., Jussila A., Jaakkola J., Pentikäinen O., Nyrönen T., Salminen T., Gyllenberg M., Johnson M. S. (2004) BODIL. A molecular modeling environment for structure-function analysis and drug design. J. Comput. Aided Mol. Des. 18, 401–419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.