

Abstract
AIM: To investigate the effect of hypoxia or hyperoxia on the progression of hepatic fibrosis and to examine the role of transforming growth factor-β (TGF-β) in the livers of rats exposed to hypoxic or hyperoxic conditions.
METHODS: Male Sprague-Dawley rats were injected intraperitoneally with thioacetamide to induce hepatic fibrosis and were randomly divided into a hypoxia group, a hyperoxia group and an untreated control group. Ten rats in the hypoxia group were exposed to an altitude of 20000 ft for 1 h/d during 7 wk. Ten rats in the hyperoxia group were exposed to a water depth of 20 m with 100% oxygen supply for 1 h/d during 7 wk. We evaluated the degree of hepatic fibrosis using Masson trichrome stain and examined the expression level of hepatic TGF-β mRNA using quantitative real-time reverse transcriptase-polymerase chain reaction analysis.
RESULTS: Eight of 10 rats exposed to hypoxia showed diffuse and confluent fibrosis with the formation of structurally abnormal parenchymal nodules involving the entire liver, consistent with hepatic cirrhosis. Nine of 10 rats exposed to hyperoxia also demonstrated obvious histological findings of hepatic cirrhosis identical to those in hypoxic rat livers. In contrast, 8 of 10 untreated rats had periportal or septal fibrosis only. The frequency of hepatic cirrhosis in hypoxic rats (P = 0.009) and hyperoxic rats (P = 0.003) was significantly higher than that in untreated rats. In addition, hepatic TGF-β mRNA levels in hyperoxic rats were significantly higher than those in untreated rats. The mean value of the normalized TGF-β mRNA/β-actin expression ratio in the hyperoxic rats was 1.9-fold higher than that in the untreated rats (P = 0.027).
CONCLUSION: We demonstrated that both hypoxia and hyperoxia accelerated the progression of hepatic fibrosis in rats. Significant up-regulation of hepatic TGF-β in hyperoxic rats suggests that TGF-β is involved in the acceleration of hepatic fibrosis under hyperoxic conditions.
Keywords: Hepatic fibrosis, Cirrhosis, Hypoxia, Hyperoxia, Transforming growth factor-β
Core tip: We observed that both hypoxic and hyperoxic rat livers exhibited significantly higher frequencies of hepatic cirrhosis than untreated rat livers. We also observed that hepatic transforming growth factor-β (TGF-β) expression in hyperoxic rats was significant higher than that in untreated rats, suggesting that TGF-β is involved in the acceleration of hepatic fibrosis under hyperoxic conditions. To the best of our knowledge, up-regulated TGF-β expression in the livers of cirrhotic rats exposed to hyperoxia has not been reported.
INTRODUCTION
Hepatic fibrosis is an essential pathophysiological consequence of various chronic liver injuries and a common underlying mechanism for hepatic insufficiency[1]. Hepatic fibrosis is currently known to be part of a dynamic process in the setting of liver injury that leads to an abnormal accumulation of extracellular matrix in the liver. The endpoint of hepatic fibrosis is cirrhosis, which is characterized by the formation of regenerative nodules of liver parenchyma separated by confluent fibrotic septa and is accompanied by significant morbidity and mortality[2]. Complex interaction between various liver cell types, such as hepatic stellate cells, hepatocytes, Kupffer cells, and liver sinusoidal endothelial cells, contributes to hepatic fibrosis. Each of these cell types releases a subset of mediators and cytokines that have diverse effects on the progression of hepatic fibrosis and the development of hepatic cirrhosis[1].
Extensive studies using animal models of hepatic fibrosis have revealed that several groups of key genes mediate liver fibrogenesis[3,4]. For example, genes regulating hepatocellular apoptosis and necrosis influence the extent of tissue damage and the subsequent profibrogenic response[5,6]. Genes regulating the generation of pro-inflammatory cytokines and reactive oxygen species (ROS) determine the profibrogenic response to injury and extracellular matrix deposition[7-10]. Among these, a particularly well-studied molecule is transforming growth factor-β (TGF-β), which is widely regarded as profibrogenic in liver injury[11]. A number of studies have identified TGF-β as the most important fibrogenesis-stimulating cytokine in the liver[1,2,12]. The findings of increased hepatic stellate cell activation and hepatic fibrosis in mice with elevated hepatic TGF-β level provide direct evidence for the critical role of TGF-β in hepatic fibrosis[13]. TGF-β is also considered profibrogenic because it inhibits hepatic stellate cell apoptosis, thereby contributing to the increased number of these cells in the setting of liver injury[14]. In addition, TGF-β stimulates extracellular matrix production by liver sinusoidal endothelial cells[15]. Strategies aimed at disrupting the TGF-β signaling pathway have markedly decreased hepatic fibrosis in experimental models[16,17].
Oxygen has important functions as a substrate for biochemical reactions and a modulator of gene expression. Impaired tissue oxygenation and cellular hypoxia are major components in the pathophysiology of a variety of clinical conditions, including infection, wounds, stroke, myocardial infarction, chronic lung disease, and hepatic fibrosis[18,19]. Hypoxia always occurs during liver injury or inflammation, in which swelling of hepatocytes, accumulation of extracellular matrix in the spaces of Disse, construction of regenerating parenchymal nodules and fibrotic septa, and the formation of abnormal vascular networks indicate that hepatocellular hypoxia is involved in the progression of hepatic fibrosis and tissue remodeling[20-25]. Conversely, exposure to hyperoxic conditions generally improves tissue oxygenation. A dramatic increase in oxygen content and arterial blood oxygen pressures accounts for the markedly facilitated diffusion of oxygen to tissues during hyperoxic exposure[26]. However, the advantage of hyperoxia in augmenting oxygen availability to tissues is challenged by the commonly accepted paradigm of cell injury, which emphasizes the role of ROS formation leading to the up-regulation of pro-inflammatory mediators and aggravation of tissue damage[27]. In particular, previous data have suggested that ROS generation is a fundamental mechanism of liver injury[28]. ROS can damage cellular macromolecules and therefore may participate in hepatocellular injury when produced in excess[29]. Moreover, increasing evidence implicates ROS in the pathogenesis of hepatic fibrosis and cirrhosis[29,30].
Although several studies have investigated the relationship between tissue fibrosis and hypoxia, they have focused on the effects of hypoxia in diseases of the heart, lung, and kidney[18,31,32]. Furthermore, the effect of hyperoxic exposure on hepatic fibrosis has not yet been reported. Therefore, the relationship between hepatic fibrosis and hypoxic or hyperoxic conditions in vivo remain to be elucidated. The aim of this study was to investigate the effects of hypoxia or hyperoxia on the progression of hepatic fibrosis and to examine the expression level of TGF-β in the livers of rats exposed to hypoxic or hyperoxic conditions.
MATERIALS AND METHODS
Experimental model
Throughout the experimental period (7 wk), 30 Sprague-Dawley adult male rats (Samtako Bio Korea Co., Ltd., Osan, Gyeonggi-do, South Korea) 6-7 wk of age and weighing between 200 and 230 g were fed standard laboratory rat chow, provided with free access to water, and maintained on a 12-h light-dark cycle under pathogen-free conditions. Temperature and moisture were controlled at 20-25 °C and 40%-45%, respectively. To induce hepatic fibrosis, the rats were injected intraperitoneally with 200 mg/kg of thioacetamide (dissolved in saline; Sigma-Aldrich Co., St. Louis, MO, United States) twice per week. The rats were randomly divided into 3 experimental groups. Ten rats in the hypoxia group were exposed to an altitude of 20000 ft for 1 h/d. Ten rats in the hyperoxia group were exposed to a water depth of 20 m with 100% oxygen supply for 1 h/d. The remaining 10 rats were used as an untreated control group. Animals were killed using diethyl ether 3 d after the last thioacetamide injection and laparotomized via a midline incision. Livers were removed from the animals and immediately preserved in a 10% formaldehyde (formalin) solution or frozen in liquid nitrogen and stored at -70 °C until quantitative real-time reverse-transcriptase polymerase chain reaction (RT-PCR) analysis was performed. The institutional animal ethics committee of the Republic of Korea Air Force Aerospace Medical Center approved all experimental procedures involving animals.
Histological evaluation
After 48 h of formalin fixation, the liver tissues were embedded in paraffin and processed for routine hematoxylin-eosin and for Masson trichrome stains. The degree of hepatic fibrosis was graded according to the standardized guideline proposed by the Korean Study Group for Pathology of Digestive Diseases (Table 1)[33].
Table 1.
Degree of hepatic fibrosis
| Descriptive diagnosis | Definition |
| No fibrosis | Normal connective tissue |
| Portal fibrosis | Fibrous portal expansion |
| Periportal fibrosis | Periportal fibrosis with short septa extending into lobules or rare porto-portal septa (intact architecture) |
| Septal fibrosis | Fibrous septa reaching adjacent portal tracts and terminal hepatic venules (architectural distortion, but no obvious cirrhosis) |
| Cirrhosis | Diffuse nodular formation |
Quantitative real-time RT-PCR analysis
Total RNA was extracted from the liver tissue samples using a NucleoSpin RNA II extraction kit (Macherey-Nagel GmbH and Co. KG, Dueren, Germany). For complementary DNA (cDNA) synthesis, 1 μg of total RNA was reverse-transcribed using a ReverTra Ace-α- reverse transcriptase kit (Toyobo Co., Ltd., Osaka, Japan). The reverse-transcribed cDNA was used for real-time RT-PCR with SsoAdvanced SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, United States). PCR was performed using a Bio-Rad CFX384 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). The primer sequences used for TGF-β were as follows: forward 5′-AGGGCTACCATGCCAACTTC-3′; reverse 5′-CCACGTAGTAGACGATGGGC-3′. The primer sequences used for β-actin were as follows: forward 5′-TCTTCCAGCCTTCCTTCCTG-3′; reverse 5′-CACACAGAGTACTTGCGCTC-3′. PCR reactions for TGF-β and β-actin were initiated with an initial PCR activation step at 95 °C for 30 s, followed by 45 cycles at 95 °C for 2 s, 58 °C for 5 s, and 65 °C for 10 s. A melting curve, ramping from 65 °C to 95 °C, was produced after each RT-PCR to test for the presence of primer dimers. When primer dimer formation was detected, the PCR was rerun using a separate aliquot of cDNA. Each measurement was repeated twice. The normalized expression ratio was calculated using the 2-ΔΔCt method[34].
Statistical analysis
Fisher’s exact test was performed to compare the degree of hepatic fibrosis between groups. The Kruskal-Wallis test was performed to examine whether the expression level of hepatic TGF-β messenger RNA (mRNA) differed significantly between the groups. Statistical analyses were performed using SPSS version 20.0 (IBM SPSS Inc., Chicago, IL, United States). A P value of less than 0.05 was deemed statistically significant.
RESULTS
Histologically, all specimens showed hepatic fibrosis, although the degree of fibrosis was variable. Eight of 10 rats exposed to hypoxia showed diffuse and confluent fibrosis with the formation of structurally abnormal parenchymal nodules involving the entire liver, consistent with hepatic cirrhosis (Figure 1A). Nine of 10 rats exposed to hyperoxia also demonstrated obvious histological findings of hepatic cirrhosis identical to those in hypoxic rat livers (Figure 1B). In contrast, only 2 of 10 untreated rats exhibited hepatic cirrhosis; the remaining 8 rats in the control group exhibited periportal (2 rats) or septal (6 rats) fibrosis only (Figure 1C). The frequency of hepatic cirrhosis in the hypoxic rats (P = 0.009) and hyperoxic rats (P = 0.003) was significantly higher than that in the untreated rats (Table 2).
Figure 1.
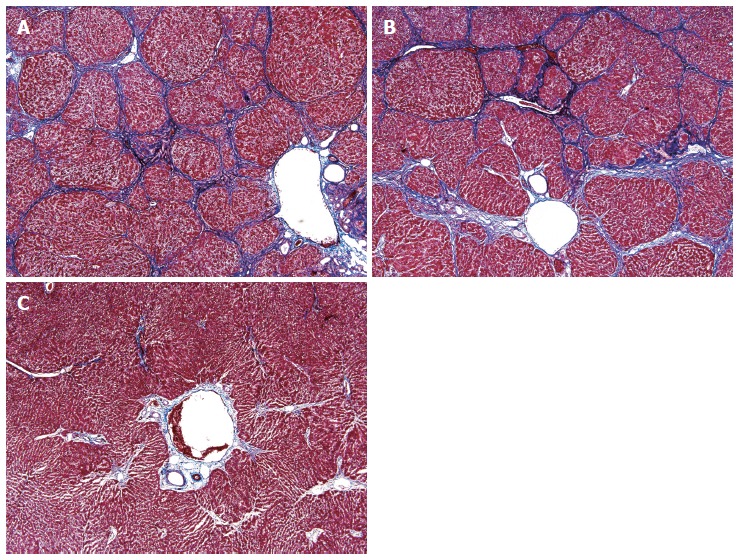
Histology of hepatic fibrosis and cirrhosis. A, B: The livers obtained from 8 of 10 hypoxic rats (A) and 9 of 10 hyperoxic rats (B) demonstrated obvious histological findings of hepatic cirrhosis, including the formation of structurally abnormal parenchymal nodules and confluent fibrotic septa; C: In contrast, 6 of 10 untreated rat livers showed periportal fibrosis only (Masson trichrome stain; original magnification, × 40).
Table 2.
Difference in the degree of hepatic fibrosis among the three groups
| Diagnosis | Number of rats |
||
| Control group | Hypoxia group | Hyperoxia group | |
| (n = 10) | (n = 10) | (n = 10) | |
| Periportal fibrosis | 2 | 0 | 0 |
| Septal fibrosis | 6 | 2 | 1 |
| Cirrhosis | 2 | 81 | 92 |
P = 0.009 vs control group;
P = 0.003 vs control group.
Quantitative real-time RT-PCR revealed that the expression level of hepatic TGF-β mRNA in the hyperoxic rats was significantly higher than that in the untreated rats (P = 0.027; Figure 2). The mean value of the normalized TGF-β mRNA/β-actin expression ratio in the hyperoxic rats was 1.9-fold higher than that in the untreated rats (Table 3). In contrast, the TGF-β mRNA levels in the livers of hypoxic rats were not significantly different from those in the untreated rats (P = 1.000; Figure 2).
Figure 2.
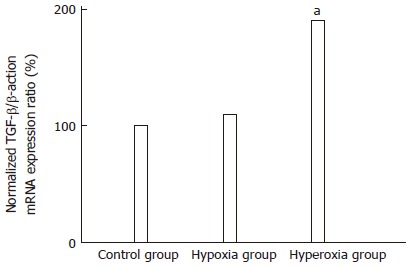
Quantitation of hepatic transformation growth factor-β mRNA expression level using real-time reverse-transcriptase polymerase chain reaction analysis. Results are mean values of 2 independent experiments with 10 rats in each group. The mean value of the normalized hepatic transformation growth factor-β (TGF-β)/β-actin mRNA expression ratio in the hyperoxic rats was significantly higher than that in the untreated rats. aP < 0.05 vs the corresponding values in the control group.
Table 3.
Quantitative real-time reverse-transcriptase polymerase chain reaction results
| Measurement | Control group | Hypoxia group | Hyperoxia group |
| TGF-β mRNA average Ct | 31.51 ± 1.49 | 30.84 ± 1.07 | 29.57 ± 0.96 |
| β-actin mRNA average Ct | 23.99 ± 1.18 | 23.35 ± 1.27 | 22.90 ± 0.72 |
| ΔCt | 7.52 ± 0.63 | 7.49 ± 0.65 | 6.67 ± 0.67 |
| ΔΔCt | 0 ± 0.63 | -0.03 ± 0.65 | -0.77 ± 0.74 |
| Normalized expression ratio | 1.0 | 1.11 | 1.92 |
P = 1.000 vs control group,
P = 0.027 vs control group.
DISCUSSION
In this study, we investigated the effect of hypoxia or hyperoxia on hepatic fibrosis progression using an animal model of hepatic fibrosis. We observed that both hypoxic and hyperoxic rat livers exhibited frequencies of hepatic cirrhosis that were significantly higher than those in untreated, normoxic rat livers. Hypoxia is known to be a key factor in tissue damage and to play a crucial role in chronic liver injury. Exposure to hypoxic conditions stimulates the release of a variety of mediators from hepatic stellate cells and affects the progression of hepatic fibrosis[21,35-37]. Hernandez-Guerra et al[38] revealed that hypoxia aggravates intrahepatic endothelial dysfunction in cirrhotic rat livers. Some in vitro studies have also reported that hypoxia activates hepatic stellate cells and accelerates hepatic fibrosis[22,23,25]. In this context, our finding supports the notion that exposure to hypoxia worsens hepatic fibrosis. In contrast, the effect of hyperoxia on the progression of hepatic fibrosis or the development of hepatic cirrhosis has very rarely been reported. Instead, some available data are sufficient to support the suggestion that hyperoxia exerts beneficial anti-inflammatory effects in models of tissue hypoxia and has protective effects on hemodynamic and metabolic parameters against splanchnic ischemia-reperfusion in rats[39,40]. However, hyperoxic exposure is generally accepted to often result in the formation of ROS in tissues directly implicated in the induction of cell injury via lipid peroxidation and up-regulation of pro-inflammatory cytokines[41-44]. In the liver, Kupffer cells and neutrophils can induce oxidative tissue damage both directly via the effect of ROS on cellular components and indirectly through the release of protease[45]. Increased oxidative stress and the formation of ROS cause extensive necrosis of the liver and ultimately contribute to the development of fibrosis and cirrhosis. Oxidative stress figures prominently in several scenarios of liver fibrogenesis[46,47]. Bhandari et al[48] demonstrated that patients with Child-Pugh class C cirrhosis have greater oxidative stress than those with class B cirrhosis, suggesting that the severity of hepatic cirrhosis is associated with the degree of oxidative stress. Given these data, our findings suggest that hyperoxic exposure accelerates the progression of hepatic fibrosis. In addition, this study can serve as a background for assessing the role of antioxidants in preventing the progression of hepatic cirrhosis.
To clarify the role of TGF-β on hepatic fibrosis under hypoxic or hyperoxic conditions, we investigated the expression of hepatic TGF-β mRNA in rats exposed to hypoxia or hyperoxia. Although a number of studies have examined the effect of TGF-β in experimental models of hepatic fibrosis, to the best of our knowledge, TGF-β expression in the livers of cirrhotic rats exposed to hypoxia or hyperoxia has not been reported. We observed that hepatic TGF-β mRNA level in hyperoxic rats was significantly higher than that in untreated rats. This finding is consistent with previous data which showed that hyperoxic exposure elevated the expression level of TGF-β[49]. Similarly, Alejandre-Alcazar et al[50] demonstrated that hyperoxia up-regulates the expression level of key components of the TGF-β signaling pathway, including type II TGF-β receptor, type I bone morphogenetic protein receptors, and Smad proteins. They further illustrated that TGF-β stimulation dramatically elevates the levels of tissue inhibitors of metalloproteinase-1 mRNA in fibroblasts, supporting the idea that dysregulation of TGF-β signaling impacts extracellular matrix deposition and tissue remodeling[50]. Together with these results, the observation in the present study that hyperoxic rats exhibited a significantly higher frequency of hepatic cirrhosis than untreated rats suggests that TGF-β is partially responsible for the acceleration of hepatic fibrosis progression in rats exposed to hyperoxia.
However, no significant difference between hepatic TGF-β mRNA levels in hypoxic rats and untreated rats was found. In fact, relevant experimental studies have provided conflicting results regarding the effect of hypoxia on TGF-β expression. An in vitro study demonstrated that hypoxic exposure induces an increase in the expression level of TGF-β mRNA[51]. A study using a model of carbon tetrachloride-induced hepatic fibrosis also showed that hypoxic rat livers release TGF-β protein[52]. Similarly, hypoxia has been shown to up-regulate TGF-β synthesis and enhance its effect in human skin and lung fibroblasts[53,54]. In contrast, a study using a cell culture method has shown that hypoxia increases the production of fibronectin, collagen I, and collagen IV, but not TGF-β, in placental fibroblasts, suggesting that increased extracellular matrix production under hypoxia is not mediated directly by increased TGF-β[55]. Our observation raises 2 possibilities. First, hepatic fibrosis might be accelerated independent of TGF-β under hypoxic conditions. Second, intermittent exposure to hypobaric hypoxia at the altitude of 20000 ft might be insufficient to affect TGF-β mRNA transcription in rat liver. Further investigations are necessary to clarify the relationship between TGF-β expression and the progression of hepatic fibrosis under hypoxic conditions.
In conclusion, we demonstrated that both hypoxia and hyperoxia accelerated the progression of hepatic fibrosis in rats. In addition, a significant up-regulation of hepatic TGF-β in hyperoxic rats suggests that TGF-β is involved in the acceleration of hepatic fibrosis under hyperoxic conditions.
COMMENTS
Background
Hepatic fibrosis is a pathophysiological consequence of chronic liver injuries and a common underlying mechanism for hepatic insufficiency. Experimental models of hepatic fibrosis have provided a means to study the cell and molecular mediators of fibrosis and revealed that several groups of key genes mediate liver fibrogenesis. In particular, extensive studies have identified transforming growth factor (TGF)-β as the most important profibrogenic cytokine in the liver. Although a number of studies have investigated the relationship between tissue fibrosis and oxygen tension, they have focused on the effects of hypoxia in diseases of the heart, lung, and kidney. The effect of exposure to hyperoxic conditions on hepatic fibrosis and the role of TGF-β in the progression of fibrosis in hyperoxic livers have not yet been reported.
Research frontiers
Hyperoxic exposure is generally accepted to result in the formation of reactive oxygen species (ROS) in a variety of tissues directly implicated in the induction of cell injury via lipid peroxidation and up-regulation of pro-inflammatory cytokines. In the liver, Kupffer cells and neutrophils can induce oxidative tissue damage both directly via the effect of ROS on cellular components and indirectly through the release of proteolytic enzymes. Furthermore, the formation of ROS and increased oxidative stress cause extensive hepatocellular necrosis and ultimately contribute to the development of hepatic fibrosis and cirrhosis. Oxidative stress figures prominently in several scenarios of liver fibrogenesis.
Innovations and breakthroughs
The authors observed that hyperoxic livers exhibited frequencies of hepatic cirrhosis that were significantly higher than those in untreated, normoxic livers. In addition, they further observed that hepatic TGF-β mRNA level in hyperoxic rats was significant higher than that in untreated rats. To the best of our knowledge, no reports have been published demonstrating a significant relationship between exposure to hyperoxia and frequencies of hepatic cirrhosis and discussing the role of TGF-β in the progression of hepatic fibrosis in hyperoxic rat livers. The observations in the present study suggest that TGF-β is partially responsible for the acceleration of hepatic fibrosis progression in rats exposed to hyperoxia.
Applications
Based on the observation that hyperoxic exposure accelerates the progression of hepatic fibrosis, this study may serve as a background for assessing the role of antioxidants in preventing the progression of hepatic cirrhosis.
Terminology
Hepatic cirrhosis is the endpoint of hepatic fibrosis and various chronic liver diseases characterized by the formation of regenerative nodules of liver parenchyma separated by confluent fibrotic septa. It is typically accompanied by significant morbidity and mortality. TGF-β is a protein that controls the cell cycle, apoptosis, proliferation and differentiation in most cells. In the liver, it plays a crucial role in inhibiting hepatic stellate cell apoptosis, thereby contributing to the increased number of these cells in the setting of liver injury. In addition, TGF-β stimulates extracellular matrix production by liver sinusoidal endothelial cells.
Peer review
This is well performed study, in which authors analyzed the frequency of hepatic cirrhosis and TGF-β expression status in rats exposed to hyperoxia. The results are interesting and suggest that TGF-β accelerates the progression of hepatic fibrosis following hyperoxic exposure. Further investigations are recommended to elucidate the molecular biological functions of TGF-β and a potential role of ROS in association with hyperoxia in the liver.
Footnotes
Supported by Aerospace Medicine Research Project funded by the Medical Division, Headquarter, Republic of Korea Air Force (2012)
P- Reviewer: Olaso E S- Editor: Zhai HH L- Editor: Webster JR E- Editor: Liu XM
References
- 1.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 2.Guyot C, Lepreux S, Combe C, Doudnikoff E, Bioulac-Sage P, Balabaud C, Desmoulière A. Hepatic fibrosis and cirrhosis: the (myo)fibroblastic cell subpopulations involved. Int J Biochem Cell Biol. 2006;38:135–151. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 3.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 5.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 6.Takehara T, Tatsumi T, Suzuki T, Rucker EB, Hennighausen L, Jinushi M, Miyagi T, Kanazawa Y, Hayashi N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol. 2004;287:G264–G273. doi: 10.1152/ajpgi.00002.2004. [DOI] [PubMed] [Google Scholar]
- 8.Streetz KL, Tacke F, Leifeld L, Wüstefeld T, Graw A, Klein C, Kamino K, Spengler U, Kreipe H, Kubicka S, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. 2003;38:218–229. doi: 10.1053/jhep.2003.50268. [DOI] [PubMed] [Google Scholar]
- 9.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA. 1997;94:10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 12.Forbes SJ, Parola M. Liver fibrogenic cells. Best Pract Res Clin Gastroenterol. 2011;25:207–217. doi: 10.1016/j.bpg.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Ueberham E, Löw R, Ueberham U, Schönig K, Bujard H, Gebhardt R. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology. 2003;37:1067–1078. doi: 10.1053/jhep.2003.50196. [DOI] [PubMed] [Google Scholar]
- 14.George J, Roulot D, Koteliansky VE, Bissell DM. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George J, Wang SS, Sevcsik AM, Sanicola M, Cate RL, Koteliansky VE, Bissell DM. Transforming growth factor-beta initiates wound repair in rat liver through induction of the EIIIA-fibronectin splice isoform. Am J Pathol. 2000;156:115–124. doi: 10.1016/s0002-9440(10)64711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnur J, Oláh J, Szepesi A, Nagy P, Thorgeirsson SS. Thioacetamide-induced hepatic fibrosis in transforming growth factor beta-1 transgenic mice. Eur J Gastroenterol Hepatol. 2004;16:127–133. doi: 10.1097/00042737-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Shek FW, Benyon RC. How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur J Gastroenterol Hepatol. 2004;16:123–126. doi: 10.1097/00042737-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Higgins DF, Kimura K, Iwano M, Haase VH. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle. 2008;7:1128–1132. doi: 10.4161/cc.7.9.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kietzmann T, Dimova EY, Flügel D, Scharf JG. Oxygen: modulator of physiological and pathophysiological processes in the liver. Z Gastroenterol. 2006;44:67–76. doi: 10.1055/s-2005-858987. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Yosef Y, Lahat N, Shapiro S, Bitterman H, Miller A. Regulation of endothelial matrix metalloproteinase-2 by hypoxia/reoxygenation. Circ Res. 2002;90:784–791. doi: 10.1161/01.res.0000015588.70132.dc. [DOI] [PubMed] [Google Scholar]
- 21.Chen PS, Zhai WR, Zhou XM, Zhang JS, Zhang YE, Ling YQ, Gu YH. Effects of hypoxia, hyperoxia on the regulation of expression and activity of matrix metalloproteinase-2 in hepatic stellate cells. World J Gastroenterol. 2001;7:647–651. doi: 10.3748/wjg.v7.i5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copple BL, Bai S, Burgoon LD, Moon JO. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int. 2011;31:230–244. doi: 10.1111/j.1478-3231.2010.02347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 24.Shi YF, Fong CC, Zhang Q, Cheung PY, Tzang CH, Wu RS, Yang M. Hypoxia induces the activation of human hepatic stellate cells LX-2 through TGF-beta signaling pathway. FEBS Lett. 2007;581:203–210. doi: 10.1016/j.febslet.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Troeger JS, Schwabe RF. Hypoxia and hypoxia-inducible factor 1α: potential links between angiogenesis and fibrogenesis in hepatic stellate cells. Liver Int. 2011;31:143–145. doi: 10.1111/j.1478-3231.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- 26.Gill AL, Bell CN. Hyperbaric oxygen: its uses, mechanisms of action and outcomes. QJM. 2004;97:385–395. doi: 10.1093/qjmed/hch074. [DOI] [PubMed] [Google Scholar]
- 27.Eppihimer MJ, Granger DN. Ischemia/reperfusion-induced leukocyte-endothelial interactions in postcapillary venules. Shock. 1997;8:16–25. doi: 10.1097/00024382-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Parks DA, Bulkley GB, Granger DN. Role of oxygen-derived free radicals in digestive tract diseases. Surgery. 1983;94:415–422. [PubMed] [Google Scholar]
- 29.Britton RS, Bacon BR. Role of free radicals in liver diseases and hepatic fibrosis. Hepatogastroenterology. 1994;41:343–348. [PubMed] [Google Scholar]
- 30.Farrell GC, Teoh NC, McCuskey RS. Hepatic microcirculation in fatty liver disease. Anat Rec (Hoboken) 2008;291:684–692. doi: 10.1002/ar.20715. [DOI] [PubMed] [Google Scholar]
- 31.Estrada KD, Chesler NC. Collagen-related gene and protein expression changes in the lung in response to chronic hypoxia. Biomech Model Mechanobiol. 2009;8:263–272. doi: 10.1007/s10237-008-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin YM, Huang SK, Wang HF, Chen LM, Tsai FJ, Hsu HH, Kuo CH, Wang PS, Huang CY, Lee SD. Short-term versus long-term intermittent hypobaric hypoxia on cardiac fibrosis and Fas death receptor dependent apoptotic pathway in rat hearts. Chin J Physiol. 2008;51:308–316. [PubMed] [Google Scholar]
- 33.Park YN, Kim H, Chon CY, Park JB, Sohn JH, Yang SH, Yu E, Lee MS, Jang JJ, Chang HK, et al. Histological grading and staging of chronic hepatitis: Standardized guideline proposed by the Korean Study Group for Pathology of Digestive Diseases. Korean J Pathol. 1999;33:337–346. [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Ankoma-Sey V, Wang Y, Dai Z. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology. 2000;31:141–148. doi: 10.1002/hep.510310122. [DOI] [PubMed] [Google Scholar]
- 36.Novo E, Cannito S, Zamara E, Valfrè di Bonzo L, Caligiuri A, Cravanzola C, Compagnone A, Colombatto S, Marra F, Pinzani M, et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol. 2007;170:1942–1953. doi: 10.2353/ajpath.2007.060887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, Kodama Y, Miura K, Ikai I, Uemoto S, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–1738. doi: 10.1053/j.gastro.2008.07.065. [DOI] [PubMed] [Google Scholar]
- 38.Hernández-Guerra M, de Ganzo ZA, González-Méndez Y, Salido E, Abreu P, Moreno M, Felipe V, Abrante B, Quintero E. Chronic intermittent hypoxia aggravates intrahepatic endothelial dysfunction in cirrhotic rats. Hepatology. 2013;57:1564–1574. doi: 10.1002/hep.26152. [DOI] [PubMed] [Google Scholar]
- 39.Bitterman H. Oxygen: an anti-inflammatory drug. Isr Med Assoc J. 2007;9:874–876. [PubMed] [Google Scholar]
- 40.Bitterman H, Bitterman N, Melamed Y, Cohen L. Effects of hyperbaric oxygen in circulatory shock induced by splanchnic artery occlusion and reperfusion in rats. Can J Physiol Pharmacol. 1989;67:1033–1037. doi: 10.1139/y89-163. [DOI] [PubMed] [Google Scholar]
- 41.Stogner SW, Payne DK. Oxygen toxicity. Ann Pharmacother. 1992;26:1554–1562. doi: 10.1177/106002809202601214. [DOI] [PubMed] [Google Scholar]
- 42.Gille JJ, van Berkel CG, Joenje H. Mutagenicity of metabolic oxygen radicals in mammalian cell cultures. Carcinogenesis. 1994;15:2695–2699. doi: 10.1093/carcin/15.12.2695. [DOI] [PubMed] [Google Scholar]
- 43.Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 44.D'Angio CT, Finkelstein JN. Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab. 2000;71:371–380. doi: 10.1006/mgme.2000.3074. [DOI] [PubMed] [Google Scholar]
- 45.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 46.Bissell DM. Chronic liver injury, TGF-beta, and cancer. Exp Mol Med. 2001;33:179–190. doi: 10.1038/emm.2001.31. [DOI] [PubMed] [Google Scholar]
- 47.Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, Foschi M, Caligiuri A, Pinzani M, Surrenti C. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology. 1997;25:361–367. doi: 10.1053/jhep.1997.v25.pm0009021948. [DOI] [PubMed] [Google Scholar]
- 48.Bhandari S, Agarwal MP, Dwivedi S, Banerjee BD. Monitoring oxidative stress across worsening Child Pugh class of cirrhosis. Indian J Med Sci. 2008;62:444–451. [PubMed] [Google Scholar]
- 49.Lecart C, Cayabyab R, Buckley S, Morrison J, Kwong KY, Warburton D, Ramanathan R, Jones CA, Minoo P. Bioactive transforming growth factor-beta in the lungs of extremely low birthweight neonates predicts the need for home oxygen supplementation. Biol Neonate. 2000;77:217–223. doi: 10.1159/000014219. [DOI] [PubMed] [Google Scholar]
- 50.Alejandre-Alcázar MA, Kwapiszewska G, Reiss I, Amarie OV, Marsh LM, Sevilla-Pérez J, Wygrecka M, Eul B, Köbrich S, Hesse M, et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L537–L549. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki A, Kusakai G, Shimojo Y, Chen J, Ogura T, Kobayashi M, Esumi H. Involvement of transforming growth factor-beta 1 signaling in hypoxia-induced tolerance to glucose starvation. J Biol Chem. 2005;280:31557–31563. doi: 10.1074/jbc.M503714200. [DOI] [PubMed] [Google Scholar]
- 52.Jeong WI, Do SH, Yun HS, Song BJ, Kim SJ, Kwak WJ, Yoo SE, Park HY, Jeong KS. Hypoxia potentiates transforming growth factor-beta expression of hepatocyte during the cirrhotic condition in rat liver. Liver Int. 2004;24:658–668. doi: 10.1111/j.1478-3231.2004.0961.x. [DOI] [PubMed] [Google Scholar]
- 53.Falanga V, Qian SW, Danielpour D, Katz MH, Roberts AB, Sporn MB. Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal fibroblasts. J Invest Dermatol. 1991;97:634–637. doi: 10.1111/1523-1747.ep12483126. [DOI] [PubMed] [Google Scholar]
- 54.Papakonstantinou E, Roth M, Tamm M, Eickelberg O, Perruchoud AP, Karakiulakis G. Hypoxia differentially enhances the effects of transforming growth factor-beta isoforms on the synthesis and secretion of glycosaminoglycans by human lung fibroblasts. J Pharmacol Exp Ther. 2002;301:830–837. doi: 10.1124/jpet.301.3.830. [DOI] [PubMed] [Google Scholar]
- 55.Chen CP, Yang YC, Su TH, Chen CY, Aplin JD. Hypoxia and transforming growth factor-beta 1 act independently to increase extracellular matrix production by placental fibroblasts. J Clin Endocrinol Metab. 2005;90:1083–1090. doi: 10.1210/jc.2004-0803. [DOI] [PubMed] [Google Scholar]