

Abstract
Objective:
The aim of this study was to identify the causal gene in a consanguineous Moroccan family with temporo-occipital polymicrogyria, psychiatric manifestations, and epilepsy, previously mapped to the 6q16–q22 region.
Methods:
We used exome sequencing and analyzed candidate variants in the 6q16–q22 locus, as well as a rescue assay in Fig4-null mouse fibroblasts and immunohistochemistry of Fig4-null mouse brains.
Results:
A homozygous missense mutation (p.Asp783Val) in the phosphoinositide phosphatase gene FIG4 was identified. Pathogenicity of the variant was supported by impaired rescue of the enlarged vacuoles in transfected fibroblasts from Fig4-deficient mice. Histologic examination of Fig4-null mouse brain revealed neurodevelopmental impairment in the hippocampus, cortex, and cerebellum as well as impaired cerebellar gyration/foliation reminiscent of human cortical malformations.
Conclusions:
This study extends the spectrum of phenotypes associated with FIG4 mutations to include cortical malformation associated with seizures and psychiatric manifestations, in addition to the previously described Charcot-Marie-Tooth disease type 4J and Yunis-Varón syndrome.
Polymicrogyria is a malformation of the cerebral cortex characterized by abnormal lamination and folding. As a result, the deeper cortical layers develop abnormally and an excessive number of small gyri form within the cortex.1 Polymicrogyria belongs to the group of cortical malformations secondary to postmigrational development.1 Clinical manifestations depend on the extent and location of the polymicrogyric cortex and range from asymptomatic to a severe phenotype variably including intractable epilepsy, cognitive impairment, and neurologic deficits.1 Causes of polymicrogyria are heterogeneous and include acquired and genetic etiology. Several genes have been identified for autosomal recessive forms of polymicrogyria: GPR562 in bilateral frontoparietal polymicrogyria; KBP 3 in polymicrogyria with microcephaly; and the RAB GTPase genes including RAB3GAP1,4 RAB3GAP2,5 and RAB186 in Warburg Micro syndrome characterized by bilateral frontal polymicrogyria and thinning of the corpus callosum. Dominant mutations in TUBA1A,7 TUBB2B,8 and TUBB39 have been associated with frontoparietal or diffuse polymicrogyria, in TUBA810 with diffuse polymicrogyria with agenesis of corpus callosum and optic nerve hypoplasia, in COL18A111 with the Knobloch syndrome, and in SRPX212 with perisylvian polymicrogyria with rolandic seizures and speech dyspraxia.
We previously reported a consanguineous Moroccan family with segregation of an autosomal recessive phenotype associating polymicrogyria, epilepsy, and psychiatric manifestations with linkage on chromosome 6q16–q22.13 In this study, we performed whole-exome sequencing and identified a variant at the homozygous state in FIG4, a gene that was previously implicated in Charcot-Marie-Tooth disease type 4J (CMT4J)14 and in Yunis-Varón syndrome (YVS).15 The causality of this mutation was reinforced by a functional assay and the study of cortical development in Fig4-deficient mice.
METHODS
Standard protocol approvals, registrations, and patient consents.
Nine family members (3 affected and 6 unaffected) gave written consent to participate in the study in accordance with the local committee of the University Mohammed V-Souissi, Rabat, Morocco (no. 108).
Whole-exome sequencing.
Exons from 3 μg of genomic DNA of patient V.11 were captured using the SureSelect kit following the manufacturer's protocols (Agilent, Santa Clara, CA). The whole-exome DNA library was sequenced on the HiSeq2000 Illumina Genome Analyzer (Illumina, San Diego, CA). Sequence reads were mapped to the reference human genome using whole genomic sequence, refgene.txt file, and reflink.txt (UCSC hg19) using the BWA v0.5.9 algorithm (http://samtools.sourceforge.net). A mean depth of 40× covered the candidate homozygosity region on chromosome 6 (chr6: 103015795–117950828) for all exons. Only one gene of the region, LOC285758 (114189178–114194512), was not covered, and was probably absent from the design in the SureSelect protocol. Variant detection was performed with the SAMTools v0.1.17 software (http://samtools.sourceforge.net) and filtered by different scripts (Perl) to fit quality threshold: depth ≥10× and a percentage of reads between 40% and 60%. Variants were annotated according to snp132.txt.
Validation of exome variants and screening of a patient cohort by Sanger sequencing.
Analysis of exome sequencing focused on the 6q16–q22 locus (chr6: 103,015,795–117,950,828; hg19), the unique region with positive logarithm of odds scores. Variants found by exome sequencing were validated, and segregation analysis was performed by Sanger sequencing using the Big-Dye Terminator kit on an ABI Prism 3730 DNA Analyzer (Applied Biosystems, Foster City, CA). Mutation interpretation was assessed with Alamut software (Interactive Biosoftware, Rouen, France). The effect of amino acid substitution on protein function was predicted using SIFT, PolyPhen-2, and MutationTaster. All 23 exons and intron-exon junctions of FIG4 (RefSeqNM_014845.5) were sequenced in 33 additional unrelated patients with polymicrogyria.
Phenotype rescue assay.
The p.Asp783Val mutation was incorporated into the mouse Fig4 complementary DNA (cDNA) clone by site-directed mutagenesis and the construct was completely sequenced. Mouse fibroblasts were isolated from P0 mouse tail biopsy by digestion with collagenase type 2 (Worthington Biochemical, Lakewood, NJ) and cultured in RPMI 1640 supplemented with 15% fetal bovine serum and containing penicillin, streptomycin, and amphotericin. Experiments were performed at passage 3 to 5.16 Fibroblasts at 50% confluence were cotransfected with Fig4 cDNA and green fluorescent protein (GFP) cDNA. GFP-positive cells were imaged 18 hours after transfection with an inverted Leica DM IRB microscope (Leica Microsystems, Buffalo Grove, IL) equipped with epifluorescence and an Olympus DP30 BW digital camera (Olympus, Center Valley, PA). Rescued cells were defined as GFP-positive cells with fewer than 6 vacuoles. The cell area occupied by vacuoles was quantitated using ImageJ software.
Caspase 3/7 activity assay.
Ten percent brain homogenates were prepared, and cleavage of aminoluciferin-tagged DEVD peptide was measured. After centrifugation of homogenates at 15,000g, protein concentration in the supernatant was adjusted to 0.02 mg/mL. An equal volume of homogenate and Caspase-Glo 3/7 reagent (Promega, Madison, WI) was added to a white-walled 96-well plate and incubated at room temperature for 60 minutes. Luminescence was read on a Promega GloMax-Multiplate luminometer.
Western blot.
Whole mouse brains were removed and lysed in 5 M urea, 2.5% sodium dodecyl sulfate, 50 mM Tris, 30 mM NaCl buffer. Protein concentration was determined by the Bradford method. Twenty-five micrograms of each sample was separated on 10% Tris-glycine polyacrylamide gels and analyzed by Western blot using monoclonal anti-FIG4 antibody (NeuroMab clone N202/7) and rabbit polyclonal anti-actin antibody (Sigma-Aldrich, St. Louis, MO).
Histochemistry.
Brains from mice aged P0 and P4 (n = 5 for each genotype) were removed and postfixed for 24 hours in 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4. Seven-micrometer-thick paraffin sections were cut. Immunohistochemistry was performed using rabbit Calbindin D-28k (Swant, Marly, Switzerland) and Nissl counterstaining using standard procedures.
Morphometry.
A drawing of the cerebellar fissures of midsagittal sections at 40× was made as previously reported17 using a Leica SCN slide scanner. We measured 6 principal fissures (precentral, preculminate, primary, prepyramidal, secondary, and posterolateral) by the Image Processing Toolbox of CaloPix software viewer (http://www.teleslide.fr). Measurements of fissures are given as mean (± standard error) and compared using a Mann-Whitney test.
RESULTS
Clinical characterization.
Clinical data of the Moroccan kindred with 6 affected siblings were previously reported.13 Since our previous publication, V.8 died of a seizure and V.9 committed suicide. In this family, 6 patients had epilepsy (complex focal and secondarily generalized seizures). Seizures consisted of visual hallucinations, sometimes followed by auditory hallucinations, as well as epigastric and thoracic oppression or neurovegetative symptoms suggesting that the epileptic discharges spread to the temporal lobe. The course and severity of epilepsy were variable from one patient to another for age at onset, ranging from birth (V.5 and V.11) to 24 years (V.1), and frequency of seizures ranging from 1 to 2 per month (V.1) to several per day (V.8 and V.9). The outcome of epilepsy largely depended on the compliance of patients with their antiepileptic medication; sudden death (SUDEP) occurred in 2 patients, V.5 and V.6, who were untreated. In addition, severe psychiatric manifestations were reported in 4 patients (V.1, V.8, V.9, and V.11). A psychotic disorder with aggressiveness and delirium was diagnosed in patients V.8 and V.11. Patient V.1 manifested chronic aggressiveness toward relatives. In addition, 2 patients (V.1, V.9) committed suicide.
The 3 patients examined by brain MRI, V.8, V.9, and V.11, exhibited a thickened irregular cortex in the lateral occipital lobes with small gyri and were diagnosed with bilateral occipital polymicrogyria.13 Upon critical re-evaluation of MRI in these 3 patients by the neurologist participating in this study (R.G.), we concluded that the cortical abnormality also involved the lateral part of the posterior temporal lobes. Patients therefore had temporo-occipital rather than occipital polymicrogyria (figure 1). Family members IV.1 (the father) and V.7 (a nonaffected sibling) had normal brain MRI.
Figure 1. Brain MRI.
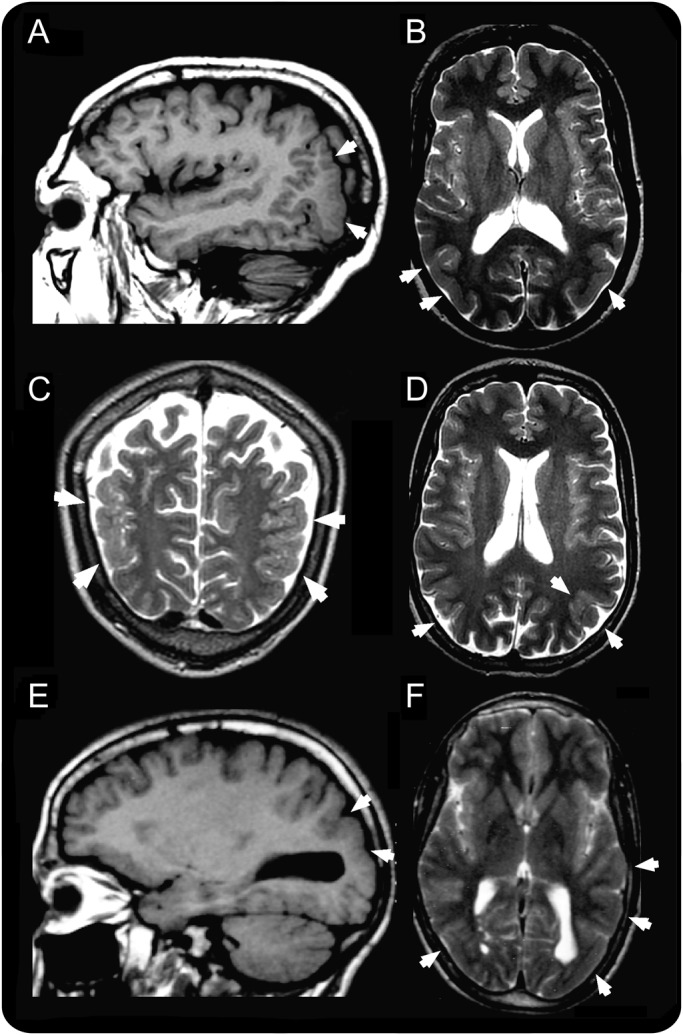
(A, B) Patient V.8, (C, D) patient V.9, and (E, F) patient V.11. Images A and E are T1-weighted sagittal sections; B, D, and F are T2-weighted axial sections; and C is a T2-weighted coronal section. The areas of abnormal cortical development in the temporo-occipital areas are indicated by the white arrows and are consistent in all 3 patients with a combination of increased cortical thickness, smooth cortex, overfolding, and microgyri.
Molecular studies.
We previously mapped the causative gene to a unique 14-Mb locus on chromosome 6q16–q22.13 Here, we performed whole-exome sequencing in one patient (V.11),13 and focused our analysis on the 6q16–q22 region (chr6: 103,015,795–117,950,828; hg19). This rare phenotype segregated as an autosomal recessive disorder in the consanguineous pedigree. Therefore, we filtered nonsynonymous and insertion/deletion variants located in this interval in the homozygous state with frequency <1% in the databases (dbSNP135, 1000 Genomes Project database, and National Heart, Lung, and Blood Institute exome variant server database). Only 2 missense variants were identified: c.716T>G/p.Leu239Arg in TUBE1 that was previously excluded because of its high frequency in a Maghrebian cohort of controls (2% allele frequency)13 and c.2348A>T/p.Asp783Val located in exon 20 of the FIG4 gene. Both parents were heterozygous for the variant in FIG4, which fully cosegregated with the phenotype in the family (figure 2A). This variant was absent from the dbSNP135, 1000 Genomes Project, and exome variant server databases. Additionally, it was not found in 750 in-house controls, including 422 ethnically matched from Maghreb. At the protein level, the Asp783 residue is located in the C-terminus of the protein (figure 2B) and is invariant in mammals and terrestrial vertebrates (figure 2C). Asp and Val have a high Grantham distance of 152 (0–215) reflecting the chemical dissimilarity between the negatively charged, polar aspartate residue and the hydrophobic valine residue. The prediction software tools MutationTaster, PolyPhen-2, and SIFT predicted that this variant was pathogenic, possibly damaging, and tolerated, respectively. Sanger sequencing of the complete coding region of FIG4 did not reveal pathogenic mutations in 33 additional patients with various types of polymicrogyria.
Figure 2. Identification of FIG4 mutation.

(A) Pedigree of the family with segregation of the FIG4 variant. Only generations IV and V are shown.13 (B) FIG4 is a multidomain protein with a protein interaction domain at the N-terminus, a SAC phosphatase domain in the central region, and poly-Pro and poly-Ser domains within the C-terminal. The mutation identified in this study is indicated in red, and missense mutations previously reported in Charcot-Marie-Tooth disease type 4J21 and Yunis-Varón syndrome15 are indicated in black and blue, respectively. (C) Multiple protein alignment showing conservation of the aspartate (D) residue in position 783 in orthologs of FIG4 across species. (D) Phenotype rescue in Fig4-null fibroblasts of plt mice. Mutant or wild-type (WT) Fig4 cDNA was cotransfected with GFP into Fig4-null fibroblasts. The large intracytoplasmic vesicles characteristic of Fig4-null fibroblasts were rescued in 60% of cells by the WT cDNA (top panels) but in only 33% of cells by the D783V mutant cDNA (bottom panels). (E) Percent of transfected (GFP-positive) fibroblasts that lack vacuoles. The results of 3 independent experiments are indicated. *p < 0.01. (F) The D783V mutant retains partial activity. In nonrescued cells from panel E, the percentage of cell area covered by vacuoles in cells expressing D783V (3.3% ± 0.6%, n = 24) is smaller than that in cells receiving vector only (10.3% ± 1.8%, n = 22) (2-tailed p value <0.001). Transfection of the mutant cDNA does not completely rescue the formation of vacuoles but does reduce the severity of vacuolization. cDNA = complementary DNA; GFP = green fluorescent protein; plt = pale tremor.
Functional studies in Fig4-null fibroblasts.
FIG4 encodes a phosphoinositide 5-phosphatase that regulates the cellular abundance of the signaling lipid PI(3,5)P2, and thus endosomal trafficking and autophagy. Complete loss of Fig4 in the null mouse mutant pale tremor (plt) results in extensive neurodegeneration of the central and peripheral nervous system.14,18 The cytoplasm of cultured fibroblasts from plt mice is filled with enlarged vacuoles.14,18 To assess the functional effect of the p.Asp783Val (D783V) variant, we performed a phenotype rescue of vacuolization in cultured cells. Transfection of Fig4-null cells with wild-type (WT) Fig4 cDNA rescues the vacuoles, while the Fig4-D783V is defective in rescue (figure 2D). In the combined data from 3 independent experiments represented in figure 2E, the WT cDNA completely corrected vacuolization in 60% of transfected cells (334/558) while the D783V cDNA corrected vacuolization in only 33% of transfected cells (262/797) (p < 0.01, t test). In cells that were not completely corrected by the mutant cDNA, the total area occupied by vacuoles was significantly reduced by expression of the D783V cDNA (p < 0.001, Yates correction χ2) (figure 2F). These data demonstrate that the D783V mutation causes partial but not complete loss of FIG4 function.
Histologic studies in Fig4-null brains.
To further investigate the role of Fig4 in cortical development, we examined the temporal expression of the protein in developing mouse brain by Western blot of whole-brain lysates between embryonic day 10.5 (E10.5) and postnatal day P14, before spongiform degeneration. FIG4 protein expression could be detected as early as E10.5, consistent with a neurodevelopmental role (figure e-1 on the Neurology® Web site at Neurology.org). We then asked whether neurodevelopmental defects of the brain were present in plt mice, as in patients with polymicrogyria. No obvious macroscopic modification was observed in brains of plt mice at postnatal age P0 and P4 (data not shown). However, histologic Nissl staining revealed increased neuronal density in plt mice in all cortical layers at P0 (figures 3A and e-2), which was no longer present at P4 (figure 3A), likely due to cell death. Indeed, during this time interval, the level of the apoptosis-associated activated caspase 3/7 was transiently elevated in the mutant brain (figure 3B). Because expression of calbindin D-28k is correlated with postnatal maturation of hippocampal dentate granule cells19 and dendritic arborization,20 we performed anti-calbindin immunohistochemistry of the WT and plt brains (n = 5 for each). Calbindin staining was drastically decreased in plt mice, indicating delayed maturation of interneurons and dentate granule cells and their processes in the hippocampus at P4 (figure 3C), in the soma and dendritic tree of Purkinje cells at P0 (figure e-3A) and P4 (figure 3D), and in cortical interneurons of plt mice (figure e-3B). Morphometric study of postnatal cerebellar development at P4 revealed a significant reduction in the depth of all fissures in plt mice, with the exception of the primary fissure, demonstrating delayed cerebellar gyration/foliation (Mann-Whitney test, p < 0.05) (figures 3E and e-4). These findings support both impaired molecular development (decreased calbindin expression) and impaired anatomical development (gyration) in Fig4-deficient mice, reflecting postmigration abnormalities that are considered to be the major mechanisms underlying polymicrogyria in human.1
Figure 3. Neurodevelopmental anomalies in plt mice.

(A) Nissl staining of cortex from plt homozygote and WT littermate at postnatal days P0 and P4. Note presence of swollen neurons in layers IV and V of plt aged P4. Cortical layers are indicated as follows: I–VI, WM (white matter), CC (corpus callosum). (B) Caspase 3/7 activity was measured in brain homogenates from 3 animals of each genotype at the indicated postnatal age. An unpaired Student t test was used to generate p values at each age. (C) Weak calbindin immunoreactivity in hippocampus of plt mouse compared with WT littermates. Higher magnification of dorsal DG (boxed regions) shows many granule cells with a well-developed dendrite tree in the WT brain, while in the plt, there is weak staining of the perikaryon and no immunostaining of dendrite trees. (D) Very weak calbindin immunoreactivity in cerebellar cortex and deep nucleus (arrows) in plt mouse compared with WT littermate at P4. Boxed areas are focused on posterolateral fissures. Scale bars: A, 100 μm; C, 300 μm (boxed area 50 μm); D, 250 μm (boxed area 50 μm). (E) Histogram representing the fissure lengths in the cerebellum at P4 (n = 5). Significant differences between lengths of fissures were observed between WT and plt. *p < 0.05; **p < 0.01. DG = dentate gyrus; n.s. = not significant; plt = pale tremor; WT = wild-type.
DISCUSSION
The present study extends to polymicrogyria the spectrum of phenotypes associated with FIG4 mutations, which were previously identified in CMT4J14,21 and YVS15 (figure 2B). A key question is how FIG4 mutations can lead to such diverse autosomal recessive disorders: (1) CMT4J, a peripheral neuropathy, (2) YVS, in which neurodegeneration and brain malformations are associated with cleidocranial dysplasia, digital anomalies, and early death, and (3) temporo-occipital polymicrogyria with seizures and psychiatric features. The major distinction is that patients with YVS are homozygous or compound heterozygous for null mutations of FIG4, and thus exhibit complete loss of function, similar to the plt mouse. In contrast, patients with CMT4J and polymicrogyria retain partial function of FIG4. Consistent with this view, the missense mutations found in patients with YVS (figure 2B) exhibit complete loss of function in the rescue of vacuolization by Fig4-null fibroblasts,15 while partial function is demonstrated for the missense mutations p.Ile41Thr16 and p.Asp783Val (this study). Spongiform neurodegeneration is only seen with complete loss of FIG4 function in the plt mouse and patients with YVS. Patients with CMT4J are compound heterozygotes carrying a null allele in combination with the partial loss-of-function allele p.Ile41Thr mutation14,21 (figure 2B). The p.Ile41Thr mutation impairs protein stability and interaction with the scaffold protein VAC14, but overexpression of the mutant protein can rescue lethality of the plt mouse, demonstrating residual function.16 Based on their location in different protein domains, the p.Asp783Val mutation causing polymicrogyria might impair interaction with a different set of proteins than p.Ile41Thr, thereby resulting in cortical malformation rather than peripheral neuropathy. There is some phenotypic overlap between the disorders caused by FIG4 mutations, because patients with YVS exhibit brain malformations including underdeveloped gyri and polymicrogyria that are the hallmark of the phenotype associated with the p.Asp783Val mutation.22,23 Interestingly, Walch et al.22 noted that in one patient with YVS, the areas of neuronal vacuolization did not overlap with areas exhibiting pachygyria and polymicrogyria, indicating that the process of neurodegeneration might mask preceding cortical abnormalities. Two features present in the family we are describing could also modulate conclusions on phenotype-genotype correlations: (1) we noted enlarged cerebral ventricles at MRI in patients V.8, V.9, and V.11,13 a feature also seen in Fig4-null mice,14 which might reflect a neurodegenerative process much milder than that occurring in YVS; and (2) an axonal sensory and motor neuropathy was diagnosed in patient V.9 that is related to the peripheral neuropathy occurring in CMT4J.13 However, in patients with CMT4J, the pathologic process is also demyelinating and associated with an age at onset that is variable, sometimes occurring in childhood. Because other patients of the family did not exhibit signs of neuropathy at age 23 (V.11), 35 (V.8), or 55 (V.1) years, it is uncertain whether the neuropathy diagnosed in patient V.9 is related to the FIG4 mutation. In addition, the electrophysiologic examination of V.8 was fully normal.
Polymicrogyria is a common cortical malformation and is associated with different morphologic patterns and syndromes. Its pathogenesis is not fully understood; brain pathology demonstrates abnormal development or loss of neurons in middle and deep cortical layers, variably associated with an unlayered cortical structure.24 Clinical, pathologic, and imaging heterogeneity suggest that polymicrogyria is not a single malformation. Different types of single gene inheritance have been reported, and causative mutations have been identified in several genes such as GPR56,2 KBP,3 NHEJ1,25 TUBB2B,8 TUBB3,9 TUBA8,10 and TUBA1A7 as well as RAB3GAP1,4 RAB3GA2,5 and RAB186 encoding Ras-related small GTPases that regulate membrane trafficking in organelles and transport of endolysosomal vesicles. The function of FIG4 is related to the Ras GTPases because it is involved in the control of intracellular PI(3,5)P2 concentration. The coordinated regulation of specific RABs and phosphoinositides is emerging as an important mechanism to ensure precision and fidelity of membrane trafficking,26 which seems to be crucial for postmigrational development of neurons.
Our findings implicate FIG4 in the pathogenesis of a temporo-occipital polymicrogyria associated with epilepsy and psychiatric manifestations. It is hoped that this discovery will shed light on the complex biology underlying the formation of cortical gyri and the role of PI(3,5)P2 signaling in this process.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Murat Gunel and Jamel Chelly for providing DNAs of patients with cortical malformations; Fiona Francis and Mihaela Vlaicu for helpful discussion; Ashley Miller and Margaret Wu for assistance with quantitation of the vacuole area in cultured cells; and Frédéric Texier and Hélène Blanquart for exome sequencing and the ICM cell imaging platform and ICM histology platform.
GLOSSARY
- cDNA
complementary DNA
- chr6
chromosome 6
- CMT4J
Charcot-Marie-Tooth disease type 4J
- GFP
green fluorescent protein
- plt
pale tremor
- WT
wild-type
- YVS
Yunis-Varón syndrome
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
S.B., B.O.A.B., P.C., J.R., E.N., K.P., C.H., S.F. collected and analyzed genetic data. G.M.L., P.A.L., C.J.F collected and analyzed data on functional studies. B.D., K.H.E.H. collected and analyzed neuropathologic data. R.G. analyzed neuroimaging and participated in the writing of the manuscript. R.O. collected and analyzed clinical data. S.B., M.H.M, E.L. supervised the work and wrote the manuscript.
STUDY FUNDING
S. Baulac received research support from Carnot Institute (CT010) and from the program Investissements d'avenir ANR-10-IAIHU-06. G.M. Lenk received research support from NIH R01 GM24872 and NIH U01 TR000433. B.O.A. Bencheikh received research support from the University Mohammed V-Souissi of Rabat in Morocco. K. Poirier received support from the Agence National de Recherche (ANR Blanc 1103 01, project R11039KK; ANR E-Rare-012-01, project E10107KP).
DISCLOSURE
S.B., G.M.L., B.D., B.O.A.B., P.C., J.R., C.J.F., E.N., K.P., R.O., K.H.E.H., M.H.M., and E.L. report no disclosures. P. Larson received research support from NIH R01 GM24872. C. Hubans and S. Ferreira are employees of GenoScreen company. R. Guerrini received honoraria from Biocodex, UCB, Eisai Inc., ValueBox, and ViroPharma, and research support from the Italian Ministry of Health, the European Community Sixth and Seventh Framework Thematic Priority Health, the Italian Ministry of Education, University and Research, the Tuscany Region, the Telethon Foundation, and the Mariani Foundation. R. Ouazzani received research support from the Programme d’Action Intégrée MA/108/04 between Morocco and France. K.H. El Hachimi reports no disclosures relevant to the manuscript. M. Meisler received research support from NIH R01 GM24872. E. Leguern received research support from Carnot Institute (CT010) and from the program Investissements d’avenir ANR-10-IAIHU-06. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012;135:1348–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piao X, Hill RS, Bodell A, et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004;303:2033–2036 [DOI] [PubMed] [Google Scholar]
- 3.Valence S, Poirier K, Lebrun N, et al. Homozygous truncating mutation of the KBP gene, encoding a KIF1B-binding protein, in a familial case of fetal polymicrogyria. Neurogenetics 2013;14:215–224 [DOI] [PubMed] [Google Scholar]
- 4.Morris-Rosendahl DJ, Segel R, Born AP, et al. New RAB3GAP1 mutations in patients with Warburg Micro syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur J Hum Genet 2010;18:1100–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borck G, Wunram H, Steiert A, et al. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet 2011;129:45–50 [DOI] [PubMed] [Google Scholar]
- 6.Bem D, Yoshimura S, Nunes-Bastos R, et al. Loss-of-function mutations in RAB18 cause Warburg Micro syndrome. Am J Hum Genet 2011;88:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poirier K, Keays DA, Francis F, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat 2007;28:1055–1064 [DOI] [PubMed] [Google Scholar]
- 8.Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet 2009;41:746–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poirier K, Saillour Y, Bahi-Buisson N, et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 2010;19:4462–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdollahi MR, Morrison E, Sirey T, et al. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet 2009;85:737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sertie AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum Mol Genet 2000;9:2051–2058 [DOI] [PubMed] [Google Scholar]
- 12.Roll P, Rudolf G, Pereira S, et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet 2006;15:1195–1207 [DOI] [PubMed] [Google Scholar]
- 13.Ben Cheikh BO, Baulac S, Lahjouji F, et al. A locus for bilateral occipital polymicrogyria maps to chromosome 6q16-q22. Neurogenetics 2009;10:35–42 [DOI] [PubMed] [Google Scholar]
- 14.Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007;448:68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campeau PM, Lenk GM, Lu JT, et al. Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013;92:781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenk GM, Ferguson CJ, Chow CY, et al. Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet 2011;7:e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahlsten D, Andison M. Patterns of cerebellar foliation in recombinant inbred mice. Brain Res 1991;557:184–189 [DOI] [PubMed] [Google Scholar]
- 18.Ferguson CJ, Lenk GM, Jones JM, et al. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet 2012;21:3525–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham H, Orsi G, Seress L. Ontogeny of cocaine- and amphetamine-regulated transcript (CART) peptide and calbindin immunoreactivity in granule cells of the dentate gyrus in the rat. Int J Dev Neurosci 2007;25:265–274 [DOI] [PubMed] [Google Scholar]
- 20.Rami A, Brehier A, Thomasset M, Rabie A. Cholecalcin (28-kDa calcium-binding protein) in the rat hippocampus: development in normal animals and in altered thyroid states—an immunocytochemical study. Dev Biol 1987;124:228–238 [DOI] [PubMed] [Google Scholar]
- 21.Nicholson G, Lenk GM, Reddel SW, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain 2011;134:1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walch E, Schmidt M, Brenner RE, et al. Yunis-Varon syndrome: evidence for a lysosomal storage disease. Am J Med Genet 2000;95:157–160 [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni ML, Vani HN, Nagendra K, et al. Yunis Varon syndrome. Indian J Pediatr 2006;73:353–355 [DOI] [PubMed] [Google Scholar]
- 24.Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci 2008;31:154–162 [DOI] [PubMed] [Google Scholar]
- 25.Cantagrel V, Lossi AM, Lisgo S, et al. Truncation of NHEJ1 in a patient with polymicrogyria. Hum Mutat 2007;28:356–364 [DOI] [PubMed] [Google Scholar]
- 26.Jean S, Kiger AA. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol 2012;13:463–470 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.