

Abstract

Combination of AChE inhibiting and histamine H3 receptor antagonizing properties in a single molecule might show synergistic effects to improve cognitive deficits in Alzheimer’s disease, since both pharmacological actions are able to enhance cholinergic neurotransmission in the cortex. However, whereas AChE inhibitors prevent hydrolysis of acetylcholine also peripherally, histamine H3 antagonists will raise acetylcholine levels mostly in the brain due to predominant occurrence of the receptor in the central nervous system. In this work, we designed and synthesized two novel classes of tri- and tetracyclic nitrogen-bridgehead compounds acting as dual AChE inhibitors and histamine H3 antagonists by combining the nitrogen-bridgehead moiety of novel AChE inhibitors with a second N-basic fragment based on the piperidinylpropoxy pharmacophore with different spacer lengths. Intensive structure–activity relationships (SARs) with regard to both biological targets led to compound 41 which showed balanced affinities as hAChE inhibitor with IC50 = 33.9 nM, and hH3R antagonism with Ki = 76.2 nM with greater than 200-fold selectivity over the other histamine receptor subtypes. Molecular docking studies were performed to explain the potent AChE inhibition of the target compounds and molecular dynamics studies to explain high affinity at the hH3R.
Keywords: Acetylcholinesterase inhibitors, histamine H3 antagonists, Alzheimer’s disease, docking, MD simulation
Alzheimer’s disease (AD) is an irreversible, progressive neurodegenerative brain disorder and the most common form of dementia. It is estimated that about 6% of the population worldwide aged over 65 currently suffer from AD.1 Worldwide, AD afflicts more than 35 million people.2,3 The occurrence of dementia increases exponentially with increasing age, and it is expected to more than triple during the next 30 years.4 This alarming prognosis demands further effort to elucidate the molecular mechanisms of AD and search for effective treatment methods.
The cholinergic hypothesis, proposed by Davies and Maloney in 1976, is one of the earliest and most extensively researched theories regarding AD pathogenesis.5 It has led to the introduction of tacrine as the first drug to be approved for the treatment of AD, and subsequently galantamine, donepezil, and rivastigmine, respectively. All these compounds represent inhibitors of acetylcholinesterase (AChE), the inhibition of which increases the amount of the neurotransmitter acetylcholine (ACh) in the synaptic cleft. Clinically this leads to a symptomatic, but significant improvement of cognitive deficits in early stages of AD. Together with the symptomatically acting N-methyl-d-aspartate (NMDA) blocker memantine, these compounds represent the only therapeutic treatment of AD currently available.
The histamine H3 receptor is postulated as a G protein-coupled receptor (GPCR) drug target for several cognitive disorders including attention deficit hyperactivity disorder (ADHD), AD, and schizophrenia.6 The H3 receptor had originally been described as a presynaptic autoreceptor that inhibits histamine release in the brain,7 and was subsequently shown also to regulate the release of other important neurotransmitters such as acetylcholine, glutamate, serotonin, dopamine, norepinephrine, gamma-aminobutyric acid (GABA), and others via a parallel role as a heteroreceptor.8,9 Therefore, blocking these receptors could be promising for potential treatment of central nervous system (CNS) disorders such as AD,9 since blocking the central hH3 receptor can induce the release of procognitive neurotransmitters like acetylcholine (ACh). Due to their multiple therapeutic possibilities including cognitive deficits, there has been a very considerable and remarkable progress in the development of hH3R antagonists both in academia and especially pharmaceutical industry.8,10 Johnson & Johnson have reported on dual-acting H3 antagonists and serotonin transporter inhibitors, and Hoffmann-LaRoche as well as Hudkines et al. on H3 ligands with piperidinylpropoxyphenyl pharmacophores (Chart 1).11−14
Chart 1. Schematic Illustration of the Merging Design Methodology for Synthesizing Dual Acting AChE inhibitors/H3 Antagonist Hybrid Structures11−13.

The complexity and multiple etiologies of AD make single-target strategies difficult to achieve desirable therapeutic effects and make the development of multitarget-directed ligands (MTDL) a more effective strategy, as intensively applied in recent years using very diverse biological targets.14−17
Recently, dual-acting AChE inhibitors and hH3 receptor antagonists have been developed, since such multitarget-directed ligands might be able to achieve a more specific cognition-enhancing level of cholinergic neurotransmission than either an AChE inhibitor or histamine H3 receptor antagonist alone. For example, such dual-acting compounds have been obtained from H3 receptor antagonists that additionally contain a tacrine-based molecular structure. These compounds act at hH3, N-methyltransferase (HMT), AChE, and butyrylcholinesterase (BChE), respectively.18 The most effective compound FUB-833 4 (Chart 2) displayed good affinity for hH3 receptors, AChE and HMT.18 Despite these exceptional activities, tacrine-based compounds always bear the risk of severe hepatotoxicity, the reason why it was withdrawn from the market.14,19,20
Chart 2. Previously Described Acetylcholinesterase Inhibitors/Histamine H3 Receptor Antagonists18,21−23.

Several groups have also reported on interesting dual AChE inhibitors and H3 antagonists.22−24 Unfortuantely, these compounds act at much lower concentration (nM) on H3 than on AChE (μM).
Significant amount of knowledge with regard to structure–activity relationships (SARs) of H3 antagonists, including the group of piperidinylpropoxyphenyl pharmacophore containing compounds, is available. In some cases, it was also possible to synthesize dual acting H3/AChE compounds, but despite the fact that the principle of combination seems to work, it has not yet been possible neither to avoid large molecular weight tacrine-containing compounds nor to describe compounds with high affinities in the same concentration range at both targets, a core problem of hybrid and dual acting compounds albeit rarely discussed and addressed in the literature.15 Gaining balanced affinities is a very demanding task, since compounds can follow completely diverse SARs at the different targets to be addressed.
Previous SARs on numerous H3 ligands described show that the phenol ethers in meta position to tertiary benzyl amine possess high H3 receptor binding affinity and antagonistic potency, for example, compounds 1, 2 (Chart 1); similarly, the presence of an anilinic amine in para position to the phenolic ether is favorable, e. g. compound 2 (Chart 1). Finally, connecting the basic cyclic amine through an alkyl spacer of variable length to a phenolic OH group of heterocyclic and/or aromatic rings is a key feature to get H3 receptor binding affinity and antagonistic potency (Charts 1 and 2).
We have previously synthesized a series of novel tri- and tetracyclic N-bridgehead AChE inhibiting lead structures with moderate activity (i.e., two digit micromolar) characterized by a phenolic hydroxyl group in the para position to a tertiary anilinic and in the meta position to the basic N-bridgehead atom and used them as starting points for the development of novel butyrylcholinesterase selective inhibitors (compound 8) (Chart 3).24 The cholinesterase inhibitory activities of these classes of compounds and their selectivity toward BChE were dramatically improved by introducing a carbamate group at the phenolic hydroxyl group (compound 9) (Chart 3).24 The chemical features of these two moieties (tri- and tetracyclic) served as the starting point for our SAR studies in order to synthesize a novel series of dual acting compounds as AChE inhibitors/H3 receptor antagonists due to their similarity to the aromatic and/or heterocyclic parts of the above-described H3 ligands (Chart 1) and then, combining the phenolic heterocycle as an ether to tertiary amines.
Chart 3. Tri- and Tetracyclic Compounds As Starting Points for H3/AChE Dual Acting Compounds’ SARs24.

With the aim to investigate SARs of this class of cholinesterase inhibitors, we synthesized novel heterocyclic templates consisting of basic tri- and tetracyclic N-bridgehead molecules with phenolic ether connected to the basic cyclic amine moieties by different carbon spacer lengths. The ability of these structural templates to antagonize selectively the H3 receptor and the effect of structural modifications on cholinesterase inhibitory activity were intensively investigated with the ultimate goal to find structures with high affinity/activity at both targets (i.e., balanced activities).
The results of SARs at AChE were used for computational docking studies of the inhibitors on AChE. Additionally, the binding mode of one of the most potent target compounds at the hH3 was investigated by molecular dynamics (MD) studies.
We intended to synthesize both tri- and tetracyclic compounds, different spacer lengths, different basicities within the heterocycle, various structures of the tertiary amines, and related structural features to cover systematically a range of chemical structures for investigating SARs at both targets.
Chemistry
Scheme 1 shows the synthesis of the tetracyclic target compounds. The starting material 6-(benzyloxy)-1H-benzo[d][1,3]oxazine-2,4-dione 11 was synthesized in four steps with almost quantitative overall yield, starting from 2-amino-5-hydroxybenzoic acid. The resulting isatoic anhydride represents an activated form of anthranilic acid with the benzylated phenolic–OH, the benzylation of the phenolic group in compound 11 is necessary to avoid methylation of the phenolic group, and to improve the solubility of the resulting isatoic anhydride.25
Scheme 1. Synthesis of Tetracyclic N-Bridgehead Compounds.

Reagents: (i) CH3I, N,N-diisopropylethylamine, N,N-dimethylacetamide, 40 °C, 24 h; (ii) toluene/reflux, 24 h; (iii) H2, Pd/C, ethanol, rt, 24 h; (iv) 1,3-dibromopropane or 1,6-dibromohexane, K2CO3, KI, acetonitrile, 90 °C, 3 h; (v) piperidine, morpholine, pyrrolidine, or azepane, K2CO3, KI, acetonitrile, 90 °C, 3 h; (vi) LiAlH4, THF, 70 °C, 3 h.
10-Hydroxy-13-methyl-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one 14 was synthesized as shown in Scheme 1: N-Methylation of 6-(benzyloxy)-1H-benzo-[d][1,3]oxazine-2,4-dione 11 in the presence of N,N-diisopropylethylamine as a base and iodomethane as an alkylating agent in N,N-dimethylacetamide as a solvent afforded compound 12.26 Compound 12 was fused with 3,4-dihydroisoquinoline in toluene under reflux overnight to afford compound 13 in an acceptable yield (40%). After benzyl deprotection by hydrogenation over Pd/C in ethanol at room temperature, compound 14 was obtained in quantitative yield. Reaction of compound 14 with 1,3-dibromopropane or 1,6-dibromohexane in the presence of potassium carbonate as a base with a catalytic amount of potassium iodide in acetonitrile as solvent for 3 h under reflux yielded compounds 15 or 16, respectively. The last synthetic step was nucleophilic substitution by reaction of bromo spacer compounds 15 or 16 with different cyclic amines using again potassium carbonate as a base with a catalytic amount of potassium iodide in acetonitrile as a solvent to yield the first series of quinazolinone target compounds 17–21. The second set of N-bridgehead target compounds was synthesized by reduction of the amide group of compounds 17–21 using LiAlH4 to give compounds 22–26.
Tricyclic target compounds were synthesized by a related synthetic procedure as for the tetracyclic compounds as shown in Scheme 2. Solventless fusion of pyrrolidin-2-one with compound 11 under microwave conditions with 200 W irradiation power at 220 °C for 1 h gave compound 27, which was then debenzylated by catalytic hydrogenation over Pd/C in ethanol at room temperature to give compound 28. Reaction of compound 28 with 1,3-dibromopropane or 1,6-dibromohexane in a Williamson ether synthesis gave compounds 29 or 30, respectively. Reaction of different cyclic amine with compounds 29 or 30 under similar reaction conditions as for compounds 17–21 yielded the series of tricyclic quinazolinone target compounds 31–34. Tricyclic compounds 35–38 were synthesized using the same reduction conditions used for the preparation of tetracyclic compounds 22–26 (LiAlH4).
Scheme 2. Synthesis of Tricyclic N-Bridgehead Compounds (quinazolinones and diamines).

Reagents: (i) MW, 200 W, 220 °C, 1 h; (ii) H2, Pd/C, ethanol, rt, 24 h; (iii) 1,3-dibromopropane or 1,6-dibromohexane, K2CO3, KI, acetonitrile, 90 °C, 3 h; (iv) pyrrolidine, piperidine, or azepane, K2CO3, KI, acetonitrile, 90 °C, 3 h; (v) LiAlH4, THF, 70 °C, 3 h.
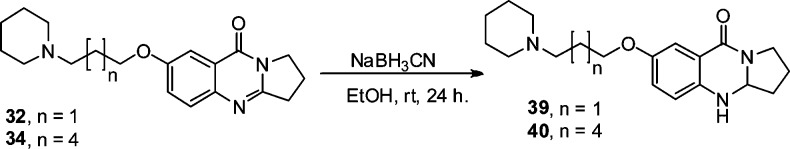
Scheme 3 shows the reaction conditions for reduction of the imine group of compounds 32 and 34 to the corresponding amines using sodium cyanoborohydride in ethanol at room temperature for 24 h to yield compounds 39 and 40, respectively.27
Scheme 3. Selective Reduction of Quinazolinone Imine Bond to the Corresponding Amine.
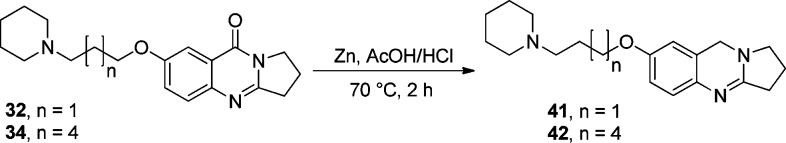
The carbonyl group of quinazolinones (compounds 32 and 34) could be selectively reduced using Zn metal powder in the presence of acetic acid as a solvent to give compounds 41 and 42, respectively. The reaction mixture was then heated at 70 °C for 2 h after a few drops of conc. hydrochloric acid had been added as shown in Scheme 4.28
Scheme 4. Selective Reduction of Quinazolinones by Clemmensen Reduction.
Results and Discussion
A novel and potent class of dual acting AChE inhibitor/H3 antagonist hybrid compounds was developed by connecting the tri- and tetracyclic N-bridgehead structural templates via a hydroxyl group, in para position to anilinic N, with different cyclic amines spaced by three or six carbon atom linkers. A detailed look at the respective SARs reveals several interesting insights, also of importance to both the development of AChE inhibitors as well as H3 antagonists individually.
All target compounds were tested for their ability to inhibit acetylcholinesterase (EeAChE, EC.3.1.1.7 from electric eel) and butyrylcholinesterase (BChE, EC.3.1.1.8 from equine serum). Both enzyme isoforms exhibit very high amino acid sequence homology with regard to the human ones (88% and 90%, respectively).29,30 Inhibitory activity at BChE, a plasma-soluble isoform of AChE, has recently gained attention for dementia-related drug development since the amount of AChE decreases during the course of the disease, whereas this seems not to be the case for BChE. Therefore, BChE inhibition might compensate for the lack of efficiency of administration of AChE inhibitors in later stages of AD.24
Additionally, selected compounds were tested for their inhibitory activity on human acetylcholinesterase hAChE (E.C.3.1.1.7, acetylcholinesterase from human erythrocytes) to confirm binding profiles and be able to compare the data with the docking results on the human enzyme.
AChE Inhibitor/H3 Antagonist Tetracyclic Hybrid Compounds
With regard to the tetracyclic template, we had previously synthesized a series of potent and selective carbamate-based (pseudo)irreversible BChE inhibitors. The free phenolic tetracyclic compound 8 displayed a low inhibitory activity at both cholinesterases (IC50 (EeAChE) = 110.3 μM, IC50 (BuChE) = 44.3 μM) (Table 1). The low activity was attributed to low basicity and poor solubility of this tetracycle moiety. However, in this project, the same template unexpectedly showed submicromolar inhibitory activity toward AChE and one digit micromolar inhibitory activity toward BChE, simply due to connection with different cyclic amines spaced by three carbon atoms (18, 20, and 21). For example, compound 18 (IC50 (EeAChE) = 0.55 μM) showed 200-fold higher inhibitory activities than those for the free phenolic tetracyclic lead structure compound 8 (Chart 3) (IC50 (EeAChE) = 110.3 μM). Interestingly, the morpholino compound 19 has an IC50 (EeAChE) = 29.81 μM and IC50 (BChE) = 8.5 μM, being the only compound with higher BChE than AChE inhibitory activity in this set of compounds (Table 1), albeit with lower inhibitory activity at AChE.
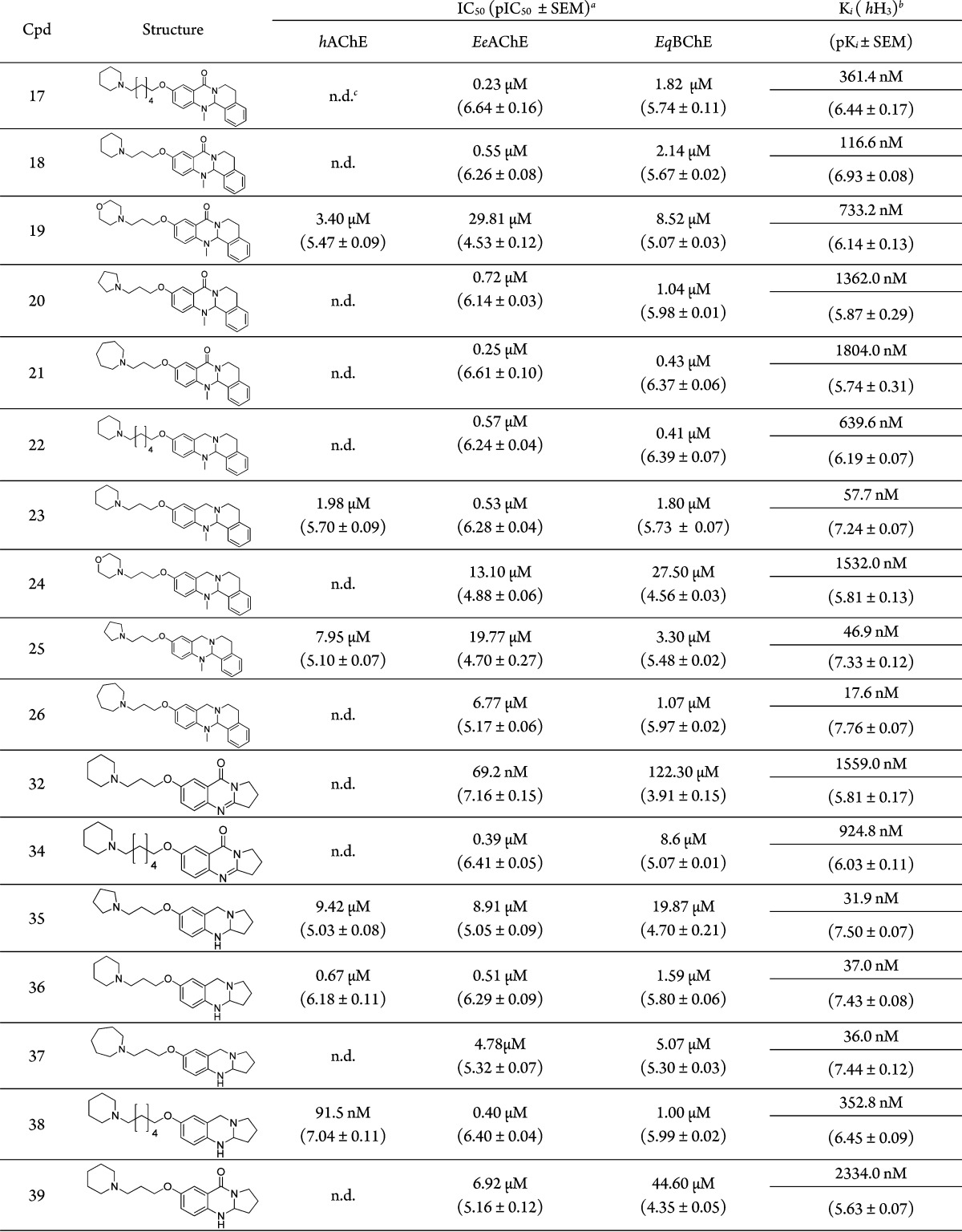
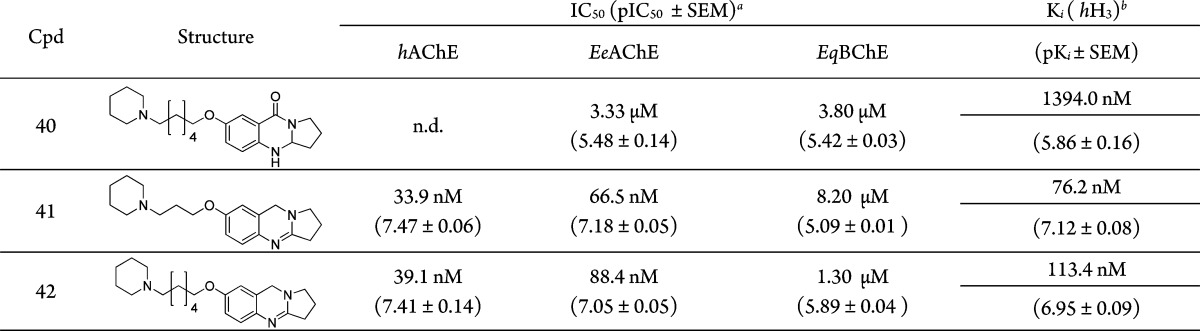
Table 1. Chemical Structures, ChE Inhibition Results, and hH3R Binding Data for Tri- and Tetracyclic Synthesized Compounds.
AChE, BChE: data are the means of three independent determinations.
hH3R binding affinity: data are means ± standard deviation of triplicate independent experiments.
n.d.: Not determined.
According to our previously published work, we expected that the inhibitory activity of this heterocycle will increase by amide reduction through increasing the basicity of the core ring, but surprisingly, the AChE inhibition activity of this set of the tetracyclic compounds (22–26) is lower than that of the same set of compounds before reduction (17–21), but still in the micromolar range (from 0.53 μM for compound 23 to 19.8 μM for compound 25). Therefore, selectivity and activity profiles for these sets of compounds depend more on the structure of the alicyclic N-ring connected to the heterocycle than the heterocycle itself. We investigated this feature in the molecular docking studies (see below) with regard to the orientation of the molecule within the enzyme.
As we had planned from the compound design, H3 binding data for these quinazolinone compounds with an amide carbonyl group (compounds 17–21) (Scheme 1), showed binding affinity in the micromolar range. The corresponding reduced tetracyclic compounds (23, 25, and 26) (Scheme 1) displayed a moderate inhibitory activity toward AChE in the micromolar range, while this set of compounds showed high binding affinity at H3 in two digit nanomolar range (Ki (hH3) = 57.7, 46.9, and 17.5 nM respectively) (Table 1).
Interestingly, the tetracyclic compounds connected with morpholine as a basic center showed poor cholinesterase inhibition affinities, compound 19 with IC50 (EeAChE) = 29.81 μM and IC50 (BChE) = 8.5 μM, and its corresponding reduced form compound 24 with IC50 (EeAChE) = 13.1 μM and IC50 (BChE) = 27.5 μM. Both compounds showed also poor binding affinities toward the H3 receptor (Table 1).
We also evaluated the influence of the carbon spacer length on the biological activity for the tetracyclic moiety by connecting the tetracyclic moiety with piperidine spaced by six carbon atoms (compound 17), and the corresponding reduced form (compound 22). Compound 22 showed inhibitory activity in submicromolar range (IC50 (EeAChE) = 0.57 μM) toward AChE, and binding toward hH3R in the same range (Ki (hH3) = 0.64 μM). In contrast, the corresponding three carbon atom spacer length compound 23 showed also submicromolar inhibition activity range toward AChE (IC50 (EeAChE) = 0.53 μM), but significantly improved the binding affinity toward hH3R (Ki (hH3) = 57.7 nM). So, a three carbon spacer length is better for H3 affinity with regard to the tetracyclic moiety (Table 1).
AChE Inhibitor/H3 Antagonist Tricyclic Hybrid Compounds
The tricyclic quinazolinone compound 10 (Chart 3) showed moderate inhibitory activities toward both enzymes (IC50 (EeAChE) = 82.5 μM and IC50 (BChE) = 25.1 μM).31 Surprisingly, the same template showed two digit nanomolar inhibitory activities toward AChE and one digit micromolar toward BChE by connecting it with different cyclic amines spaced by three C atoms (compounds 31, 32, and 33 depicted in Scheme 2). This fact and the fact that these compounds did not show high binding affinity toward the H3 receptor (compound 32 with Ki (hH3) = 1.56 μM) (Table 1) intensively made us study the binding mode of this class of compounds toward AChE in a separate project by synthesis of additional compounds and computational studies. Results will be reported separately.
We also evaluated the influence of the spacer length for this moiety, and again three carbon spacer length is better for achieving dual acting compound from the tricyclic moiety, when compound 32 with three carbon spacer length showed IC50 (EeAChE) = 69.2 nM and (hH3) = 1.6 μM, while the same moiety with six carbon spacer length compound 34 showed IC50 (EeAChE) = 390 nM and (hH3) = 0.92 μM (Table 1).
For this moiety, we evaluated the effect and biological consequences of the amide oxygen, the double bond, and the resulting basicity differences of the core ring of tricyclic quinazolinone compounds (31–34). First, total reduction of the core carbonyl and imine groups using LiAlH4 afforded the diamine class of compounds 35–38; these amines showed good inhibitory activity toward AChE (submicromolar to one digit micromolar range) and showed a high binding affinity at the H3 receptor (two digit nanomolar range) for compounds 35, 36, and 37 (Ki (hH3) = 31.9, 37.0, 36.0 nM), respectively. Second, reducing the imine group in the quinazolinone core ring using NaBH3CN yielded its corresponding amine (39, 40), but these compounds lost their AChE inhibitory activity and showed very weak binding at H3 receptor (compound 39 with IC50 (EeAChE) = 6.92 μM and (hH3) = 2.3 μM, and compound 40 with IC50 (EeAChE) = 3.33 μM and (hH3) = 1.4 μM). Third, selective reduction of the carbonyl group was achieved by Zn/AcOH reduction (Clemmensen reaction) and left the imine double bond as it is (41, 42). This set of compounds showed a balanced activity toward our two targets; they act as balanced acetylcholinesterase inhibitors/hH3 antagonists, with compound 41 showing IC50 (hAChE) = 34 nM, IC50 (EeAChE) = 67 nM, and Ki (hH3) = 76 nM. Similarly, compound 42 showed IC50 (hAChE) = 39 nM, IC50 (EeAChE) = 88 nM, and Ki (hH3) = 113 nM (Table 1).
Balanced activity, that is, compound activity and affinity, respectively, at the relevant targets in the same or at least a similar concentration range, is a therapeutically highly relevant issue, that is very rarely properly addressed in research efforts on hybrid and multitarget compounds. This is not surprising, since two, often distinct and independent, SARs have to be investigated at each target, and even if a balanced activity is reached for one compound, it is rarely the highest affinity/activity of the set of compounds described.
In general, all synthesized target compounds represent moderate or potent AChE inhibitors with inhibitory activities ranging from 30 μM (compound 19) down to the two digit nanomolar range (67 nM for compound 41) with roughly 500-fold higher activity. This compound is in the range of the “gold-standard” of AChE inhibition, tacrine.
Affinity at the H3 receptor turned out to be very high as planned and desired from the compounds’ design. Since none of our structures had been tested for H3 affinity before, not even our ChE inhibiting lead structures, this fact proved our assumptions concerning ligand design. With regard to the H3 affinities, they ranged from the (this time one digit) micromolar range (Ki (hH3) = 2.3 μM for compound 39) to the nanomolar range (Ki (hH3) = 18 nM for compound 26) covering a range of 2 orders of magnitude. Not surprisingly, SARs follow different routes than for AChE inhibition as described in detail above.
Pharmacology
In addition to determination of inhibitory activities, compound 41 was selected for kinetic measurements, in order to understand the mode of inhibition and binding of this inhibitor at EeAChE. The mechanism of inhibition was analyzed by recording substrate–velocity curves in the presence of different concentrations of compound 41 using 50–450 μM acetylthiocholine (ATC) substrate EeAChE. Figure 1 shows the Lineweaver–Burk plot (reciprocal rates versus reciprocal substrate concentrations for the different inhibitor concentrations resulting from the substrate–velocity curves for EeAChE). In this Lineweaver–Burk plot, the Km values differ with the different inhibitor concentrations, while the vmax value remains unchanged. The Lineweaver–Burk plot for EeAChE shows reversible and competitive inhibition by compound 41.
Figure 1.
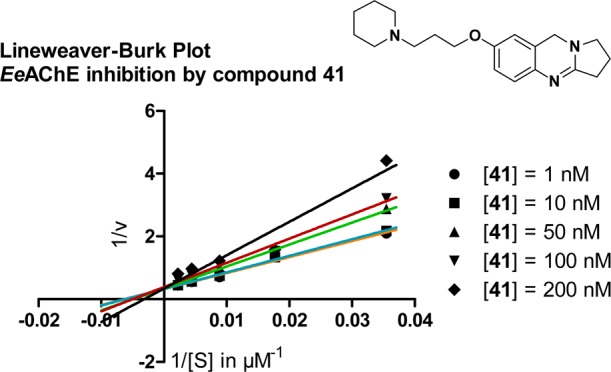
Lineweaver–Burk plot resulting from substrate–velocity curves of EeAChE activity with different substrate (ATC) concentrations (50–450 μM) in the presence of 1, 10, 50, 100, and 200 nM compound 41.
Affinity toward the human histamine H3 receptor was determined by displacement of specific [3H]-histamine binding to H3 receptor binding sites in Sf9 insect cell membranes32 (Table 1).
Putative affinity at other hH receptor subtypes, that is, H1, H2, and H4, respectively, was determined by testing high concentration (10 μM) of the most potent compounds at hH3R for displacement of [3H]mepyramine (hH1R), [3H]UR-DE257 (hH2R),33 and [3H]-histamine (hH4R) (Figure 2). Generally, all of the tested compounds exhibited a very good selectivity profile, since none of the tested compounds have affinity at hH4R and hH2R even at a concentration of 10 μM. At 10 μM, some of the tested compounds showed to slightly interact with hH1R (Figure 2).
Figure 2.
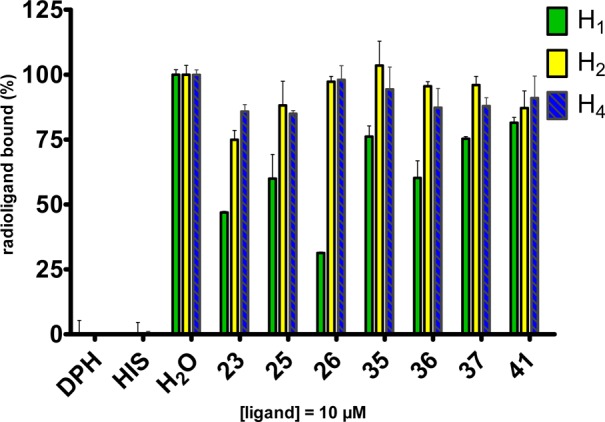
Binding assay results toward hH1R, hH2R, and hH4R, applying a 10 μM concentration of the most potent hH3R antagonist compounds. Diphenhydramine (DPH) as hH1R standard ligand and histamine (HIS) as hH2R, and hH4R standard ligand.
Determination of whether the synthesized compounds possess antagonist, agonist, or inverse agonist properties was investigated by steady-state GTPase activity assay. The functional assay (GTPase) was performed at 10 μM concentration of all synthesized compounds. As a reference hH3R antagonist, 10 μM concentration of thioperamide was used (Figure 3). Almost all the tri- and tetracyclic synthesized compounds are hH3R antagonists or inverse agonists; only compound 24, which is connected with morpholine by three carbon spacer length, showed slight agonist properties at hH3R.
Figure 3.
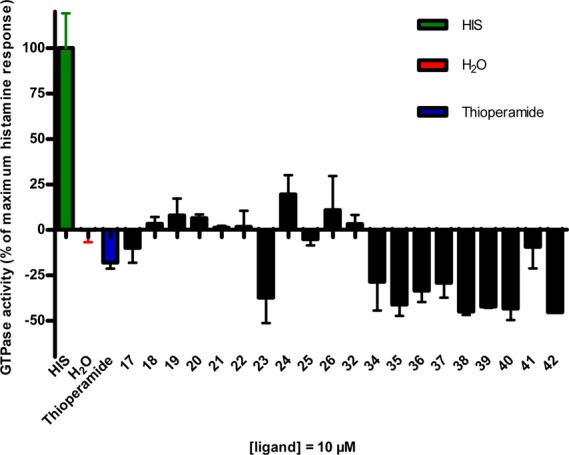
Functional assay (GTPase) results at the hH3 receptor applying a 10 μM concentration of all synthesized compounds with water as a baseline and thioperamide as control (antagonist).34
Computational Docking Studies on Human AChE
Docking studies were carried out on AChE to investigate the putative preferred binding mode of the compounds in the inhibitor library. Docking studies with the structure of human AChE (PDB 4EY7)36 and consideration of conserved water molecules (cf. Methods) suggest two possible modes of inhibition: a “classical” binding mode corresponding to the placement of the tri- or tetracyclic scaffold near the catalytic active site (CAS) at the bottom of the binding gorge; and an “inverted” binding mode showing the protonated tertiary amine instead of the aromatic scaffold deep in the binding gorge. In the latter, cation−π interactions are formed with mostly Trp86 or Tyr337 (hAChE numbering), residues known for attracting the tertiary amine of the natural substrate acetylcholine.21
The ligands were docked in their expected ionization state at pH 8. This means protonation at the tertiary amine of all compounds, consistent with their pKa values between 10.13 and 9.58 as calculated with MoKa.37 A pKa of 7.66 was calculated for the morpholine substituent. The aminal structure in compounds 35–38 is protonated due to a calculated pKa of 8.73. Similarly, the amidine moiety was protonated in the inhibitors 41 and 42 with a calculated pKa of 10.72 for both scaffolds resulting in double protonation of these compounds.
A very clear preference for the inverted binding mode is shown by the most potent compounds, for which good clustering of the docking results in the top poses was obtained. This includes the nonchiral tricyclic inhibitors 32, 34, 41, and 42, but also the chiral tricyclic compounds 36 and 38, which are closely related to 41 and 42, respectively (Figure 4). The quinazolinone compounds 31 and 33 also fit excellently in here; the binding as well as the docking results for a whole set of quinazolines will be presented in a subsequent publication. The piperidinyl ring of 41 and 42 is placed near the key residue of the choline binding site, Trp86. The alkyl chains point away from the water molecules in the binding site, so that the tricyclic ring systems are placed near the peripheral anionic site (PAS) interacting with the Tyr72 hydroxyl group or the backbone carbonyl of Tyr341. All of the mentioned compounds (except 34: 3.8) achieve a per-atom-score (GoldScore/no. heavy atoms) of >4, indicating influential contributions of most atoms to the overall interaction. In contrast, the weaker inhibitors generally show lower per-atom-scores and/or inconsistent binding-orientation preferences for the two enantiomeric forms. Yet, for the most active tetracyclic compounds with a carbonyl function (17 and 21) the inverted binding mode appears to be preferred over the classical binding mode for both enantiomers with comparable score. In this binding mode, a 3.0–3.6 Å hydrogen distance bond can be observed between the carbonyl oxygen and a conserved water molecule (Figure 4).
Figure 4.
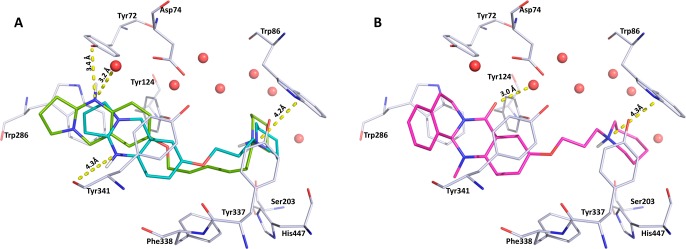
(A) Docking pose showing the inverted binding mode of inhibitor 41 (cyan) with a 4.2 Å cation−π interaction to Trp86 and potential hydrogen bonds to a water molecule (3.2 Å) and Tyr72 (3.4 Å). Inhibitor 42 (green) shows the same placement of the piperidine ring near the CAS (4.2 Å to Trp86) and a distance of 4.3 Å to the Tyr341 backbone carbonyl. (B) Selected docking pose of the tetracyclic inhibitor 21 (pink, R-enantiomeric form) showing the inverted binding mode. The inhibitor is stabilized by a 3.0 Å interaction of the scaffold carbonyl oxygen with a water molecule and a 4.3 Å cation−π interaction of the piperidine with Trp86 near the CAS. Color code: red, conserved water molecules; gray, hAChE binding site; catalytic active site, Ser203, His447 (Glu334 not shown here); peripheral anionic site (PAS), Tyr72, Trp286. Both figures were created using Pymol.35
The docking results suggest that the inverted binding mode is favored for most of the inhibitors tested in this study. Especially for strong inhibitors (IC50 < 0.1 μM), the scores show a clear preference for this binding mode. The main interactions consist of a cation−π interaction with Trp86 and hydrogen bonds with conserved water molecules. Weaker inhibitors show lower per-atom-scores and in most cases less clear preference of the inverted binding mode versus the classical binding mode for the two enantiomeric forms.
The Binding Mode of Compound 41 at hH3R
The molecular dynamics (MD) simulation revealed a stable binding mode of the double protonated form of compound 41 (the most potent compound at both targets investigated) in the binding pocket of the inactive hH3R (Figures 5, 6A). During the whole productive phase of the MD simulation, a stable electrostatic interaction between the positively charged piperidine moiety of compound 41 and the highly conserved Asp3.32 was observed (Figure 5). The mean distances between the hydrogen of the piperidine nitrogen of compound 41 and the oxygens of the Asp3.32 side chain were fairly well maintained over time at about 0.22 nm (Figure 6A). A second stable interaction during the MD simulation was observed between Glu5.46 and the positively charged amidine moiety of 41 (Figures 5, 6B). Here, the mean distance between the hydrogen and the oxygens of Glu5.46 is about 0.20 nm.
Figure 5.

Compound 41 embedded in the binding pocket of the inactive hH3R, obtained as a snapshot of MD simulation. The translucent surface of the ligand is colored according to the electrostatic potential.
Figure 6.
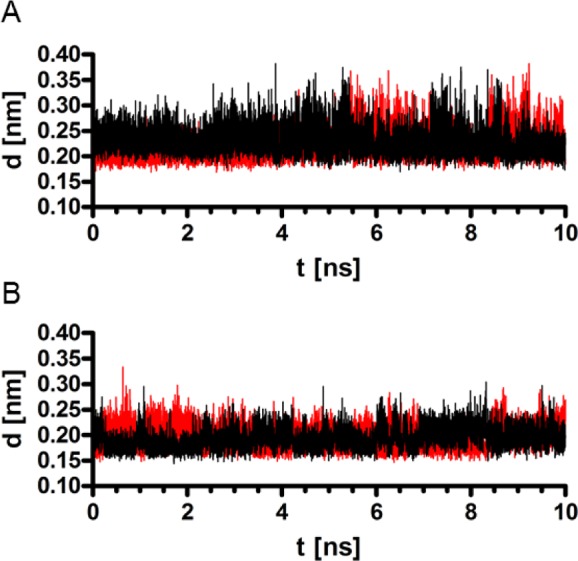
(A) Time evolution of the distances between the hydrogen of the protonated piperidine of 41 and the oxygens of the Asp3.32 side chain during the 10 ns productive MD. (B) Time evolution of the distances between the hydrogen of the protonated amidine of 41 and the oxygens of the Glu5.46 side chain during the 10 ns productive MD.
The tricyclic partial structure of compound 41 remained stable in a pocket formed by the amino acids Leu3.29, Tyr3.33, Cys3.36, Trp6.48, Tyr6.51, Met6.55, and Phe193 (E2-loop) (Figure 5). Furthermore, the hydrophobic part of the piperidine moiety of compound 41 is in close contact to Tyr2.61 and Leu7.42 (Figure 5). Besides, the MD simulations revealed that water molecules occupy parts of the binding pocket in presence of compound 41. An aromatic interaction was observed between Tyr3.33 and the aromatic partial structure of compound 41.
The observed binding mode based on modeling studies of compound 41 at hH3R is similar to the binding mode of the hH3R ligands FUB836,38 imoproxifan39,40 (Chart 4), and compound 6(41) (Chart 2). The binding mode described for compound 6 at hH3R is very similar to that we observed for compound 41 within this study: both positively charged moieties were observed to interact electrostatically with Asp3.32 and Glu5.46.
Chart 4. FUB83638 and Imoproxifan39,40.
The positively charged imidazole moiety of imoproxifan was found to interact electrostatically with Asp3.32, and the remaining part of imoproxifan, including the aromatic moiety, was embedded in a pocket, similar as described for compound 41. Surprisingly, imoproxifan is described to act, in contrast to compound 41, as an agonist at hH3R.40 A reason for that difference may be the imidazole moiety (as present in the full agonist histamine) in imoproxifan, instead of the piperidine partial structure in compound 41. The pharmacological data suggest that the introduction of an amide moiety into the tricycle leads to a decrease in affinity to hH3R, as shown by direct comparison of compound 41 with compound 32 (Table 1). This may be explained by a sterical hindrance of the amide with the hH3R or with the differences in electronic distribution between the two compounds. Phe193, located in the E2-loop, points into the binding pocket and shows an interaction with the ligand. This is in good accordance to data obtained for the histamine H4 receptor.42 For the histamine H4 receptor, it was described that Phe169, which corresponds to Phe193 in the hH3R, is responsible for differences in binding of agonists between hH4R and mH4R.42 As presented in Figure 2, compound 41 shows no affinity to hH1R, hH2R, and hH4R. It is not surprising that compound 41 shows no affinity to hH1R or hH2R, because the structural profile of this compound does not fit to the typical hH1R or hH2R ligand structure.43 A comparison of the most important amino acids in the binding pocket of the four human histamine receptor subtypes (Figure 7) shows that only the highly conserved Asp3.32, Trp6.48, and Tyr6.51 are identical, which corresponds to an identity of only 20% (with respect to the 15 compared amino acids, Figure 7). A complete comparison of the different histamine receptor subtypes and species can be found in literature.44 In contrast, between hH3R and hH4R about 60% of the amino acids (comparison of 15 amino acids, Figure 7) are identical. A large number of ligands with affinity or potency at hH3R and hH4R are known, for example, proxifan, thioperamide, UR-PI294, and UR-AK381.43 Arylbenzimidazols were described to show affinity to hH4R.45 The 1-methyl-4-(3-phenoxypropyl) piperazine partial structure of these compounds is similar to the 1-(3-phenoxypropyl) piperidine partial structure of compound 41. However, a reason for selectivity of compound 41 toward hH3R (compared to hH4R) may be the exchange of the N-methylpiperazine moiety into a piperidine moiety. The N-methylpiperazine moiety is typical for a large number of H4R ligands,46 its basic amine is relevant for interaction with Asp3.32, and it is located at a different position compared to the piperidine. It might be suggested that the sequence difference at position 7.42 (hH3R, Leu; hH4R, Gln) (Figure 7) would be responsible, since this amino acid side chain is in direct neighborhood to compound 41. For the hH3R and rH3R, it was shown that species differences between hH3R and rH3R at positions 3.37 and 3.40 (hH3R: Thr3.37, Ala3.40; rH3R: Ala3.37, Val3.40) are responsible for pharmacological differences of improxifan.39 Thus, it could also be speculated that the differences in amino acids between hH3R and hH4R at position 3.40 (Figure 7) may be involved in subtype differences. However, in order to obtain a more detailed insight into the interactions with hH3R, site-directed mutagenesis studies combined with further molecular modeling studies have to be performed.
Figure 7.
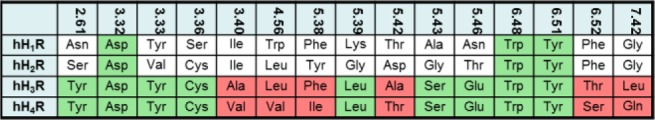
Comparison of selected amino acids, located in the binding pocket of the four human histamine receptor subtypes; green, highly conserved amino acids within all subtypes or identical amino acids between hH3R and hH4R, red: amino acids being different between hH3R and hH4R.
In summary, merging the piperidinylpropoxy phenyl and related hH3 pharmacophores into heterocyclic templates with AChE inhibiting properties yielded reversible and competitive AChE inhibitors with antagonizing properties at hH3R. SARs led to highly potent compounds with balanced two digit nanomolar affinities at both targets. The compounds show excellent selectivity over all other hHR receptor subtypes. Docking studies could explain the pronounced influence of the tertiary amine on SARs onto AChE inhibitory activity by an “inverted” binding mode locating the tertiary amine into the CAS. MD simulation confirmed the compound design strategy for hH3R affinity by identifying a binding mode related to other highly potent hH3R antagonists.
Methods
Construction of Inactive-State Model of hH3R
The crystal structure of hH1R 3RZE47 representing the inactive conformation of the hH1R, was used as a template to construct the inactive state of hH3R. Lysozyme which was artificially cocrystallized in 3RZE was deleted. The N-terminus, which is missing in the crystal structure, was added using SYBYL 7.0 (Tripos Inc.). The “Loop-Search” module of SYBYL 7.0 was used to complete the E2-loop. Due to the lack of information concerning the conformation of the long I3-loop of the hH3R (more than 100 amino acids) the amino acids Ala239 to Arg347 were not included in the model. To close the gap between the intracellular parts of TM V and TM VI eight alanines were inserted instead. Since internal water molecules were shown to play an important role in stabilization or activation of aminergic GPCRs these internal water molecules were included into the model of inactive hH3R.48,49 Using SYBYL7.0 the compound 41 was docked manually into the binding pocket of hH3R in such manner that the positively charged piperidine moiety interacts electrostatically with the highly conserved Asp3.32, similarly to previous studies addressing other H3R ligands.38,40 The tricyclic partial structure of compound 41 was embedded in a pocket between TM III, TM V and TM VI. The resulting complex was energetically minimized. Afterward, the minimized ligand–receptor complex was embedded in a POPC lipid bilayer. Intracellular and extracellular water, as well as an appropriate number of sodium and chloride ions were added into the simulation box and the molecular dynamics simulation, using GROMACS 4.0.2 (http://www.gromacs.org), was performed as described.50,51 The parametrization for the ligand was obtained from the PRODRG server (http://davapc1.bioch.dundee.ac.uk/prodrg/), but the partial charges were adopted based on the Gasteiger-Hückel partial charges, calculated by SYBYL 7.0. For the POPC lipid, the force field parameters available at an appropriate online source (http://moose.bio.ucalgary.ca/index.php?page=Structures_and_Topologies) were used. Subsequent to a 5 ns equilibration phase (force constants (250 kJ/(mol nm2) for the first 2.5 ns) and 100 kJ/(mol nm2) for the second 2.5 ns were put onto the backbone atoms of the TM domains of hH3R), a 10 ns productive phase was performed.
Computational Docking of Inhibitors to hAChE Binding Site
Docking was carried out with GOLD v5.1 and GOLD v5.2 (GOLDSUITE 5.1 and 5.2, CCDC Software, http://www.ccdc.cam.ac.uk)52 in the 4EY7 crystal structure of human AChE. The protein was set up for docking by removing all water molecules and nonprotein atoms and protonation of the amino acids was performed with MOE 2011.1053 using Protonate 3D at standard conditions. The search region was focused on the binding area comprising the CAS, the binding gorge, and the PAS. The binding site was defined by the following residues: Tyr72, Asp74, Trp86, Asn87, Gly120, Gly121, Tyr124, Ser125, Gly126, Ala127, Tyr133, Glu202, Ser203, Trp236, Trp286, Phe295, Phe297, Glu334, Tyr337, Phe338, Tyr341, His447, Gly448, And Tyr449. The ligand structures were either built in MOE (compounds 17–26, 32 and 34–42 and the chiral compounds in their R- and S-forms) or extracted from the crystal structure (ligands for redocking experiments, cf. below). The protonation for the ligands was set according to calculations performed with the program MoKa.37 The inhibitor structures were energy minimized in MOE with the MMFF94x force field to an rms-gradient of 0.001 kcal/(mol·Å). Using default settings in GOLD, the best-suited scoring function was selected based on redocking experiments in the Torpedo californica AChE binding site, with the galanthamine derivative of PDB structure 1W4L and donepezil of 1EVE. Another redocking experiment was performed using the donepezil ligand of 4EY7 and (−)-huperzine A of 4EY5, both human AChE structures. With each of the four scoring functions available in GOLD (ASP, CHEMPLP, ChemScore, GoldScore), 50 ligand poses were generated with the default number of operations of 100 000. In all cases, the top-ranked docking pose was deviating less than 1.07 Å with GoldScore.54,55 By raising the number of operations in the GOLD GA settings to 500 000, thereby prolonging the optimization time, the top-pose showed a significantly lower rmsd to the crystal pose of 0.55 Å (compared to 1.04 Å with GA 100 000) for redocking using 4EY7 and 0.63 Å for 1EVE (compared to 0.65 Å with GA 100 000). In all cases, a top-ranked docking pose deviating less than 0.91 Å from the crystal structure was obtained with GoldScore. Based on a docking study for AChE selective compounds [manuscript in preparation], the GoldScore function54,55 was best able to reflect the affinity and, hence, chosen for this project, too. All docking poses were clustered with a 1.5 Å cluster-cutoff by applying the complete linkage method. Of the five best-scored poses, the pose associated with the biggest cluster was selected for further pose analysis. The described redocking experiments showed that the largest cluster always contained the pose with the lowest rmsd to the crystal structure and the top-scoring pose. Seven conserved water molecules (HOH722, 729, 731, 737, 881, 952, 954 form the structure 4EY7) were chosen from an alignment of 1EVE, 4EY7, and the apoprotein structure 4EY4. The usage of these selected water molecules was validated using the water-“toggle” and water-“on” mode in the docking program. In 9 out of 12 analyzed docking runs, all water molecules were accepted in the “toggle”-mode and kept for generating ligand binding modes. In only three cases, one water molecule, located at the entrance of the binding site, was excluded from the docking process. Docking studies were thus carried out with the selected conserved water molecules in the water-“on” mode. For the donepezil ligand in 1EVE, the redocking with water gave an rmsd of 0.52 Å for the pose most similar to the crystal structure and 47 poses in the cluster. For the ligand in 4EY7, an rmsd of 0.35 Å was found (49 poses in the cluster).
Enzyme Inhibition
Acetyl- and Butyrylcholinesterase Inhibition Assay
The assay has been previously described in detail:14,24 AChE (E.C.3.1.1.7, Type VI-S, from electric eel), BChE (E.C.3.1.1.8, from equine serum), and hAChE (E.C.3.1.1.7, acetylcholinesterase from human erythrocytes) were purchased from Sigma-Aldrich (Steinheim, Germany); DTNB (Ellman’s reagent), ATC, and BTC iodides were obtained from Fluka (Buchs, Switzerland).
The assay was performed as described in the following procedure: stock solutions of the tested compounds were prepared in ethanol/water (1:4 ratio), 100 μL of which gave a final concentration of 10–3 M when diluted to the final volume of 3.32 mL. The highest concentration of the tested compounds applied in the assay was 10–4 M (10% EtOH in the stock solution did not influence enzyme activity). In order to obtain an inhibition curve, at least five different concentrations (normally 10–4–10–9 M) of the test compound were measured at 25 °C and 412 nm, each concentration in triplicate.
For buffer preparation, 1.36 g of potassium dihydrogen phosphate (10 mmol) was dissolved in 100 mL of water and adjusted with NaOH to pH = 8.0 ± 0.1. Enzyme solutions were prepared to give 2.5 units mL–1 in 1.4 mL aliquots. Furthermore, 0.01 M DTNB solution and 0.075 M ATC and BTC solutions were used. A cuvette containing 3.0 mL of phosphate buffer, 100 μL of the respective enzyme, and 100 μL of the test compound solution was allowed to stand for 4.5 min, then 100 μL of DTNB was added, and the reaction was started by addition of 20 μL of the substrate solution (ATC/BTC). The solution was mixed immediately, and exactly 2.5 min after substrate addition the absorption was measured. For the reference value, 100 μL of water replaced the test compound solution. For determining the blank value, additionally 100 μL of water replaced the enzyme solution. The inhibition curve was obtained by plotting the percentage enzyme activity (100% for the reference) versus logarithm of test compound concentration.
Radioligand Binding Assays
For the binding experiments, three different batches of Sf9 insect cell membranes were generated and prepared as described in literature.33,56−59 The accordant membranes were thawed and sedimented by a 10 min centrifugation at 4 °C and 13 000g. Membranes were resuspended in binding buffer (12.5 mM MgCl2, 1 mM EDTA, 75 mM Tris/HCl, pH 7.4). Each well (total volume 100 μL) contained 40 μg (hH1R), 60 μg (hH2R), 50 μg (hH3R), or 90 μg (hH4R) of protein. Competition binding experiments were performed in attendance of 5 nM [3H]mepyramine (KD = 4.49 nM)56 (hH1R) (American Radiolabeled Chemicals, St. Louis, MO), 30 nM [3H]UR-DE257 (KD = 31.33 nM)33 (hH2R), or 15 nM [3H]histamine (KD = 39.53 nM (hH3R); KD = 15.88 nM (hH4R))60 (American Radiolabeled Chemicals, St. Louis, MO), increasing concentrations of unlabeled ligands in hH3R binding assay and for hH1R, hH2R, and hH4R experiments, a single concentration (10 μM) of unlabeled ligand was used. The assays were conducted in 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany) and incubated for 60 min at 25 °C under shaking at 250 rpm using a Heidolph Titramax 101 instrument (Heidolph Instruments GmbH & Co. KG, Schwabach, Germany). Bound radioligand was separated from free radioligand by filtration through GF/C filters, which were pretreated with 0.3% polyethyleneimine solution. Subsequently, three washing steps with cold binding buffer (4 °C) using a Brandel Harvester instrument (Brandel, Gaithersburg, MD) were followed. Filter-bound radioactivity was detected after an equilibration phase of at least 12 h by liquid scintillation counting in 96-well plates (PerkinElmer, Waltham, MA).
The stock solutions of all tested compounds were prepared by dissolving the free base compound in equivalent ethanolic HCl and water. The dilution series of all stock solutions were prepared by using water (the highest concentration (10 μM) of all tested compounds contains less than 0.01% ethanol (v/v)).
Steady-State GTPase Activity Assay
GTPase activity assays were operated as described for the hH1R, hH2R, hH3R, and hH4R33,56−59hH1R assays: Sf9 insect cell membranes coexpressing the hH1R and RGS4 were employed; hH2R assays: Sf9 insect cell membranes expressing the hH2R-GsαS fusion protein were employed; hH3R assays: Sf9 insect cell membranes coexpressing the hH3R, mammalian Giα2, and Gβ1γ2 were employed; hH4R assays: Sf9 insect cell membranes coexpressing the hH4R-RGS19 fusion protein, mammalian Giα2, and Gβ1γ2 were employed. The relative membranes were thawed and then centrifuged for 10 min at 4 °C and 13 000g.
Membranes were resuspended in 10 mM Tris/HCl, pH 7.4. Each assay tube contained Sf9 membranes expressing the respective histamine-receptor subtype (10 μg protein/tube), MgCl2 (hH1R, hH2R assays: 1.0 mM, hH3R, hH4R assays: 5.0 mM), 100 μM EDTA, 100 μM ATP, 100 nM GTP, 100 μM adenylyl imidophosphate, 5 mM creatine phosphate, 40 μg creatine kinase, 0.2% (w/v) bovine serum albumin in 50 mM Tris/HCl (pH 7.4), and the investigated ligands at various concentrations. All hH4R assays additionally included 100 mM NaCl.
Reaction mixtures (80 μL) were incubated for 2 min at 27.5 °C. After the addition of 20 μL of [γ-33P]GTP (0.05 μCi/tube), reaction mixtures were incubated for 20 min at 27.5 °C. The reactions were stopped by the addition of 900 μL of slurry consisting of 5% (w/v) activated charcoal and 50 mM NaH2PO4, pH 2.0. Charcoal absorbs nucleotides but not 33Pi. Charcoal-quenched reaction mixtures were centrifuged for 7 min at 4 °C and 13 000g. A volume of 600 μL of the supernatant was taken, and the activity of 33Pi was determined by liquid scintillation counting.
Spontaneous [γ-33P]GTP degradation was determined in tubes containing all components described above, plus a high concentration of unlabeled GTP (1 mM) that, due to competition with [γ-33P]GTP, prevents [γ-33P]GTP hydrolysis by enzymatic activities in the presence of Sf9 membranes. Spontaneous [γ-33P]GTP degradation was <1% of the total amount of radioactivity added. The experimental conditions chosen ensured that less than 10% of the total amount of [γ-33P]GTP added was converted to 33Pi. For all the experiments, three different batches of Sf9 membranes were prepared and used. All experimental data were analyzed by nonlinear regression using Prism 5 program (GraphPad Software, San Diego, CA).
Chemistry
General Methods
Melting points are uncorrected and were measured in open capillary tubes, using a Barnstead Electrothermal IA9100 melting point apparatus. 1H and 13C NMR spectral data were obtained from a Bruker Advance spectrometer (300 and 75 MHz, respectively). TLC was performed on silica gel on aluminum foils with fluorescent indicator 254 nm (Merck). For detection, iodine vapor or UV light (254 nm) was used. ESI-MS samples were analyzed using electrospray ionization ion-trap mass spectrometry in nanospray mode using a Thermo Finnigan LCQ Deca instrument . The CHN analyses were undertaken using Perkin-Elmer elemental analyzer PE2400CHNS. In case purity was determined by elemental analysis, small amounts of solvent had to be taken into account which had been applied in previous column chromatography and appeared in the corresponding NMR spectra. For column chromatography, silica gel 60, 230–400 mesh (Merck) was used.
Benzyl 2-Amino-5-benzyloxy-N-ethoxycarbonylbenzoate (I)25
2-Amino-5-hydroxybenzoic acid (0.765 g, 5.0 mmol) and ethyl chloroformate (0.814 g, 0.72 mL, 7.5 mmol) were heated in dry dioxane (25 mL) at reflux temperature for 2 h. The solvent was evaporated, the residue dissolved in dry DMF (10 mL), K2CO3 (2.07 g, 0.015 mol), and benzyl bromide (1.88 g, 1.31 mL, 0.011 mol) added and the reaction stirred under N2 for 2 h at ambient temperature. The reaction was diluted with water (50 mL) and extracted with ether (3 × 20 mL). The ether extract was concentrated and purified by column chromatography using 10:1 (EtOAc/hexanes) as eluent system to afford the title compound (1.63 g, 80%) as a white solid, mp 65–67 °C (lit. mp 64 °C). 1H NMR (300 MHz, CDCl3) δ: 10.17 (s, 1H, NH), 8.37 (d, J = 9.3 Hz, 1H, arom.), 7.63 (d, J = 3.1 Hz, 1H, arom.), 7.50–7.27 (m, 10H, arom.), 7.19 (dd, J = 9.2, 3.1 Hz, 1H, arom.), 5.35 (s, 2H, OCH2Ph), 5.04 (s, 2H, OCH2Ph), 4.22 (d, J = 7.1 Hz, 2H, OCH2CH3), 1.37–1.25 (m, 3H, OCH2CH3). Lit. 1H NMR (CDCl3) δ: 10.2 (s, 1H, NH), 8.35 (d, 1H, J = 9 Hz, H3), 7.62 (d, 1H, J = 3 Hz, H6), 7.40 (m, 10H, OCH2Ph), 7.19 (dd, 1H, J = 3 and 9 Hz, H4), 5.34 (s, 2H, OCH2Ph), 5.04 (s, 2H, OCH2Ph), 4.21 (q, 2H, J = 7 Hz, OCH2CH3), 1.31 (t, 3H, J = 7 Hz, OCH2CH3).
6-Amino-3-benzyloxy-N-ethoxycarbonylbenzoic Acid (II)25
The benzyl ester I (0.405 g, 1.0 mmol) and KOH (0.112 g, 2.0 mmol) were heated at reflux in MeOH/H2O (10 mL, 3:1) for 2 h. The reaction was cooled and acidified with 1 N HCl (2 mL). The precipitate was filtered and air-dried to afford the title compound (0.281 g, 89%) as a white solid, mp 157–160 °C (lit. mp 159–160 °C). 1H NMR (300 MHz, CDCl3) δ: 9.94 (s, 1H, CO2H), 8.40 (d, J = 9.3 Hz, 1H, arom.), 7.69 (d, J = 3.1 Hz, 1H, arom.), 7.51–7.18 (m, 7H, arom.), 5.08 (s, 2H, OCH2Ph), 4.24 (q, J = 7.1 Hz, 2H, OCH2CH3), 1.34 (s, 3H, OCH2CH3). Lit. 1H NMR (CDCl3) δ: 9.94 (s, 1H, CO2H), 8.39 (d, 1H, J = 9 Hz, H3), 7.69 (d, 1H, J = 3 Hz, H6), 7.37 (m, 5H, OCH2Ph), 7.24 (dd, 1H, J = 3 and 9 Hz, H4), 5.07 (s, 2H, OCH2Ph), 4.24 (q, 2H, J = 7 Hz, OCH2CH3), 1.34 (t, 3H, J = 7 Hz, OCH2CH3).
6-Benzyloxyisatoic Anhydride (11)25
The acid II (0.472 g, 1.5 mmol) was dissolved in dry THF (15 mL), oxalyl chloride (1 mL) added, and the reaction heated at reflux temperature for 2 h. The solvent was evaporated, ether (10 mL) added to the residue, and the mixture heated under reflux for 10 min. The precipitate was filtered and washed with ether (10 mL) to afford the title compound (0.350 g, 87%) as a white solid, mp 220–222 °C (lit. mp 218–220 °C). 1H NMR (300 MHz, DMSO) δ: 11.62 (s, 1H), 7.39 (ddd, J = 19.4, 13.6, 7.6 Hz, 7H), 7.11 (d, J = 8.6 Hz, 1H), 5.17 (s, 2H). Lit. 1H NMR (DMSO-d6) δ: 11.60 (s, 1H, NH), 7.40 (m, 7H, CH2Ph, H7 and H5), 7.12 (d, 1H, J = 8.5 Hz, H4), 5.16 (s, 2H, OCH2Ph).
6-(Benzyloxy)-1-methyl-1H-benzo[d][1,3]oxazine-2,4-dione (12)
To a solution of 6-benzyloxyisatoic anhydride compound 11 (500 mg, 1.86 mmol) in 5 mL of DMAc (N,N-dimethyl acetamide), diisopropylethylamine (370 μL, 2.23 mmol) was added. The solution was stirred for 10 min prior to addition of methyl iodide (140 μL, 2.23 mmol). The solution was heated at 60 °C for 6 h. The solution was allowed to cool to room temperature, and 15 mL of cooled water was added. The resulting suspension was stirred vigorously for 30 min, filtered, and washed with 20 mL of water and 20 mL of n-hexane to afford the title compound (490 mg, 93%) as a yellow solid, mp 248–251 °C. ESI-MS: 284.18 m/z [M]+. 1H NMR (300 MHz, DMSO) δ: 7.61–7.29 (m, 8H, arom.), 5.22 (s, 2H, CCH2C), 3.44 (s, 3H, NCH3). 13C NMR (75 MHz, DMSO) δ: 158.73 (CO), 153.85 (CO), 147.68 (arom.), 136.42 (arom.), 136.39 (arom.), 128.39 (arom.), 127.87 (arom.), 127.58 (arom.), 125.61 (arom.), 116.52 (arom.), 112.08 (arom.), 112.02 (arom.), 69.68 (CCH2C), 31.65 (NCH3).
10-(Benzyloxy)-13-methyl-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (13)
A suspension of 6-(benzyloxy)-1-methyl-1H-benzo[d][1,3]-oxazine-2,4-dione 12 (300 mg, 1.45 mmol) and 3,4-dihydroisoquinoline (228 mg, 1.74 mmol) in 50 mL of toluene was refluxed for 24 h. After cooling, the solvent was removed under reduced pressure and the residue was purified by column chromatography using (100:1) CH2Cl2/MeOH as eluent system to afford the title compound (150 mg, 35%) as yellow thick oil. HRESIMS (C24H22N2O2+H)+, m/z calcd: 371.1754; found: 371.1758. 1H NMR (300 MHz, CDCl3) δ: 7.68 (d, J = 2.7 Hz, 1H), 7.51–7.21 (m, 9H), 7.18–7.07 (m, 2H), 5.73 (s, 1H, NCHN), 5.19–5.05 (m, 2H, OCH2C), 4.66 (ddd, J = 12.8, 4.8, 3.1 Hz, 1H), 3.26 (ddd, J = 12.8, 11.4, 3.9 Hz, 1H), 3.10–2.79 (m, 2H), 2.37 (d, J = 4.5 Hz, 3H, NCH3). 13C NMR (75 MHz, CDCl3) δ: 164.12 (CO), 155.04 (arom.), 145.30 (arom.), 136.93 (arom.), 136.86 (arom.), 132.02 (arom.), 128.62 (arom.), 128.57 (arom.), 128.42 (arom.), 128.25 (arom.), 128.05 (arom.), 127.65 (arom.), 127.12 (arom.), 124.11 (arom.), 123.65 (arom.), 122.06 (arom.), 111.82 (arom.), 71.80 (NCHN), 70.46 (OCH2C), 38.85 (NCH2CH2), 36.29 (NCH3), 28.72 (CH2CH2C).
10-Hydroxy-13-methyl-13,13a-dihydro-5H-isoquinolino-[1,2-b]quinazolin-8(6H)-one (14)
To a mixture of compound 13 (300 mg, 0.81 mmol) and 10% Pd–C (20 mg) was added 10 mL of ethanol. The mixture was stirred at room temperature and under an atmosphere of H2 for 4 h. The mixture was filtered over celite to afford the title compound (190 mg, 84%) as greenish yellow solid, mp 189–192 °C. ESI-MS: 281.0 m/z [M]+. 1H NMR (300 MHz, CDCl3) δ: 2.33 (s, 3H, NCH3). 2.86 (m, 1H, CCH2CH2), 2.99 (m, 1H, CCH2CH2), 3.37–3.23 (m, 1H, NCH2CH2), 4.66 (m, 1H, NCH2CH2), 5.75 (s, 1H, NCHN), 7.13–7.02 (m, 2H, arom.), 7.25–7.19 (m, 1H, arom.), 7.39–7.27 (m, 2H, arom.), 7.55–7.41 (m, 1H, arom.), 7.97 (s, 1H, arom.) ppm. 13C NMR (75 MHz, CDCl3) δ: 28.69 (CCH2CH2), 36.31 (NCH3), 38.99 (NCH2CH2), 71.98 (NCHN), 114.37 (arom.), 121.72 (arom.), 123.85 (arom.), 123.88 (arom.), 127.19 (arom.), 128.27 (arom.), 128.51 (arom.), 128.51 (arom.), 131.78 (arom.), 136.75 (arom.), 144.44 (arom.), 153.43 (COH), 164.63 (CO) ppm.
10-(3-Bromopropoxy)-13-methyl-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (15)
1,3-Dibromopropane (505 mg, 2.5 mmol), compound 14 (350 mg, 1.25 mmol), potassium carbonate (260 mg, 1.9 mmol), and potassium iodide (21 mg, 0.125 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl, and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (400 mg, 80%) as a yellow thick oil. HRESIMS (C20H21BrN2O2+H)+, m/z calcd: 401.0859; found: 401.0855. 1H NMR (300 MHz, CDCl3) δ: 7.60–7.53 (m, 1H), 7.47 (dd, J = 7.9, 5.1 Hz, 1H), 7.36–7.26 (m, 2H), 7.24–7.18 (m, 1H), 7.12–7.01 (m, 2H), 5.72 (s, 1H), 4.75–4.56 (m, 1H), 4.14–4.06 (m, 2H), 3.67–3.54 (m, 2H), 3.33–3.16 (m, 1H), 3.07–2.77 (m, 2H), 2.36 (s, 3H), 1.31–1.20 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 164.10 (CO), 154.92 (arom.), 145.27 (arom.), 136.90 (arom.), 132.00 (arom.), 128.55 (arom.), 128.37 (arom.), 128.24 (arom.), 127.10 (arom.), 124.09 (arom.), 123.62 (arom.), 121.67 (arom.), 111.58 (arom.), 71.82 (NCHN), 65.83, 38.86, 36.29 (NCH3), 32.34, 29.96, 28.69.
10-((6-Bromohexyl)oxy)-13-methyl-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (16)
1,6-Dibromohexane (610 mg, 2.5 mmol), compound 14 (350 mg, 1.25 mmol), potassium carbonate (260 mg, 1.9 mmol) and potassium iodide (21 mg, 0.125 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl, and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over sodium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (390 mg, 70%) as a yellow thick oil. HRESIMS (C23H27BrN2O2+H)+, m/z calcd: 443.12829; found: 443.1283. 1H NMR (300 MHz, CDCl3) δ: 7.54 (d, J = 2.5 Hz, 1H), 7.48 (dd, J = 7.9, 5.3 Hz, 1H), 7.37–7.26 (m, 2H), 7.21 (dd, J = 7.8, 5.1 Hz, 1H), 7.12–7.00 (m, 2H), 5.72 (s, 1H), 4.65 (ddd, J = 12.8, 4.8, 3.1 Hz, 1H), 4.11–3.90 (m, 2H), 3.42 (t, J = 6.8 Hz, 2H), 3.34–3.16 (m, 1H), 3.07–2.77 (m, 2H), 2.35 (s, 3H), 2.01–1.73 (m, 4H), 1.57–1.43 (m, 4H). 13C NMR (75 MHz, CDCl3) δ: 164.17 (CO), 155.36 (arom.), 145.01 (arom.), 136.91 (arom.), 132.05 (arom.), 128.53 (arom.), 128.4 (arom.), 128.20 (arom.), 127.10 (arom.), 124.13 (arom.), 123.65 (arom.), 121.78 (arom.), 111.32 (arom.), 71.82 (NCHN), 68.25, 38.81, 36.29 (NCH3), 33.84, 32.72, 29.07, 28.73, 27.95, 25.30.
13-Methyl-10-((6-(piperidin-1-yl)hexyl)oxy)-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (17)
Piperidine (64 mg, 0.75 mmol), compound 16 (222 mg, 0.50 mmol), potassium carbonate (105 mg, 0.75 mmol), and potassium iodide (8 mg, 0.05 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl, and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (160 mg, 71%) as a yellow thick oil. HRESIMS (C28H37N3O2+H)+, m/z calcd: 448.2959; found: 448.2960. 1H NMR (300 MHz, CDCl3) δ: 7.53 (d, J = 2.4 Hz, 1H), 7.49–7.43 (m, 1H), 7.36–7.27 (m, 2H), 7.24–7.18 (m, 1H), 7.10–7.00 (m, 2H), 5.71 (s, 1H), 4.64 (ddd, J = 12.8, 4.8, 3.1 Hz, 1H), 4.18–3.82 (m, 2H), 3.24 (ddd, J = 12.8, 11.4, 3.9 Hz, 2H), 3.12–2.68 (m, 2H), 2.47–2.23 (m, 9H), 1.88–1.70 (m, 2H), 1.64–1.30 (m, 12H). 13C NMR (75 MHz, CDCl3) δ: 164.19 (CO), 155.45 (arom.), 144.95 (arom.), 136.91 (arom.), 132.05 (arom.), 128.51 (arom.), 128.41 (arom.), 128.18 (arom.), 127.09 (arom.), 124.13 (arom.), 123.66 (arom.), 121.78 (arom.), 111.31 (arom.), 71.81 (NCHN), 68.42, 59.54, 54.65, 38.78 (NCH3), 36.27, 29.18, 28.73, 27.52, 26.84, 26.03, 25.95, 24.48. Elem. Anal. (C28H37N3O2 + 0.13 CH2Cl2) Calcd.: C, 73.66; H, 8.19; N, 9.16; O, 6.98. Found: C, 73.85; H, 8.12; N, 9.14.
13-Methyl-10-(3-(piperidin-1-yl)propoxy)-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (18)
Piperidine (64 mg, 0.75 mmol), compound 15 (200 mg, 0.50 mmol), potassium carbonate (105 mg, 0.75 mmol), and potassium iodide (8 mg, 0.05 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (136 mg, 67%) as a colorless thick oil. HRESIMS (C25H31N3O2+H)+, m/z calcd: 406.2495; found: 406.2501. 1H NMR (300 MHz, MeOD) δ: 7.50–7.36 (m, 2H), 7.36–7.18 (m, 3H), 7.17–7.08 (m, 2H), 5.73 (s, 1H), 4.66–4.47 (m, 1H), 4.18–3.96 (m, 2H), 3.32–3.17 (m, 2H), 3.08–2.83 (m, 2H), 2.77–2.55 (m, 6H), 2.40 (s, 3H), 2.06 (qd, J = 11.8, 6.8 Hz, 2H), 1.70 (ddd, J = 16.2, 11.8, 5.9 Hz, 4H), 1.53 (d, J = 5.0 Hz, 2H). 13C NMR (75 MHz, MeOD) δ: 166.00 (CO), 156.25 (arom.), 146.48 (arom.), 138.26 (arom.), 133.47 (arom.), 129.73 (arom.), 129.57 (arom.), 129.21 (arom.), 128.17 (arom.), 124.26 (arom.), 124.23 (arom.), 122.88 (arom.), 112.65 (arom.), 73.38 (NCHN), 67.68, 57.00, 55.40, 49.92, 49.64, 49.36, 49.07, 48.79, 48.51, 48.22, 40.44, 36.71 (NCH3), 29.37, 27.07, 26.18, 24.82. Elem. Anal. (C25H31N3O2 + 0.3 CHCl3) Calcd.: C, 68.85; H, 7.15; N, 9.52; O, 7.25. Found: C, 68.74; H, 7.54; N, 9.29.
13-Methyl-10-(3-morpholinopropoxy)-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (19)
Morpholine (65 mg, 0.75 mmol), compound 15 (200 mg, 0.50 mmol), potassium carbonate (105 mg, 0.75 mmol), and potassium iodide (8 mg, 0.05 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (144 mg, 71%) as a colorless thick oil. HRESIMS (C24H29N3O3+H)+, m/z calcd: 408.2287; found: 408.2286. 1H NMR (300 MHz, MeOD) δ: 7.47–7.38 (m, 2H), 7.36–7.22 (m, 3H), 7.14–7.07 (m, 2H), 5.73 (s, 1H), 4.57–4.49 (m, 1H), 4.14–3.98 (m, 2H), 3.73–3.68 (m, 4H), 3.24 (ddd, J = 12.7, 10.9, 4.5 Hz, 1H), 3.06–2.81 (m, 2H), 2.61–2.46 (m, 6H), 2.41 (s, 3H), 2.07–1.91 (m, 2H). 13C NMR (75 MHz, MeOD) δ: 166.03 (CO), 156.40 (arom.), 146.44 (arom.), 138.27 (arom.), 133.46 (arom.), 129.72 (arom.), 129.56 (arom.), 129.24 (arom.), 128.17 (arom.), 124.31 (arom.), 124.27 (arom.), 122.93 (arom.), 112.59 (arom.), 73.38 (NCHN), 67.70, 67.67, 56.79, 54.83, 40.42, 36.69 (NCH3), 29.39, 27.24. Elem. Anal. (C24H29N3O3+ 0.13 CHCl3) Calcd.: C, 68.51; H, 6.94; N, 9.93; O, 11.35. Found: C, 68.58; H, 7.13; N, 9.90.
13-Methyl-10-(3-(pyrrolidin-1-yl)propoxy)-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (20)
Pyrrolidine (53 mg, 0.75 mmol), compound 15 (200 mg, 0.50 mmol), potassium carbonate (105 mg, 0.75 mmol), and potassium iodide (8 mg, 0.05 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (130 mg, 66%) as a yellow thick oil. HRESIMS (C24H29N3O2+H)+, m/z calcd: 392.2333; found: 392.2336. 1H NMR (300 MHz, CDCl3) δ: 7.55 (d, J = 2.4 Hz, 1H), 7.50–7.44 (m, 1H), 7.38–7.28 (m, 2H), 7.25–7.17 (m, 1H), 7.12–7.01 (m, 2H), 5.73 (s, 1H), 4.65 (m, 1H), 4.22–3.97 (m, 2H), 3.34–3.20 (m, 1H), 2.84 (m, 6H), 2.36 (s, 3H), 2.16 (d, J = 6.7 Hz, 4H), 1.91 (s, 4H). 13C NMR (75 MHz, MeOD) δ: 166.03 (CO), 156.23, 146.52, 146.13, 138.26, 129.71, 129.56, 129.19, 128.15, 124.23, 122.87, 113.68, 112.68, 73.41 (NCHN), 67.49, 55.17, 54.23, 40.45, 36.70 (NCH3), 29.37, 29.03, 24.19. Elem. Anal. (C24H29N3O2 + 0.8 CH2Cl2 + 0.5 H2O) Calcd.: C, 63.58; H, 6.80; N, 8.97; O, 8.54. Found: C, 63.46; H, 7.14; N, 9.06.
10-(3-(Azepan-1-yl)propoxy)-13-methyl-13,13a-dihydro-5H-isoquinolino[1,2-b]quinazolin-8(6H)-one (21)
Azepane (75 mg, 0.75 mmol), compound 15 (200 mg, 0.50 mmol), potassium carbonate (105 mg, 0.75 mmol), and potassium iodide (8 mg, 0.05 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving the title compound (145 mg, 69%) as a yellow thick oil. HRESIMS (C26H33N3O2+H)+, m/z calcd: 420.2646; found: 420.2650. 1H NMR (300 MHz, CDCl3) δ: 7.55 (d, J = 2.1 Hz, 1H), 7.47 (d, J = 6.2 Hz, 1H), 7.36–7.18 (m, 3H), 7.13–6.99 (m, 2H), 5.73 (s, 1H), 4.78–4.52 (m, 1H), 4.21–3.92 (m, 2H), 3.26 (td, J = 12.6, 3.8 Hz, 1H), 3.06–2.67 (m, 8H), 2.33 (s, 3H), 2.12–1.95 (m, 2H), 1.67 (d, J = 28.3 Hz, 8H). 13C NMR (75 MHz, CDCl3) δ: 166.93 (CO), 156.29, 147.52, 147.23, 138.46, 129.91, 129.76, 129.39, 128.65, 124.27, 122.99, 113.75, 112.75, 71.83 (NCHN), 55.35, 54.80, 38.82, 36.29 (NCH3), 32.50, 30.96, 28.72, 27.16, 27.02. Elem. Anal. (C26H33N3O2 + 1.75 CH2Cl2) Calcd.: C, 58.66; H, 6.47; N, 7.40; O, 5.63. Found: C, 58.45; H, 6.84; N, 7.24.
13-Methyl-10-((6-(piperidin-1-yl)hexyl)oxy)-6,8,13,13a-tetrahydro-5H-isoquinolino[1,2-b]quinazoline (22)
A solution of compound 17 (116 mg, 0.26 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (30 mg, 0.78 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (20:1:0.1) as an eluent system to afford the title compound (95 mg, 84%) as a dark yellow oil. HRESIMS (C28H39N3O+H)+, m/z calcd: 434.3166; found: 434.3160. 1H NMR (300 MHz, CDCl3) δ: 7.53–7.43 (m, 1H), 7.25–7.06 (m, 3H), 6.94 (d, J = 8.8 Hz, 1H), 6.75 (dd, J = 8.8, 2.8 Hz, 1H), 6.56 (d, J = 2.8 Hz, 1H), 4.71 (d, J = 8.5 Hz, 1H), 4.02–3.78 (m, 4H), 3.35–3.05 (m, 2H), 2.84–2.61 (m, 2H), 2.49 (m, 5H), 2.42–2.35 (m, 2H), 1.84–1.32 (m, 16H). 13C NMR (75 MHz, CDCl3) δ: 153.64 (arom.), 142.72(arom.), 136.29 (arom.), 134.23 (arom.), 128.61 (arom.), 128.46 (arom.), 127.14 (arom.), 126.84 (arom.), 126.00 (arom.), 122.73 (arom.), 114.02 (arom.), 112.04 (arom.), 77.43 (HCHN), 68.23, 59.27, 57.02, 54.44, 48.91, 38.77 (NCH3), 29.31, 28.90, 27.43, 26.39, 26.00, 25.48, 24.16. Elem. Anal. (C28H39N3O + 1.1 CHCl3) Calcd.: C, 61.87; H, 7.15; N, 7.44; O, 2.83. Found: C, 61.90; H, 7.26; N, 7.16.
13-Methyl-10-(3-(piperidin-1-yl)propoxy)-6,8,13,13a-tetrahydro-5H-isoquinolino[1,2-b]quinazoline (23)
A solution of compound 18 (105 mg, 0.26 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (30 mg, 0.78 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (20:1:0.1) as an eluent system to afford the title compound (30 mg, 41%) as yellow thick oil. HRESIMS (C25H33N3O+H)+, m/z calcd: 392.2696; found: 392.2697. 1H NMR (300 MHz, CDCl3) δ: 7.54–7.42 (m, 1H), 7.28–7.07 (m, 3H), 6.93 (dd, J = 8.8, 3.5 Hz, 1H), 6.75 (dd, J = 8.8, 2.9 Hz, 1H), 6.54 (dd, J = 12.9, 2.8 Hz, 1H), 4.72 (s, 1H), 4.05–3.80 (m, 4H), 3.31–3.02 (m, 2H), 2.84–2.41 (m, 11H), 2.04 (td, J = 12.4, 6.2 Hz, 2H), 1.76–1.59 (m, 4H), 1.49 (s, 2H). 13C NMR (75 MHz, CDCl3) δ: 136.28 (arom.), 128.54 (arom.), 127.15 (arom.), 126.84 (arom.), 125.99 (arom.), 122.67 (arom.), 114.05 (arom.), 112.06 (arom.), 77.43 (NCHN), 66.69, 56.99, 56.02, 54.49, 48.87, 38.74 (NCH3), 28.89, 26.50, 25.44, 24.08. Elem. Anal. (C25H33N3O + 0.9 H2O + 1.8 CH3CH2OH) Calcd.: C, 57.24; H, 8.16; N, 7.00; O, 9.87. Found: C, 57.57; H, 7.81; N, 6.72.
4-(3-((13-Methyl-6,8,13,13a-tetrahydro-5H-isoquinolino-[1,2-b]quinazolin-10yl)oxy)propyl)morpholine (24)
A solution of compound 19 (106 mg, 0.26 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (30 mg, 0.78 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (20:1:0.1) as an eluent system to afford the title compound (38 mg, 42%) as a yellow thick oil. HRESIMS (C24H31N3O2+H)+, m/z calcd: 394.2489; found: 394.2490. 1H NMR (300 MHz, CDCl3) δ: 7.51–7.44 (m, 1H), 7.25–7.18 (m, 2H), 7.17–7.10 (m, 1H), 6.94 (d, J = 8.8 Hz, 1H), 6.76 (dd, J = 8.8, 2.9 Hz, 1H), 6.57 (d, J = 2.8 Hz, 1H), 4.72 (s, 1H), 4.04–3.85 (m, 4H), 3.73 (dd, J = 8.5, 3.9 Hz, 4H), 3.31–3.05 (m, 2H), 2.85–2.60 (m, 2H), 2.56–2.43 (m, 9H), 2.01–1.87 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 153.52 (arom.), 142.85 (arom.), 136.28 (arom.), 134.21 v, 128.61 (arom.), 128.46 (arom.), 127.16 (arom.), 126.85 (arom.), 126.00 (arom.), 122.69 (arom.), 114.03 (arom.), 112.08 (arom.), 77.40 (NCHN), 67.03, 66.52, 57.00, 55.66, 53.78, 48.89, 38.76 (NCH3), 28.89, 26.59. Elem. Anal. (C24H31N3O2 + 1.15 CH2Cl2) Calcd.: C, 50.30; H, 6.09; N, 7.00; O, 5.33. Found: C, 49.91; H, 6.50; N, 6.72.
13-Methyl-10-(3-(pyrrolidin-1-yl)propoxy)-6,8,13,13a-tetrahydro-5H-isoquinolino[1,2-b]quinazoline (25)
A solution of compound 20 (100 mg, 0.26 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (30 mg, 0.78 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (20:1:0.1) as an eluent system to afford the title compound (78 mg, 79%) as a light yellow oil. HRESIMS (C24H31N3O+H)+, m/z calcd: 379.2571; found: 379.2570. 1H NMR (300 MHz, MeOD) δ: 7.52–7.32 (m, 1H), 7.28–7.04 (m, 3H), 6.92 (d, J = 8.8 Hz, 1H), 6.80–6.67 (m, 1H), 6.60 (t, J = 4.6 Hz, 1H), 4.67 (s, 1H), 4.03–3.75 (m, 4H), 3.27–3.15 (m, 1H), 3.15–3.00 (m, 1H), 2.77–2.61 (m, 8H), 2.48 (s, 3H), 1.98 (dt, J = 15.8, 6.1 Hz, 2H), 1.91–1.79 (m, 4H). 13C NMR (75 MHz, MeOD) δ: 155.00 (arom.), 143.44 (arom.), 137.37 (arom.), 135.22 (arom.), 129.87 (arom.), 129.41 (arom.), 128.62 (arom.), 127.31 (arom.), 123.27 (arom.), 115.37 (arom.), 113.10 (arom.), 78.65 (NCHN), 67.46, 57.48, 55.14, 54.40, 38.99 (NCH3), 29.44, 24.23. Elem. Anal. (C24H31N3O + 0.57 CH2Cl2) Calcd.: C, 69.28; H, 7.61; N, 9.87; O, 3.76. Found: C, 69.28; H, 7.65; N, 9.68.
10-(3-(Azepan-1-yl)propoxy)-13-methyl-6,8,13,13a-tetrahydro-5H-isoquinolino[1,2-b]quinazoline (26)
A solution of compound 21 (110 mg, 0.26 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (30 mg, 0.78 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (20:1:0.1) as an eluent system to afford the title compound (85 mg, 81%) as a light yellow oil. HRESIMS (C26H35N3O+H)+, m/z calcd: 406.2853; found: 406.2851. 1H NMR (300 MHz, MeOD) δ: 7.51–7.38 (m, 1H), 7.30–7.09 (m, 3H), 6.92 (d, J = 8.9 Hz, 1H), 6.81–6.68 (m, 1H), 6.61 (t, J = 4.9 Hz, 1H), 4.69 (s, 1H), 3.98–3.77 (m, 4H), 3.28–3.17 (m, 1H), 3.17–2.99 (m, 1H), 2.81–2.68 (m, 8H), 2.49 (s, 3H), 2.02–1.86 (m, 2H), 1.75–1.60 (m, 8H). 13C NMR (75 MHz, MeOD) δ: 155.03 (arom.), 143.41 (arom.), 137.36 (arom.), 135.21 (arom.), 129.87 (arom.), 129.40 (arom.), 128.62 (arom.), 127.40 (arom.), 127.18 (arom.), 123.23 (arom.), 115.40 (arom.), 113.12 (arom.), 78.65 (NCHN), 67.59, 64.97, 57.46, 56.63, 56.16, 49.67, 38.95 (NCH3), 29.37, 27.99, 27.90, 27.79. Elem. Anal. (C26H35N3O + 0.24 CH2Cl2) Calcd.: C, 73.99; H, 8.40; N, 9.86; O, 3.76. Found: C, 74.03; H, 8.45; N, 9.66.
7-(Benzyloxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (27)
Pyrrolidin-2-one (1.00 g, 12.25 mmol) and 6-benzyloxyisatoic anhydride 11 (1.00 g, 3.71 mmol) were placed in a 10 mL crimp-sealed thick-walled glass tube equipped with a pressure sensor and a magnetic stirrer. The sealed reaction tube was placed inside the cavity of a CEM Discover focused microwave synthesis system, operated at 130 °C (temperature monitored by a built-in infrared sensor), power 10–200 W, and pressure 50–100 psi for 1 h. The residue was purified by column chromatography using (2:1) CH2Cl2/acetone as eluent system to afford the title compound (450 mg, 78%) as a yellow solid, mp 188–190 °C. ESI-MS: 293.0 m/z [MH]+. 1H NMR (300 MHz, CDCl3) δ: 2.39–2.20 (m, 2H, CH2CH2CH2), 3.25–3.08 (m, 2H, CCH2CH2), 4.30–4.11 (m, 2H, NCH2CH2), 5.17 (s, 2H, COCH2), 7.44–7.29 (m, 5H, arom.), 7.47 (dd, J = 8.2, 1.2 Hz, 1H, arom.), 7.65–7.58 (m, 1H, arom.), 7.75 (d, J = 2.9 Hz, 1H, arom.). 13C NMR (75 MHz, CDCl3) δ: 19.69 (CH2CH2CH2), 32.25 (CCH2CH2), 46.57 (NCH2CH2), 70.51(COCH2), 107.14 (arom.), 124.90 (arom.), 127.71 (arom.), 128.20 (arom.), 128.24 (arom.), 128.66 (arom.), 131.25 (arom.), 136.34 (arom.), 149.10 (arom.), 157.18 (arom.), 157.41 (NCN), 160.76 (CO).
7-Hydroxy-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (28)24
7-(Benzyloxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one 27 (750 mg, 2.57 mmol) and 10% Pd–C (80 mg) were added to 50 mL of ethanol. The mixture was stirred at room temperature and under an atmosphere of H2 for 4 h. The mixture was filtered over celit to afford the title compound (450 mg, 87%) as beige solid, mp 284–287 °C (lit. mp 289–290 °C). 1H NMR (300 MHz, DMSO) δ: 2.21–2.10 (m, 2H, CH2CH2CH2), 3.08–2.94 (m, 2H, CCH2CH2), 4.12–3.94 (m, 2H, NCH2CH2), 7.23 (dd, J = 8.8, 2.9 Hz, 1H, arom.), 7.39 (d, J = 2.8 Hz, 1H, arom.), 7.47 (d, J = 8.8 Hz, 1H, arom.), 9.95 (s, 1H, COH). Lit. 1H NMR (DMSO-d6, 250 MHz) δ: 10.50 (br s, OH), 7.65 (1H, d, J = 8.8 Hz, H4), 7.45 (1H, d, J = 2.5 Hz, H1), 7.38 (1H, dd, J = 8.5, 2.5 Hz, H3), 4.09 (2H, t, J = 7.0 Hz), 3.26 (2H, d, J = 7.6 Hz), 2.23 (2H, quintet, J = 7.6 Hz).
7-(3-Bromopropoxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (29)
1,3-Dibromopropane (810 mg, 4.0 mmol), compound 28 (400 mg, 2.0 mmol), and potassium carbonate (415 mg, 3.0 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography to afford the title compound (430 mg, 67%) as a yellow thick oil. HRESIMS (C14H15BrN2O2+H)+, m/z calcd: 323.0390; found: 323.0392. 1H NMR (300 MHz, CDCl3) δ: 7.55 (d, J = 2.5 Hz, 1H), 7.49 (dd, J = 8.9, 1.4 Hz, 1H), 7.29–7.16 (m, 1H), 4.23–4.02 (m, 4H), 3.66–3.47 (m, 2H), 3.08 (t, J = 7.9 Hz, 2H), 2.36–2.24 (m, 2H), 1.97 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 160.71 (NCN), 157.41 (COCH2), 156.96 (CO), 143.67 (arom.), 128.20 (arom.), 124.38 (arom.), 121.12 (arom.), 106.71 (arom.), 65.81, 46.50, 32.16, 32.14, 29.82, 19.59.
7-((6-Bromohexyl)oxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (30)
1,6-Dibromohexane (980 mg, 4.0 mmol), compound 28 (400 mg, 2.0 mmol), and potassium carbonate (415 mg, 3.0 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography to afford the title compound (420 mg, 58%) as a yellow solid, mp 76–79 °C. HRESIMS (C17H21BrN2O2+H)+, m/z calcd: 365.0859; found: 365.0864. 1H NMR (300 MHz, CDCl3) δ: 7.59 (dd, J = 12.3, 5.9 Hz, 2H), 7.31 (dd, J = 8.9, 2.9 Hz, 1H), 4.30–4.14 (m, 2H), 4.06 (t, J = 6.4 Hz, 2H), 3.48–3.37 (m, 2H), 3.16 (t, J = 7.9 Hz, 2H), 2.36–2.21 (m, 2H), 1.97–1.76 (m, 4H), 1.60–1.45 (m, 4H). 13C NMR (75 MHz, CDCl3) δ: 160.83 (NCN), 157.48 (COCH2), 157.20 (CO), 143.43 (arom.), 128.16 (arom.), 124.68 (arom.), 121.17 (arom.), 106.55 (arom.), 68.33, 46.54, 33.80, 32.68, 32.24, 28.96, 27.93, 25.30, 19.69.
7-(3-(Pyrrolidin-1-yl)propoxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (31)
Pyrrolidine (66 mg, 0.93 mmol), compound 29 (200 mg, 0.62 mmol), and potassium carbonate (130 mg, 0.93 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography to afford the title compound (150 mg, 77%) as a yellow thick oil. HRESIMS (C18H23N3O2+H)+, m/z calcd: 314.1863; found: 314.1871. 1H NMR (300 MHz, CDCl3) δ: 7.58 (d, J = 2.9 Hz, 1H), 7.50 (t, J = 6.8 Hz, 1H), 7.30–7.22 (m, 1H), 4.12 (dt, J = 12.6, 6.7 Hz, 4H), 3.10 (dd, J = 10.4, 5.5 Hz, 2H), 2.76–2.67 (m, 2H), 2.67–2.58 (m, 4H), 2.34–2.17 (m, 2H), 2.17–2.01 (m, 2H), 1.86–1.76 (m, 4H). 13C NMR (75 MHz, CDCl3) δ: 160.79 (NCN), 157.23 (COCH2), 157.19 (CO), 143.62 (arom.), 128.20 (arom.), 124.45 (arom.), 121.14 (arom.), 106.70 (arom.), 66.69, 54.19, 53.07, 46.49, 32.22, 28.25, 23.44, 19.65. Elem. Anal. (C18H23N3O2 + 0.3 CHCl3) Calcd.: C, 62.94; H, 6.73; N, 12.03; O, 9.16. Found: C, 62.69; H, 6.93; N, 12.15.
7-(3-(Piperidin-1-yl)propoxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (32)
Piperidine (80 mg, 0.93 mmol), compound 29 (200 mg, 0.62 mmol), and potassium carbonate (130 mg, 0.93 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography to afford the title compound (120 mg, 59%) as a white solid, mp 167–169 °C. HRESIMS (C19H25N3O2+H)+, m/z calcd: 328.202; found: 328.202. 1H NMR (300 MHz, CDCl3) δ: 1.42–1.52 (m, 2H), 1.61–1.73 (m, 4H), 2.05–2.14 (m, 2H), 2.22–2.33 (m, 2H), 2.48–2.64 (m, 6H), 3.15 (t, J = 7.8 Hz, 2H), 4.11 (t, J = 6.3 Hz, 2H), 4.20 (t, J = 7.5 Hz, 2H), 7.30 (dd, J1 = 9 Hz, J2 = 3 Hz, 1H), 7.55 (d, J = 9.0 Hz, 1H), 7.62 (d, J = 3 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 19.7, 22.4, 23.1, 24.4, 32.3, 46.5, 53.7, 55.4, 65.7, 107.1 (arom.), 121.2 (arom.), 124.0 (arom.), 128.5 (arom.), 144.0 (arom.), 156.6 (CO), 157.5 (COCH2), 160.7 (NCN). Elem. Anal. (C19H25N3O2 + 1.33 CHCl3) Calcd.: C, 50.22; H, 5.46; N, 8.64; O, 6.58. Found: C, 49.90; H, 5.82; N, 8.93.
7-(3-(Azepan-1-yl)propoxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (33)
Azepane (92 mg, 0.93 mmol), compound 29 (200 mg, 0.62 mmol), and potassium carbonate (130 mg, 0.93 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography giving afford the title compound (164 mg, 77%) as a yellow thick oil. HRESIMS (C20H27N3O2+H)+, m/z calcd: 342.2182; found: 342.2176. 1H NMR (300 MHz, CDCl3) δ: 7.61 (d, J = 2.9 Hz, 1H), 7.54 (d, J = 8.9 Hz, 1H), 7.33–7.25 (m, 1H), 4.23–4.14 (m, 2H), 4.10 (t, J = 6.3 Hz, 2H), 3.21–3.05 (m, 2H), 2.76–2.61 (m, 6H), 2.36–2.19 (m, 2H), 2.07–1.92 (m, 2H), 1.61 (ddd, J = 8.7, 6.6, 3.2 Hz, 8H). 13C NMR (75 MHz, CDCl3) δ: 160.85 (NCN), 157.43 (COCH2), 157.11 (CO), 143.59 (arom.), 128.20 (arom.), 124.54 (arom.), 121.18 (arom.), 106.70 (arom.), 66.9̀1, 55.49, 54.68, 46.49, 36.49, 32.24, 31.42, 27.78, 27.24, 26.99, 19.68. Elem. Anal. (C20H27N3O2 + 0.56 CHCl3) Calcd.: C, 60.48; H, 6.80; N, 10.29; O, 7.84. Found: C, 60.36; H, 6.95; N, 10.02.
7-((6-(Piperidin-1-yl)hexyl)oxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one (34)
Piperidine (80 mg, 0.93 mmol), compound 30 (226 mg, 0.62 mmol), and potassium carbonate (130 mg, 0.93 mmol) were refluxed in absolute acetonitrile (50 mL) for 2 h. After cooling, the inorganic salts were filtered off and the filtrate was concentrated under reduced pressure. The remaining residue was taken up in aq. solution of NaCl and extracted with methylene chloride. The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography to afford the title compound (184 mg, 80%) as a yellow solid, mp 183–187 °C. HRESIMS (C22H31N3O2+H)+, m/z calcd: 370.2489; found: 370.2494. 1H NMR (300 MHz, CDCl3) δ: 7.51 (dd, J = 8.4, 5.9 Hz, 2H), 7.29–7.20 (m, 1H), 4.15 (dd, J = 17.6, 10.5 Hz, 2H), 3.98 (t, J = 6.3 Hz, 2H), 3.09 (t, J = 7.9 Hz, 2H), 2.97 (m, 4H), 2.89–2.74 (m, 2H), 2.32–2.14 (m, 2H), 2.03–1.89 (m, 4H), 1.80 (ddt, J = 20.9, 14.3, 7.1 Hz, 4H), 1.61 (d, J = 4.4 Hz, 2H), 1.53–1.29 (m, 4H). 13C NMR (75 MHz, CDCl3) δ: 160.84 (NCN), 157.20 (COCH2), 157.33 (CO), 143.56 (arom.), 128.23 (arom.), 124.51 (arom.), 121.11 (arom.), 106.59 (arom.), 68.14, 57.71, 53.37, 46.52, 32.21, 28.83, 26.69, 25.59, 24.06, 23.03, 22.32, 19.65. Elem. Anal. (C22H31N3O2+ 1.09 CHCl3) Calcd.: C, 55.51; H, 6.47; N, 8.41; O, 6.40. Found: C, 55.29; H, 6.67; N, 8.73.
7-(3-(Pyrrolidin-1-yl)propoxy)-1,2,3,3a,4,9-hexahydro-pyrrolo[2,1-b]quinazoline (35)
A solution of compound 31 (100 mg, 0.32 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (36 mg, 1.0 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (10:1:0.1) as an eluent system to afford the title compound (72 mg, 75%) as a light yellow oil. HRESIMS (C18H27N3O+H)+, m/z calcd: 302.2227; found: 302.2227. 1H NMR (300 MHz, MeOD) δ: 6.63 (dt, J = 6.4, 3.2 Hz, 1H), 6.61–6.55 (m, 2H), 3.99–3.72 (m, 5H), 3.17–2.99 (m, 1H), 2.77–2.61 (m, 6H), 2.51 (td, J = 8.9, 6.9 Hz, 1H), 2.18–2.03 (m, 1H), 2.03–1.88 (m, 4H), 1.88–1.81 (m, 4H), 1.76–1.62 (m, 1H). 13C NMR (75 MHz, MeOD) δ: 153.57 (COCH2), 138.48 (arom.), 122.29 (arom.), 118.43 (arom.), 115.55 (arom.), 114.15 (arom.), 73.95 (NHCHN), 67.73, 55.14, 54.43, 53.06, 51.94, 31.55, 29.52, 24.21, 21.73. Elem. Anal. (C18H27N3O + 0.2 CH2Cl2) Calcd.: C, 68.65; H, 8.67; N, 13.20; O, 5.02. Found: C, 68.99; H, 8.65; N, 12.75.
7-(3-(Piperidin-1-yl)propoxy)-1,2,3,3a,4,9-hexahydropyrrolo[2,1-b]quinazoline (36)
A solution of compound 32 (100 mg, 0.31 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (35 mg, 0.92 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (10:1:0.1) as an eluent system to afford the title compound (75 mg, 77%) as a light yellow oil. HRESIMS (C19H29N3O+H)+, m/z calcd: 316.2383; found: 316.2379. 1H NMR (300 MHz, CDCl3) δ: 1.42–1.47 (m, 2H), 1.57–1.68 (m, 6H), 1.80–2.00 (m, 5H), 2.08–2.17 (m, 2H), 2.42–2.60 (m, 8H), 3.05–3.12 (m,1H), 3.89–3.94 (m, 5H), 6.54–6.56 (m, 2H), 6.63 (dd, J1 = 9 Hz, J2 = 3 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 152.38, 122.08, 117.51, 114.09, 113.27, 72.41, 67.08, 56.11, 54.59, 51.68, 50.91, 31.47, 26.82, 25.82, 24.34, 21.00. Elem. Anal. (C19H29N3O + 0.08 CH2Cl2) Calcd.: C, 71.11; H, 9.12; N, 13.04; O, 4.96. Found: C, 71.27; H, 8.75; N, 12.82.
7-(3-(Azepan-1-yl)propoxy)-1,2,3,3a,4,9-hexahydropyrrolo[2,1-b]quinazoline (37)
A solution of compound 33 (100 mg, 0.29 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (33 mg, 0.88 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq. NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (10:1:0.1) as an eluent system to afford the title compound (79 mg, 83%) as a light yellow oil. HRESIMS (C20H31N3O+H)+, m/z calcd: 330.2540; found: 330.2541. 1H NMR (300 MHz, MeOD) δ: 6.60 (ddd, J = 8.7, 6.1, 1.7 Hz, 3H), 3.82 (tt, J = 9.1, 5.8 Hz, 5H), 3.17–2.92 (m, 1H), 2.71 (ddd, J = 9.8, 7.6, 5.3 Hz, 6H), 2.50 (td, J = 8.9, 6.9 Hz, 1H), 2.19–2.03 (m, 1H), 2.03–1.84 (m, 4H), 1.76–1.58 (m, 9H). 13C NMR (75 MHz, MeOD) δ: 153.60 (COCH2), 138.48 (arom.), 122.29 (arom.), 118.42 (arom.), 115.58 (arom.), 114.19 (arom.), 73.93 (NHCHN), 67.89, 56.64, 56.19, 53.06, 51.94, 31.57, 28.01, 27.88, 21.74. Elem. Anal. C20H31N3O + 0.11 CH2Cl2) Calcd.: C, 71.29; H, 9.29; N, 12.40; O, 4.72. Found: C, 71.39; H, 9.16; N, 12.05.
7-((6-(Piperidin-1-yl)hexyl)oxy)-1,2,3,3a,4,9-hexahydro- pyrrolo[2,1-b]quinazoline (38)
A solution of compound 24 (100 mg, 0.27 mmol) in dry THF was added dropwise to a suspension of lithium aluminum hydride (31 mg, 0.81 mmol) in dry THF under nitrogen atmosphere. The mixture was heated to reflux for 3 h, and then the reaction mixture was poured into ice water, basified with 20% aq NaOH, and extracted with ethyl acetate. The combined extracts were washed with saturated aq. NaCl solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product, which was column chromatographed on a silica gel using CH2Cl2/MeOH/ammonia solution (10:1:0.1) as an eluent system to afford the title compound (69 mg, 71%) as a light yellow oil. HRESIMS (C22H35N3O+H)+, m/z calcd: 358.2853; found: 358.2848. 1H NMR (300 MHz, MeOD) δ: 6.75–6.68 (m, 2H), 6.66 (d, J = 0.8 Hz, 1H), 4.26–4.19 (m, 1H), 3.91 (q, J = 6.4 Hz, 2H), 3.33–2.88 (m, 10H), 2.34–2.18 (m, 1H), 2.10 (dt, J = 14.5, 8.3 Hz, 3H), 1.92–1.70 (m, 10H), 1.59–1.38 (m, 5H). 13C NMR (75 MHz, MeOD) δ 154.47 (arom.), 137.10 (arom.), 120.26 (arom.), 119.28 (arom.), 116.55 (arom.), 113.97 (arom.), 75.57, 69.40, 58.32, 54.29, 52.18, 51.98, 30.55, 30.25, 27.52, 26.77, 25.06, 24.35, 22.87, 21.57. Elem. Anal. (C22H35N3O + 0.65 CH2Cl2) Calcd.: C, 65.91; H, 8.86; N, 10.18; O, 3.88. Found: C, 66.16; H, 9.01; N, 10.08.
7-(3-(Piperidin-1-yl)propoxy)-2,3,3a,4-tetrahydropyrrolo[2,1-b]quinazolin-9(1H)-one (39)
To a stirred solution of compound 32 (340 mg, 1.04 mmol) in 40 mL of ethanol, NaBCNH3 (220 mg, 3.11 mmol), 1 N HCl in isopropanol (5 drops), was added. The reaction mixture was stirred at room temperature for 24 h, TLC showed incomplete reaction, and we added NaBCNH3 (136 mg, 2.16 mmol), 1 N HCl in isopropanol (5 drops) to the reaction mixture until TLC showed complete reaction. The reaction mixture was quenched by iced-water and basified using 1 N sodium hydroxide solution, extracted by CH2Cl2. The organic phase was extracted by CH2Cl2 (3 × 50 mL). The combined organic phase was dried over sodium sulfate; the solvents were removed under reduced pressure. The residue was purified by column chromatography using (20:1:0.1) CH2Cl2/MeOH/ammonia as eluent system to afford the title compound (310 mg, 90%) as a white solid, mp 277–279 °C. HRESIMS (C19H27N3O2+H)+, m/z calcd: 330.2176; found: 330.2177. 1H NMR (300 MHz, CDCl3): δ 7.28 (d, J = 3 Hz, 1H), 6.96 (dd, J = 9 Hz, 3 Hz, 1H), 6.75 (d, J = 9 Hz, 1H), 4.91 (t, J = 6.3 Hz, 2H), 4.01 (t, J = 6.0 Hz, 2H), 3.58–3.72 (m, 2H), 2.80–2.92 (m, 6H), 2.31–2.42 (m, 1H), 1.93–2.15 (m, 5H), 1.70–1.78 (m, 4H), 1.54–1.65 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 164.46, 153.65, 122.90, 117.88, 112.58, 71.87, 67.35, 56.66, 55.07, 49.89, 49.60, 49.32, 49.04, 48.75, 48.47, 48.19, 45.48, 26.45, 25.53, 24.05, 22.85. Elem. Anal. (C19H27N3O2 + 0.36 CH2Cl2) Calcd.: C, 64.59; H, 7.76; N, 11.67; O, 8.89. Found: C, 64.97; H, 7.73; N, 11.27.
7-((6-(Piperidin-1-yl)hexyl)oxy)-2,3,3a,4-tetrahydropyrrolo[2,1-b]quinazolin-9(1H)-one (40)
To a stirred solution of compound 34 (200 mg, 0.54 mmol) in 50 mL of ethanol, NaBCNH3 (68 mg, 1.8 mmol), 1 N HCl in isopropanol (5 drops), was added. The reaction mixture was stirred at room temperature for 24 h, TLC showed incomplete reaction, and we added NaBCNH3 (68 mg, 1.8 mmol), 1 N HCl in isopropanol (5 drops) to the reaction mixture until TLC showed complete reaction. The reaction mixture was quenched by iced-water, basified using 1 N sodium hydroxide solution, and extracted by CH2Cl2. The organic phase was extracted by CH2Cl2 (3 × 50 mL). The combined organic phase was dried over sodium sulfate; the solvents were removed under reduced pressure. The residue was purified by column chromatography using (20:1:0.1) CH2Cl2/MeOH/ammonia as eluent system to afford the title compound (160 mg, 80%) as yellow solid, mp 141–144 °C. HRESIMS (C22H33N3O2 +H)+, m/z calcd: 372.2646; found: 372.2648. 1H NMR (300 MHz, CDCl3) δ: 7.11 (dd, J = 13.2, 2.9 Hz, 1H), 6.71 (dd, J = 8.7, 2.9 Hz, 1H), 6.53 (d, J = 8.7 Hz, 1H), 4.74 (dd, J = 7.1, 5.7 Hz, 1H), 3.85–3.65 (m, 2H), 3.62–3.36 (m, 2H), 3.32–3.02 (m, 2H), 3.04–2.83 (m, 4H), 2.85–2.69 (m, 2H), 2.14 (ddq, J = 16.5, 15.3, 8.2 Hz, 2H), 1.97–1.81 (m, 2H), 1.73–1.61 (m, 4H), 1.61–1.48 (m, 4H), 1.40–1.21 (m, 4H). 13C NMR (75 MHz, CDCl3) δ: 166.96 (CO), 156.65 (arom.), 146.10 (arom.), 125.72 (arom.), 122.47 (arom.), 120.85 (arom.), 115.55 (arom.), 74.41 (NHCHN), 72.16, 61.34, 57.30, 48.26, 36.63, 32.65, 30.13, 29.37, 27.87, 27.05, 25.73, 25.61. Elem. Anal. (C22H33N3O2 + 1.584 CH2Cl2) Calcd.: C, 50.53; H, 6.22; N, 7.50; O, 5.71. Found: C, 50.15; H, 6.51; N, 7.90.
7-(3-(Piperidin-1-yl)propoxy)-1,2,3,3a,4,9-hexahydropyrrolo[2,1-b]quinazoline (41)
Zn dust (400 mg) was added to compound 32 (100 mg, 0.3 mmol) dissolved in 10 mL of acetic acid, and to the reaction mixture 2 mL of HCl (10 M) was then added. The reaction mixture was then heated up to 70 °C for 2 h. The reaction mixture was allowed to cool to room temperature. The solvent was concentrated under vacuum, and the reaction mixture basified by NaOH (4 N) and extracted by ethylacetate (3 × 100 mL). The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography using 5:1:0.1 CH2Cl2/MeOH/ammonia solution mixture as eluent system to afford the title compound (36 mg, 38%) as a yellow thick oil. HRESIMS (C19H27N3O +H)+, m/z calcd: 314.2232; found: 314.2236. 1H NMR (300 MHz, CDCl3) δ: 6.95 (t, J = 17.1 Hz, 1H), 6.67 (ddd, J = 28.4, 8.6, 2.7 Hz, 1H), 6.37 (d, J = 2.7 Hz, 1H), 4.45 (s, 2H), 3.87 (t, J = 6.3 Hz, 2H), 3.27 (t, J = 6.9 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.41 (m, 6H), 1.99 (dd, J = 14.9, 7.4 Hz, 2H), 1.92–1.82 (m, 2H), 1.60–1.48 (m, 4H), 1.45–1.31 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 161.49 (N=C), 155.96 (arom.), 134.84 (arom.), 124.08 (arom.), 119.74 (arom.), 114.16 (arom.), 111.95 (arom.), 66.68, 55.85, 54.46, 51.55, 47.01, 30.88, 26.59, 25.69, 24.25, 18.98. Elem. Anal. (C19H27N3O + 1.0 CH3COOH+ 0.1CH2Cl2) Calcd.: C, 66.34; H, 8.23; N, 11.00; O, 12.57. Found: C, 66.34; H, 8.01; N, 11.25.
7-((6-(Piperidin-1-yl)hexyl)oxy)-1,2,3,9-tetrahydropyrrolo[2,1-b]quinazoline (42)
Zn dust (400 mg) was added to compound 34 (100 mg, 0.27 mmol) dissolved in 10 mL acetic acid, and to the reaction mixture 2 mL of HCl (10 M) was then added. The reaction mixture was heated up to 70 °C for 2 h. The reaction mixture was allowed to cool to room temperature, the solvent was concentrated under vacuum, and the reaction mixture basified by NaOH (4 N) and extracted by ethylacetate (3 × 100 mL). The combined organic extracts were washed with brine and dried over magnesium sulfate. The organic solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography using 5:1:0.1 CH2Cl2/MeOH/ammonia solution mixture as eluent system to afford to afford the title compound (43 mg, 45%) as a yellow semisolid. HRESIMS (C22H33N3O +H)+, m/z calcd: 356.2702; found: 356.2701. 1H NMR (300 MHz, CDCl3) δ: 6.94 (d, J = 8.6 Hz, 1H), 6.64 (dd, J = 8.6, 2.8 Hz, 1H), 6.39 (d, J = 2.7 Hz, 1H), 4.48 (s, 2H), 3.92–3.78 (m, 2H), 3.27 (t, J = 6.9 Hz, 2H), 2.73–2.51 (m, 2H), 2.38 (s, 4H), 2.34–2.20 (m, 2H), 2.11–1.90 (m, 2H), 1.82–1.62 (m, 2H), 1.54 (m, 6H), 1.45–1.34 (m, 4H), 1.33–1.25 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 161.11 (N=C), 155.96 (arom.), 135.80 (arom.), 124.50 (arom.), 119.97 (arom.), 114.04 (arom.), 112.07 (arom.), 68.14, 59.31, 54.48, 51.42, 47.18, 30.98, 29.24, 27.44, 26.52, 25.99, 25.63, 24.26, 19.09. Elem. Anal. (C22H33N3O + 1.0 CH3COOH) Calcd.: C, 69.36; H, 8.97; N, 10.11; O, 11.55. Found: C, 69.53; H, 9.05; N, 10.18.
Acknowledgments
Appreciation is expressed to Professors S. Elz, A. Buschauer, and G. Bernhardt at Regensburg University for providing their expertise and facilities to determine the binding affinities, selectivities, and functional properties at hH receptors.
Glossary
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AChEIs
acetylcholinesterase inhibitors
- AD
Alzheimer’s disease
- ADHD
attention deficit hyperactivity disorder
- ATC
acetylthiocholine
- BTC
butyrylthiocholine
- CAS
catalytic active site
- ChEs
cholinesterases
- ChEIs
cholinesterase inhibitors
- CNS
central nervous system
- DPH
diphenhydramine
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- EeAChE
Electrophorus electricus acetylcholinesterase
- eqBuChE
horse serum butyrylcholinesterase
- HIS
histamine
- hAChE
human recombinant acetylcholinesterase
- hH1R
human histamine receptor type 1
- hH2R
human histamine receptor type 2
- hH3R
human histamine receptor type 3
- hH4R
human histamine receptor type 4
- GABA
gamma-aminobutyric acid
- GPCR
G protein-coupled receptor
- MTDL
multitarget-directed ligands
- MD
molecular dynamics
- NMDA
N-methyl-d-aspartate
- PAS
peripheral anionic site
- RGS4
regulator of G protein signaling 4
- SARs
structure–activity relationships
Supporting Information Available
Additional docking data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
M. Decker was responsible for the supervision and development of the whole project. F. H. Darras performed the chemical syntheses of the majority of the synthesized compounds, performed the kinetic analysis and the EeChE and eqBChE inhibition studies; G. Huang synthesized compounds 32 and 36 and performed the hAChE inhibition study; S. Pockes and M. Nimczick performed the hH3 binding affinity test. S. Pockes performed the selectivity study over hH1R, hH2R, and hH4, as well as the functional GTPase assay; S. Wehle and C. Sotriffer conducted the computational studies on AChE. A. Strasser and H. Wittmann performed the molecular simulation analysis at hH3R.
M. Decker gratefully acknowledges the German Science Foundation (DFG) for financial support (DFG DE 1546/6-1) as well as the German Academic Exchange Service (DAAD) for awarding a Ph.D. scholarship to F. H. Darras.
The authors declare no competing financial interest.
Dedication
This article is dedicated to Professor John L. Neumeyer on the occasion of his 84th birthday.
Supplementary Material
References
- Maslow K. (2010) 2010 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 6, 158–194. [DOI] [PubMed] [Google Scholar]
- Qiu C.; De Ronchi D.; Fratiglioni L. (2007) The epidemiology of the dementias: an update. Curr. Opin. Psychiatry 20, 380–385. [DOI] [PubMed] [Google Scholar]
- Reitz C.; Brayne C.; Mayeux R. (2011) Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R.; Johnson E.; Ziegler-Graham K.; Arrighi H. M. (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dementia 3, 186–191. [DOI] [PubMed] [Google Scholar]
- Davies P.; Maloney A. J. F. (1976) Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 308, 1403. [DOI] [PubMed] [Google Scholar]
- Esbenshade T. A.; Browman K. E.; Bitner R. S.; Strakhova M.; Cowart M. D.; Brioni J. D. (2008) The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol. 154, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang J.-M.; Garbarg M.; Schwartz J.-C. (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302, 832–837. [DOI] [PubMed] [Google Scholar]
- Berlin M.; Boyce C. W.; de Lera Ruiz M. (2011) Histamine H3 Receptor as a Drug Discovery Target. J. Med. Chem. 54, 26–53. [DOI] [PubMed] [Google Scholar]
- Esbenshade T. A.; Fox G. B.; Cowart M. D. (2006) Histamine H3 receptor antagonists: preclinical promise for treating obesity and cognitive disorders. Mol. Interventions 6, 77–88. [DOI] [PubMed] [Google Scholar]
- Singh M.; Jadhav H. R. (2013) Histamine H3 Receptor Function and Ligands: Recent Developments. Mini-Rev. Med. Chem. 13, 47–57. [PubMed] [Google Scholar]
- Keith J. M.; Gomez L. A.; Wolin R. L.; Barbier A. J.; Wilson S. J.; Boggs J. D.; Mazur C.; Fraser I. C.; Lord B.; Aluisio L.; Lovenberg T. W.; Carruthers N. I. (2007) Pyrrolidino-tetrahydroisoquinolines as potent dual H3 antagonist and serotonin transporter inhibitors. Bioorg. Med. Chem. Lett. 17, 2603–2607. [DOI] [PubMed] [Google Scholar]
- Roche O.; Rodríguez Sarmiento R. M. (2007) A new class of histamine H3 receptor antagonists derived from ligand based design. Bioorg. Med. Chem. Lett. 17, 3670–3675. [DOI] [PubMed] [Google Scholar]
- Tao M.; Aimone L. D.; Huang Z.; Mathiasen J.; Raddatz R.; Lyons J.; Hudkins R. L. (2012) Optimization of 5-pyridazin-3-one phenoxypropylamines as potent, selective histamine H3 receptor antagonists with potent cognition enhancing activity. J. Med. Chem. 55, 414–423. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zenger K.; Lupp A.; Kling B.; Heilmann J.; Fleck C.; Kraus B.; Decker M. (2012) Tacrine-Silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J. Med. Chem. 55, 5231–5242. [DOI] [PubMed] [Google Scholar]
- Chen X.; Decker M. (2013) Multi-target compounds acting in the central nervous system designed from natural products. Curr. Med. Chem. 20, 1673–1685. [DOI] [PubMed] [Google Scholar]
- Decker M. (2007) Recent advances in the development of hybrid molecules/designed multiple compounds with antiamnesic properties. Mini-Rev. Med. Chem. 7, 221–229. [DOI] [PubMed] [Google Scholar]
- Bajda M.; Guzior N.; Ignasik M.; Malawska B. (2011) Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem. 18, 4949–4975. [DOI] [PubMed] [Google Scholar]
- Petroianu G.; Arafat K.; Sasse B. C.; Stark H. (2006) Multiple enzyme inhibitions by histamine H3 receptor antagonists as potential procognitive agents. Pharmazie 61, 179–182. [PubMed] [Google Scholar]
- Blackard W. G. J.; Sood G. K.; Crowe D. R.; Fallon M. B. (1998) Tacrine: A cause of fatal hepatotoxicity?. J. Clin. Gastroenterol. 26, 57–59. [DOI] [PubMed] [Google Scholar]
- Melo T.; Videira R. A.; André S.; Maciel E.; Francisco C. S.; Oliveira-Campos A. M.; Rodrigues L. M.; Domingues M. R. M.; Peixoto F.; Manuel Oliveira M. (2012) Tacrine and its analogues impair mitochondrial function and bioenergetics: a lipidomic analysis in rat brain. J. Neurochem. 120, 998–1013. [DOI] [PubMed] [Google Scholar]
- Bembenek S. D.; Keith J. M.; Letavic M. A.; Apodaca R.; Barbier A. J.; Dvorak L.; Aluisio L.; Miller K. L.; Lovenberg T. W.; Carruthers N. I. (2008) Lead identification of acetylcholinesterase inhibitors–histamine H3 receptor antagonists from molecular modeling. Bioorg. Med. Chem. 16, 2968–2973. [DOI] [PubMed] [Google Scholar]
- Morini G.; Comini M.; Rivara M.; Rivara S.; Bordi F.; Plazzi P. V.; Flammini L.; Saccani F.; Bertoni S.; Ballabeni V.; Barocelli E.; Mor M. (2008) Synthesis and structure–activity relationships for biphenyl H3 receptor antagonists with moderate anti-cholinesterase activity. Bioorg. Med. Chem. 16, 9911–9924. [DOI] [PubMed] [Google Scholar]
- Bajda M.; Kuder K. J.; Łażewska D.; Kieć-Kononowicz K.; Więckowska A.; Ignasik M.; Guzior N.; Jończyk J.; Malawska B. (2012) Dual-Acting Diether Derivatives of Piperidine and Homopiperidine with Histamine H3 Receptor Antagonistic and Anticholinesterase Activity. Arch. Pharm. 345, 591–597. [DOI] [PubMed] [Google Scholar]
- Darras F. H.; Kling B.; Heilmann J.; Decker M. (2012) Neuroprotective tri- and tetracyclic BChE inhibitors releasing reversible inhibitors upon carbamate transfer. ACS Med. Chem. Lett. 3, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos G. H. P.; Drastlik K. A. (2003) Approach to the synthesis of (+)-Ifforestine. Model studies directed at the tetracyclic framework. Heterocycles 60, 2023–2044. [Google Scholar]
- Beutner G. L.; Kuethe J. T.; Yasuda N. (2007) A practical method for preparation of 4-hydroxyquinolinone esters. J. Org. Chem. 72, 7058–7061. [DOI] [PubMed] [Google Scholar]
- Takeuchi H.; Matsushita Y.; Eguchi S. (1991) Novel ring enlargement of lactams via quinazolinone annelation. A facile route to benzoannelated large-membered cyclic 1,5-diamines. J. Org. Chem. 56, 1535–1537. [Google Scholar]
- Decker M. (2005) Novel inhibitors of acetyl- and butyrylcholinesterase derived from the alkaloids dehydroevodiamine and rutaecarpine. Eur. J. Med. Chem. 40, 305–313. [DOI] [PubMed] [Google Scholar]
- Yücel Y. Y.; Tacal Ö.; Özer I. (2008) Comparative effects of cationic triarylmethane, phenoxazine and phenothiazine dyes on horse serum butyrylcholinesterase. Arch. Biochem. Biophys. 478, 201–205. [DOI] [PubMed] [Google Scholar]
- Karlsson D.; Fallarero A.; Brunhofer G.; Mayer C.; Prakash O.; Mohan C. G.; Vuorela P.; Erker T. (2012) The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 47, 190–205. [DOI] [PubMed] [Google Scholar]
- Decker M.; Krauth F.; Lehmann J. (2006) Novel tricyclic quinazolinimines and related tetracyclic nitrogen bridgehead compounds as cholinesterase inhibitors with selectivity towards butyrylcholinesterase. Bioorg. Med. Chem. 14, 1966–1977. [DOI] [PubMed] [Google Scholar]
- Schnell D.; Burleigh K.; Trick J.; Seifert R. (2010) No Evidence for Functional Selectivity of Proxyfan at the Human Histamine H3 Receptor Coupled to Defined Gi/Go Protein Heterotrimers. J. Pharmacol. Exp. Ther. 332, 996–1005. [DOI] [PubMed] [Google Scholar]
- Baumeister P. E. D.; Bernhardt G.; Buschauer A. (2011) [3H]UR-DE257: A new tritium-labeled histamine H2 receptor antagonist. Poster at Frontiers in Medicinal Chemistry, a Joint German-Swiss Meeting, Saarbrücken, Germany. [Google Scholar]
- Schnell D.; Seifert R. (2010) Modulation of histamine H3 receptor function by monovalent ions. Neurosci. Lett. 472, 114–118. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.6.0.0, Schrödinger, LLC.
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. (2012) Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 55, 10282–10286. [DOI] [PubMed] [Google Scholar]
- Milletti F.; Storchi L.; Sforna G.; Cruciani G. (2007) New and Original pKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 47, 2172–2181. [DOI] [PubMed] [Google Scholar]
- Schlegel B.; Laggner C.; Meier R.; Langer T.; Schnell D.; Seifert R.; Stark H.; Höltje H.-D.; Sippl W. (2007) Generation of a homology model of the human histamine H3 receptor for ligand docking and pharmacophore-based screening. J. Comput.-Aided Mol. Des.. 21, 437–453. [DOI] [PubMed] [Google Scholar]
- Yao B. B.; Hutchins C. W.; Carr T. L.; Cassar S.; Masters J. N.; Bennani Y. L.; Esbenshade T. A.; Hancock A. A. (2003) Molecular modeling and pharmacological analysis of species-related histamine H3 receptor heterogeneity. Neuropharmacology 44, 773–786. [DOI] [PubMed] [Google Scholar]
- Schnell D.; Strasser A.; Seifert R. (2010) Comparison of the pharmacological properties of human and rat histamine H3-receptors. Biochem. Pharmacol. 80, 1437–1449. [DOI] [PubMed] [Google Scholar]
- Morini G.; Comini M.; Rivara M.; Rivara S.; Lorenzi S.; Bordi F.; Mor M.; Flammini L.; Bertoni S.; Ballabeni V.; Barocelli E.; Plazzi P. V. (2006) Dibasic non-imidazole histamine H3 receptor antagonists with a rigid biphenyl scaffold. Bioorg. Med. Chem. Lett. 16, 4063–4067. [DOI] [PubMed] [Google Scholar]
- Lim H. D.; Jongejan A.; Bakker R. A.; Haaksma E.; de Esch I. J. P.; Leurs R. (2008) Phenylalanine 169 in the Second Extracellular Loop of the Human Histamine H4 Receptor Is Responsible for the Difference in Agonist Binding between Human and Mouse H4 Receptors. J. Pharmacol. Exp. Ther. 327, 88–96. [DOI] [PubMed] [Google Scholar]
- Seifert R.; Strasser A.; Schneider E. H.; Neumann D.; Dove S.; Buschauer A. (2013) Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol. Sci. 34, 33–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A.; Wittmann H.-J.; Buschauer A.; Schneider E. H.; Seifert R. (2013) Species-dependent activities of G-protein-coupled receptor ligands: lessons from histamine receptor orthologs. Trends Pharmacol. Sci. 34, 13–32. [DOI] [PubMed] [Google Scholar]
- Lee-Dutra A.; Arienti K. L.; Buzard D. J.; Hack M. D.; Khatuya H.; Desai P. J.; Nguyen S.; Thurmond R. L.; Karlsson L.; Edwards J. P.; Breitenbucher J. G. (2006) Identification of 2-arylbenzimidazoles as potent human histamine H4 receptor ligands. Bioorg. Med. Chem. Lett. 16, 6043–6048. [DOI] [PubMed] [Google Scholar]
- Marson C. M. (2011) Targeting the Histamine H4 Receptor. Chem. Rev. 111, 7121–7156. [DOI] [PubMed] [Google Scholar]
- Shimamura T.; Shiroishi M.; Weyand S.; Tsujimoto H.; Winter G.; Katritch V.; Abagyan R.; Cherezov V.; Liu W.; Han G. W.; Kobayashi T.; Stevens R. C.; Iwata S. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel T. E.; Chance M. R.; Palczewski K. (2009) Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 106, 8555–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. (2012) Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straßer A.; Striegl B.; Wittmann H.-J.; Seifert R. (2008) Pharmacological Profile of Histaprodifens at Four Recombinant Histamine H1 Receptor Species Isoforms. J. Pharmacol. Exp. Ther. 324, 60–71. [DOI] [PubMed] [Google Scholar]
- Igel P.; Geyer R.; Strasser A.; Dove S.; Seifert R.; Buschauer A. (2009) Synthesis and Structure–Activity Relationships of Cyanoguanidine-Type and Structurally Related Histamine H4 Receptor Agonists. J. Med. Chem. 52, 6297–6313. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. (2003) Improved protein–ligand docking using GOLD. Proteins 52, 609–623. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2011.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2011.
- Jones G.; Willett P.; Glen R. C. (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Seifert R. W.-S. K.; Bürckstümmer T.; Pertz H. H.; Schunack W.; Dove S.; Buschauer A.; Elz S. (2003) Multiple differences in agonist and antagonist pharmacology between human and guinea pig histamine H1-receptor. J. Pharmacol. Exp. Ther. 305, 1104–1115. [DOI] [PubMed] [Google Scholar]
- Kelley M. T.; Bürckstümmer T.; Wenzel-Seifert K.; Dove S.; Buschauer A.; Seifert R. (2001) Distinct interaction of human and guinea pig histamine H2-receptor with guanidine-type agonists. Mol. Pharmacol. 60, 1210–1225. [DOI] [PubMed] [Google Scholar]
- Schnell D. B. I.; Ladova K.; Schneider E.; Igel P.; Dove S.; Buschauer A.; Seifert R. (2011) Expression and functional properties of canine, rat and murine histamine H4 receptors in Sf9 insect cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 383, 457–470. [DOI] [PubMed] [Google Scholar]
- Birnkammer T. (2011) Highly potent and selective acylguanidinetype histamine H2 receptor agonists: synthesis and structure-activity relationships of mono and bivalent ligands. Ph.D. Dissertation, University of Regensburg, Germany. [Google Scholar]
- Geyer R.; Kaske M.; Baumeister P.; Buschauer A. (2013) Synthesis and Functional Characterization of Imbutamine Analogs as Histamine H3 and H4 Receptor Ligands. Arch. Pharm. 10.1002/ardp.201300316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.