

Abstract
Particulate matter (PM) air pollution poses a formidable public health threat to the city of Beijing. Among the various hazards of PM pollutants, microorganisms in PM2.5 and PM10 are thought to be responsible for various allergies and for the spread of respiratory diseases. While the physical and chemical properties of PM pollutants have been extensively studied, much less is known about the inhalable microorganisms. Most existing data on airborne microbial communities using 16S or 18S rRNA gene sequencing to categorize bacteria or fungi into the family or genus levels do not provide information on their allergenic and pathogenic potentials. Here we employed metagenomic methods to analyze the microbial composition of Beijing’s PM pollutants during a severe January smog event. We show that with sufficient sequencing depth, airborne microbes including bacteria, archaea, fungi, and dsDNA viruses can be identified at the species level. Our results suggested that the majority of the inhalable microorganisms were soil-associated and nonpathogenic to human. Nevertheless, the sequences of several respiratory microbial allergens and pathogens were identified and their relative abundance appeared to have increased with increased concentrations of PM pollution. Our findings may serve as an important reference for environmental scientists, health workers, and city planners.
Introduction
As an international megacity with a population of over 20 million, Beijing has been suffering from frequent smog events in recent years.1−5 Since the official daily monitoring data became available in 1999, particulate matter has been shown to be a major air pollutant in Beijing,1 and its impact to the public health may be profound.1,4 Categorized by PM2.5 and PM10 (particulate matter with nominal mean aerodynamic diameters of ≤2.5 and ≤10 μm, respectively), PM pollutants of different sizes deposit and affect different regions of the respiratory tract: when inhaled, coarse particles (PM2.5–10) deposit primarily in the head airways, while fine particles (PM2.5) are more likely to penetrate and deposit deeper in the tracheobronchial and alveolar regions.6 Historical data suggest that exposure to such atmospheric particulate matter is linked to increases in morbidity and mortality, and decreases in life expectancy.7,8
During the period of January 10–14, 2013, the city of Beijing, along with the rest of the mideastern region of China experienced a massive, severe smog event.2 The highest daily average PM2.5 concentration in Beijing measured greater than 500 μg/m3 at times (20-fold higher than the WHO guideline value), raising serious public health concerns. Wide-spread respiratory irritation symptoms (e.g., “Beijing cough”) and significant increases of outpatient cases related to respiratory diseases have been reported.2 Here we asked the question of what microorganisms, particularly inhalable allergens and pathogens, are in Beijing’s PM2.5 and PM10 pollutants and what potential effects they may have on the public health during severe smog events like this.
The public health effects of PM, particularly those of PM2.5, have been well documented in the literature.9,10 While the physical and chemical properties of PM2.5 and PM10 pollutants have been extensively studied, relatively less is known about inhalable biological particles such as bacteria, fungi, viruses, pollens, and cell debris in the micrometer to submicrometer size range. It has been suggested that materials of biological origin may contribute as much as 25% to the atmospheric aerosol,11 and they are responsible for various diseases and allergies. The abundance of airborne bacteria measured from 104 to 106 cells per m3, depending on the environment.12 While culture-based methods have been used to detect airborne microorganisms,13 they are constrained to the identification of a limited number of cultivatable species. Although the use of amplicon-based (e.g., 16S or 18S ribosomal RNA (rRNA) gene) sequencing and related techniques have allowed us to detect both cultivatable and noncultivatable microorganisms (although DNA from cell debris may also be detected) and categorize the microbial populations in airborne particles,12,14−17 it has been challenging to sequence the fine, inhalable PM2.5 samples (which are more relevant to human health) due to the low DNA yield, unless with amplification of the extracted DNA.18 Yet amplicon-based sequencing methods often result in biases,19 and most importantly, they are generally limited to categorizing bacteria or fungi at the family or genus level (without the use of marker genes).16,20 Because microbial species within the same family or genus may differ significantly in pathogenic potential, the discovery of microbial allergens and pathogens requires the identification of bacteria, fungi, and viruses at the species or even strain level.21 Thus, microbial metagenomic sequencing represents a powerful alternative for studying complex microbial communities,22 particularly for its ability to discover clinically relevant microbes at the species level.23
Materials and Methods
Particulate Matter Collection
PM2.5 and PM10 samples were collected from the roof top of the Environmental Science Building (40°0′17″N, 116°19′34″E, ∼10 m above the ground, ∼20 m and ∼690 m from the nearest river and hospital, respectively) at Tsinghua University, an area without major pollution sources nearby. This site has been used to monitor PM2.5 pollution in Beijing since 1999.24−26 Sampling was conducted by three high volume air samplers (Thermo Electron Corp., MA, U.S.), two of which were equipped with PM2.5 fractionating inlets, the third one being equipped with a PM10 fractionating inlet. Ambient air was drawn at an average flow rate of 1.13 m3/min for 23 h (10:00 AM to 9:00 AM the next day) per sampling day from January 8 to January 14 (including January 8 as a nonsmog control, according to the Chinese Class II Standard), resulting in approximately 1559 m3 of air flow-through per sampling day. Particulate matter with aerodynamic diameters of ≤2.5 and ≤10 μm were collected on 20.32 × 25.4 cm2 Tissuquartz filters (PALL, NY, U.S.) with 99.9% typical aerosol retention. All the filters were sterilized by baking in a Muffle furnace at 500 °C for 5 h prior to sampling. Each sterilized filter was packaged in sterilized aluminum foil and stored in a sealed bag until being loaded into the filter cartridge. The filter holder and all the tools used for changing new filters were cleaned with 75% ethanol or autoclaved every day to avoid contamination. The net weight of each filter was recorded at mg accuracy before and after sampling. The concentrations of PM2.5 and PM10 at our sampling site were estimated by the net weight of each sample (average weight of the two PM2.5 samples) divided by the 23 h flow-through volume per sampling day (to avoid microbial contamination, samples were not kept under 45% relative humidity at 20 °C as typically required for PM measurements). A 47 mm diameter filter punch was taken from the PM10 sample and one of the PM2.5 samples each day for chemical component and elemental analyses. The filter punches were kept in size adaptive chambers and stored at −20 °C. All other samples were stored at −80 °C until downstream analyses were performed.
DNA Extraction
To overcome the issue of low yield during genomic DNA extraction, several technical improvements were made to optimize the extraction of high-quality DNA from PM samples. Considering the different DNA yield of PM2.5 and PM10 samples, 1/4 of PM10 filter (a total of ∼103.04 cm2) and 1 and 3/4 of the PM2.5 filters (a total of ∼721.28 cm2) from each sampling day were used for DNA extraction. The filters were cut into 8.96 ×11.5 cm2 pieces and were placed in 50 mL centrifuge tubes filled with sterilized 1X PBS buffer. The PM samples were then pelleted at 4 °C by centrifugation at 200g for 2 h. After gentle vortexing, the resuspension was filtered with a 0.2 μm Supor 200 PES Membrane Disc Filter (PALL, NY, U.S.), which was then cut into small pieces and used for DNA extraction using the MO-BIO PowerSoil DNA isolation kit (Carlsbad, CA, U.S.). All the steps mentioned above were carried out in a clean bench. Scissors, forceps, and filter funnels were all sterilized before use. The samples were then heated to 65 °C in PowerBead Tubes for 10 min followed by vortexing for 2 min. The remaining steps of the extraction were performed according to the standard MO-BIO PowerSoil DNA isolation protocol except for the column purification step, which was replaced with magnetic bead purification (Agencourt AMPure XP, Beckman, CA, U.S.) for improved yield. Genomic DNA quality and concentration were analyzed by gel electrophoresis and a fluorescent dsDNA-binding dye assay (Qubit Fluorometer, Life Technologies, CA, U.S.). Blank control samples were collected by placing a sterilized filter inside of the sampler without operation for 23 h, and treated similarly as above. DNA extraction of blank control samples resulted in DNA concentrations below the detection limit of our instruments, and library generation efforts failed to generate useable sequencing libraries. All the extracted DNA samples were stored at −80 °C until further use.
Sequencing and Phylogenetic Analysis
The Illumina MiSeq (for library validation) and HiSeq 2000 sequencing systems (Illumina, CA, U.S.) were used for sequencing, and the library preparation kits were purchased from New England Biolabs (MA, U.S.). Sequencing library construction and template preparation were performed according to the NEB library preparation protocols. We constructed a paired-end library with insert size of ∼500 bp for each sample. An aliquot of 5 ng DNA from each sample was used as the starting amount (except for 3 samples, the total quantities of DNA of which were less than 5 ng, Supporting Information (SI), Table S1) for library preparation in order to ensure sample consistency. In order to minimize possible bias introduced by PCR, 12 cycles were performed during PCR amplification. Each sample was barcoded and equal quantities of barcoded libraries were used for sequencing (for index sequences, see SI Table S1). Adaptor contamination and low-quality reads were discarded from the raw data. In total, ∼98 Gb sequence with a uniform read length of 90 bp was obtained and an average of ∼7 Gb high-quality HiSeq sequences were generated from each sample (SI Table S1). The rarefaction curve (analyzed by the Metagenomics RAST server (MG-RAST, release 3.3))27 suggested that the sequencing depth of the HiSeq data was sufficient to capture most of the microorganisms but not the MiSeq data (average data set of 683Mb) (SI Figure S1). MetaPhlAn (Metagenomic Phylogenetic Analysis)28 was used to estimate the relative abundance of bacteria and archaea with unique clade-specific genes at the species level (SI Figures S2 and S3). The Illumina HiSeq reads were aligned to a cohort of nonredundant NCBI complete genomes (2637 complete genomes, including bacteria, fungi, archaea, and viruses) using the Short Oligonucleotide Analysis Package (SOAP) alignment tool (release 2.21t)29 to profile the common core species. We used a 90% identity threshold for bacteria, archaea, and fungi, and 100% identity for viruses due to their smaller genome sizes. Genome coverage was calculated using the SOAP.coverage package (version 2.7.7). Only uniquely aligned reads were used in the analysis. Bacterial, fungal, and viral species with coverage of ≥5%, ≥0.5% (average alignment of all chromosomes), and ≥1%, respectively, in either PM2.5 or PM10 samples of 7 consecutive sampling days were listed in SI Table S2. The genome-size-normalized relative abundance of these species was calculated based on the number of aligned reads normalized by the species’ genome size (SI Figure S4 and Table S3). The variations of the hit abundance of species across 7 sampling days were estimated based on the hit numbers normalized by number of total aligned reads (SI Figure S5). The Greengenes 16S rRNA gene database30 was used for 16S rRNA phylogenetic analysis with the following alignment parameters: >97% identity, minimal alignment 40 bp. DNA sequence data have been deposited in MG-RAST (http://metagenomics.anl.gov/) at the following URL: http://metagenomics.anl.gov/linkin.cgi?project=3756.
Analysis of Original Bacteria Habitats
The Greengenes 16S rRNA gene database30 was used for assigning the HiSeq reads at >97% identity threshold (only uniquely aligned reads were used for following calculations). All of the Greengenes database sequences with available information of bacteria habitats were classified into the four categories without overlaps. In addition, the 16S sequences of previous studies on high-altitude16 and urban airborne bacteria of Milan14 and New York17 were assigned to the same habitat categories and compared to our results (SI Figure S6).
Results
Sequence of the Airborne Metagenome
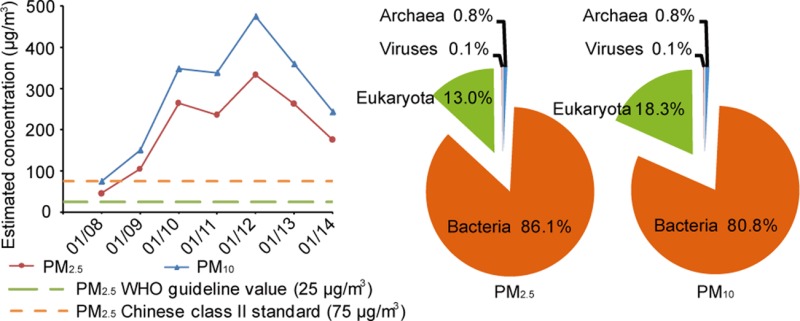
We sought to sequence the metagenome of inhalable airborne microorganisms in Beijing’s PM2.5 and PM10 pollutants, after having overcome the technical issues involved in high-volume PM2.5 and PM10 sample collection, DNA extraction, and library generation (for details of the DNA extraction methods, see Materials and Methods). PM2.5 and PM10 samples collected at a Beijing Tsinghua PM monitoring site (40°0′17″N, 116°19′34″E) from January 8–14, 2013, during which period Beijing’s PM2.5 and PM10 pollution indexes rapidly deteriorated from healthy to record-high hazardous levels, were used for sequencing (Figure 1A, SI Figures S7–S9 and Table S4; air temperatures are typically low in January in Beijing, creating a unique high PM, low temperature environment). The Illumina HiSeq data from a total of 7 daily PM2.5 and 7 PM10 samples is more than 1000-fold larger than those of three previous studies on airborne bacteria combined,14,16,17 from which original sequence data were publicly available (SI Table S1). By aligning to the Greengenes 16S rRNA gene database30 at a 97% identity threshold, we discovered 255 more bacteria genera than those identified by the three previous studies (SI Figure S10 and Table S5). Overall, the PM2.5 samples contained 86.1% bacterial, 13.0% eukaryotic, 0.8% archaeal, and 0.1% viral reads, while the PM10 samples contained 80.8% bacterial, 18.3% eukaryotic, 0.8% archaeal, and 0.1% viral reads (Figure 1B). The higher relative abundance of eukaryotic reads (which included those from fungi, plants, algae, and animal debris), as well as the higher alpha diversity (a measurement of species diversity) found in PM10 compared with those of PM2.5 samples (Figure 1C), may in part be attributed to the fact that the aerodynamic diameters of many fungal spore agglomerates were between 2.5 and 10 μm.31−33 Principal component analysis (PCA) of the microbial relative abundance (Figure 1D and SI Table S6) and dinucleotide frequency (SI Figure S11) suggested that the metagenomes of airborne microbes were distinct from those of other environments, though relatively more related to the soil metagenomes.
Figure 1.

Characteristics of the collected PM samples and sequenced metagenomes. (A) Daily average PM2.5 and PM10 concentrations estimated from the collected samples during January 8–14, 2013. (B) Relative abundance of the MG-RAST taxonomic hits at the domain level in PM2.5 and PM10 samples. (C) Estimated average alpha diversity of the PM2.5 and PM10 samples (error bars represent SD of the 7 daily PM2.5 and 7 PM10 samples, respectively). (D) Principal component analysis of the relative abundance of microorganisms at the phylum level of the 14 sequenced PM metagenomes (red) compared to those of other environments (other colors).
Bacteria Were the Most Abundant Airborne Prokaryotic Microorganisms and the Majority of Them Were Terrestrial-Related
Bacteria appeared to be the most abundant prokaryotic microorganisms in PM2.5 and PM10 pollutants. To identify the prokaryotic species and to estimate their relative abundance, we used the Metagenomic Phylogenetic Analysis (MetaPhlAn) toolbox28 to reveal a picture of complex bacteria and archaea community (Figure 2A and SI Figure S2). We show that the most abundant phyla were Actinobacteria, Proteobacteria, Chloroflexi, Firmicutes, Bacteroidetes, and Euryarchaeota (relative abundance ≥1%). At the species level, 1315 distinct bacterial and archaeal species were identified from the 14 samples. An unclassified bacterium in the nitrogen fixing, filamentous bacteria genus Frankia appeared to be the most abundant (Figure 2A and SI Figure S3). The most abundant classified bacterial species appeared to be Geodermatophilus obscurus, a bacterium commonly found in dry soil environments (SI Table S7). By aligning to the Greengenes 16S database, categorized by terrestrial, fecal, freshwater, and marine-associated bacteria (see Materials and Methods), we show that the majority (>85%) of the categorized bacteria in the collected PM2.5 and PM10 samples were related to fecal and terrestrial sources (Figure 2B and SI Figure S6). The proportion of bacteria from terrestrial-related sources appeared to be higher than those identified from the three previous studies (Figure 2C and SI Figure S6).14,16,17 This may in part be attributed to the lack of vegetation coverage and abundance of dry, exposed soil, and construction sites in the city of Beijing and its surrounding areas, especially during the winter seasons. In addition, while the proportion of freshwater and marine-associated bacteria remained relatively constant, the fraction of fecal-associated bacteria appeared to have increased (from 4.5% to as high as 11.4% in PM2.5 samples) (Figure 2B) with progressively increased concentrations of PM pollution.
Figure 2.

Bacterial and archaeal species in PM samples and their original habitats. (A) Phylogenetic tree of the bacteria and archaea identified from PM2.5 samples, analyzed by MetaPhlAn. The sizes of the nodes correspond to the relative abundance at the corresponding levels in the cohort. The family, genus, and species levels of the most abundant order Actinomycetales are plotted. Only nodes with ≥1% relative abundance are labeled. (B) Original habitats of the identified bacteria in daily PM2.5 and PM10 samples, categorized by terrestrial, fecal, freshwater, and marine sources. (C) Bacterial and archaeal species in Beijing's PM samples were pooled and compared with those identified from the GRIP high-altitude, Milan urban, and New York subway studies.
Most Abundant Bacterial, Fungal, and Viral Species in PM2.5 and PM10
Since not only bacteria, but also fungi and viruses are responsible for various human allergies and diseases, we sought to identify the microbial species including fungi and viruses (which are currently not supported by the MetaPhlAn toolbox) in PM2.5 and PM10 pollutants. We employed the Short Oligonucleotide Analysis Package (SOAP) alignment tool29 to align the HiSeq reads from each sample to a cohort of 2637 nonredundant species of NCBI complete genomes, including bacteria, archaea, fungi, and viruses. At a 90% identity threshold and ≥5% coverage of the complete bacterial genomes (for a typical bacterial genome of 4 Mb, it corresponds to a minimal alignment length of ∼200 kb, >100-fold longer than the 16S rRNA gene and thus provides more confidence) or ≥0.5% coverage for fungal genomes, the 48 most abundant bacterial and 2 fungal species were identified (SI Table S2). Because of the smaller genome size of viruses, we used a more stringent alignment strategy (i.e., 100% identity and ≥1% coverage), and 3 most abundant viral species were identified. We next estimated the genome-size-normalized relative abundance (defined as the number of unique hit reads normalized by genome size) of each species within the most common ones (SI Table S3) and analyzed the daily variations of their relative abundance during the 7 sampling days (Figure 3). Consistent with the MetaPhlAn results, the soil-associated bacteria G. obscurus appeared to be the most abundant classified bacterial species (with an average genome coverage of ∼42.7% and relative abundance of ∼14.6% in the PM2.5 samples), followed by Modestobacter marinus, Blastococcus saxobsidens, Kocuria rhizophila, and Micrococcus luteus, all of which are commonly found in soil habitats and some with the abilities to survive under “tough” (e.g., UV radiation) conditions (SI Table S7). Although the relative abundance of most of the bacterial species remained stable during the 7 sampling days, as was found in previous studies,12,34 some showed considerable variations. For example, the relative abundance of Thermobifida fusca, an important bacterial degrader of plant cell walls and commonly found in decaying organic matter (SI Table S7), increased ∼5-fold from an average of 0.7% during the first 2 less polluted days to an average of 3.7 ± 2.5% in the 5 heavily polluted days in PM2.5 samples (SI Figures S4 and S12).
Figure 3.

Box plot of the daily variations of the relative abundance of 48 most common bacterial, 2 fungal, and 3 viral species in PM samples. Boxes correspond to the interquartile range between the 25th and 75th percentiles, and the central lines represent the 50th percentile. Whiskers correspond to the lowest and highest values no more than 1.5 times the interquartile range from the box, while dots are the outliers beyond the whiskers. PM2.5 samples are labeled pink and PM10 are black.
Microbial Allergens and Pathogens in the PM2.5 and PM10 Samples
Among the identified microbial species, several are known to cause human allergies and respiratory diseases, including Streptococcus pneumoniae, Aspergillus fumigatus, and human adenovirus C (with average genome coverage of 2.0%, 14.5%, and 6.5%, respectively). Among them, S. pneumoniae is the most common cause for community-acquired pneumonia (CAP), having been isolated from nearly 50% of CAP cases.35 Its representation within the entire bacteria community (analyzed by MetaPhlAn) was 0.012% in PM2.5 samples and 0.017% in PM10 samples, and the normalized number of hit reads (hit abundance) appeared to have increased by ∼2 fold from an average of 0.024% during the first 2 less polluted days to an average of 0.05 ± 0.02% in the 5 heavily polluted days in PM2.5 samples (Figure 4A). A. fumigatus, likely collected in the form of spores, is known as a major fungal allergen and opportunistic pathogen that causes airway or lung invasion in immunodeficient patients.36 Its average hit abundance was found to be higher in PM10 than in PM2.5 samples (4.5% vs 1.7%), most likely because the aerodynamic diameters of the fungal spore agglomerates are between 2.5 and 10 μm.37 The hit abundance of A. fumigates also appeared to be correlated with the increase of PM pollution levels, increasing ∼4-fold from an average of 1.5% during the first 2 less polluted days to an average of 5.8 ± 1.8% in the 5 heavily polluted days in PM10 samples (Figure 4B). To confirm the existence of A. fumigatus in our samples, we cultured the fungus and validated its existence by sequencing the 18S rRNA gene and a species-specific gene (gliI) (SI Table S8), as well as SEM imaging (SI Figure S13). Human adenovirus, a dsDNA virus that accounts for 5–10% of upper and lower respiratory tract infections in children,38 was also found (with 100% sequence matched to human adenovirus C in all 14 samples). The hit abundance of adenovirus in our samples also appeared to have increased during the heavily polluted days, though with more daily variations than those of S. pneumonia and A. fumigates (SI Figure S5).
Figure 4.

Daily variations of the normalized hit abundance of microbial pathogens and allergens in the collected PM2.5 and PM10 samples. (A) S. pneumoniae and (B) A. fumigatus.
Chemical Composition Analysis of the PM Pollutants
To put our findings in the context of aerosol chemistry, we analyzed the organic and elemental carbon, water-soluble ions, and elemental composition of the collected samples (SI Tables S9–S11). We found that sulfate, ammonium, nitrate, and organic matter were among the most abundant, and altogether their relative abundance in PM2.5 and PM10 samples were 80% and 71% (w/w), respectively. These results, as well as the high weight ratio of PM2.5 to PM10 (∼0.7), suggested that secondary formation of fine particles likely led to the high PM concentrations during this smog event.26 Additionally, the high relative humidity (SI Figure S8) during the period may have contributed to particle growth through water uptake and promoting aqueous redox chemistry (e.g., the oxidation of sulfur dioxide to sulfate). This also suggested that most of the particles were rich in water content during the polluted days and thus would favor the survival of microbes.39
Discussion
PM pollution has been studied extensively in the context of aerosol chemistry and physics,24,26 and statistical correlations between PM pollution and decreased life expectancy have been made.8 So far, no specific components of PM have been conclusively shown to be harmless.40 In particular, much less is understood about what microorganisms are in the PM pollutants. Previous studies have shown that bioaerosols containing pathogens are responsible for the spread of respiratory diseases,41,42 and thus it is crucial to understand the composition of airborne microbes at the species level and to identify the potential microbial allergens and pathogens. Most of the clinically relevant studies on inhalable pathogens were conducted in hospital environments,42−45 yet in Beijing, a significant increase of outpatient cases related to respiratory diseases during the same severe PM pollution period studied here has been reported.2 Our results have provided sequence-based evidence for the existence of inhalable microbial allergen and pathogen species in an open environment, and suggested that high PM pollution may pose health threats to the susceptible population (e.g., the elderlies and the immunodeficient). Besides, information on the original habitats of airborne bacteria provides important insights for understanding the source of the biological particles, and may be used as a reference for future urban planning efforts to reduce PM pollution and the spread of airborne microbial allergens and pathogens. In future studies, clinical samples (e.g., sputum samples from respiratory disease patients) during severely polluted and unpolluted days can be obtained, and the sequence information can be compared to those from collected PM samples for comparison. Furthermore, PM exposure studies on animal models can be performed to characterize the effects of PM-associated allergens and pathogens, leading to better understandings of their pathogenicity.
Using the current methods, we were able to identify bacterial, archaeal, fungal, and dsDNA viral species in the collected PM samples. Cultivation was used to verify the existence of A. fumigatus. We also attempted to culture other bacteria and fungi species, but not all were successfully cultured (data not shown) since some species were slow-growing or difficult to culture, and the samples were stored at −80 °C before use. RNA viruses such as rhinovirus and influenza virus are undoubtedly important viral agents that affect the public health. Yet in our experience, it appeared to be technically challenging to extract sufficient quantities of RNA for reverse transcription and sequencing from PM samples containing various RNA-degrading containments such as divalent cations. Low-bias preamplification techniques may be used to generate sufficient libraries for the sequencing of RNA viruses in PM samples in future studies.46 As for human dsDNA viruses, human adenovirus C appeared to be the most abundant in our PM samples based on our sequencing results. More importantly, the current study was limited by the daily sampling capacity and availability of sampling sites, as well as the trade-off between obtaining high-depth sequence data for species-level characterization vs more sampling days (98 Gb data from 14 samples). In particular, though PM pollution levels are typically high in the winter of Beijing, low temperatures are often associated with lower overall microbial abundance compared to warmer seasons. Thus, future longitudinal and multiple location studies to identify airborne microorganisms should be performed to compare with our current results and to provide better insights on the increased incidences of respiratory diseases during urban smog events, and to correlate with meteorological data, chemical components, and clinically obtained pathogen samples. Additionally, the establishment of a monitoring network for airborne microbes can be invaluable during outbreaks of deadly respiratory diseases. Information on the abundance of particular airborne pathogens and their regional and seasonal variations will be of particular importance for the prevention of respiratory diseases at a public scale, in areas such as vaccine design and distribution, as well as for understanding the spread of drug resistant respiratory pathogens.
Acknowledgments
We thank Gong Cheng, Lei Huang, Babak Javid, Masood Kayani, Yigong Shi, Melody Toosky, Hongwei Wang, Zhen Xie, Nieng Yan, Li Yu, Jingren Zhang, and Michael Q. Zhang for helpful discussions and comments on the manuscript, and Qingran Bai, He Chen, Siyu Chen, Zhen Cheng, Hongliang Fu, Ru He, Long Hu, Dongfang Li, Junxiang Li, Cuihua Liu, Kaigui Luo, Peng Liang, Yu Liang, Yongbin Li, Zhixun Shen, Shuxiao Wang, Tingting Wang, Zhiying Xie, Siyin Zhang, and Wei Zhou for their assistance with the experiments. BGI-Shenzhen provided the sequencing platforms used in this study and assistance in preliminary sequence analysis. NSCC-TJ (D.L.) and Tsinghua University School of Information Science and Technology provided computational facilities and assistance with the data analysis. This work was supported in part by funding from the Tsinghua University—Peking University Center for Life Sciences (CLS), Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, the Science and Technology Major Project of the Ministry of Science and Technology of China (Grant No. 2013ZX10003003 to Z.Z.), National Natural Science Foundation of China (Grant No. 21190054, 21107060, and 21221004), Center for Marine Medicine and Rescue of Tsinghua University, and Tsinghua Qian Ren Tuan Dui funding (to M.Q.Z.).
Supporting Information Available
Detailed methods: PM and meteorological data; scanning electron microscopy; OC and EC analysis; elemental analysis; water soluble ion analysis; (Figure S1) rarefaction curves of PM2.5 and PM10 samples sequenced by MiSeq and HiSeq platforms; (Figure S2) phylogenetic tree of bacteria and archaea identified from PM2.5 and PM10 samples; (Figure S3) heatmaps of the relative abundance of bacteria and archaea identified by MetaPhlAn; (Figure S4) variations of the genome-size-normalized relative abundance of the most common bacterial, fungal, and viral species; (Figure S5) daily variations of the normalized hit abundance of human adenovirus C in the collected PM2.5 and PM10 samples; (Figure S6) original habitats of the bacteria identified from the Tsinghua PM2.5 and PM10 samples compared with three previous studies; (Figure S7) PM2.5 and PM10 concentration of Beijing, Haidian district and Tsinghua monitoring site; (Figure S8) meteorological data plotted with PM2.5 and PM10 concentration in January 2013; (Figure S9) filter coloration and SEM images of PM2.5 and PM10 samples; (Figure S10) number of overlapping airborne microbes at the genus level between Tsinghua PM study and three previous studies; (Figure S11) principle component analysis of dinucleotide frequency of pooled PM2.5 samples and PM10 samples; (Figure S12) percentage increase of 53 common species identified from PM samples; (Figure S13) images of cultured Aspergillus fumigatus from PM samples; (Table S1) summary of sequencing data; (Table S2) genome coverage of the 48 most common bacterial, 2 fungal, and 3 viral species; (Table S3) genome-size-normalized relative abundance of the 48 most common bacterial, 2 fungal, and 3 viral species; (Table S4) summary of PM and meteorological data; (Table S5) airborne bacteria identified at the genus level from Tsinghua PM study and three previous studies combined; (Table S6) public data of metagenomic studies used in PCA analysis; (Table S7) annotations of the 48 most common bacterial, 2 fungal, and 3 viral species; (Table S8) primers and Sanger sequencing results; (Table S9) estimated concentrations of organic carbon and elemental carbon measured in PM2.5 and PM10 samples; (Table S10) estimated concentrations of anions and cations measured in PM2.5 and PM10 samples; (Table S11) estimated concentrations of elements measured in PM2.5 and PM10 samples; and additional references. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
▽ These authors contributed equally to this work.
The authors declare no competing financial interests.
Supplementary Material
References
- Cheng Z.; Jiang J.; Fajardo O.; Wang S.; Hao J. Characteristics and health impacts of particulate matter pollution in China (2001–2011). Atmos. Environ. 2013, 650186–194. [Google Scholar]
- Ouyang Y. China wakes up to the crisis of air pollution. Lancet Respir. Med. 2013, 1112. [DOI] [PubMed] [Google Scholar]
- Parrish D. D.; Zhu T. Climate change. Clean air for megacities. Science 2009, 3265953674–675. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Mauzerall D. L.; Zhu T.; Liang S.; Ezzati M.; Remais J. V. Environmental health in China: Progress towards clean air and safe water. Lancet 2010, 37597201110–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; He K.; Huo H. Policy: Cleaning China’s air. Nature 2012, 4847393161–162. [DOI] [PubMed] [Google Scholar]
- Brook R. D.; Franklin B.; Cascio W.; Hong Y.; Howard G.; Lipsett M.; Luepker R.; Mittleman M.; Samet J.; Smith S. C. Jr.; Tager I. Air pollution and cardiovascular disease: A statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation 2004, 109212655–2671. [DOI] [PubMed] [Google Scholar]
- Hunt A.; Abraham J. L.; Judson B.; Berry C. L. Toxicologic and epidemiologic clues from the characterization of the 1952 London smog fine particulate matter in archival autopsy lung tissues. Environ. Health Perspect. 2003, 11191209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C. A. 3rd; Ezzati M.; Dockery D. W. Fine-particulate air pollution and life expectancy in the United States. N. Engl. J. Med. 2009, 3604376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzalova K.; Rossner P.; Sram R. J. Oxidative damage induced by carcinogenic polycyclic aromatic hydrocarbons and organic extracts from urban air particulate matter. Mutat. Res.-Gen. Tox. En. 2010, 6962114–121. [DOI] [PubMed] [Google Scholar]
- Valavanidis A.; Fiotakis K.; Vlachogianni T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Heal. Part C 2008, 264339–362. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. Abundance of cellular material and proteins in the atmosphere. Science 2005, 308571873. [DOI] [PubMed] [Google Scholar]
- Bowers R. M.; McLetchie S.; Knight R.; Fierer N. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 2011, 54601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.; Grinshpun S. A.; Martuzevicius D.; Adhikari A.; Crawford C. M.; Luo J.; Reponen T. Relationship between indoor and outdoor bioaerosols collected with a button inhalable aerosol sampler in urban homes. Indoor Air 2006, 16137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini V.; Gandolfi I.; Ambrosini R.; Bestetti G.; Innocente E.; Rampazzo G.; Franzetti A. Temporal variability and effect of environmental variables on airborne bacterial communities in an urban area of Northern Italy. Appl. Microbiol. Biotechnol. 2013, 97146561–6570. [DOI] [PubMed] [Google Scholar]
- Brodie E. L.; DeSantis T. Z.; Parker J. P.; Zubietta I. X.; Piceno Y. M.; Andersen G. L. Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. U.S.A. 2007, 1041299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon-Rodriguez N.; Lathem T. L.; Rodriguez R. L.; Barazesh J. M.; Anderson B. E.; Beyersdorf A. J.; Ziemba L. D.; Bergin M.; Nenes A.; Konstantinidis K. T. Microbiome of the upper troposphere: species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc. Natl. Acad. Sci. U.S.A. 2013, 11072575–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson C. E.; Baumgartner L. K.; Harris J. K.; Peterson K. L.; Stevens M. J.; Frank D. N.; Pace N. R. Culture-independent analysis of aerosol microbiology in a metropolitan subway system. Appl. Environ. Microbiol. 2013, 79113485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers R. M.; Clements N.; Emerson J. B.; Wiedinmyer C.; Hannigan M. P.; Fierer N. Seasonal variability in bacterial and fungal diversity of the near-surface atmosphere. Environ. Sci. Technol. 2013, 472112097–12106. [DOI] [PubMed] [Google Scholar]
- Acinas S. G.; Sarma-Rupavtarm R.; Klepac-Ceraj V.; Polz M. F. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 2005, 71128966–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers R. M.; Lauber C. L.; Wiedinmyer C.; Hamady M.; Hallar A. G.; Fall R.; Knight R.; Fierer N. Characterization of airborne microbial communities at a high-elevation site and their potential to act as atmospheric ice nuclei. Appl. Environ. Microbiol. 2009, 75155121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoling A.; Wiklund G.; Savarino S. J.; Cohen D. I.; Svennerholm A. M. Comparative analyses of phenotypic and genotypic methods for detection of enterotoxigenic Escherichia coli toxins and colonization factors. J. Clin. Microbiol. 2007, 45103295–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Li R.; Raes J.; Arumugam M.; Burgdorf K. S.; Manichanh C.; Nielsen T.; Pons N.; Levenez F.; Yamada T.; Mende D. R.; Li J.; Xu J.; Li S.; Li D.; Cao J.; Wang B.; Liang H.; Zheng H.; Xie Y.; Tap J.; Lepage P.; Bertalan M.; Batto J. M.; Hansen T.; Le Paslier D.; Linneberg A.; Nielsen H. B.; Pelletier E.; Renault P.; Sicheritz-Ponten T.; Turner K.; Zhu H.; Yu C.; Jian M.; Zhou Y.; Li Y.; Zhang X.; Qin N.; Yang H.; Wang J.; Brunak S.; Dore J.; Guarner F.; Kristiansen K.; Pedersen O.; Parkhill J.; Weissenbach J.; Bork P.; Ehrlich S. D. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464728559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Li Y.; Cai Z.; Li S.; Zhu J.; Zhang F.; Liang S.; Zhang W.; Guan Y.; Shen D.; Peng Y.; Zhang D.; Jie Z.; Wu W.; Qin Y.; Xue W.; Li J.; Han L.; Lu D.; Wu P.; Dai Y.; Sun X.; Li Z.; Tang A.; Zhong S.; Li X.; Chen W.; Xu R.; Wang M.; Feng Q.; Gong M.; Yu J.; Zhang Y.; Zhang M.; Hansen T.; Sanchez G.; Raes J.; Falony G.; Okuda S.; Almeida M.; LeChatelier E.; Renault P.; Pons N.; Batto J. M.; Zhang Z.; Chen H.; Yang R.; Zheng W.; Li S.; Yang H.; Wang J.; Ehrlich S. D.; Nielsen R.; Pedersen O.; Kristiansen K.; Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490741855–60. [DOI] [PubMed] [Google Scholar]
- He K.; Yang F.; Ma Y.; Zhang Q.; Yao X.; Chan C. K.; Cadle S.; Chan T.; Mulawa P. The characteristics of PM2.5 in Beijing, China. Atmos. Environ. 2001, 35294959–4970. [Google Scholar]
- Wang S.; Zhao M.; Xing J.; Wu Y.; Zhou Y.; Lei Y.; He K.; Fu L.; Hao J. Quantifying the air pollutants emission reduction during the 2008 Olympic games in Beijing. Environ. Sci. Technol. 2010, 4472490–2496. [DOI] [PubMed] [Google Scholar]
- Yang F.; Tan J.; Zhao Q.; Du Z.; He K.; Ma Y.; Duan F.; Chen G.; Zhao Q. Characteristics of PM2.5 speciation in representative megacities and across China. Atmos. Chem. Phys. 2011, 11115207–5219. [Google Scholar]
- Meyer F.; Paarmann D.; D’Souza M.; Olson R.; Glass E. M.; Kubal M.; Paczian T.; Rodriguez A.; Stevens R.; Wilke A.; Wilkening J.; Edwards R. A. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N.; Waldron L.; Ballarini A.; Narasimhan V.; Jousson O.; Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 98811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Yu C.; Li Y.; Lam T. W.; Yiu S. M.; Kristiansen K.; Wang J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25151966–1967. [DOI] [PubMed] [Google Scholar]
- DeSantis T. Z.; Hugenholtz P.; Larsen N.; Rojas M.; Brodie E. L.; Keller K.; Huber T.; Dalevi D.; Hu P.; Andersen G. L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 7275069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. A.; Liao C. H. Size-selective assessment of agricultural workers’ personal exposure to airborne fungi and fungal fragments. Sci. Total Environ. 2014, 466–467, 725–732. [DOI] [PubMed] [Google Scholar]
- Reponen T.; Willeke K.; Ulevicius V.; Reponen A.; Grinshpun S. A. Effect of relative humidity on the aerodynamic diameter and respiratory deposition of fungal spores. Atmos. Environ. 1996, 30233967–3974. [Google Scholar]
- Brook R. D.; Franklin B.; Cascio W.; Hong Y. L.; Howard G.; Lipsett M.; Luepker R.; Mittleman M.; Samet J.; Smith S. C.; Tager I. Air pollution and cardiovascular disease—A statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation 2004, 109212655–2671. [DOI] [PubMed] [Google Scholar]
- Bowers R. M.; Sullivan A. P.; Costello E. K.; Collett J. L. Jr.; Knight R.; Fierer N. Sources of bacteria in outdoor air across cities in the midwestern United States. Appl. Environ. Microbiol. 2011, 77186350–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim W. S.; Macfarlane J. T.; Boswell T. C.; Harrison T. G.; Rose D.; Leinonen M.; Saikku P. Study of community acquired pneumonia aetiology (SCAPA) in adults admitted to hospital: implications for management guidelines. Thorax 2001, 564296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierman W. C.; Pain A.; Anderson M. J.; Wortman J. R.; Kim H. S.; Arroyo J.; Berriman M.; Abe K.; Archer D. B.; Bermejo C.; Bennett J.; Bowyer P.; Chen D.; Collins M.; Coulsen R.; Davies R.; Dyer P. S.; Farman M.; Fedorova N.; Fedorova N.; Feldblyum T. V.; Fischer R.; Fosker N.; Fraser A.; Garcia J. L.; Garcia M. J.; Goble A.; Goldman G. H.; Gomi K.; Griffith-Jones S.; Gwilliam R.; Haas B.; Haas H.; Harris D.; Horiuchi H.; Huang J.; Humphray S.; Jimenez J.; Keller N.; Khouri H.; Kitamoto K.; Kobayashi T.; Konzack S.; Kulkarni R.; Kumagai T.; Lafon A.; Latge J. P.; Li W.; Lord A.; Lu C.; Majoros W. H.; May G. S.; Miller B. L.; Mohamoud Y.; Molina M.; Monod M.; Mouyna I.; Mulligan S.; Murphy L.; O’Neil S.; Paulsen I.; Penalva M. A.; Pertea M.; Price C.; Pritchard B. L.; Quail M. A.; Rabbinowitsch E.; Rawlins N.; Rajandream M. A.; Reichard U.; Renauld H.; Robson G. D.; Rodriguez de Cordoba S.; Rodriguez-Pena J. M.; Ronning C. M.; Rutter S.; Salzberg S. L.; Sanchez M.; Sanchez-Ferrero J. C.; Saunders D.; Seeger K.; Squares R.; Squares S.; Takeuchi M.; Tekaia F.; Turner G.; Vazquez de Aldana C. R.; Weidman J.; White O.; Woodward J.; Yu J. H.; Fraser C.; Galagan J. E.; Asai K.; Machida M.; Hall N.; Barrell B.; Denning D. W. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 2005, 43870711151–1156. [DOI] [PubMed] [Google Scholar]
- Deacon L. J.; Pankhurst L. J.; Drew G. H.; Hayes E. T.; Jackson S.; Longhurst P. J.; Longhurst J. W. S.; Liu J.; Pollard S. J. T.; Tyrrel S. F. Particle size distribution of airborne Aspergillus fumigatus spores emitted from compost using membrane filtration. Atmos. Environ. 2009, 43355698–5701. [Google Scholar]
- Shike H.; Shimizu C.; Kanegaye J.; Foley J. L.; Burns J. C. Quantitation of adenovirus genome during acute infection in normal children. Pediatr. Infect. Dis. J. 2005, 24129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier C. O.; Khlystov A. Y.; Chan W. R.; Mandiro M.; Pandis S. N. A method for the in situ measurement of fine aerosol water content of ambient aerosols: The dry-ambient aerosol size spectrometer (DAASS). Aerosol Sci. Technol. 2004, 38, 215–228. [Google Scholar]
- Fowler D.; Brunekreef B.; Fuzzi S.; Monks P. S.; Sutton M. A.; Brasseur G. P.; Friedrich R.; Passante L. G.; Jimenez Mingo J. M.. Research Findings in support of the EU Air Quality. 2013. http://www.accent-network.org/EuAirQualityReview/docs/PDF%20BASSA.pdf (accessed Jan 10, 2014)
- Clifton I. J.; Peckham D. G. Defining routes of airborne transmission of Pseudomonas aeruginosa in people with cystic fibrosis. Exp. Rev. Respir. Med. 2010, 44519–529. [DOI] [PubMed] [Google Scholar]
- Eames I.; Tang J. W.; Li Y.; Wilson P. Airborne transmission of disease in hospitals. J. R. Soc. Interface 2009, 6, S697–S702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies D.; Fanning A.; Yuan L.; FitzGerald J. M. Hospital ventilation and risk for tuberculous infection in Canadian health care workers. Ann. Intern. Med. 2000, 13310779–789. [DOI] [PubMed] [Google Scholar]
- Haddad S. H.; Arabi Y. M.; Memish Z. A.; Al-Shimemeri A. A. Nosocomial infective endocarditis in critically ill patients: A report of three cases and review of the literature. Int. J. Infect. Dis. 2004, 84210–216. [DOI] [PubMed] [Google Scholar]
- Ferroni A.; Werkhauser-Bertrand A.; Le Bourgeois M.; Beauvais R.; Vrielynck S.; Durand C.; Lenoir G.; Berche P.; Sermet-Gaudelus I. Bacterial contamination in the environment of hospitalised children with cystic fibrosis. J. Cyst. Fibros. 2008, 76477–482. [DOI] [PubMed] [Google Scholar]
- Duhaime M. B.; Deng L.; Poulos B. T.; Sullivan M. B. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environ. Microbiol. 2012, 1492526–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.