

Abstract
Background
Systemic lupus erythematosus (SLE) is associated with premature atherosclerosis but the mechanisms underlying this association are not understood. The role of endothelial dysfunction is hypothesized.
Methods
In predominantly non-Caucasian patients with SLE (n=119) and controls (n=71), carotid ultrasonography was performed and circulating endothelial cells (CECs), soluble endothelial protein C receptor and gene polymorphism at A6936G, soluble E-selectin (sE-selectin), and adiponectin were assessed.
Results
Carotid plaque was more prevalent among patients than controls (43% vs 17%, p = 0.0002). Mean CCA IMT was greater in patients compared to controls (0.59mm ± 0.19 vs 0.54mm ± 0.11, p=0.03). Among SLE patients, plaque was not associated with smoking, body mass index, LDL, triglycerides, homocysteine, C-reactive protein, anti-ds DNA antibody, C3, C4, SLE activity, or medications. Age and levels of soluble E-selectin and adiponectin were significantly higher in the SLE patients with plaque compared to those without plaque in univariate and multivariate analyses. sE-selectin and adiponectin were found to serve as independent predictors of carotid plaque and that elevations were persistent over more than one visit. Unexpectedly, these biomarkers were present despite clinical quiescence.
Conclusion
Premature atherosclerosis is a consistent feature of SLE and extends across ethnicities. Higher levels of adiponectin may represent a physiological attempt to limit further endothelial damage already reflected by the elevation in sE-selectin and the observed increase in plaque represents overwhelming of this reparative process by atherogenic stimuli.
Keywords: Systemic Lupus Erythematosus, atherosclerosis, Carotid Stenosis, Adiponectin, E-Selectin
Introduction
Systemic Lupus Erythematosus (SLE) is a disease state posing several challenges to the clinician, including heterogeneity of presentation, undulating course, and an extraordinary risk for premature cardiovascular disease [1, 2]. Over two decades ago it was noted that the majority of deaths in patients with longer disease duration were attributed to atherosclerosis [1]. Indeed, the rate of myocardial infarction in women aged 35–44 years is 50 times greater than expected [2]. Patients with SLE have an increased atherosclerotic risk despite adjustment for traditional Framingham risk factors [2, 3]. Risk factors among SLE patients are somewhat controversial but may include longer duration of disease and lower likelihood of treatment with prednisone, cyclophosphamide, or hydroxychloroquine [4]. Thus, inflammation related to underlying disease is likely to be contributory. McMahon and coworkers have recently shown that plasma from SLE patients with premature atherosclerosis is enriched in proinflammatory HDL [5]. However, the inflammation may be clinically subtle since detectable cardiovascular events have been unexpectedly reported in SLE patients with extended periods of quiescence [6] and subclinical atherosclerosis has not correlated with disease activity index scores [4]. Thus, identification of biomarkers associated with atherogenic injury which might be present, independent of recognized lupus activity, should advance understanding of the pathology and guide prophylaxis for at risk subgroups.
The endothelium merits focus since it provides the physiologic boundary which limits extravasation and diapedesis of inflammatory cells. The generation of nitric oxide by the endothelium also promotes relaxation of the contractile elements of the smooth muscle of the arterial blood vessels. In atherogenic disease, endothelial protection is subverted perhaps because of a loss of boundary function via detachment of endothelial cells into the circulation and/or a change in the endothelial cell phenotype. Increased levels of circulating endothelial cells (CECs) have been observed in patients with active disease [7] and apoptotic CECs have been reported in SLE patients with diminished flow mediated dilatation (FMD) [8]. Soluble E-selectin (sE-selectin), likely shed from an abnormally activated endothelium, has been recently associated with atherosclerosis as defined by an abnormal coronary artery calcium assessed using electron beam computed tomography EBCT [9]. An integral membrane protein expressed on endothelial cells, endothelial protein C receptor (EPCR) has both anti-inflammatory and anti-thrombotic properties via its role in generating activated Protein C. A recent study has linked increased atherosclerotic risk to shedding of the EPCR from the endothelial cell surface [10].
While these studies have highlighted the potential association of markers directly reflecting injury, absent is a focus on protective (anti-injury) molecules such as the adipocyte-derived protein, adiponectin. Germane to the consideration of protection is the relationship of adiponectin to atherosclerosis. Adiponectin, when elevated in peripheral blood is reported to promote a wide range of benefit at the site of blood vessel injury, such as the downregulation of adhesion molecules by attenuating the nuclear factor kB pathway [11, 12]. Adiponectin accumulates in the sub-endothelium of injured human arteries where it inhibits monocyte adhesion to endothelial cells and ultimately inhibits the migration and proliferation of vascular smooth muscle, which contributes to the atherosclerotic process [13]. In addition, adiponectin was found in blood vessel walls after experimental endothelial injury [14], and was strongly expressed around infarcted but not normal myocardium [15], supporting a role in vascular and endothelial remodeling.
Accordingly, this study was initiated to explore the endothelial contribution to cardiovascular risk in SLE. This was approached by evaluation of carotid ultrasonography in 119 SLE patients and 71 age and sex matched controls. An assessment of the endothelial contribution was made by evaluating a panel of biomarkers chosen to reflect pro– and anti-atherogenic injury.
METHODS
Study Population
This study and its informed consent form were approved by the Institutional Review Board of the New York University School of Medicine. During the period from August 2005 through July 2007, 119 patients were enrolled from the Lupus Clinic at the NYU-Hospital for Joint Diseases and the private practices of NYU physicians. All patients fulfilled at least four of the revised American College of Rheumatology criteria for the diagnosis of SLE [16]. Seventy-one healthy employees of the NYU Langone Medical Center were matched to the SLE patients based on age, sex, and race/ethnicity. The only exclusion criterion was an unwillingness to participate.
Clinical Assessments
SLE patients and controls underwent a complete medical and physical exam. For the SLE patients, disease activity was measured by the SELENA-SLEDAI [17], an adaptation of the SLE Disease Activity Index [16], a validated instrument for the assessment of current disease activity. Patients were given the option to return within twelve months for the purpose of a repeat exam and blood workup. Sixty-two patients elected to participate in a second visit.
Carotid Ultrasound
A linear array transducer (Philips model 11-7L) was used by experienced monographers to obtain high resolution images of the far wall of the right and left common carotid arteries, internal carotid arteries and carotid bulbs, in accordance with the recommendations of the American Society of Echocardiography Carotid Intima-Media Thickness Task Force [18]. Sonographers and interpreting physicians were blinded to disease status. At least three measurements were taken over a one centimeter length of each common carotid arterial (CCA) segment and these measurements on both sides were averaged to obtain the mean IMT in that segment; these values were then averaged to obtain mean CCA IMT. The presence, location, and thickness of any plaques were recorded. Plaque was defined as ≥50% increase over background IMT in any arterial segment.
Determination of sE-selectin, sEPCR and adiponectin
Levels of sE-selectin, sEPCR, and adiponectin were measured using an enzyme-linked immunosorbent assay according to the instructions of the manufacturers (R&D Biosystems; Diagnostica Stago; Linco Research Inc, respectively). For each assay, the inter– and intra-assay variations were less than 9%. In a substudy, using sE-selectin as a phenotype, patients were stratified based on a threshold level of sE-selectin as previously described, high (patients with >40 ng/ml) and low (patients ≤ 40 ng/ml) [19]. A similar subgroup analysis was also performed for adiponectin, high (>13 ug/ml; patients who fell above the median value of entire cohort herein) and low (≤13 ug/ml; median value, patients at median and below).
Isolation of Circulating Endothelial Cells (CECs)
Evaluation of EPCR polymorphism
For genetic analysis, gene polymorphism at A6936G was assessed as previously described [20].
Statistical analysis
Categorical variables were compared between groups with Fisher’s exact test and continuous variables with the two sample T-test. Multivariable logistic regression models were also fit to the data using a stepwise variable selection approach to identify independent predictors of plaque status. All variables with p<0.20 for the bivariate association with plaque status were considered for inclusion in the model, and only those which remained significant at the p<0.05 level were retained in the final model. Results are reported as odds ratios with 95% confidence intervals. Two sided p-values < 0.05 were considered to be statistically significant.
RESULTS
Comparison of patients with SLE and controls
The demographics of the 119 patients with SLE and 71 healthy controls are shown in Table 1. The cohort was ethnically diverse, including a minority of Caucasian subjects (41%). Hypertension was more common in SLE patients (36% vs 12% of controls; p = 0.0006). HDL and total cholesterol were lower in patients (patients versus controls; 57.2 mg/dL vs 63.6 mg/dL, p=0.03; 177 mg/dL vs 188 mg/dL, p = 0.039, respectively), consistent with findings from prior studies [4]. There were no differences in LDL, TG, homocysteine and dyslipidemia. A higher proportion of patients had elevated CRP levels (>10 mg/dL) compared to controls (12% vs 3%, respectively; p= 0.05). Abnormal titers of anti-ds DNA antibodies were present in over half the patients (and none in controls). C3 and C4 were significantly lower in patients than controls (94.7 mg/dL vs 111.2 mg/dL; p = 0.0002; 18 mg/dL vs 25 mg/dL; p < 0.0001). Twelve patients with SLE were known to have prior cardiovascular disease, including coronary revascularization (percutaneous in three patients, surgical in one), MI (two patients) or stroke (six patients).
Table 1.
Lupus patients versus controls*
| Pts (N=119) | Control (N=71) | P value | |
|---|---|---|---|
| Age (yr) | 42.6 (12.4) | 41.3 (11.9) | 0.49 |
| Caucasian (%) | 41 | 47 | 0.81 |
| Postmenopausal (%) | 38 | 31 | 0.47 |
| Female (%) | 91 | 91 | 0.95 |
| Body-mass index | 25.9 (6.3) | 24.8 (5.3) | 0.29 |
| Plaque (%) | 43 | 17 | 0.0002 |
| IMT (mm) | 0.59 (0.19) | 0.54 (0.11) | 0.03 |
| Current Smoker (%) | 9.7 | 15.4 | 0.33 |
| Hypertension (%) | 37 | 12 | 0.0006 |
| Diabetes (%) | 3 | 2 | 1.00 |
| Systolic blood pressure | 117.7 (19.6) | 113.2 (16.9) | 0.13 |
| Diastolic blood pressure | 74.2 (11.5) | 73.6 (11.0) | 0.74 |
| Total Cholesterol (mg/dl) | 177.2 (38.6) | 189.0 (33.4) | 0.04 |
| HDL Cholesterol (mg/dl) | 57.2 (18.8) | 63.6 (17.3) | 0.03 |
| LDL Cholesterol (mg/dl) | 100.0 (31.0) | 107.0 (28.6) | 0.13 |
| Triglycerides (mg/dl) | 101.8 (63.8) | 91.6 (55.6) | 0.27 |
| Homocysteine (μM) | 10.2 (4.0) | 9.2 (2.7) | 0.07 |
| Dyslipidemia** (%) | 40.3 | 38.0 | 0.76 |
| Cardio CRP (% >10 mg/L) | 12 | 3 | 0.05 |
| Cardio CRP (% >3 mg/L) | 36 | 23 | 0.09 |
| anti-ds DNA (%) | 0.46 | 0 | <0.0001 |
| C3 (mg/dl) | 94.7 (27.6) | 111.2 (27.5) | 0.0002 |
| C4 (mg/dl) | 18.5 (9.9) | 25.3 (8.2) | <0.0001 |
| sE-selectin (ng/ml) | 64.1 (45.3) | 36.5 (16.2) | <0.0001 |
| CECs (cells/ml) | 19.4 (14.7) | 3.8 (3.3) | <0.0001 |
| Genotype EPCR (%) | 26 | 17 | 0.46 |
| sEPCR (ng/ml) | 237.3 (158.0) | 232.2 (99.2) | 0.79 |
| Adiponectin (μg/ml) | 16.0 (9.2) | 11.4 (6.1) | 0.0001 |
For continuous variables, mean value (S.D.)
Dyslipidemia was calculated as previously reported [5]
Compared to matched healthy controls, the prevalence of carotid plaque was significantly higher in SLE patients than controls (51/119 =43% vs 12/71 = 17%; p=0.0002). Mean CCA IMT was greater in patients compared to controls (0.59±0.19mm vs 0.54± 0.11mm, p = 0.03). There was no difference in prevalence of plaque by race/ethnicity [24/48 (50%) among Caucasians, 12/29(41%) Hispanics, 13/26 (50%) African Americans, and 2/14 (14%) Asians (p=0.10)] among lupus only. The highest frequency of plaque (32/51 = 63%) was found in the age group 40–59 years as compared to two patients (1%) and 17 patients (33 %) in age groups <40 years and ≥60 years, respectively (Figure 1, lupus only).
Figure 1.
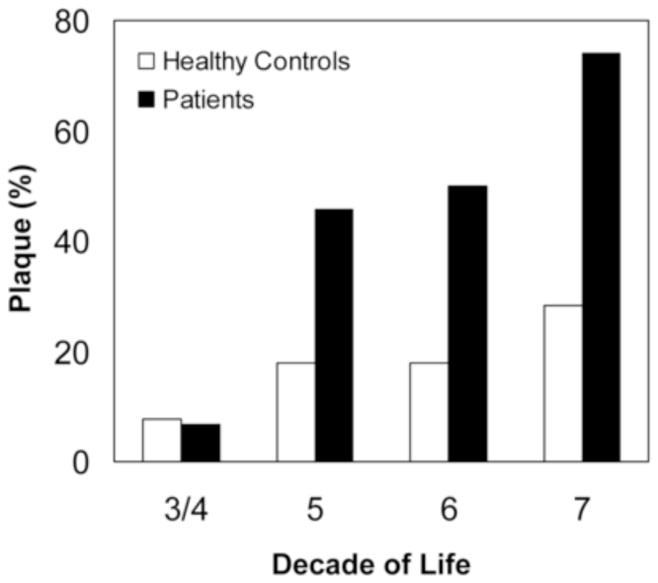
Prevalence of atherosclerotic plaque among control subjects and patients with systemic lupus erythematosis according to decade of life.
Evaluation of the endothelial biomarkers revealed significant differences between patients and controls. The levels of CEC and sE-selectin were shown to be significantly elevated in patients compared to controls [19.4 CECs/mL vs 3.8 CECs/mL (p<0.0001); 64 ng/ml vs 36 ng/ml (p<0.0001)] (Table 1). Unexpectedly, adiponectin was also significantly higher in patients compared to healthy controls (16 ug/mL vs 11 ug/mL; p = 0.0001). There were no differences between patients and controls regarding the levels of sEPCR or the distribution of the SNP at EPCR. Logistic regression models were fit to the data to identify independent predictors of plaque status in the combined population of SLE and control subjects using a stepwise variable selection approach. The variables which were retained in the final model included age (OR = 2.1 per 10 year increase; 95% CI: 1.5–3.0; p<0.0001), SLE status (OR = 3.4 for SLE vs control; 95% CI: 1.3–9.1); p =0.01, sE-selectin (OR = 1.2 per 10 unit increase; 95% CI: 1.0–1.4; p=0.02) and adiponectin (OR = 1.5 per 10 unit increase; 95% CI: 1.0–2.4; p = 0.05). Adiponectin and sE-selectin remained significantly associated with plaque status after further adjustment for established risk factors for cardiovascular disease, including hypertension and smoking, which were not significant in the multivariate model (results not shown).
After eliminating the 12 SLE and 3 control subjects who had previously diagnosed cardiac disease, the results were unchanged. The incidence of plaque, E-selectin, adiponectin and CEC levels remained significantly elevated in SLE patients compared to controls (p=0.0003 for plaque; p < 0.0001 for E-selectin, adiponectin, CEC).
Correlates of plaque among SLE patients
Patients with plaque identified on carotid ultrasound were older and more likely to be postmenopausal (Table 2). Patients with plaque were older at diagnosis and had longer disease duration. A higher proportion of patients with plaque had hypertension compared to patients without plaque (56% vs 22%; p=0.0009). More patients with plaque were taking beta blockers; however, there was no difference between patients with and without plaque with regard to their current use of aspirin, statins, angiotensin converting enzyme inhibitors, or Warfarin. Nor was there a difference in the rates of current use of various anti-inflammatory agents. No differences were found for levels of cardiac CRP, complement or presence/absence of anti-dsDNA antibody. Furthermore, SELENA-SLEDAI scores were similar in patients with and without plaque and the proportion of patients with inactive or stable/mildly disease (SLEDAI ≤ 4) did not segregate with the presence of plaque. Neither past (not shown) nor current medications hydroxychloroquine, glucocorticoids, cylophosphamide, azathioprine, and mycophenolate mofetil (Table 2) were significantly associated with plaque. In addition, in our study, E-selectin and adiponectin were not found to be significantly associated with use of prednisone, hydroxychloroquine, azathioprine, myclophenolate mofetil, or methotrexate (p>0.10 for all comparisons).
Table 2.
Lupus patients: plaque versus no plaque*
| Plaque (N=51) | No Plaque (N=68) | P value | |
|---|---|---|---|
| Age (yr) | 49.5 (10.6) | 37.5 (11.2) | <0.0001 |
| Caucasian (%) | 47 | 37 | 0.12 |
| Postmenopausal (%) | 60 | 24 | 0.0004 |
| Female (%) | 94 | 88 | 0.35 |
| Body-mass index | 26.3 (6.1) | 25.5 (6.5) | 0.54 |
| IMT (mm) | 0.65 (0.24) | 0.54 (0.11) | 0.0039 |
| Δ vascular age (yrs) | 15.8 (22.6) | 16.1 (10.2) | 0.93 |
| Current smoker (%) | 10.2 | 9.2 | 1.00 |
| Hypertension (%) | 56 | 22 | 0.0009 |
| Diabetes (%) | 6 | 0 | 0.07 |
| Systolic blood pressure | 124.5 (21.0) | 112.2 (16.7) | 0.0009 |
| Diastolic blood pressure | 76.6 (11.7) | 72.2 (11.0) | 0.045 |
| Total Cholesterol (mg/dl) | 186.5 (43.7) | 170.0 (32.6) | 0.02 |
| HDL Cholesterol (mg/dl) | 58.3 (22.5) | 56.4 (15.4) | 0.61 |
| LDL Cholesterol (mg/dl) | 105.1 (34.8) | 96.1 (27.3) | 0.13 |
| Triglycerides (mg/dl) | 111.5 (74.5) | 94.2 (53.2) | 0.15 |
| Homocysteine (μM) | 10.6(4.5) | 9.9 (3.7) | 0.39 |
| Dyslipidemia** (%) | 54.9 | 29.4 | 0.0079 |
| Cardio CRP (% >10 mg/L) | 13 | 12 | 1.00 |
| Cardio CRP (% >3 mg/L) | 38 | 35 | 0.84 |
| anti-ds DNA (%) | 0.41 | 0.50 | 0.33 |
| C3 (mg/dl) | 97.3 (29.8) | 92.8 (26.0) | 0.39 |
| C4 (mg/dl) | 19.6 (10.0) | 17.7 (9.8) | 0.31 |
| SLEDAI >4 (%) | 19 | 31 | 0.14 |
| Age at onset of disease (yrs) | 32.9 (12.9) | 26.1 (8.4) | 0.002 |
| Duration of disease (yrs) | 16.9 (10.1) | 11.1 (9.3) | 0.002 |
| sE-selectin (ng/ml) | 78.5 (58.6) | 52.9 (27.1) | 0.006 |
| CECs (cells/ml) | 18.7 (15.4) | 19.9 (14.3) | 0.72 |
| Genotype EPCR (%) | 22 | 28 | 0.64 |
| sEPCR (ng/ml) | 259.6 (187.9) | 220.8 (130.7) | 0.22 |
| Adiponectin (μg/ml) | 18.1 (8.7) | 14.4 (9.3) | 0.033 |
| Prednisone use (%) | 46 | 48 | 0.85 |
| Hydroxychloroquine (%) | 65 | 77 | 0.20 |
| AZA (%) | 15 | 11 | 0.58 |
| MTX (%) | 12 | 8 | 0.74 |
| CYC (%) | 0 | 5 | 0.27 |
| MMF (%) | 13 | 20 | 0.58 |
| Statin (%) | 13 | 8 | 0.52 |
| Beta blocker (%) | 26 | 5 | 0.0019 |
| ACE I (%) | 23 | 30 | 0.52 |
| ASA (%) | 36 | 25 | 0.23 |
| Warfarin (%) | 8 | 11 | 0.76 |
For continuous variables, mean value (S.D.)
Dyslipidemia was calculated as previously reported [5]
sE-selectin and adiponectin levels were significantly higher in SLE patients with plaque compared to those without plaque [mean sE-selectin: 78.5 ng/ml versus 52.9 (p=0.006); mean adiponectin: 18.1 ug/ml versus 14.4 (p=0.033)] (Table 2). Numbers of CEC, sEPCR levels, and EPCR genotype were not associated with plaque.
In the multiple logistic regression analysis, age (OR = 2.6 per 10 year increase; 95% CI: 1.7–4.0; p < 0.0001), sE-selectin (OR = 1.3 per 10 unit increase; 95% CI: 1.1–1.5; p=0.009), and adiponectin (OR = 1.8 per 10 unit increase; 95% CI: 1.1–3.0; p=0.02) were significantly associated with plaque status. After eliminating patients with cardiac disease, the dominant predictors of plaque status maintained significance in lupus patients. The results of the logistic regression analysis remained unchanged after further adjustment for BMI and prednisone use.
In the subgroup of clinically inactive or stable/mildly active subjects (SELENA-SLEDAI ≤ 4), age (p=0.007), sE-selectin (p=0.02), and adiponectin (p=0.02) were remained significantly associated with risk of plaque. This same subgroup was also stratified by levels of sE-selectin (< 40 vs ≥ 40 ng/ml) and adiponectin (< 13 vs ≥ 13 ug/ml). As shown in Table 3, the prevalence of plaque was greatest in the group of stable lupus patients with both high sE-selectin and adiponectin (55.3%; p-value: 0.0009) confirming the findings of the multivariable analyses.
Table 3.
Incidence of plaque in patients with quiescent disease (SLEDAI ≤ 4) and varied distribution of soluble E-selectin and adiponectin.
| Group | N | Distribution of biomarkers | Plaque (%) |
|---|---|---|---|
| 1 | 8 | Low selectin, low adiponectin | 2.6 |
| 2 | 14 | Low selectin, high adiponectin | 15.8 |
| 3 | 29 | High selectin, low adiponectin | 26.3 |
| 4 | 27 | High selectin, high adiponectin | 55.3 |
Sixty–two patients elected to donate blood at a second visit. High levels of sE-selectin were sustained in 20 of 26 (80%) plaque patients compared to 12 of 36 (33%) of non-plaque patients (p=0.0009) (Figure 2). Longitudinal assessments across visits for adiponectin also demonstrated a significant difference between sustained high levels in patients with plaque compared to those without plaque (p=0.0011).
Figure 2.
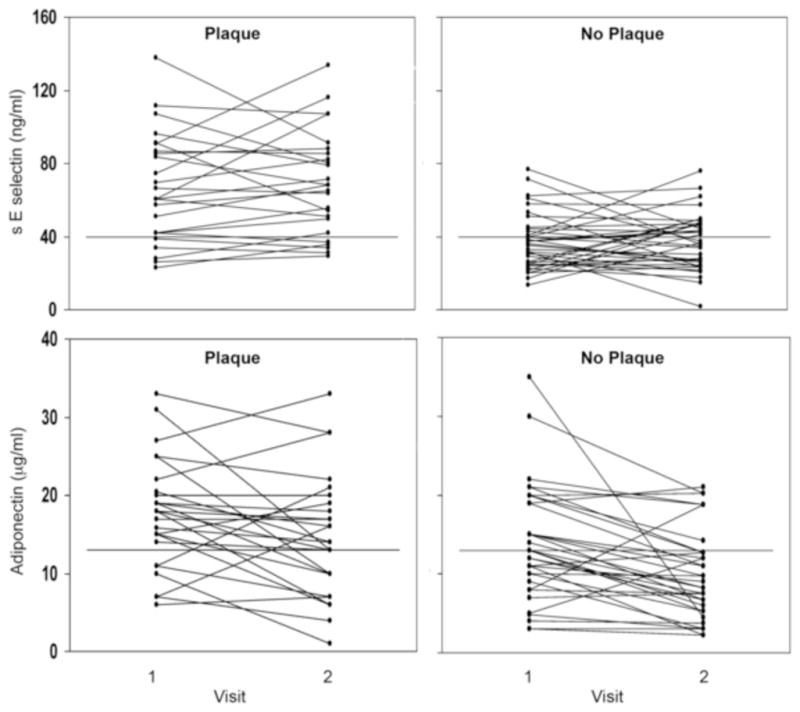
Longitudinal evaluation of levels of sE-selectin and adiponectin in sixty-two patients. Upper panel: plasma levels of sE-selectin versus visit (40 ng/ml, the threshold for high sE selectin is indicted by a solid line). Lower panel: plasma levels of adiponectin versus visit (Note solid line at 13 μg/mL, threshold for high adiponectin).
DISCUSSION
Premature atherosclerosis is a consistent feature of SLE and herein extends across all ethnicities. Carotid plaque was observed in over twice the proportion of lupus patients compared to age-, sex- and race/ethnicity-matched controls. Excess prevalence of plaque was seen beginning in the 5th decade of life. On multivariate analysis, age, SLE disease duration, sE-selectin, and adiponectin were the only independent predictors of plaque. Among lupus patients with plaque, elevations in these biomarkers were persistent over more than one visit in those with multiple measurements. Elevation of sE-selectin was anticipated, because this biomarker reflects activation of the endothelium and has been previously associated with atherosclerosis and cardiovascular risk in both SLE and non-SLE cohorts [9, 21, 22]. The association with elevated adiponectin was unexpected since adiponectin is generally considered to be vasoprotective. Perhaps the elevated adiponectin represents a continued but unsuccessful attempt at vascular repair.
Inflammation has long been appreciated to contribute to the accrual of atherosclerosclerotic disease. Thus, many have speculated that inflammation, the central feature of SLE pathogenesis and clinical flares, is linked to the increased atherosclerotic risk among these patients in addition to higher rates of traditional risk factors. The absence of association of carotid plaque with overt disease activity in this study and others runs counter to the “inflammation” hypothesis. One simple explanation is that the SELENA-SLEDAI instrument may underestimate disease activity. However, this instrument does capture current inflammation in the cutaneous, renal, serosal, hematologic, neurologic, and serologic systems. In fact, it is generally in these clinical settings of activity, that the traditional markers of endothelial (luminal) injury have been previously identified. sE-selectin and adiponectin have been reported in patients with active disease [19, 23, 24]. The elevation of sE-selectin in patients with plaque despite clinical quiescence and the stability of that elevation over time likely indicates that a low level of inflammation or atherogenic injury independent of inflammation does contribute to atherosclerosis in lupus [25]. Further precedent in SLE for the demonstration of endothelial activation in the absence of an inflammatory infiltrate has been reported for the kidney [26] and the skin[27], in which increased expression of adhesion molecules including E-selectin were observed in the microvasculature of non lesional areas.
Adiponectin has pleiotropic biological activities including improving insulin sensitivity [28, 29]. Beyond these metabolic actions, there is also considerable preclinical evidence that adiponectin exerts direct anti-atherosclerotic and cardioprotective effects [30]. Epidemiological studies evaluating the association of adiponectin with cardiovascular events have reported somewhat conflicting results and have been limited largely to men, especially older men. [31, 32, 33, 34, 35]. Based on a recent biomarker study from Vanderbilt, performed in a retrospective cross-sectional analysis of 65 patients with SLE and 69 controls, there was no association between adiponectin levels and coronary calcium scores [36, 37, 38]. While a definitive explanation for the disparate findings between this study and the one presented herein await larger longitudinal cohorts, several differences are worthy of mention. The Vanderbilt cohort comprised a greater percentage of Caucasians, the adiponectin levels were higher in controls than most reported series, subset analysis of patients with low SLEDAI scores was not performed, and assessment was limited to one sampling. In the New York cohort, evaluation across visits in 60% of patients also demonstrated a significant difference between sustained high levels in patients with plaque compared to those without plaque. This is of relevance since cross-sectional evaluation of a biomarker for a condition that represents accrued insult has limitations. Most interesting, in both cohorts the adiponectin levels were significantly higher overall in the SLE patients compared to healthy controls, yet these patients are at risk for premature atherosclerosis. This alone distinguishes SLE from most reports of men in which lower levels of adiponectin are associated both with atherosclerosis and risk for progression [39]. We hypothesize that for as yet unknown reasons the endothelial dysfunction characteristic of SLE (reflected by elevated e-selectin in both studies) drives a higher adiponectin level which is nevertheless not effective in protection. Therefore, increased adiponectin concentration could represent a compensatory mechanism to existing vascular damage. It is notable that children with diabetes (another high risk group for premature atherosclerosis) have been recently reported to have a higher concentration of adiponectin than healthy matched controls [40].
Other biomarkers which have been recently linked to endothelial damage per se or inflammatory luminal injury include CECs as well as sEPCR [7, 20]. Neither tracked with plaque suggesting that inflammation may not be the sole explanation.
Limitations of our study include the challenges in precise quantification of the activity, severity, and disease treatment over the lifetime of the patient with SLE. Atherosclerosis develops over years. Because this study was only limited to one year, it is acknowledged that activity at the time of evaluation is not necessarily reflective of the total burden of undulating activity which might lead to plaque. Reliable indices of the cumulative burden of inflammatory disease activity in SLE do not exist. The SLE Damage Index is the closest approximation but that instrument assesses damage accrued since diagnosis, some of which may not be directly related to the disease process per se such as avascular necrosis or premature ovarian failure [41]. Although cardiovascular event rates in SLE are markedly higher than matched healthy controls, the rates are too low to permit correlation with biomarkers, thus carotid ultrasound studies are used as proxies of true events.
In sum, this study confirms the atherogenic risk observed in SLE patients including those of non-Caucasian race, and highlights two potential vascular markers of potential clinical applicability in monitoring patients. These findings may be particularly useful since these markers extend beyond current instruments of disease activity. Future studies are needed to determine whether screening of SLE patients with these biomarkers identify those who would benefit from anti-atherosclerotic and/or anti-inflammatory therapy to avoid cardiovascular events.
Acknowledgments
We thank Drs. ER Gehrie, H Comerci, and E. Karis who worked on this project during their residencies/fellowships.
FUNDING
This work was supported by a research grant to R.M.C. from the Lupus Research Institute, New York, NY. P.M.I. was supported by SLE Foundation, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Urowitz MB, Bookman AA, Koehler BE, et al. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976;60:221–225. doi: 10.1016/0002-9343(76)90431-9. [DOI] [PubMed] [Google Scholar]
- 2.Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997;145:408–415. doi: 10.1093/oxfordjournals.aje.a009122. [DOI] [PubMed] [Google Scholar]
- 3.Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–2337. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 4.Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–2406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- 5.McMahon M, Grossman J, FitzGerald J, et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006;54:2541–2549. doi: 10.1002/art.21976. [DOI] [PubMed] [Google Scholar]
- 6.Doherty NE, Siegel RJ. Cardiovascular manifestations of systemic lupus erythematosus. Am Heart J. 1985;110:1257–1265. doi: 10.1016/0002-8703(85)90023-7. [DOI] [PubMed] [Google Scholar]
- 7.Clancy R, Marder G, Martin V, et al. Circulating activated endothelial cells in systemic lupus erythematosus: further evidence for diffuse vasculopathy. Arthritis Rheum. 2001;44:1203–1208. doi: 10.1002/1529-0131(200105)44:5<1203::AID-ANR204>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 8.Rajagopalan S, Somers EC, Brook RD, et al. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood. 2004;103:3677–3683. doi: 10.1182/blood-2003-09-3198. [DOI] [PubMed] [Google Scholar]
- 9.Rho YH, Chung CP, Oeser A, et al. Novel cardiovascular risk factors in premature coronary atherosclerosis associated with systemic lupus erythematosus. J Rheumatol. 2008;35:1789–1794. [PMC free article] [PubMed] [Google Scholar]
- 10.Ireland H, Konstantoulas CJ, Cooper JA, et al. EPCR Ser219Gly: Elevated sEPCR, prothrombin F1+2, risk for coronary heart disease, and increased sEPCR shedding in vitro. Atherosclerosis. 2005 doi: 10.1016/j.atherosclerosis.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Ouchi N, Kihara S, Arita Y, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 12.Ouchi N, Kihara S, Arita Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 13.Kumada M, Kihara S, Sumitsuji S, et al. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol. 2003;23:85–89. doi: 10.1161/01.atv.0000048856.22331.50. [DOI] [PubMed] [Google Scholar]
- 14.Okamoto Y, Arita Y, Nishida M, et al. An adipocyte-derived plasma protein, adiponectin, adheres to injured vascular walls. Horm Metab Res. 2000;32:47–50. doi: 10.1055/s-2007-978586. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa Y, Akasaka Y, Ishii T, et al. Changes in the distribution pattern of gelatin-binding protein of 28 kDa (adiponectin) in myocardial remodelling after ischaemic injury. Histopathology. 2003;42:43–52. doi: 10.1046/j.1365-2559.2003.01518.x. [DOI] [PubMed] [Google Scholar]
- 16.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 17.Petri M, Kim MY, Kalunian KC, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. 2005;353:2550–2558. doi: 10.1056/NEJMoa051135. [DOI] [PubMed] [Google Scholar]
- 18.Stein JH, Korcarz CE, Hurst RT, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr. 2008;21:93–111. doi: 10.1016/j.echo.2007.11.011. quiz 189–190. [DOI] [PubMed] [Google Scholar]
- 19.Egerer K, Feist E, Rohr U, et al. Increased serum soluble CD14, ICAM-1 and E-selectin correlate with disease activity and prognosis in systemic lupus erythematosus. Lupus. 2000;9:614–621. doi: 10.1191/096120300678828749. [DOI] [PubMed] [Google Scholar]
- 20.Sesin CA, Yin X, Esmon CT, et al. Shedding of endothelial protein C receptor contributes to vasculopathy and renal injury in lupus: In vivo and in vitro evidence. Kidney Int. 2005;68:110–120. doi: 10.1111/j.1523-1755.2005.00385.x. [DOI] [PubMed] [Google Scholar]
- 21.Hwang SJ, Ballantyne CM, Sharrett AR, et al. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation. 1997;96:4219–4225. doi: 10.1161/01.cir.96.12.4219. [DOI] [PubMed] [Google Scholar]
- 22.Rohde LE, Lee RT, Rivero J, et al. Circulating cell adhesion molecules are correlated with ultrasound-based assessment of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 1998;18:1765–1770. doi: 10.1161/01.atv.18.11.1765. [DOI] [PubMed] [Google Scholar]
- 23.Rovin BH, Song H, Hebert LA, et al. Plasma, urine, and renal expression of adiponectin in human systemic lupus erythematosus. Kidney Int. 2005;68:1825–1833. doi: 10.1111/j.1523-1755.2005.00601.x. [DOI] [PubMed] [Google Scholar]
- 24.Panes J, Perry M, Granger DN. Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention. Br J Pharmacol. 1999;126:537–550. doi: 10.1038/sj.bjp.0702328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roldan V, Marin F, Lip GY, et al. Soluble E-selectin in cardiovascular disease and its risk factors. A review of the literature. Thromb Haemost. 2003;90:1007–1020. doi: 10.1160/TH02-09-0083. [DOI] [PubMed] [Google Scholar]
- 26.Izmirly PM, Barisoni L, Buyon JP, et al. Expression of endothelial protein C receptor in cortical peritubular capillaries associates with a poor clinical response in lupus nephritis. Rheumatology (Oxford) 2009 doi: 10.1093/rheumatology/kep034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belmont HM, Buyon J, Giorno R, et al. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus. The Shwartzman phenomenon revisited. Arthritis Rheum. 1994;37:376–383. doi: 10.1002/art.1780370311. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto Y, Kihara S, Ouchi N, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 29.Shibata R, Ouchi N, Kihara S, et al. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J Biol Chem. 2004;279:28670–28674. doi: 10.1074/jbc.M402558200. [DOI] [PubMed] [Google Scholar]
- 30.Hopkins TA, Ouchi N, Shibata R, et al. Adiponectin actions in the cardiovascular system. Cardiovasc Res. 2007;74:11–18. doi: 10.1016/j.cardiores.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pischon T, Girman CJ, Hotamisligil GS, et al. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 32.Frystyk J, Berne C, Berglund L, et al. Serum adiponectin is a predictor of coronary heart disease: a population-based 10-year follow-up study in elderly men. J Clin Endocrinol Metab. 2007;92:571–576. doi: 10.1210/jc.2006-1067. [DOI] [PubMed] [Google Scholar]
- 33.Sattar N, Wannamethee G, Sarwar N, et al. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation. 2006;114:623–629. doi: 10.1161/CIRCULATIONAHA.106.618918. [DOI] [PubMed] [Google Scholar]
- 34.Kanaya AM, Wassel Fyr C, Vittinghoff E, et al. Serum adiponectin and coronary heart disease risk in older Black and White Americans. J Clin Endocrinol Metab. 2006;91:5044–5050. doi: 10.1210/jc.2006-0107. [DOI] [PubMed] [Google Scholar]
- 35.Kizer JR, Barzilay JI, Kuller LH, et al. Adiponectin and risk of coronary heart disease in older men and women. J Clin Endocrinol Metab. 2008;93:3357–3364. doi: 10.1210/jc.2008-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung CP, Long AG, Solus JF, et al. Adipocytokines in systemic lupus erythematosus: relationship to inflammation, insulin resistance and coronary atherosclerosis. Lupus. 2009;18:799–806. doi: 10.1177/0961203309103582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–2415. doi: 10.1056/NEJMoa035611. [DOI] [PubMed] [Google Scholar]
- 38.Chung CP, Oeser A, Raggi P, et al. Increased coronary-artery atherosclerosis in rheumatoid arthritis: relationship to disease duration and cardiovascular risk factors. Arthritis Rheum. 2005;52:3045–3053. doi: 10.1002/art.21288. [DOI] [PubMed] [Google Scholar]
- 39.Maahs DM, Ogden LG, Kinney GL, et al. Low plasma adiponectin levels predict progression of coronary artery calcification. Circulation. 2005;111:747–753. doi: 10.1161/01.CIR.0000155251.03724.A5. [DOI] [PubMed] [Google Scholar]
- 40.Heilman K, Zilmer M, Zilmer K, et al. Elevated plasma adiponectin and decreased plasma homocysteine and asymmetric dimethylarginine in children with type 1 diabetes. Scand J Clin Lab Invest. 2009;69:85–91. doi: 10.1080/00365510802419454. [DOI] [PubMed] [Google Scholar]
- 41.Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39:363–369. doi: 10.1002/art.1780390303. [DOI] [PubMed] [Google Scholar]