

Abstract
Natural products remain the best sources of drugs and drug leads and serve as outstanding small-molecule probes to dissect fundamental biological processes. A great challenge for the natural product community is to discover novel natural products efficiently and cost effectively. Here we report the development of a practical method to survey biosynthetic potential in microorganisms, thereby identifying the most promising strains and prioritizing them for natural product discovery. Central to our approach is the innovative preparation, by a two-tiered PCR method, of a pool of pathway-specific probes, thereby allowing the survey of all variants of the biosynthetic machineries for the targeted class of natural products. The utility of the method was demonstrated by surveying 100 strains, randomly selected from our actinomycete collection, for their biosynthetic potential of four classes of natural products, aromatic polyketides, reduced polyketides, nonribosomal peptides, and diterpenoids, identifying 16 talented strains. One of the talented strains, Streptomyces griseus CB00830, was finally chosen to showcase the discovery of the targeted classes of natural products, resulting in the isolation of three diterpenoids, six nonribosomal peptides and related metabolites, and three polyketides. Variations of this method should be applicable to the discovery of other classes of natural products.
Natural products occupy tremendous chemical structural space that is unmatched by any other small-molecule libraries. As such, they remain the best sources of drugs and drug leads and serve as outstanding small-molecule probes to dissect fundamental biological processes.1−4 Bacteria of both terrestrial and marine origin have proven to be excellent sources of novel natural products. Recent advances in microbial genomics have unequivocally demonstrated that the potential of natural product biosynthesis in bacteria is much higher than previously appreciated.5−8 A great challenge for the natural product community is to discover novel natural products efficiently and cost effectively.
Traditional microbial natural product discovery programs start from fermenting each strain individually, often in multiple media, followed by preparation of crude extracts. There are two primary approaches to search for novel natural products from extracts: bioactivity-guided fractionation and chemical profiling of metabolites possessing novel structural features.9−13 In both cases a molecule of interest must be produced in sufficient quantities to permit isolation, purification, characterization, and dereplication in a reasonable time frame. The ultimate success in discovering a new natural product typically requires three principal steps: dereplication of known compounds at an early stage to avoid the duplication of effort, isolation and purification of the targeted molecules from a highly complex matrix, and, finally, structure elucidation of the purified natural product. This traditional sequence of steps still characterizes new compound discovery from crude extracts today. While successful, it is a tedious and laborious process. This process could be significantly shortened and much more cost-effective if the biosynthetic potentials of the strain collection were known in advance so that the resources could be devoted preferentially to interrogate only the strains that hold the greatest promise in producing novel natural products or classes of natural products of high interest.
Complementary to the traditional approaches, the progress made in the last two decades in connecting natural products to the genes that encode their biosynthesis has fundamentally changed the landscape of natural products research and sparked the emergence of a suite of contemporary approaches to natural product discovery. Thus, genes have become as important as chemistry in categorizing known natural products and identifying likely new ones yet to be discovered. Advances in microbial genomics have unequivocally demonstrated that we are missing ∼90% of the natural product biosynthetic capacity of even the workhorse producers, the actinomycetes.5 To gain access to this untapped reservoir of potentially new natural products, two principal strategies have been applied to induce these cryptic biosynthetic pathways.5−8,14−22 The so-called epigenetic-related approaches include challenging the microorganisms through culture conditions, nutritional or environmental factors, external cues, and stress, as well as exploiting interspecies crosstalk. The genomics-based approaches include mining the genomes to predict metabolite structures, engineering the pathways by manipulating global and/or pathway-specific regulators, and expressing the cryptic pathways in selected heterologous hosts. While each of the various approaches has different strengths and weaknesses, they have been successful in yielding cryptic natural products but only on a case-by-case basis; currently they are far from being of practical use for natural product discovery and generally are low throughput. Thus, in spite of the rapid advances in DNA sequencing technologies and bioinformatics, sequencing all strains and annotating all genomes within a collection is still not a practical means to discover new natural products. Furthermore, it is also unlikely that expedient genetic systems can be rapidly developed for every strain within a collection, without which any attempt of metabolic pathway engineering will not be possible.
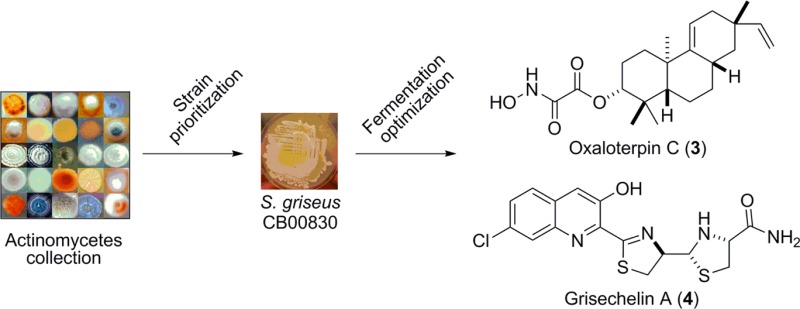
Here we report the development of a practical method to survey biosynthetic potential in microorganisms, thereby identifying the most promising strains and prioritizing them for natural product discovery. Central to our approach is the innovative preparation, by a two-tiered PCR method, of a pool of pathway-specific probes, thereby allowing the survey of all variants of the biosynthetic machineries for the targeted class of natural products. We first used 16 representative strains, known to produce the selected four major classes of natural products, to develop and validate the method. We then applied the method to 100 randomly selected strains from our actinomycete collection to survey their biosynthetic potential for the four targeted classes of natural products. We finally selected one lead strain, Streptomyces griseus CB00830, to showcase the discovery of diterpenoids, resulting in the isolation of viguiepinol (1), oxaloterpin E (2), and oxaloterpin C (3), together with nine additional natural products. Variations of this method should be applicable to the discovery of other classes of natural products.
Results and Discussion
Rationale, Design, and Validation of the Method Surveying Natural Product Biosynthetic Potential in Microorganisms
In our effort to assemble a natural product library, actinomycetes from various unexplored and underexplored ecological niches were fermented for natural product isolation. Each strain was fermented in two media (Table S1), and the resultant extracts were subjected to HPLC-photodiode array-based chemical profiling and titer estimation for all major metabolites presented.23−30 The lessons we learned from the initial effort included the following: (i) the metabolite profile of a given strain was medium dependent, (ii) increases in the number of media used for each strain correlated well with the total number of metabolites detected, (iii) with the two media under the fermentation conditions examined, only 25–30% of the strains showed promising metabolite profiles deemed worthy of pursuing subsequent natural product isolation, (iv) given the limited fermentation media and conditions examined, there was little correlation between the metabolite profile of the extract and the intrinsic biosynthetic potential of the strain, and (v) it may not be productive to prioritize the strains for subsequent natural product discovery based on chemical profiling of the extracts alone. Clearly, a general, high-throughput method is needed to rapidly survey the biosynthetic potential of a strain collection. Strains that harbor the highest biosynthetic potential can then be identified, prioritized, and subjected to epigenetic- and/or genomics-based approaches5−8,14−22 to induce all cryptic biosynthetic pathways for novel natural product discovery. This could fundamentally change the way in which microbial natural products are discovered.
Polyketides and nonribosomal peptides are two large families of microbial natural products.5,8,18,19,22 While the majority of diterpenoids are of plant origin, recent advances in genomics have revealed that the biosynthetic capacity of diterpenes in bacteria may be underestimated.31,32 Since the biosynthetic machineries for polyketides, nonribosomal peptides, and diterpenoids in actinomycetes have been extensively studied, it is now possible to survey the biosynthetic potential of these natural products by scanning the bacterial genomes for the trademark genes encoding their respective biosynthetic machineries.5−8,14−22 Thus, elements of the biosynthetic machineries that are highly conserved across the entire family of natural products, as exemplified by the type I polyketide synthases (PKSs) for reduced polyketides, type II PKSs for aromatic polyketides, ribosomal peptide synthetases (NRPSs) for nonribosomal peptides, and diterpene synthases (DTSs) for diterpenoids, respectively, provide unified paradigms for the synthesis of the characteristic molecular scaffolds for each of these families. On the other hand, variations among the biosynthetic machineries for a given class of natural products, as exemplified by the various accessory domains for type I PKSs and NRPSs and the myriad of tailoring enzymes associated with each of the biosynthetic machineries, afford the remarkable structural diversity within each of the natural product scaffolds. These features could be exploited to survey the intrinsic biosynthetic potential, thereby rapidly identifying the most promising strains and prioritizing them for natural product discovery.
As a proof of concept study, we therefore reasoned that (i) genes encoding biosynthetic enzymes of these four classes of natural products could be amplified from pools of genomic DNA by PCR using pathway-specific primers, (ii) the resultant products could be uniformly labeled by PCR using unbiased “tag primers” to afford pools of pathway-specific DNA probes, (iii) the biosynthetic potential for the four targeted classes of natural products could then be surveyed by probing a genomic DNA array of the strains using pools of the pathway-specific probes, and (iv) strains with the greatest biosynthetic potential could be identified, prioritized, and subjected to an emerging suite of technologies to awaken the cryptic pathways for production and subsequent isolation and characterization of the targeted classes of natural products (Figure 1).
Figure 1.
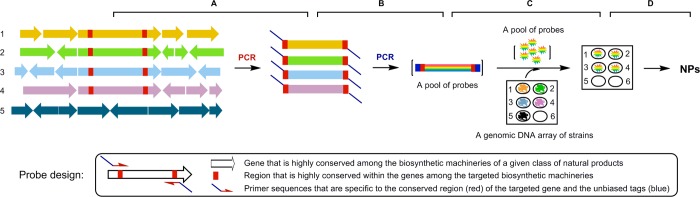
Biosynthetic potential-based strain prioritization for natural product discovery: (A) amplification of genes characteristic of representative biosynthetic machinery from a pool of genomic DNA by PCR using pathway-specific primers, (B) uniformly labeling the first-round PCR products by the second-round PCR using unbiased “tag primers” to afford a pool of pathway-specific DNA probes, (C) survey of the biosynthetic machinery of the targeted class of natural products by screening a genomic DNA array of the strains with the pool of pathway-specific probes, (D) fermentation optimization of “hit” or “talented” stains for production and isolation of the targeted natural products. Five strains are depicted, with strains 1–4 harboring the biosynthetic machineries of the targeted class of natural products. Position 6 of the genomic DNA array depicts a negative control.
Figure 1 depicts the design of the method to survey biosynthetic potential in microorganisms and application of the method to prioritize strains for targeted natural product discovery. We first selected 16 representative strains, known to produce the four major classes of natural products, i.e., reduced polyketides (six), aromatic polyketides (four), nonribosomal peptides (four), and diterpenoids (two), to develop the method (Figure S1). The gene clusters encoding the biosynthetic machineries for each of the 16 natural products have been cloned and characterized (Table S2). PCR methods to amplify the genes characteristic of each of the four representative biosynthetic machineries, i.e., type I PKSs33 for reduced polyketides, type II PKSs34 for aromatic polyketides, NRPSs33 for nonribosomal peptides, and DTSs35 for ditepenoids, have all been developed. Figure S2A summarizes the sequence alignments of genes encoding the selected type I PKSs, type II PKSs, NRPSs, and DTSs, and the biosynthetic machinery-specific primers were designed according to the conserved protein sequences (Table 1). Most importantly, we fused an AT-rich tag, taking advantage of the high GC content of the actinomycete genome,5 to the pathway-specific primers (Table 1), allowing us to prepare by a two-tiered PCR method a pool of pathway-specific probes enriched with all possible variants of the targeted biosynthetic machinery (Figure 1). Thus, genomic DNAs from the 16 strains were pooled and used as templates to prepare each pool of the pathway-specific probes by the two-tiered PCR method, yielding distinct products with the expected sizes (Figure S2B). Ten randomly selected clones from each pool of the probes were sequenced, and all of them were confirmed to be the targeted pathway-specific genes, with eight variants each for type I and type II PKSs, six variants for NRPSs, and two variants for DTSs, respectively. Finally, the genomic DNAs of the 16 strains were arrayed on a nylon membrane and screened with each pool of pathway-specific probes. Six of the six reduced polyketide producers, four of the four aromatic polyketide producers, three of the four nonribosomal peptide producers, and two of the two diterpenoid producers were readily identified. While the DTS pool of probes hybridized only to the two known diterpenoid producers, hybridizations using the other three pools of probes revealed that many of the 16 strains, in addition to being the producers of the known class of natural products, are potential producers of the other two classes of natural products as well (Figure S2C). These hybridization results were further correlated with PCR amplification, using the same set of primers but each of the 16 genomic DNAs individually as a template (Figure S2D).
Table 1. Primers and Conditions for the Two-Tiered PCR Method to Prepare a Pool of Pathway-Specific Probes and Use Them to Survey Strains for Biosynthetic Potential of the Targeted Class of Natural Products.
| natural product class | pathway-specific probe | primera | 1st round PCR | 2nd round PCR | hybridization temp (°C) |
|---|---|---|---|---|---|
| reduced polyketides | type I PKS | 5′-ATGTAGTGAGCTGCTTCGGCGATGGACCCGCAGCAGCG-3′ | 95 °C/30
s; 60 °C/1 min; 72 °C/40 s; 10 cycles |
95 °C/30 s; 48 °C/30 s; 72 °C/40 s; 35 cycles |
55 |
| 5′-AATCAGCATCCTTCGACCGCGCCGTCCTGGTTSACSGC-3′ | |||||
| aromatic polyketides | type II PKS | 5′-ATGTAGTGAGCTGCTTCGGGCAGCGGITTCGGCGGITTCCAG-3′ | 95 °C/30
s; 65 °C/1 min; 72 °C/30 s; 10 cycles |
95 °C/30 s; 48 °C/30 s; 72 °C/30 s; 35 cycles |
55 |
| 5′-AATCAGCATCCTTCGACCCGITGTTIACIGCGTAGAACCAGGCG-3′ | |||||
| nonribosomal peptides | NRPS | 5′-ATGTAGTGAGCTGCTTCGACCTCSGGCWCCACCGGC-3′ | 95 °C/30
s; 65 °C/1 min; 72 °C/50 s; 10 cycles |
95 °C/30 s; 48 °C/30 s; 72 °C/50 s; 35 cycles |
57 |
| 5′-AATCAGCATCCTTCGACCGCCGSIGATCTTSACCTG-3′ | |||||
| diterpenoids | DTS | 5′-ATGTAGTGAGCTGCTTCGACGCTCAGTGCGGTSGAG-3′ | 95 °C/30
s; 65 °C/1 min; 72 °C/1 min; 10 cycles |
95 °C/30 s; 48 °C/30 s; 72 °C/1 min; 35 cycles |
58 |
| 5′-AATCAGCATCCTTCGACCGGIGAGGCGTGCCACTTGTC-3′ |
The designer primers consisted of the pathway-specific sequences and the unbiased AT-rich tag (underlined). S, C/G; W, A/T; I, inosine.
Survey of 100 Strains for Biosynthetic Potential Yielding 16 Talented Natural Product Producers
We next randomly selected 100 strains from our actinomycete collection and applied the method described above to survey their biosynthetic potential for the four targeted classes of natural products. The genomic DNAs for each of the 100 strains were individually prepared and arrayed on a nylon membrane. Each pool of the four pathway-specific probes was similarly prepared by the two-tiered PCR method (Figure 1), using the same set of primers (Table 1) but with the pooled genomic DNAs from the 100 strains as templates. Screening of the 100 strains with each pool of the probes revealed great biosynthetic potentials for these strains to produce the four targeted classes of natural products (Figure S3). As summarized in Figure 2, 36, 39, 38, and 25 out the 100 strains were identified as potential producers of reduced polyketides, aromatic polyketides, nonribosomal peptides, and diterpenoids, respectively, with 16 of them as “talented” strains that could have the potential to produce all four classes of natural products.
Figure 2.
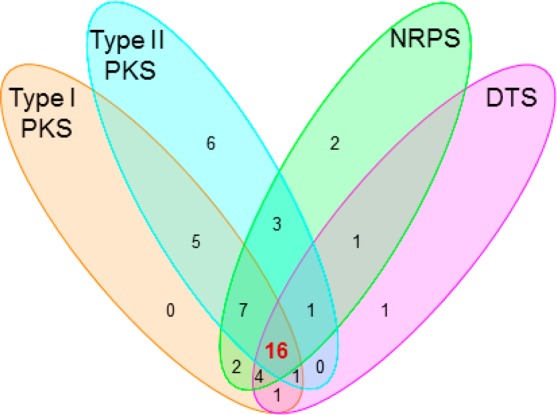
Venn diagram summarizing the biosynthetic potential of the four targeted classes of natural products in the selected 100 strains upon screening with each pool of probes. Sixteen “talented” strains (highlighted in red) were found having the potential to produce all four classes of natural products, and S. griseus CB00830 is one of the 16 talented strains.
S. griseus CB00830 Showcasing the Discovery of Three Diterpenoids and Nine Additional Natural Products
S. griseus CB00830 was first cultured, on a small scale (50 mL), in 10 different media for fermentation optimization (Table S1), and crude extracts of the resultant cultures were then subjected to TLC and HPLC analysis. On the basis of metabolite profiles, which varied significantly among the 10 media examined, medium E was then selected for large-scale fermentation (5 L) and natural product isolation. The crude extract was subjected to a series of chromatographies, affording 12 natural products, and their structures were elucidated on the basis of MS and NMR spectroscopic analyses, unveiling three diterpenoids (1–3), four nonribosomal peptides (4–7), two chloroanthranilates (8 and 9), and three polyketides (10–12) (Figure 3).
Figure 3.

Structures of the 12 natural products isolated from S. griseus CB00830: the three diterpenoids viguiepinol (1) and oxaloterpins E (2) and C (3), the four nonribosomal peptides grisechelins A (4), B (5), C (6), and D (7), the two biosynthetically associated metabolites 4-chloroanthranilamide (8) and methyl 4-chloroanthranilate (9), and the three polyketides dinactin (10), feigrisolide C (11), and seco-dinactin (12).
Compound 1 was obtained as a colorless oil. High-resolution electrospray ionization (HRESI)MS analysis afforded an [M – H2O + H]+ ion at m/z 271.2421, establishing the molecular formula of 1 as C20H32O (calculated [M – H2O + H]+ ion at m/z 271.2425). The 1H NMR spectrum showed resonances attributed to (i) four tertiary methyl groups at δH 1.07 (s, 3H, H3-18), δH 0.99 (s, 3H, H3-20), δH 0.97 (s, 3H, H3-17), and δH 0.87 (s, 3H, H3-19), (ii) aliphatic moieties between δH 1.20 and 2.50, and (iii) double bonds or oxygen-linked methine groups between δH 4.00 and 6.00 (Table S3). These chemical shifts and coupling patterns, in combination with 20 carbon signals in the 13C NMR spectrum (Table S3), suggested a characteristic diterpenoid chemical skeleton of 1, whose identity as viguiepinol (Figure 3) was finally confirmed by 1H–1H COSY, HSQC, and HMBC experiments (Figure S4).36,37
Compound 2 was obtained as a white powder. HRESIMS analysis afforded an [M + Na]+ ion at m/z 354.2400, establishing the molecular formula of 2 as C21H33NO2 (calculated for [M + Na]+ ion at m/z 354.2408). The 1H, 13C, 1H–1H COSY, HSQC, and HMBC spectra showed that the only difference between 1 and 2 was an extra carbamoyl group attached at the C-3 oxygen atom in 2 (Table S3 and Figure S4), and 2 was thus identified as oxaloterpin E (Figure 3).36
Compound 3 was obtained as a white powder. HRESIMS analysis afforded an [M + Na]+ ion at m/z 398.2297, establishing the molecular formula of 3 as C22H33NO4 (calculated [M + Na]+ ion at m/z 398.2306). Similarly, the 1H, 13C, 1H–1H COSY, HSQC, and HMBC spectra revealed that the only difference between 2 and 3 was an N-hydroxyoxalyl amide group attached at the C-3 oxygen atom in 3, replacing the carbamoyl group in 2 (Table S3 and Figure S4), and 3 was hence identified as oxaloterpin C (Figure 3).26
Compound 4 was obtained as a yellow powder. HRESIMS analysis of 4 afforded an [M + H]+ ion at m/z 395.0406, with the characteristic 3:1 ratio for the isotope ions at m/z 395.0406 and m/z 397.0374 for one chlorine atom, establishing the molecular formula of 4 as C16H15ClN4O2S2 (calculated for [M + H]+ ion at m/z 395.0402). The 1H and 1H–1H COSY spectra of 4 showed resonances attributed to (i) an aliphatic −CH2CHCH– coupling system at δH 3.62 (1H, dd, J = 11.0, 9.4 Hz, H-3′a), δH 3.32 (1H, t, J = 10.9 Hz, H-3′b), δH 5.29 (1H, dt, J = 9.2, 5.6 Hz, H-4′), and δH 4.98 (1H, dd, J = 11.6, 5.7 Hz, H-6′) and (ii) an aliphatic ABX coupling system at δH 3.21 (1H, dd, J = 10.0, 6.5 Hz, H-8′a), δH 2.80 (1H, dd, J = 9.8, 9.0 Hz, H-8′b), and δH 3.74 (1H, ddd, J = 11.7, 8.8, 6.4 Hz, H-9′) (Figure 4A and Table 2). The chemical shifts and coupling patterns of these two coupling systems in the 1H NMR spectrum, in combination with the correlated carbon signals at δC 175.2 (C-1′), δC 32.5 (C-3′), δC 79.7 (C-4′), δC 72.5 (C-6′), δC 37.3 (C-8′), δC 65.9 (C-9′), and δC 171.9 (C-11′) established by HSQC and HMBC experiments (Figure 4A and Table 2), suggested a C-1′ and C-9′ disubstituted thiazolinylthiazolidinyl moiety in 4, similar to that in the siderophore pyochelin produced in Pseudomonas aeruginosa.38−42 However, the carboxylic acid moiety at C-9′ in pyochelin was substituted by an amide moiety in 4, as supported by the two amide signals at δH 7.58 (1H, br s) and δH 7.32 (1H br s), and the −N-CH3 group in pyochelin was replaced by −N-H at δH 3.12 (1H, t, J = 11.6 Hz, 10′-NH) in 4 as supported by the correlations between H-6′ and 10′-N-H and between H-9′ and 10′-N-H in the 1H–1H COSY spectrum (Figure 4A).
Figure 4.

Key COSY, HMBC, and NOESY correlations supporting (A) the structures of grisechelins A (4), B (5), C (6), and D (7), 4-chloroanthranilamide (8), and methyl 4-chloroanthranilate (9) and (B) the relative configuration of grisechelin A (4).
Table 2. 1H (700 MHz) and 13C (175 MHz) NMR Data for Grisechelins A (4) and D (7) in d6-DMSO and Grisechelins B (5) and C (6) in CDCl3a.
| grisechelin
A (4) |
grisechelin
B (5) |
grisechelin
C (6) |
grisechelin
D (7) |
|||||
|---|---|---|---|---|---|---|---|---|
| position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 2 | 138.0, C | 138.4, C | 139.2, C | 139.6, C | ||||
| 3 | 151.5, C | 152.0, C | 150.2, C | 150.2, C | ||||
| 4 | 119.4, CH | 7.96, br s | 119.1, CH | 7.67, s | 119.3, CH | 7.70, s | 120.3, CH | 8.03, s |
| 4a | 128.9, C | 132.7, C | 128.7, C | 129.2, C | ||||
| 5 | 128.5, CH | 7.95, d (8.9) | 127.5, CH | 7.68, d (8.8) | 127.6, CH | 7.68, d (8.8) | 129.1, CH | 7.97, d (8.8) |
| 6 | 129.3, CH | 7.65, dd (8.9, 2.1) | 129.6, CH | 7.50, dd (8.8, 2.1) | 128.8, CH | 7.47, dd (8.8, 2.1) | 129.3, CH | 7.65, dd (8.5, 1.4) |
| 7 | 131.9, C | 129.0, C | 132.8, C | 132.5, C | ||||
| 8 | 127.3, CH | 8.04, d (2.1) | 128.5, CH | 8.11, d (2.0) | 127.9, CH | 8.07, d (2.0) | 127.5, CH | 8.08, br s |
| 8a | 141.7, C | 142.6, C | 142.8, C | 142.5, C | ||||
| 1′ | 175.2, C | 177.0, C | 170.5, C | 169.8, C | ||||
| 3′ | 32.5, CH2 | 3.62, dd (11.0, 9.4) | 32.0, CH2 | 3.50, dd (10.9, 9.3) | 117.8, CH | 7.49 br s | 134.9, CH | 9.01, s |
| 3.32, t (10.9) | 3.40, dd (10.9, 8.3) | |||||||
| 4′ | 79.7, CH | 5.29, dt (9.2, 5.6) | 79.0, CH | 5.08, m | 156.3, C | 154.4, C | ||
| 6′ | 72.5, CH | 4.98, dd (11.6, 5.7) | 64.3, CH2 | 4.08, br d (11.5) | 61.0, CH2 | 4.94 br s | 185.1, CH | 10.07, s |
| 3.96, br d (11.5) | ||||||||
| 8′ | 37.3, CH2 | 3.21, dd (10.0, 6.5) | ||||||
| 2.80, dd (9.8, 9.0) | ||||||||
| 9′ | 65.9, CH | 3.74, ddd (11.7, 8.8, 6.4) | ||||||
| 11′ | 171.9, C | |||||||
| 3-OH | 12.20, s | 11.90, br s | 11.72, br s | 11.54, br s | ||||
| 10′-NH | 3.12, t (11.6) | |||||||
| CONH2 | 7.58, br s | |||||||
| 7.32, br s | ||||||||
Assignments were based on COSY, HMBC, HSQC, and NOESY experiments.
After excluding the thiazolinylthiazolidinyl carboxamide moiety, there were nine additional carbon signals in the 13C spectrum, all of which were characteristic of aromatic carbons (Table 2). HSQC and HMBC experiments established these nine carbon signals correlated with the aromatic ABX coupling systems at δH 7.95 (1H, d, J = 8.9 Hz, H-5), δH 7.65 (1H, dd, J = 8.9, 2.1 Hz, H-6), and δH 8.04 (1H, d, J = 2.1 Hz, H-8) and the aromatic singlet signal at δH 7.96 (1H, br s, H-4) in the 1H NMR spectrum (Figure 4A). These chemical shifts and coupling patterns, in combination with the yellow color of the compound, suggested a C-2, C-3, and C-7 trisubstituted quinoline moiety in 4. The substitution of the thiazolinylthiazolidinyl carboxamide moiety at C-2 was supported by the correlation of H-4′ with C-2, the substitution of the hydroxyl group at C-3 was supported by correlations of 3-OH with C-2, C-3, and C-4, and this finally enabled the assignment of the Cl at C-7, consistent with the chemical shifts of C-7 at δC 131.9, C-6 at δC 129.3, and C-8 at δC 127.3, as well as correlations of C-7 with H-5 and H-8 (Figure 4A and Table 2). Taken together, 4 was identified as a new natural product, named grisechelin A, featuring a chlorinated quinoline and a thiazolinylthiazolidinyl carboxamide (Figure 3).
The stereochemistry of 4 was established by a NOESY experiment. The correlation of H-6′ with H-9′ indicated H-6′ was on the same side of the reduced thiazole ring as H-9′. Due to the considerable spatial steric effect between the thiazoline and thiazolidine rings, the angle between the H-6′–C-6′–C-4′ plane and the H-4′–C-4′–C-6′ plane should be nearly 180°, positioning H-4′ and H-6′ in an opposite orientation (Figure 4B). Accordingly, H-6′ correlated only with H-3′b, while H-4′ yielded stronger correlations with H-3′a than H-3′b. This was also consistent with the lack of correlations between 10′-N-H and H-3′a or H-3′b, indicating 10′-N-H took different sides of the H-6′–C-6′–C-4′ plane from H-3′a and H-3′b (Figure 4B). Taken together, the relative configuration at C-4′, C-6′, and C-9′ in 4 was assigned as R, R, and R, respectively (Figure 3).
Compound 5 was obtained as a yellow powder. HRESIMS analysis of 5 afforded an [M + H]+ ion at m/z 295.0306, with the similar 3:1 ratio for the isotope ions at m/z 295.0306 and m/z 297.0275, establishing the molecular formula of 5 as C13H11ClN2O2S (calculated for the [M + H]+ ion at m/z 295.0307). The 1H and 1H–1H COSY spectra of 5 showed similar resonances to 4 attributed to one aromatic ABX coupling system at δH 7.68 (1H, d, J = 8.8 Hz, H-5), δH 7.50 (1H, dd, J = 8.8, 2.1 Hz, H-6), and δH 8.11 (1H, d, J = 2.0 Hz, H-8) and one aromatic singlet signal at δH 7.67 (1H, s, H-4) (Figure 4A and Table 2). While the aliphatic ABX coupling system of 4 disappeared in 5, the aliphatic −CH2CHCH– coupling system in 4 was replaced by a −CH2CHCH2– coupling system at δH 3.50 (1H, dd, J = 10.9, 9.3 Hz, H-3′a), δH 3.40 (1H, dd, J = 10.9, 8.3 Hz, H-3′b), δH 5.08 (1H, m, H-4′), δH 4.08 (1H, br d, J = 11.5 Hz, H-6′a), and δH 3.96 (1H, br d, J = 11.5 Hz, H-6′b) (Table 2). These comparisons between 4 and 5 indicated 5 had the same chlorinated quinoline moiety as 4 but had only the C-1′ and C-4′ disubstituted thiazolidine moiety in the structure. The substitution of the thiazolidine moiety at C-2 was similarly supported by the correlation of H-4′ with C-2, while the assignment of a hydroxymethyl group at C-4′ was based on its chemical shift at δC 64.3 (C-6′) and correlations between H-3′ and C-4′ and between H-3′ and C-6′ (Figure 4). Taken together, 5 was identified as a new natural product, named grisechelin B, featuring a chlorinated quinoline and a thiazolidine, whose configuration at C-4′ was not assigned but assumed to be the same as 4 (Figure 3).
Compound 6 was obtained as a yellow powder. HRESIMS analysis of 6 afforded an [M + H]+ ion at m/z 293.0152 with the similar 3:1 ratio for the isotope ions at m/z 293.0152 and m/z 295.0121. This established the molecular formula of 6 as C13H9ClN2O2S (calculated for the [M + H]+ ion at m/z 293.0151), which differed from 5 by two hydrogens. Comparisons between 5 and 6 in the 1H and 1H–1H COSY spectra showed that the only difference was that the −CH2CHCH2– coupling system in 5 was replaced by one aromatic methine signal at δH 7.49 (1H, br s, H-3′) and one aliphatic methylene signal at δH 4.94 (2H, br s, H-6′) in 6, hence the assignment of a C-4′ hydroxymethyl-substituted thiazole moiety to 6 (Table 2). HSQC and HMBC experiments (Figure 4) finally established 6 as a new natural product, named grisechelin C, featuring a chlorinated quinoline and a thiazole (Figure 3).
Compound 7 was obtained as a yellow powder. HRESIMS analysis of 7 afforded an [M + H]+ ion at m/z 290.9998 with the similar 3:1 ratio for the isotope ions at m/z 290.9998 and m/z 292.9969. This established the molecular formula of 7 as C13H7ClN2O2S (calculated for the [M + H]+ ion at m/z 290.9994), which differed from 6 by two hydrogens. Comparisons of the 1H and 1H–1H COSY spectra of 6 and 7 showed that the only differences were that the aromatic methine signal in thiazole ring of 6 was shifted downfield to δH 9.01 (1H s, H-3′) and the aliphatic methylene signal in 6 was replaced by an aldehyde signal at δH 10.07 (1H, s, H-6′) in 7, respectively, indicating that the C-6′ hydroxymethyl group in 6 was replaced by an aldehyde group in 7 (Table 2). HSQC and HMBC experiments (Figure 4) finally confirmed the assignment of 7 as a new natural product, named grisechelin D, featuring a chlorinated quinoline and a thiazole carbaldehyde (Figure 3).
Compounds 8, 9, 10, and 11 were identified as 4-chloroanthranilamide (8),43 methyl 4-chloroanthranilate (9),43 dinactin (10),44,45 and feigrisolide C (11)46 on the basis of MS and NMR spectroscopic analysis (Tables S4 and S5). Although 8 and 9 have been synthesized previously,43 this is the first time to our knowledge that they have been isolated as microbial natural products.
Compound 12 was obtained as a yellow oil. HRESIMS analysis of 12 yielded the [M + H]+ and [M + Na]+ ions at m/z 783.4871 and 805.4688, respectively, establishing the molecular formula of 12 as C42H70O13 (calculated for the [M + H]+ and [M + Na]+ ions at m/z 783.4892 and m/z 805.4712). Comparisons between 10 and 12 in the 1H, 13C, and 1H–1H COSY spectra showed that they were very similar except the overlapped proton or carbon chemical shifts between the two symmetrical units in 10 were split in 12 (Table S5). The most significant split signals occurred at C-1/C-1′, C-16/C-16′, and C-20/C-20′, as exemplified by C-1/C-1′ at δC 174.3 in 10 to C-1 at δC 175.9 and C-1′ at δC 174.5 in 12, H-16/H-16′ at δH 4.92 (1H, m) in 10 to H-16 at δH 4.87 (1H, m) and H-16′ at δH 3.78 (1H, m) in 12, C-16/C-16′ at δC 73.1 in 10 to C-16 at δC 73.5 and C-16′ at δC 70.3 in 12, and C-20/C-20′ at δC 27.3 in 10 to C-20 at δC 30.0 and C-20′ at δC 27.5 in 12, respectively. These spectroscopic data indicated the ester linkage at C-1 in 10 was hydrolyzed in 12, which was supported by the disappearance of the HMBC correlation between H-16′ and C-1 in 12 (Figure S5). Taken together, 12 was identified as seco-dinactin, a new analogue of the macrotetrolide family,44,45 resulting from hydrolysis of 10 at the C-1 position (Figure 3).
Streptomyces griseus CB00830 Demonstrating the Utility of Biosynthetic Potential-Based Strain Prioritization for Natural Product Discovery
We chose S. griseus CB00830 to showcase natural product discovery on the basis that (i) it was identified as a diterpenoid producer and diterpenoids of bacterial origin are underrepresented among known natural products and (ii) it was one of the talented strains that could produce all four classes of the targeted natural products. Upon fermentation optimization in 10 different media, we indeed isolated three diterpenoids, 1–3, from S. griseus CB00830 (Figure 3). All diterpenoids are derived from geranylgeranyl diphosphate (GGDP), and DTSs catalyze the critical steps in diterpenoid biosynthesis by morphing GGDP into one of the many diterpenoid scaffolds, further transformations of which by pathway-specific tailoring enzymes afford vast structural diversity known for diterpenoid natural products.31,32 The three diterpenoids 1–3 had been isolated previously from Streptomyces sp. KO-3988, the biosynthesis of which from GGDP had been confirmed to be catalyzed by two DTSs, an ent-copalyl diphosphate (ent-CPP) synthase and a pimaradiene synthase.36,47 We have cloned the genes that encode the GGDP synthase, ent-CPP synthase, and pimaradiene synthase from S. griseus CB00830, which show high sequence identities to their homologues from Streptomyces sp. KO-3988 (Figure S6). Taken together, these findings support our DTS-based strain prioritization strategy for diterpenoid discovery. Fermentation optimization of the other talented strains already identified in this study or application of our strategy to identify additional diterpenoid producers holds great promise in accelerating the discovery and isolation of additional bacterial diterpenoid natural products.32
S. griseus CB00830 as a nonribosomal peptide and polyketide producer is supported by the isolation of compounds 4–9 and 10–12, respectively (Figure 3). Thus, the biosynthesis of 4, in analogy to pyrochelin,48,49 could be proposed to be catalyzed by an NRPS consisting of at least three modules: (i) an initiation module that specifies 7-chloro-3-hydroxyquinaldic acid as the starter unit and (ii) two extension modules that incorporate two molecules of l-cysteine as the extender units to afford the nonribosomal peptide backbone of 4 (Figure 5). Compounds 5–7 could be viewed as shunt metabolites of 4, terminated from the NRPS biosynthetic machinery prematurely before the incorporation of the second l-cysteine residue. Quinaldic acid-containing nonribosomal peptide natural products are rare but known, and at least two distinct pathways have been proposed for their biosynthesis, both of which originated from tryptophan.50,51 To our knowledge, however, 4 is the first natural product that contains a 7-chloro-3-hydroxyquinaldic acid moiety, hence providing an exciting opportunity to investigate its biosynthesis. It is tempting to speculate that the 7-chloro-3-hydroxyquinaldic acid moiety of 4 could originate from 7-chlorotryptophan, in biosynthetic analogy to the 3-hydroxyquinaldic acid moiety of thiocoraline from tryptophan in Micromonospora sp. ML1 (Figure 5).50 Compounds 8 and 9 could be then viewed as degradation products of 7-chlorotryptophan, though they could equally be shunt metabolites of chorismate metabolism via the intermediacy of anthranilic acid (Figure 5).52 Similarly, isolation of compounds 10–12 is consistent with the prediction of S. griseus CB00830 as a polyketide producer. We have previously established the polyketide origin of 10 with 11 and 12 as putative biosynthetic intermediates.44,45 Both 11 and 12 therefore could be viewed as shunt metabolites for 10 biosynthesis, although 12 could also be a degradation metabolite of 10.
Figure 5.

Proposed pathway for grisechelin A (4) biosynthesis in S. griseus CB00830, featuring 7-chlorotryptophan and 7-chloro-3-hydroxyquinaldic acid as key intermediates and a NRPS biosynthetic machinery incorporating 7-chloro-3-hydroxyquinaldic acid and two molecules of l-cysteine to afford the nonribosomal peptide backbone, as supported by the isolation of shunt metabolites grisechelins B (5), C (6), and D (7), 4-chloroanthranilamide (8), and methyl 4-chloroanthranilate (9) from the same strain. “–S-Enz” depicts biosynthetic intermediates covalently tethered to the peptidyl carrier protein of the NRPS.
Our method was effective in identifying potential producers of aromatic polyketides (39), reduced polyketides (36), and nonribosomal peptides (38) from the 100 strains examined (Figure 2), and these findings were consistent with the actinomycete genomes sequenced to date, each of which was shown to encode multiple type I and II PKSs as well as NRPSs.5,8 In retrospect, type I and II PKSs and NRPSs might not be the best choice of probes to survey biosynthetic potential of a strain due to their universal distribution in actinomycetes, hence the likelihood of high hit rates. In contrast, 25 of the 100 strains were identified as potential diterpenoid producers (Figure 2), and this was unexpected because only a very small number of diterpenoids have been characterized from actinomycetes.31,32 The latter finding highlights that variations and future application of this method may be most productive in surveying strains for potential producers of natural products of underrepresented scaffolds or with specifically defined structural features.
Experimental Section
General Experimental Procedures
Optical rotations were measured with an AUTOPOL IV automatic polarimeter (Rudolph Research Analytical). UV spectra were collected with a NanoDrop 2000C spectrophotometer (Thermo Scientific). Circular dichroism spectrum was measured with a J-815 CD spectrometer (JASCO). NMR data were recorded on a Bruker Ultra Shield 700 instrument. HRESIMS analysis was carried out on a Thermo Finnigan LTQ Orbitrap mass spectrometer. MPLC separation was conducted on Biotage Isolera One using a Biotage SNAP Cartridge HP-Sil column (25 g). HPLC was carried out on a Varian semipreparative HPLC system using a Prostar 330 detector and a GRACE Apollo C18 column (250 mm × 4.6 mm, 5 μm) for analysis and an Alltima C18 column (250 mm × 10.0 mm, 5 μm) for purification. Fermentation was carried out in a New Brunswick Scientific Innova 44 incubator shaker for small scale (50 mL in 250 mL baffled Erlenmeyer flasks) or a New Brunswick BioFlo/CelliGen 115 fermentor for large scale (5 L in a 14 L vessel).
Bacterial Strains, Plasmids, Biochemicals, Chemicals, and Media
Escherichia coli DH5α was used as the host for common subcloning and plasmid preparation.53 The 16 strains used to develop and validate the method are summarized in Table S2. The actinomycete collection at The Scripps Research Institute consists of strains isolated from various unexplored and underexplored ecological niches as exemplified in early publications.23−30 Actinomycete strain cultivation and genomic DNA preparation followed standard protocols.54 Taq 2X Master Mix (New England BioLabs Inc.) was used for PCR amplification, and QIAquick Gel extraction kit and QIAprep Spin miniprep kit (Qiagen) were used for PCR product recovery and plasmid extraction, respectively. Common biochemical and chemicals were purchased from standard commercial sources and used directly. Diaion HP-20 resins were purchased from Mitsubishi Chemical Corporation. Sephadex LH-20 was from GE Healthcare.
Primer Design and Preparation of a Pool of Probes by the Two-Tiered PCR Method
The designer primers, consisting of the pathway-specific sequences for the targeted classes of natural products and unbiased AT-rich tags, are summarized in Table 1. The pathway-specific PCR primers for type I PKSs,33 type II PKSs,34 NRPSs,33 and DTSs35 were similarly designed according to literature protocols (Figure S2A), and an 18 bp AT-rich tag sequence was added at the 5′ end of each pathway-specific primer to form the designer primers for the first round of PCR. The primers for the second round of PCR were identical to the 18 bp AT-rich tag sequences (Table 1).
For the first round of PCR, 1 μg of genomic DNA mixture containing an equal amount for each genome was used as a template in a 50 μL PCR reaction. Only 10 cycles were used for the first round of PCR to retain product diversity (Table 1). The resultant products were recovered by 1% agar gel and used as the template of the second round of PCR. The second-round PCR used 35 cycles to ensure that all variants of the first-round PCR products were well represented as a pool of probes. The final PCR products were cloned into pGEM-T and pGEM-T Easy Vector Systems for sequencing to validate the accuracy and diversity.
Survey of Biosynthetic Potential within a Collection of Strains
The genomic DNA of individual strains was denatured in boiling water for 5 min and quickly chilled in ice to obtain single-stranded DNA. Approximately 2 μg of resultant DNA was dotted onto an Amersham Hybond-N+ membrane (GE Healthcare), dried, and cross-linked according to standard protocols.53 The probe labeling and hybridization procedures followed DIG-High Prime DNA labeling and detection starter kit I (Roche). Approximately 500 ng of PCR products was used for each labeling, and a final concentration of 25 ng/mL of the probes was used in each hybridization process (Table 1). Upon completion of the hybridization process, the final blots were documented by photography.
Fermentation Optimization and Natural Product Isolation from S. griseus CB00830
The CB00830 strain, collected from the Nagqu prefecture of Tibet, China, was assigned as an S. griseus species on the basis of polyphasic taxonomy (Figure S7) and preserved as a spore suspension in 20% glycerol at −80 °C. S. griseus CB00830 was first cultured in TSB at 28 °C and 250 rpm for 2 days to prepare the seed inoculum. This seed culture was then used to inoculate 10 different media (50 mL in 250 mL baffled Erlenmeyer flasks) (Table S1), each of which was fermented at 28 °C and 250 rpm for 7 days. After fermentation, Diaion HP-20 resins (5%) were added to each of the cultures, and the resultant slurries of resins and cell mass were agitated for 4 h at room temperature. Upon centrifugation, the supernatants were discarded, and the pelleted resins and cell mass were collected, washed with water, and extracted with MeOH to recover all natural products present in the fermentation. These crude extracts were finally subjected to TLC and HPLC analysis to compare metabolite profiles.
For large-scale fermentation, the same seed culture (500 mL) was used to inoculate 5 L of medium E (Table S1), and the fermentation was carried out in a 14 L vessel fermentor at 250 rpm, 28 °C for 7 days. After fermentation, 200 g of Diaion HP-20 resins was added into the culture and stirred overnight, and the resin and cell mass then were harvested by centrifugation. The resin and cell pellet were washed with water and extracted with 95% ethanol, and the ethanol extracts were concentrated in a vacuum to afford the crude extract.
The crude extract was resuspended in water, followed by extraction with EtOAc. The EtOAc extract was subjected to MPLC, using a Biotage SNAP Cartridge HP-Sil column (25 g), eluted with a linear gradient of MeOH in CH2Cl2 from 0% to 90%, to afford 90 fractions. Fractions 4–6 were combined and subjected to preparative TLC (SiO2), eluted with CHCl3, to yield 9 (7.9 mg). Fractions 11–13 were combined and subjected sequentially to preparative TLC (SiO2), eluted with CHCl3, and Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), to afford 1 (3.6 mg) and 7 (2.5 mg), respectively. Fractions 22–26 were combined and subjected to Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), to afford compound 2 (3.9 mg). Fractions 27–31 were combined and subjected sequentially to Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), and preparative HPLC (C18), eluted with CH3CN/H2O (67:33), to afford compounds 5 (0.9 mg) and 6 (1.1 mg), respectively. Fraction 33 was subjected to Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), to afford compound 10 (140.1 mg). Fraction 39, at room temperature for 3 days, precipitated out a yellow powder, which was collected and washed with CHCl3 to afford 4 (5.5 mg). Fractions 43 and 44 were combined and subjected sequentially to Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), and preparative HPLC (C18), eluted with CH3CN/H2O (85:15), to afford 3 (4.4 mg). Fractions 64–67 were combined and subjected to Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), to afford compounds 8 (0.7 mg) and 12 (4.2 mg). Finally, fractions 73–79 were combined and subjected sequentially to SiO2 column chromatography, eluted with CHCl3/MeOH (30:1), and Sephadex LH-20 column chromatography, eluted with CHCl3/MeOH (1:1), to afford 11 (6.5 mg).
Grisechelin A (4):
yellow powder; UV (CHCl3) λmax (log ε) 248 (4.41), 301 (3.73), 377 (3.50) nm; circular dichroism spectrum (see Figure S13); 1H and 13C NMR (see Table 2); HRESIMS for the [M + H]+ ion at m/z 395.0406 (calculated [M + H]+ ion for C16H15ClN4O2S2 at m/z 395.0402).
Grisechelin B (5):
yellow powder; UV (CHCl3) λmax (log ε) 248 (4.33), 306 (3.66), 375 (3.51) nm; 1H and 13C NMR (see Table 2); HRESIMS for the [M + H]+ ion at m/z 295.0306 (calculated [M + H]+ ion for C13H11ClN2O2S at m/z 295.0307).
Grisechelin C (6):
yellow powder; UV (CHCl3) λmax (log ε) 247 (4.08), 325 (3.69), 377 (3.70) nm; 1H and 13C NMR (see Table 2); HRESIMS for the [M + H]+ ion at m/z 293.0152 (calculated [M + H]+ ion for C13H9ClN2O2S at m/z 293.0151).
Grisechelin D (7):
yellow powder; UV (CHCl3) λmax (log ε) 242 (4.51), 322 (4.10), 372 (4.08) nm; 1H and 13C NMR (see Table 2); HRESIMS for the [M + H]+ ion at m/z 290.9998 (calculated [M + H]+ ion for C13H7ClN2O2S at m/z 290.9994).
seco-Dinactin (12):
yellow oil; [α]27D +4.5 (c 0.4, CHCl3); 1H and 13C NMR (see Table S5); HRESIMS for the [M + H]+ and [M + Na]+ ions at m/z 783.4871 and m/z 805.4688 [M + Na]+, respectively (calculated [M + H]+ and [M + Na]+ ions for C42H70O13 at m/z 783.4892 and m/z 805.4712, respectively).
Acknowledgments
This work is supported in part by the Natural Products Library Initiative at The Scripps Research Institute, NIH grant GM086184 (to B.S.), the Chinese Ministry of Education 111 Project B08034 (to Y.D.), and National High Technology Joint Research Program of China grant 2011ZX09401-001 (to Y.D.).
Supporting Information Available
Table S1 provides the 10 media used for fermentation optimization of actinomycete strains in The Scripps Research Institute collection. Table S2 lists the 16 strains known to produce aromatic polyketides, reduced polyketides, nonribosomal peptides, and diterpenoids, respectively, which were used to develop and optimize the biosynthetic potential-based strain prioritization method. Tables S3, S4, and S5 summarize the 1H and 13C NMR data for diterpenoids 1–3, chloroanthranilates 8 and 9, and polyketides 10–12, respectively. Table S6 summarizes the primers used for amplifying and sequencing the putative DTS and housekeeping genes from S. griseus CB00830. Figure S1 shows the structures of the 16 natural products whose producing strains are summarized in Table S2. Figure S2 summarizes primer design and method development and validation to survey the biosynthetic potential of the four targeted classes of natural products using the 16 known producers (Table S2). Figure S3 shows the survey of the randomly selected 100 strains for their biosynthetic potential of the four targeted classes of natural products. Figures S4 and S5 depict key COSY and HMBC correlations supporting the structural confirmation of diterpenoids 1, 2, and 3 and polyketides 10–12, respectively. Figure S6 compares the biosynthetic loci of diterpenoids 1, 2, and 3 between S. griseus CB00830 and S. sp. KO-3988. Figure S7 summarizes the taxonomic analysis supporting the assignment of strain CB00830 as a S. griseus species. Figures S8–S12 show the 1H and 13C NMR spectra of 4, 5, 6, 7, and 12, respectively. Figure S13 provides the CD spectrum of 4. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ P. Xie and M. Ma contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Li J. W.-H.; Vederas J. C. Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdy J. J. Antibiot. 2012, 65, 385–395. [DOI] [PubMed] [Google Scholar]
- Schmitt E. K.; Moore C. M.; Krastel P.; Petersen F. Curr. Opinion Chem. Biol. 2011, 15, 497–504. [DOI] [PubMed] [Google Scholar]
- Nett M.; Ikeda H.; Moore B. S. Nat. Prod. Res. 2009, 26, 1362–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan S. Nat. Biotechnol. 2012, 30, 385–387. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Phillips J. W.; Wang J. Curr. Opinion Drug Discovery Dev. 2007, 10, 160–166. [PubMed] [Google Scholar]
- Craney A.; Ahmed S.; Nodwell J. J. Antibiot. 2013, 66, 387–400. [DOI] [PubMed] [Google Scholar]
- Cordell G. A.; Shin Y. G. Pure Appl. Chem. 1999, 71, 1089–1094. [Google Scholar]
- Wolf D.; Siems K. Chimia 2007, 61, 339–345. [Google Scholar]
- Bradshaw J.; Butina D.; Dunn A. J.; Green R. H.; Hajek M.; Jones M. M.; Lindon J. C.; Sidebottom P. J. J. Nat. Prod. 2001, 64, 1541–1544. [DOI] [PubMed] [Google Scholar]
- Konishi Y.; Kiyota T.; Draghici C.; Gao J.-M.; Yeboah F.; Acoca S.; Jarussophon S.; Purisima E. Anal. Chem. 2007, 79, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Genilloud O.; Gonzalez I.; Salazar O.; Martin J.; Tormo J. R.; Vicente F. J. Ind. Microbiol. Biotechnol. 2011, 38, 375–389. [DOI] [PubMed] [Google Scholar]
- Khosla C.; Keasling J. D. Nat. Rev. Drug Discovery 2003, 2, 1019–1025. [DOI] [PubMed] [Google Scholar]
- Bode H. B.; Muller R. Angew. Chem., Int. Ed. 2005, 44, 6828–6846. [DOI] [PubMed] [Google Scholar]
- Van Lanen S. G.; Shen B. Curr. Opin. Microbiol. 2006, 9, 252–260. [DOI] [PubMed] [Google Scholar]
- Galm U.; Shen B. Exp. Opin. Drug Discovery 2006, 1, 409–437. [DOI] [PubMed] [Google Scholar]
- Wilkinson B.; Micklefield J. Nat. Chem. Biol. 2007, 3, 379–386. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Fischbach M. A. J. Am. Chem. Soc. 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Curr. Opin. Chem. Biol. 2011, 15, 5–10. [DOI] [PubMed] [Google Scholar]
- Winter J. M.; Behnken S.; Hertweck C. Curr. Opin. Chem. Biol. 2011, 15, 22–31. [DOI] [PubMed] [Google Scholar]
- Davies J. J. Antibiot. 2013, 66, 361–364. [DOI] [PubMed] [Google Scholar]
- Powell E.; Huang S.-X.; Xu Y.; Rajski S. R.; Wang Y.; Peters N.; Guo S.; Xu H. E.; Hoffmann F. M.; Shen B.; Xu W. Biochem. Pharmacol. 2010, 80, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-X.; Powell E.; Rajski S. R.; Zhao L.; Jiang C.; Duan Y.; Xu W.; Shen B. Org. Lett. 2010, 12, 3525–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.-X.; Huang S.-X.; Tang S.-K.; Jiang C.-L.; Duan Y.; Beutler J. A.; Henrich C. J.; McMahon J. B.; Schmid T.; Blees J. S.; Colburn N. H.; Rajski S. R.; Shen B. J. Nat. Prod. 2011, 74, 1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Vodanovic-Jankovic S.; Ledeboer N.; Huang S.-X.; Rajski S. R.; Kron M.; Shen B. Org. Lett. 2011, 13, 2034–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Vodanovic-Jankovic S.; Kron M.; Shen B. Org. Lett. 2012, 14, 4946–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Zhao L.-X.; Jiang C.-L.; Duan Y.; Wong L.; Carver K. C.; Schuler L. A.; Shen B. J. Antibiot. 2011, 64, 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-X.; Yu Z.; Robert F.; Zhao L.-X.; Jiang Y.; Duan Y.; Pelletier J.; Shen B. J. Antibiot. 2011, 64, 164–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbets B.; Yu Z.; Stewart D.; Zhao L.-X.; Jiang Y.; Xu L.-H.; Shen B.; Klein B. Med. Mycol. 2013, 51, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smanski M. J.; Yu Z.; Casper J.; Lin S.; Peterson R. M.; Chen Y.; Wendt-Pienkowski E.; Rajski S. R.; Shen B. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smanski M. J.; Peterson R. M.; Huang S.-X.; Shen B. Curr. Opin. Chem. Biol. 2012, 16, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso-Sacido A.; Genilloud O. Microbiol. Ecol. 2005, 49, 10–24. [DOI] [PubMed] [Google Scholar]
- Wawrik B.; Kerkhof L.; Zylstra G. J.; Kukor J. J. Appl. Environ. Microbiol. 2005, 71, 2232–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyomasu T.; Tsukahara M.; Kaneko A.; Niida R.; Mitsuhashi W.; Dairi T.; Kato N.; Sassa T. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 3084–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi K.; Ueno R.; Sue M.; Furihata K.; Matsumoto T.; Dairi T.; Omura S.; Seto H. J. Nat. Prod. 2007, 70, 1712–1717. [DOI] [PubMed] [Google Scholar]
- Soriano-Garcia M.; Guerrero C.; Toscano R. A. Acta Crystallogr. 1986, C42, 729–731. [Google Scholar]
- Cox C. D.; Rinehart K. L.; Moore M. L.; Cook J. C. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 4256–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankenbauer R. G.; Staley A. L.; Rinehart K. L.; Cox C. D. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 1878–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitte J. J.; Cox C. D.; Rasmussen G. T.; Britigan B. E. Arch. Biochem. Biophys. 2001, 393, 236–244. [DOI] [PubMed] [Google Scholar]
- Schlegel K.; Lex J.; Taraz K.; Budzikiewicz H. Z. Naturforsch. 2006, 61c, 263–266. [DOI] [PubMed] [Google Scholar]
- Yoganathan S.; Sit C. S.; Vederas J. C. Org. Biomol. Chem. 2011, 9, 2133–2141. [DOI] [PubMed] [Google Scholar]
- Theeraladanon C.; Arisawa M.; Nishida A.; Nakagawa M. Tetrahedron 2004, 60, 3017–3035. [Google Scholar]
- Kwon H.-J.; Smith W. C.; Xiang L.; Shen B. J. Am. Chem. Soc. 2001, 123, 3385–3386. [DOI] [PubMed] [Google Scholar]
- Kwon H.-J.; Smith W. C.; Scharon A. J.; Hwang S. H.; Kurth M. J.; Shen B. Science 2002, 297, 1327–1330. [DOI] [PubMed] [Google Scholar]
- Kim W. H.; Jung J. H.; Lee E. J. Org. Chem. 2005, 70, 8190–8192. [DOI] [PubMed] [Google Scholar]
- Ikeda C.; Hayashi Y.; Itoh N.; Seto H.; Dairi T. J. Biochem. 2007, 141, 37–45. [DOI] [PubMed] [Google Scholar]
- Thomas M. S. Biometals 2007, 20, 431–452. [DOI] [PubMed] [Google Scholar]
- Youard Z. A.; Wenner N.; Reimmann C. Biometals 2011, 24, 513–522. [DOI] [PubMed] [Google Scholar]
- Lombo F.; Velasco A.; Castro A.; de la Calle F.; Brana A. F.; Sanchez-Puelles J. M.; Mendez C.; Salas J. A. ChemBioChem 2006, 7, 366–376. [DOI] [PubMed] [Google Scholar]
- Duan L.; Wang S.; Liao R.; Liu W. Chem. Biol. 2012, 19, 443–448. [DOI] [PubMed] [Google Scholar]
- Van Lanen S. G.; Lin S.; Shen B. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J.; Russell D. W.. Molecular Cloning; CSHL Press: Woodbury, NY, 2001. [Google Scholar]
- Kieser T.; Bibb M. J.; Buttner M. J.; Chater K. F.; Hopwood D. A.. Practical Streptomyces Genetics; The John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.