

Abstract
A tandem dienone-photorearrangement-cycloaddition (DPC) reaction of novel cyclohexadienone substrates tethered with various 2π and 4π reaction partners resulted in the formation of polycyclic, bridged frameworks. In particular, use of alkynyl ether-tethered substrates led to (3+2) cycloaddition to afford strained alkenes which could be further elaborated by intra- and intermolecular cycloaddition chemistry to produce complex, polycyclic chemotypes.
Introduction
Photochemistry is a useful tool for the generation of architecturally interesting small molecules1 and is often used as a key step in the synthesis of natural products.2 In light of these developments, it is surprising that photochemistry is underutilized in the development of novel scaffolds for drug development.3 As part of a program directed towards the synthesis of novel chemotypes, we have been interested in leveraging the powerful transformations enabled by photochemical excitation for the rapid generation of molecular diversity from simple, readily available scaffolds.4 Moreover, photochemical transformations are easily parallelized with simple reactor designs5 and scale up can be readily accomplished using continuous processing technology.6 Equally important, photochemical transformations provide access to products with rich topological complexity and stereochemical diversity.
We wished to investigate the photochemical rearrangement of substituted cyclohexadienones to oxyallyl cations,7 versatile intermediates which have found broad applicability in reaction development and complex molecule synthesis.8 In 1985, Schultz and coworkers demonstrated that dienone photorearrangement of cyclohexadienone 1 led to the formation of an oxyallyl cation intermediate which underwent dipolar cycloaddition with a pendant furan to afford the (4+3) cycloadduct 2 (Scheme 1a).9 While the majority of examples conducted were intramolecular cycloadditions involving carbon-tethered alkenes, Schultz and coworkers also observed intermolecular reactions, albeit in lower chemical yields.10 A limited study on the factors controlling the mode of cycloaddition showed that substituent effects (both steric and electronic) appear to play dominant roles in determining product distributions.11
Scheme 1.

(a) Tandem Dienone Photorearrangement by Schultz et al.9 (b) Possible Reaction Pathways for Intramolecular (3+2) or (5+2) Cycloaddition with Tethered Alkene Substrates
With the advent of powerful strategies for the oxidative dearomatization of phenols,12 we were interested in examining the potential of a modular dearomatization-photorearrangement-cycloaddition strategy to generate topologically rich molecular architectures from readily available materials (phenols and ω-alkenols or ω-alkynols). Specifically, we were interested in determining the factors responsible for divergent, intramolecular (3+2) and (5+2) photocycloaddition pathways of oxyallyl cations 3 derived from substrates 4 (Scheme 1b) to produce bridged (5, 6, and 7) or fused frameworks (e.g. 8).
Herein, we report use of tandem dienone photorearrangement-intramolecular cycloaddition (DPC) of alkenyl and alkynyl ether-tethered cyclohexadienone substrates to produce a variety of structurally complex cycloadducts. In particular, use of alkynyl ether-tethered substrates led to (3+2) cycloaddition to afford strained alkenes which could be further elaborated to polycyclic chemotypes.
Results and Discussion
Our initial investigations focused on substituted cyclohexadienone substrate 9, readily available via oxidative dearomatization of 2,4,6-trimethylphenol.13 Having optimized reaction parameters (solvent/concentration/reaction time) for the conversion of 1→2 in a multi-welled photoreactor (Scheme 1),13 we examined dienone-photorearrangement-cycloaddition of alkene-tethered substrate 9 (Scheme 2). Indeed, 9 was smoothly converted to a 10:1 mixture of the 5,1-and 1,5-bonded products (10 and 11, respectively) in 83% isolated yield upon irradiation (λ > 300 nm) in benzene at 20 °C for 20 min. Interestingly, 1,3- and 1,6- cycloaddition products were not observed upon irradiation of substrate 9. In addition, cycloadduct 10 was observed as the major regioisomeric cycloadduct. Although the reason for this outcome was unclear at the time, we observed that the selectivity was attenuated (10/11 = 6:1) when the reaction mixture was not degassed prior to irradiation, presumably due to degradation of the major product under the reaction conditions. The use of polar solvents to potentially stabilize the oxyallyl cation did not change the product distribution.13, 14 Structural assignment of the major isomer was confirmed by X-ray crystallographic analysis of alcohol 12, obtained as a single diastereomer from reduction of 10:11 with NaBH4 in methanol (Scheme 2).13
Scheme 2.

Dienone-Photorearrangement-Cycloaddition of Alkene-Tethered Substrate 9
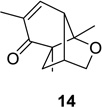
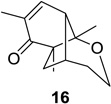
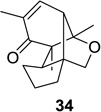
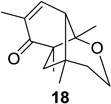
We next focused our efforts on evaluating the scope of the reaction (Table 1). Irradiation of substrate 9 with >300 nm light in degassed benzene for 20 min afforded the DPC product in a 10:1 ratio of 5,1:1,5-bonded product in 83% isolated yield (Table 1, entry 1). In most cases, isolation of pure 5,1-products such as 10 was accomplished by a reduction/oxidation sequence.13 Switching from methallyl- to allyl-substituted cyclohexadienone substrate 13 provided similar regioselectivity to afford the 5,1-product 14 in 80% isolated yield (Table 1, entry 2). Elongation of the carbon spacer with an extra methylene group led to complete regioselectivity in which case products 16 and 18 could be obtained in 85% and 78% yield, respectively (Table 1, entries 3–4).
Table 1.
Substrate Scope for the Dienone-Photorearrangement-Cycloadditiona
| Entry | Substrate | Major Product | Yield [%]b, c | Entry | Substrate | Major Product | Yield [%]b,c |
|---|---|---|---|---|---|---|---|
| 1 | 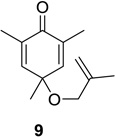 |
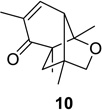 |
83 (10:1) | 7 | 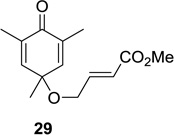 |
 |
80 (>25:1) |
| 2 | 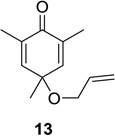 |
 |
80 (9:1) | 8 | 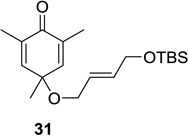 |
 |
53 (12:1) |
| 3 | 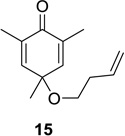 |
 |
85 (>25:1) | 9 | 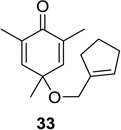 |
 |
67 (4:1) |
| 4 | 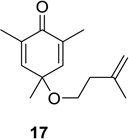 |
 |
78 (>25:1) | 10 |  |
 |
73 (4:1) |
| 5 | 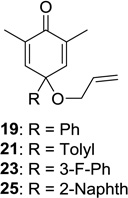 |
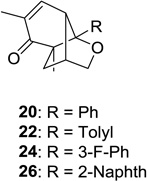 |
20: 75 (10:1) 22: 63 (4:1)d 24: 60 (4:1)d 26: 60 (4:1)d |
11 | 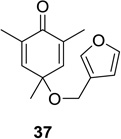 |
 |
51 (7:1) |
| 6 | 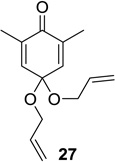 |
 |
52 (9:1) | 12 | 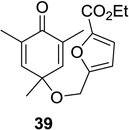 |
 |
81 (>25:1) |
Reaction conditions: substrate (0.25 mmol) in benzene (10 mL) degassed for 30 min followed by irradiation (>300 nm) at room temperature for 20 min.
Isolated yield.
Regioselectivity determined by 1H NMR analysis of the crude reaction mixture in parenthesis.
Isolated yield of the 5,1-product.
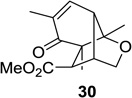
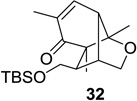
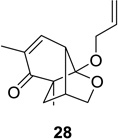
Upon replacement of the methyl group in the 4-position of the cyclohexadienone substrate with aromatic substituents, a decrease in regioselectivity was observed in some cases (cf. Table 1, entry 5). Irradiation of diallyl acetal 27 provided the DPC product in 52% yield as a 9:1 mixture of regioisomers (Table 1, entry 6). Substitution of the terminal position of the alkene with an ester moiety led to selective formation of cycloadduct 30 in 80% yield (Table 1, entry 7). Photoirradiation of substrate 31 provided silyl ether product 32 in 53% yield as a 12:1 mixture of 5,1:1,5-product (Table 1, entry 8), whereas its corresponding Z-isomer gave an indiscernible mixture. Upon irradiation of cyclic alkene-substituted cyclohexadienone substrates, a decrease in yield and regioselectivity was observed (Table 1, entries 9–10).
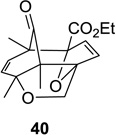
As depicted in Scheme 1, irradiation of cyclohexadienone substrate 1 bearing a tethered furan led solely to the formation of (4+3) cycloadduct 2 in excellent yield. Substrate 37 bearing a 3-furyl ether connected to the cyclohexadienone moiety did not lead to the formation of the corresponding (4+3) cycloadduct. In this case, (5+2) cycloaddition was observed to provide a 7:1 mixture of the 5,1- and the 1,5-bonded products in 71% isolated yield (Table 1, entry 11). Substitution of the 5-position of the furyl group of substrate 1 with an ester moiety cleanly afforded the (4+3) cycloadduct 40 in 81% isolated yield (Table 1, entry 12).
In order to rationalize the observed preference for 5,1- vs 1,5-bonded products, we examined molecular models of the zwitterionic intermediate formed after dienone photorearrangement (Scheme 3).15 Formation of the 1,5-bonded product appears to be disfavored due to steric hindrance between one of the methyl groups on the cyclohexadienone core and the tethered alkene. This disfavored steric interaction is particularly apparent in the case of substrate 15 which provided exclusively the 5,1-product 16 in 85% isolated yield.
Scheme 3.

Mechanistic Rationale for Selective Formation of 5,1-Bonded Cycloaddition Products
Based on these results, we surmised that the use of a tethered alkyne in the cycloaddition may provide a surrogate for the formation of the 1,3-bonded cycloaddition product. Examination of transition state models for dienone photorearrangement of substrate 41 (Scheme 4) shows that as a consequence of the linear geometry of the alkyne, cycloaddition to form 5,1- or 1,5-adducts is geometrically impossible.
Scheme 4.

Mechanistic Proposal for Selective Formation of 1,3-Bonded Cycloaddition Products
In order to test this hypothesis, we prepared alkyne-tethered cyclohexadienone substrate 4116 in analogous fashion to the alkene counterparts.13 Photoirradiation of substrate 41 led to the formation of the (3+2) cycloaddition product 42 as predicted by our hypothetical transition state model (Scheme 4). Although adduct 42 was cleanly formed during the photochemical reaction as observed by 1H-NMR analysis of the crude reaction mixture, the product proved to be unstable to attempted chromatographic purification and other isolation attempts (Scheme 5). This instability can be explained by the strained nature of the system which was also confirmed by DFT calculations (B3LYP/6-31g**) predicting torsion angles for strained alkene 42 up to45.9° out of the plane (χC’ = 45.9° and χC” = 10.8°, see Scheme 5).17, 18 We were able to leverage this high energy intermediate in cycloaddition chemistry by successfully trapping the alkene in an intramolecular (4+2) cycloaddition with furan providing a 3:2 mixture of regioisomers (43:44) in 70% isolated yield (Scheme 5).19, 20 Although the two diastereomeric cycloadducts were inseparable, the major isomer could be assigned on the basis of coupling constant analysis (43: Ha, singlet, calculated dihedral angle Ha:Hb = 87.5° and 44: Ha’, doublet, J = 4.9 Hz, calculated dihedral angle Ha’:Hb’ = 39.7°).21 Intramolecular dipolar cycloaddition with in situ22-generated benzonitrile oxide afforded a 5.5:1 mixture of tetracyclic regioisomers 45:46.
Scheme 5.

Trapping of Unstable, Strained Alkene 42 by Cycloaddition with Furan or a Nitrile Oxide
Inspired by the successful intermolecular transformations utilizing strained alkene intermediate 42, we envisioned that the use of alkynol ether substrates tethered with furfuryl ethers (Scheme 6, 48–49) would lead to the formation of highly complex, polycyclic structures. Oxidative dearomatization of trimethylphenol with 1,4-butynediol using PIDA followed by etherification of the resulting alkynol 47 provided the substituted dienone substrates 48–49 in two steps. Photoirradiation of 48–49 for 20 min. cleanly afforded polycyclic compounds 50–51 in good isolated yield as single diastereomers. The structural assignment of 51 was confirmed by X-ray crystallographic analysis (Figure 1).13 It is noteworthy that four new bonds are formed in the tandem DPC/(4+2) sequence which produces a highly complex architecture containing six rings and seven stereogenic centers.
Scheme 6.

Tandem DPC / Intramolecular (4+2)-Cycloaddition to Afford Complex Polycyclic Chemotypes
Figure 1.

X-ray crystal structure of polycyclic compound 50.13
Conclusion
In summary, we have employed a dienone photorearrangement cycloaddition (DPC) sequence for the synthesis of highly functionalized, bridged tri- and tetracyclic frameworks. Photoirradiation of alkenyl ether-tethered cyclohexadienones led to the favored production of 5,1-bonded (5+2) cycloaddition products. Alternatively, use of alkynyl ether-tethered substrates led to (3+2) cycloaddition to afford strained alkenes which could be further elaborated by both inter- and intramolecular cycloaddition chemistry to produce complex polycyclic adducts. Further studies on reactions of strained alkenes produced by DPC as well as biological studies of the chemotypes and derived adducts are currently in progress and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the NIH-NIGMS (P50 GM067041) is gratefully acknowledged. We thank Prof. Aaron Beeler (Boston University) for assistance with photochemistry equipment, Drs. Kiel Lazarski (Boston University) and Alexander Grenning (Boston University) for helpful discussions, and Dr. Jeffrey Bacon (Boston University) for X-ray crystal structure analysis. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the NSF.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures and spectral data including 1H NMR, 13C NMR, and HRMS for all new compounds and X-ray structure analysis of compounds 12 and 50. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest
REFERENCES
- 1.Maskill KG, Knowles JP, Elliott LD, Alder RW, Booker-Milburn KI. Angew. Chem. Int. Ed. 2013;52:1499. doi: 10.1002/anie.201208892. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on the use of photochemistry in natural product synthesis, see: Iriondo-Alberdi J, Greaney MF. Eur. J. Org. Chem. 2007:4801. Ciana C, Bochet CG. Chimia. 2007;61:650. Hoffmann N. Chem. Rev. 2008;108:1052. doi: 10.1021/cr0680336. Hoffmann N, Gramain J, Bouas-Laurent H. Actual Chim. 2008;317:6. Bach T, Hehn JP. Angew. Chem. Int. Ed. 2011;50:1000. doi: 10.1002/anie.201002845.
- 3.Coyle EE, Oelgemöller M. Photochem. Photobiol. Sci. 2008;7:1313. doi: 10.1039/b808778d. [DOI] [PubMed] [Google Scholar]
- 4.a) Goodell JR, Poole JL, Beeler AB, Aubé J, Porco JA., Jr J. Org. Chem. 2011;76:9792. doi: 10.1021/jo2018922. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tucker JW, Stephenson CRJ. Org. Lett. 2011;13:5468. doi: 10.1021/ol202178t. [DOI] [PubMed] [Google Scholar]; c) Scully SS, Porco JA., Jr Org. Lett. 2012;14:2646. doi: 10.1021/ol3010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Xia B, Gerard B, Solano DM, Wan J, Jones G, II, Porco JA., Jr Org. Lett. 2011;13:1346. doi: 10.1021/ol200032f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yavorskyy A, Shvydkiv O, Hoffmann N, Nolan K, Oelgemöler M. Org. Lett. 2012;14:4342. doi: 10.1021/ol301773r. [DOI] [PubMed] [Google Scholar]
- 6.For select, recent examples of photochemical transformations conducted on flow, see: Hook BDA, Dohle W, Hirst PR, Pickworth M, Berry MB, Booker-Milburn KI. J. Org. Chem. 2005;70:7558. doi: 10.1021/jo050705p. Goodell JR, McMullen JP, Zaborenko N, Maloney JR, Ho C–H, Jensen KF, Porco JA, Jr, Beeler AB. J. Org. Chem. 2009;74:6169. doi: 10.1021/jo901073v. Shvydkiv O, Yavorskyy A, Nolan K, Youssef A, Riquet E, Hoffmann N, Oelgemöller M. Photochem. Photobiol. Sci. 2010;9:1601. doi: 10.1039/c0pp00223b. Fukuyama T, Kajihara Y, Hino Y, Ryu I. J. Flow Chem. 2011;13:5008. Gutierrez AC, Jamison TF. J. Flow Chem. 2011;1:24. Gutierrez AC, Jamison TF. Org. Lett. 2011;13:6414. doi: 10.1021/ol2027015. Pimparkar K, Yen B, Goodell JR, Martin VI, Lee W–H, Porco JA, Jr, Beeler AB, Jensen KF. J. Flow Chem. 2011;2:53. Tucker JW, Zhang Y, Jamison TF, Stephenson CRJ. Angew. Chem. Int. Ed. 2012;51:4144. doi: 10.1002/anie.201200961. Nguyen JD, Reiβ B, Dai C, Stephenson CRJ. Chem. Commun. 2013;19:4352. doi: 10.1039/c2cc37206a.
- 7.For early studies on the dienone photorearrangement process, see: Barton DHR, De Mayo P, Shafiq M. J. Chem. Soc. 1957:929. Arigoni D, Bosshard H, Bruderer H, Büchi G, Jeger O, Krebaum LJ. Helv. Chim. Acta. 1957;40:1732. Barton DHR, De Mayo P, Shafiq M. J. Chem. Soc. 1958:3314. Barton DHR, De Mayo P, Shafiq M. J. Chem. Soc. 1958:140. Piers E, Cheng KF. Can. J. Chem. 1970;48:2234. Zimmerman HE, Jones G. J. Am. Chem. Soc. 1970;92:2753. Caine D, Tuller FN. J. Org. Chem. 1973;38:3663. Cowan DO, Drisko RL. Elements of Organic Photochemistry. New York: Plenum press; 1976. pp. 307–336.
- 8.a) Mann J. Tetrahedron. 1986;42:4611. [Google Scholar]; b) Cha JK, Oh J. Curr. Org. Chem. 1998;2:217. [Google Scholar]; c) Harmata M. Chem. Commun. 2010;46:8886. doi: 10.1039/c0cc03620j. [DOI] [PubMed] [Google Scholar]; d) Lohse AG, Hsung RP. Chem. Eur. J. 2011;17:3812. doi: 10.1002/chem.201100260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz AG, Puig S, Wang Y. J. Chem. Soc. Chem. Commun. 1985:785. [Google Scholar]
- 10.Schultz AG, Reilly J. J. Am. Chem. Soc. 1992;114:5068. [Google Scholar]
- 11.a) Schultz AG, Lavieri FP, Macielag M, Plummer M. J. Am. Chem. Soc. 1987;109:3991. [Google Scholar]; b) Schultz AG, Plummer M. J. Org. Chem. 1989;54:2112. [Google Scholar]; c) Schultz AG, Lockwood LO., Jr J. Org. Chem. 2000;65:6354. doi: 10.1021/jo000209v. [DOI] [PubMed] [Google Scholar]
- 12.For reviews on oxidative dearomatization, see: Quideau S, Pouységu L, Deffieux D. Synlett. 2008:467. Pouységu L, Deffieux D, Quideau S. Tetrahedron. 2010;66:2235. Pouységu L, Sylla T, Garnier T, Rojas LB, Charris J, Deffieux D, Quideau S. Tetrahedron. 2010;66:5908. Roche SP, Porco JA., Jr Angew. Chem. Int. Ed. 2011;50:4068. doi: 10.1002/anie.201006017. Zhuo C–X, Zheng W, You S–L. Angew. Chem. Int. Ed. 2011;51:12662. doi: 10.1002/anie.201204822.
- 13.See the Supporting Information for complete experimental details.
- 14.a) Huisgen R. Acc. Chem. Res. 1977;10:117. [Google Scholar]; b) Huisgen R. Pure Appl. Chem. 1980;52:2283. [Google Scholar]; c) Brückner R, Huisgen R. Tetrahedron Lett. 1990;31:2557. [Google Scholar]; d) Brückner R, Huisgen R. Tetrahedron Lett. 1990;31:2561. [Google Scholar]
- 15.a) Schuster DI, Abraitys VY. J. Chem. Soc. D. 1969:419. [Google Scholar]; b) Schuster DI. Acc. Chem. Res. 1978;11:65. [Google Scholar]; c) Samuel CJ. J. Chem Soc. Perkin Trans. 1981;2:736. [Google Scholar]
- 16.For metal-catalyzed reactions of alkyne-tethered cyclohexadienones, see: Tello-Aburto R, Harned AM. Org. Lett. 2009;11:3998. doi: 10.1021/ol901642w. Cai S, Liu Z, Zhang W, Zhao X, Wang DZ. Angew. Chem. Int. Ed. 2011;50:11133. doi: 10.1002/anie.201104028.
- 17.For a review on the chemistry of strained/pyrimidalized alkenes, see: Vásquez S, Camps P. Tetrahedron. 2005;61:5147.
- 18.Winkler FK, Dunitz JD. J. Mol. Biol. 1971:59. doi: 10.1016/0022-2836(71)90419-0. [DOI] [PubMed] [Google Scholar]
- 19.For a review on Diels-Alder chemistry of furans, see: Kappe CO, Murphree SS, Padwa A. Tetrahedron. 1997;53:14179.
- 20.For a review on strained alkenes in natural product synthesis, see: Wilson MR, Taylor RE. Angew. Chem. Int. Ed. 2013;52:4078. doi: 10.1002/anie.201207712.
- 21.Dihedral angles were calculated using Schrödinger Maestro, Version 9 at the B3LYP/6-31g** level of theory.
- 22.Gutsmiedl K, Wirges CT, Ehmke V, Carell T. Org. Lett. 2009;11:2405. doi: 10.1021/ol9005322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.