


Abstract
Searching for Darwinian selection in natural populations has been the focus of a multitude of studies over the last decades. Here we present the 1000 Genomes Selection Browser 1.0 (http://hsb.upf.edu) as a resource for signatures of recent natural selection in modern humans. We have implemented and applied a large number of neutrality tests as well as summary statistics informative for the action of selection such as Tajima’s D, CLR, Fay and Wu’s H, Fu and Li’s F* and D*, XPEHH, ΔiHH, iHS, FST, ΔDAF and XPCLR among others to low coverage sequencing data from the 1000 genomes project (Phase 1; release April 2012). We have implemented a publicly available genome-wide browser to communicate the results from three different populations of West African, Northern European and East Asian ancestry (YRI, CEU, CHB). Information is provided in UCSC-style format to facilitate the integration with the rich UCSC browser tracks and an access page is provided with instructions and for convenient visualization. We believe that this expandable resource will facilitate the interpretation of signals of selection on different temporal, geographical and genomic scales.
INTRODUCTION
Initiatives such as the 1000 Genomes Project (1,2) are generating resequencing data from world-wide human populations on a genome-wide scale. Resequencing data constitutes a major leap for population genomic analysis due to its higher information density and limited SNP ascertainment bias compared to genotyping data. Therefore such data is appropriate to calculate summary statistics that are based on the site frequency spectrum like CLR or Tajima’s D. Using the neutral evolutionary model as a null hypothesis, diverse statistics can be applied to genetic data to identify deviations from neutrality (Table 1). These statistical tests show varying degrees of robustness to demographic events (e.g. population bottlenecks and expansions) and sensitivity to different types of selection (e.g. positive, purifying or balancing). For instance, population bottlenecks, can lead to footprints that are similar to those caused by positive selection (21). Therefore, outlier approaches, which are commonly used to identify non-neutral loci in the extremes of a genome-wide distribution, are likely to contain a number of false positives in their extremes. Likewise, a number of false negatives, hence misidentified truly selected loci, are expected in a grey zone near the (arbitrary) outlier threshold (22). Outlier approaches in genome scans have proven powerful, but certainly they should be interpreted carefully in order to avoid storytelling (23). Even more, a profound understanding of adaptive evolution requires the integration of biological function (24) and if possible, validation on an experimental basis (25). Molecular network approaches can also give a functional context to the specific genes under adaptive selection (26,27). In all studies, care should be taken in communicating putative loci under selection to the public in order to avoid racist misinterpretation (28). Despite of these limitations and the fact that complete selective sweeps may not be extremely widespread in humans (29), a large number of regions under strong positive selection can be expected in the genome (30).
Table 1.
List of available summary statistics
| Method family | Method | Reference | Window size | Rank scores tail |
|---|---|---|---|---|
| Allele frequency spectrum | Tajima’s D | Tajima (3) | 30 kb | Lower |
| CLR | Nielsen et al. (4) | Variable size | Upper | |
| Fay and Wu’s H | Fay and Wu (5) | 30 kb | Lower | |
| Fu and Li’s F* | Fu and Li (6) | 30 kb | Lower | |
| Fu and Li’s D* | Fu and Li (6) | 30 kb | Lower | |
| R2 | Ramos-Onsins and Rozas (7) | 30 kb | Lower | |
| Linkage disequilibrium structure | XP-EHH | modified from Sabeti et al. (8) | SNP-specific | Upper |
| AiHH | modified from Voight et al. (9) | SNP-specific | Upper | |
| his | modified from Voight et al. (9) | SNP-specific | Upper | |
| EHH_average | modified from Sabeti et al. (10) | 30 kb | Upper | |
| EHH_max | modified from Sabeti et al. (10) | 30 kb | Upper | |
| Wall’s B | Wall (11) | 30 kb | Upper | |
| Wall’s Q | Wall (12) | 30 kb | Upper | |
| Fu’s F | Fu (13) | 30 kb | Lower | |
| Dh | Nei (14) | 30 kb | Upper | |
| Za | Rozas et al. (15) | 30 kb | Upper | |
| ZnS | Kelly (16) | 30 kb | Upper | |
| ZZ | Rozas et al. (15) | 30 kb | Upper | |
| Population differentiation | Fst (global and pairwise) | Weir and Cockerham (17) | SNP-specific | Upper |
| ΔDAF (standard and absolute) | Hofer et al. (18) | SNP-specific | Upper | |
| XP-CLR | Chen et al. (19) | 0.1 cM (maximum window) | Upper | |
| Descriptive statistics | Segregating sites | 30 kb | NA | |
| Singletons | 30 kb | NA | ||
| pi (nucleotide diversity) | Nei and Li (20) | 30 kb | NA | |
| DAF (derived allele frequency) | SNP-specific | NA | ||
| MAF (minor allele frequency) | SNP-specific | NA |
DESCRIPTION OF APPLIED STATISTICAL TESTS
Due to linkage, neutral alleles in the surrounding region hitchhike with the selected allele. Maynard Smith and Haigh (31) described this process of genetic hitchhiking and the so-called selective sweep. More recent studies showed that genetic hitchhiking generates distinct polymorphism signatures on the genome such as: (i) reduction of polymorphism level and excess of low- and high-frequency derived variants (32), (ii) spatial patterns of linkage-disequilibrium (33) and (iii) increased genetic differentiation among populations (34). Taking advantage of these three theoretical expectations, several methods to detect positive selection have been developed in the last two decades. This makes reference to the fact that no single statistic is enough to describe selection under various demographic models and modes of selection (22).
Here, we implemented a large number of statistical tests (Table 1) in order to allow for a more comprehensive analysis of natural selection, especially, positive selection. In brief, we have assigned the statistical tests to different method families (Table 1). Within the first family which is based on the allele frequency spectrum, Tajima’s D (3) is a classical neutrality test that compares estimates of the number of segregating sites and the mean pair-wise difference between sequences. CLR is a multi-locus, composite likelihood ratio test (4,35). Fay and Wu’s H (5) uses another facet of the site-frequency spectrum, by comparing the number of derived segregating sites at high frequencies to the number of variants at intermediate frequencies. Fu and Li’s F* compares the number of singletons to the mean pair-wise difference between sequences and Fu and Li’s D* compares it to the total number of nucleotide variants in a genomic region (6). R2 (7) is a statistical test for detecting population growth based on the comparison of the difference between the number of singletons per sequence and the average number of nucleotide differences.
Among the linkage disequilibrium structure methods, XP-EHH (8) is a cross-population test based on extended haplotype homozygosity (EHH). ΔiHH considers the difference between the integrated haplotype homozygosity scores for each allele in a single population while iHS (9) is defined as their log ratio. EHH average and EHH maximum (36); modified from (10) are based on the extended haplotype homozygosity. Wall’s B (11) counts the number of pairs of adjacent segregating sites that are congruent (if the subset of the data consisting of the two sites contains only two different haplotypes), while Wall’s Q (12) adds the number of partitions (two disjoint subsets whose union is the set of individuals in the sample) induced by congruent pairs to Wall’s B. Fu’s F (13) takes into account the haplotype diversity in the sample. Dh (14) is a summary statistic based on the number of different haplotypes in the sample.
The third family of methods is based on population differentiation. FST (37); calculated following the diploid method in Weir 1996 (p. 178) and ΔDAF (18) are estimates of population differentiation based on derived allele frequencies. XP-CLR (19) is a multi-locus allele-frequency-differentiation statistic between two populations. Additional statistics like segregating sites per 30-kb window and the nucleotide diversity and others (Table 1) are listed as descriptive statistics. A thorough description of the tests is given in the original literature (see Table 1) and in diverse excellent reviews on the topic (38,39).
COMPUTATIONAL FRAMEWORK AND DESCRIPTION OF 1000 GENOMES SOURCE DATA
A framework to calculate diverse summary statistics (Table 1) from 1000 genomes data was developed (Figure 1). A detailed description of how the statistics were implemented is given (Supplementary Material). A genome-wide overview of the results stored in the database for selected summary statistics is given (Supplementary Table S1). As described in the 1000 genomes Phase 1 paper (1), the quality of the 1000 genomes low coverage data has improved considerably over the pilot phase (2), but a number of limitations need to be kept in mind for population genomic analysis: (i) singletons and other rare variants are still underrepresented, (ii) the accessibility of the genome with the used short-read-sequencing technologies ∼94% and (iii) the reported phasing switch error every 250 kb (median, Supplementary Figure S5 in (1)) likely underestimates the length of long-shared haplotypes expected to occur around recent selective sweeps. Despite of these drawbacks which are mainly due to the nature of the low coverage approach, the short-read technology and differences in read depth (40), this dataset has important advantages over genotyping data, most importantly (i) a higher SNP density, (ii) the overcoming of ascertainment bias and (iii) a larger number of individuals per population, when compared to previous datasets (HapMap II and HGDP). We used phased data from the CEU, the CHB and the YRI populations from the integrated Phase 1 variant set (April 2012), with 97, 85 and 88 individuals, respectively. From the input vcf (variant call format) file we extracted exclusively the low-coverage VSQR SNP calls in order to avoid any bias that might result from differences between low-coverage calls and high-coverage exome SNP calls. Indels were not used. Ancestral states in this data set were identified using a 4-way alignment of humans, chimp, orangutan and rhesus macaque, provided by the 1000 genomes consortium (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/analysis_results/supporting/ancestral_alignments/).
Figure 1.
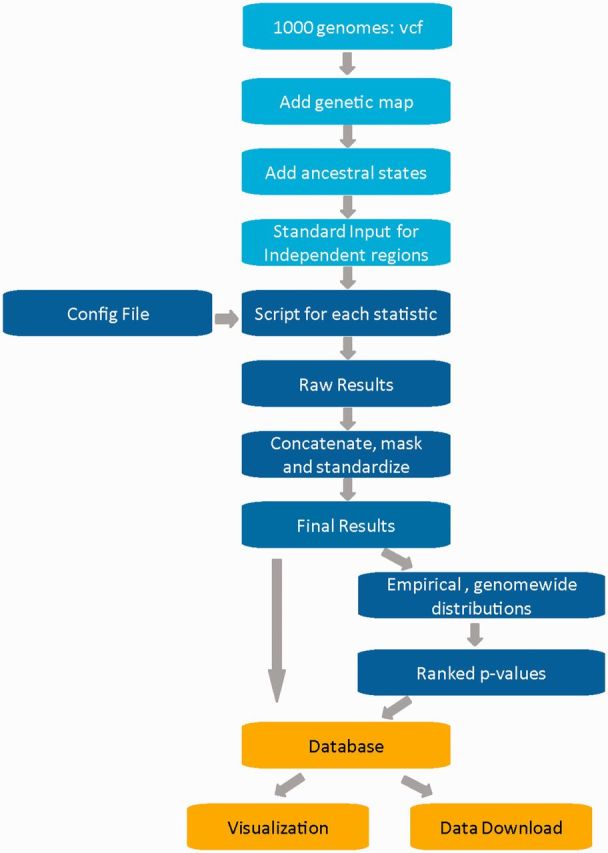
Schematic workflow developed in order to calculate diverse genome-wide summary statistics informative for the action of selection and to build a database in order to share and visualize the results.
AVAILABILITY OF DATABASE
All data is available via our entry page: http://hsb.upf.edu. A search mask gives the user easy access to the results for a specific gene or a genomic region of choice. The ‘submit’ button leads the user to a UCSC-style genome browser (http://pgb.ibe.upf.edu/) which is a custom installation of the UCSC Genome Browser (41,42). This UCSC Genome Browser installment allows for a visual inspection of the data, and for an integration of our data with many other available datasets. The raw scores of the tracks can be conveniently downloaded using the UCSC Table function (43) and is integrated with the Galaxy platform (galaxyproject.org). Using the ‘configure’ function on the browser page, the tracks can be further customized and using ‘right click’ the visualized genomic regions can be downloaded as a picture in .png format. For every statistical test, we provide two tracks, one for the raw scores and one for ranked scores. The purpose of the rank score tracks is to provide a comparison to the rest of the genome. Conveniently, the rank scores are presented in such a way that they present a peak (instead of a valley) in regions under positive selection. They are calculated using an outlier approach (22,44) by sorting all the scores genome-wide and determining the −log10 of the rank divided by the number of values in the distribution, taking the upper tail for most of the tests, or the lower tail for Tajima’s D, Fay and Wu’s H, Fu and Li’s F and D, R2 and Fu’s F (see Table 1 and a more detailed description on the entry page). The main purpose of the entry page is to provide a channel of communication with users, following the guidelines in (45). It serves as a platform for updates, questions and feedback (46). Therefore the page also provides documentation on the tracks and on the tests implemented as well as a FAQ and a feedback section.
EXAMPLE APPLICATIONS
First, we exemplify the use of the database by extracting results for a number of established loci under selection: EDAR (47), LCT (46), SLC45A2 (48), CD36 (49), HERC2 (50), SLC24A5 (51), CD5 (52) and APOL1 (53). A loci-specific summary of statistical tests is given (Supplementary Table S2). Interestingly, for any given locus, only a subset of statistical tests shows an extreme outlier score. This is consistent with differences in the architecture of selective sweeps. iHS scores near to certain very pronounced selective sweeps (e.g. LCT and SLC24A5) failed to compute due to inherent properties of the statistics, because either (i) the selected haplotype was near fixation or (ii) the EHH did not drop below the defined threshold in a given window. Examples for both positive (SLC45A2) and balancing (HLA region) selection are visualized in Figure 2. As expected, Tajima’s D scores around HLA (54) as well as the ABO locus (55) (data not shown) were pronouncedly elevated in all three analyzed populations, a pattern which is compatible with the action of balancing selection.
Figure 2.
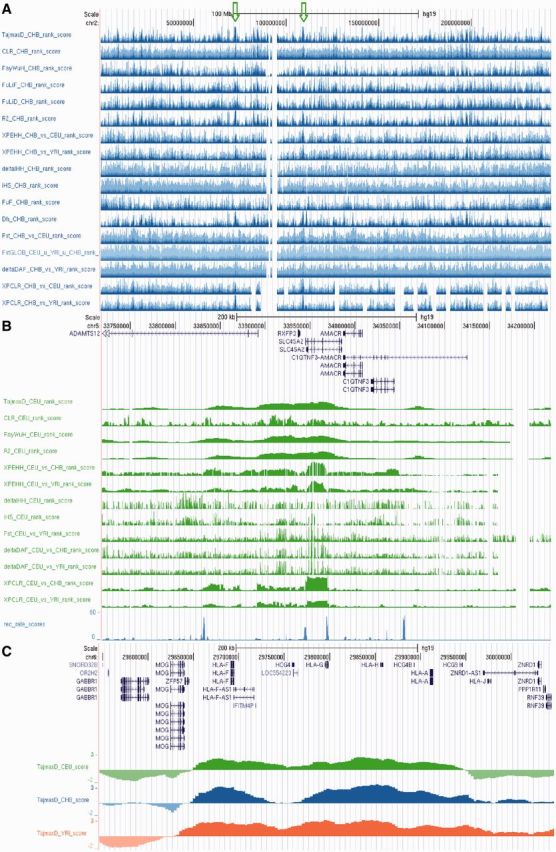
Examples of genomic regions under selection in the 1000 genomes selection browser. Tracks of statistics from different populations are visualized in colour (CEU in green, CHB in red and YRI in blue). Additional examples are given at http://hsb.upf.edu (A) The p- and q-arms of chromosome 2 (−log10 of empirically ranked scores). Recurrent peaks at around 72.5 Mb (left green arrow) and 109.5 Mb (right green arrow) indicate the loci CYP26B1/EXOC6B and EDAR, respectively. (B) Signature of positive selection around SLC45A2, another established skin colour gene, in the CEU population (0.5-Mb window; −log10 of empirically ranked scores). (C) Widespread balancing selection in the HLA region indicated by strongly positive scores for Tajima’s D in all three analysed human populations (0.5-Mb window).
COMPARISON TO OTHER WEB RESOURCES
As for positive selection based on between-species comparisons, the Selectome database (http://bioinfo.unil.ch/selectome/; (56)) presents results based on the dN/dS method using a branch-site specific likelihood test. As for recent natural selection within modern humans, a number of web resources are available. For previous datasets, e.g. the HapMap 2 and HGDP projects, several positive selection statistics are available in form of the haplotter tool (http://haplotter.uchicago.edu/; (24)) and in form of the HGDP selection browser (http://hgdp.uchicago.edu/; (57)). For the 1000 genomes project data, the online tool ENGINES (http://spsmart.cesga.es; (58)) is useful for the analysis of allele frequencies and a recent study presented a method to calculate corrected summary statistics from low coverage sequencing data (40). dbPSHP (http://jjwanglab.org/dbpshp) offers a large number of statistical tests in a SNP-specific manner for HapMap 3 and 1000 genomes datasets. Complementary to these databases, our database gives a large number of region- and SNP-specific scores (depending on the test statistic) based on resequencing data (1000 genomes Phase 1), with a special focus on genome-wide significance (by the ranked scores) and the visualization of several statistics in parallel (Figure 2).
CONCLUSIONS
By applying a large number of summary statistics to data from the 1000 genomes project, we have built a timely and expandable resource for the population genomics research community. An associated user-friendly genome browser gives a visual impression of the genetic variation in a genomic region of interest and offers functionality for an array of down-stream analyses. While this resource will not replace a thorough, case by case analysis of selection, we expect that it will prove useful for the research community through the large number of test statistics and the fine-grained character of resequencing data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thankfully acknowledge contributions from Anna Ramírez-Soriano, Arcadi Navarro, Francesc Calafell, Elena Bosch, Chris Tyler-Smith, Gonçalo Abecasis, Roger Bartomeus Peñalver and the 1000 Genomes Project (1000genomes.org). The authors also thank Txema Heredia and the National Institute of Bioinformatics (http://www.inab.org) for computational support.
FUNDING
Ministerio de Ciencia y Tecnología (Spain); Direcció General de Recerca, Generalitat de Catalunya (Grup de Recerca Consolidat 2009 SGR 1101); Subprogram BMC [BFU2010-19443 awarded to J.B.]; Post-doctoral scholarship from the Volkswagenstiftung [Az: I/85 198 to J.E.]; Spanish government [BFU-2008-01046; SAF2011-29239]; The Spanish government FPI scholarships [BES-2009-017731 and BES-2011-04502 to G.M.D. and M.P., respectively]; PhD fellowship from ‘Acción Estratégica de Salud, en el marco del Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica 2008-2011’ from Instituto de Salud Carlos III (to P.L.). Funding for open access charge: Prof. Jaume Bertranpetit.
Conflict of interest statement. None declared.
REFERENCES
- 1.The 1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002;19:2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- 8.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald JG, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 11.Wall JD. Recombination and the power of statistical tests of neutrality. Genet. Res. 1999;74:65–79. [Google Scholar]
- 12.Wall JD. A comparison of estimators of the population recombination rate. Mol. Biol. Evol. 2000;17:156–163. doi: 10.1093/oxfordjournals.molbev.a026228. [DOI] [PubMed] [Google Scholar]
- 13.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nei M. Molecular Evolutionary Genetics. New York, NY: Columbia University Press; 1987. [Google Scholar]
- 15.Rozas J, Gullaud M, Blandin G, Aguadé M. DNA variation at the rp49 gene region of Drosophila simulans: evolutionary inferences from an unusual haplotype structure. Genetics. 2001;158:1147–1155. doi: 10.1093/genetics/158.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly JK. A test of neutrality based on interlocus associations. Genetics. 1997;146:1197–1206. doi: 10.1093/genetics/146.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 18.Hofer T, Ray N, Wegmann D, Excoffier L. Large allele frequency differences between human continental groups are more likely to have occurred by drift during range expansions than by selection. Ann. Hum. Genet. 2009;73:95–108. doi: 10.1111/j.1469-1809.2008.00489.x. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20:393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barton NH. The effect of hitch-hiking on neutral genealogies. Genet. Res. 1998;72:123–133. [Google Scholar]
- 22.Akey JM. Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 2009;19:711–722. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol. Biol. Evol. 2012;29:3237–3248. doi: 10.1093/molbev/mss136. [DOI] [PubMed] [Google Scholar]
- 24.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 25.Barrett RDH, Hoekstra HE. Molecular spandrels: tests of adaptation at the genetic level. Nat. Rev. Genet. 2011;12:767–780. doi: 10.1038/nrg3015. [DOI] [PubMed] [Google Scholar]
- 26.Dall’olio GM, Laayouni H, Luisi P, Sikora M, Montanucci L, Bertranpetit J. Distribution of events of positive selection and population differentiation in a metabolic pathway: the case of asparagine N-glycosylation. BMC Evol. Biol. 2012;12:98. doi: 10.1186/1471-2148-12-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luisi P, Alvarez-Ponce D, Dall’olio GM, Sikora M, Bertranpetit J, Laayouni H. Network-level and population genetics analysis of the insulin/TOR signal transduction pathway across human populations. Mol. Biol. Evol. 2012;29:1–40. doi: 10.1093/molbev/msr298. [DOI] [PubMed] [Google Scholar]
- 28.Vitti JJ, Cho MK, Tishkoff SA, Sabeti PC. Human evolutionary genomics: ethical and interpretive issues. Trends Genet. 2012;28:137–145. doi: 10.1016/j.tig.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, Sella G, Przeworski M. Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, Park DJ, Griesemer D, Karlsson EK, Wong SH, et al. Identifying recent adaptations in large-scale genomic data. Cell. 2013;152:703–713. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 32.Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Nielsen R. Linkage disequilibrium as a signature of selective sweeps. Genetics. 2004;167:1513–1524. doi: 10.1534/genetics.103.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 35.Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramírez-Soriano A, Ramos-Onsins SE, Rozas J, Calafell F, Navarro A. Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics. 2008;179:555–567. doi: 10.1534/genetics.107.083006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weir BS, Hill WG. Estimating F-statistics. Annu. Rev. Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen R. Population genetic analysis of ascertained SNP data. Hum. Genomics. 2004;1:218–224. doi: 10.1186/1479-7364-1-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat. Rev. Genet. 2003;4:99–111. doi: 10.1038/nrg999. [DOI] [PubMed] [Google Scholar]
- 40.Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinform. 2013;14:289. doi: 10.1186/1471-2105-14-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler AD. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Res. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dall’Olio GM, Marino J, Schubert M, Keys KL, Stefan MI, Gillespie CS, Poulain P, Shameer K, Sugar R, Invergo BM, et al. Ten simple rules for getting help from online scientific communities. PLoS Comput. Biol. 2011;7:e1002202. doi: 10.1371/journal.pcbi.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Järvelä I. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 47.Bryk J, Hardouin E, Pugach I, Hughes D, Strotmann R, Stoneking M, Myles S. Positive selection in East Asians for an EDAR allele that enhances NF-kappaB activation. PLoS One. 2008;3:e2209. doi: 10.1371/journal.pone.0002209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Branicki W, Brudnik U, Draus-Barini J, Kupiec T, Wojas-Pelc A. Association of the SLC45A2 gene with physiological human hair colour variation. J. Hum. Genet. 2008;53:966–971. doi: 10.1007/s10038-008-0338-3. [DOI] [PubMed] [Google Scholar]
- 49.Fry AE, Ghansa A, Small KS, Palma A, Auburn S, Diakite M, Green A, Campino S, Teo YY, Clark TG, et al. Positive selection of a CD36 nonsense variant in sub-Saharan Africa, but no association with severe malaria phenotypes. Hum. Mol. Genet. 2009;18:2683–2692. doi: 10.1093/hmg/ddp192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duffy DL, Montgomery GW, Chen W, Zhao ZZ, Le L, James MR, Hayward NK, Martin NG, Sturm RA. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am. J. Hum. Genet. 2007;80:241–252. doi: 10.1086/510885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamason RL, Mohideen M-APK, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 52.Carnero-Montoro E, Bonet L, Engelken J, Bielig T, Martínez-Florensa M, Lozano F, Bosch E. Evolutionary and functional evidence for positive selection at the human CD5 immune receptor gene. Mol. Biol. Evol. 2012;29:811–823. doi: 10.1093/molbev/msr251. [DOI] [PubMed] [Google Scholar]
- 53.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hedrick PW, Thomson G. Evidence for balancing selection at HLA. Genetics. 1983;104:449–456. doi: 10.1093/genetics/104.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calafell F, Roubinet F, Ramírez-Soriano A, Saitou N, Bertranpetit J, Blancher A. Evolutionary dynamics of the human ABO gene. Hum. Genet. 2008;124:123–135. doi: 10.1007/s00439-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 56.Proux E, Studer RA, Moretti S, Robinson-Rechavi M. Selectome: a database of positive selection. Nucleic Acids Res. 2009;37:D404–D407. doi: 10.1093/nar/gkn768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amigo J, Salas A, Phillips C. ENGINES: exploring single nucleotide variation in entire human genomes. BMC Bioinformatics. 2011;12:105. doi: 10.1186/1471-2105-12-105. [DOI] [PMC free article] [PubMed] [Google Scholar]