

Abstract
Bifenthrin, a relatively stable type I pyrethroid that causes tremors and impairs motor activity in rodents, is broadly used. We investigated whether nanomolar bifenthrin alters synchronous Ca2+ oscillations (SCOs) necessary for activity-dependent dendritic development. Primary mouse cortical neurons were cultured 8 or 9 days in vitro (DIV), loaded with the Ca2+ indicator Fluo-4, and imaged using a Fluorescence Imaging Plate Reader Tetra. Acute exposure to bifenthrin rapidly increased the frequency of SCOs by 2.7-fold (EC50 = 58 nM) and decreased SCO amplitude by 36%. Changes in SCO properties were independent of modifications in voltage-gated sodium channels since 100 nM bifenthrin had no effect on the whole-cell Na+ current, nor did it influence neuronal resting membrane potential. The L-type Ca2+ channel blocker nifedipine failed to ameliorate bifenthrin-triggered SCO activity. By contrast, the metabotropic glutamate receptor (mGluR)5 antagonist MPEP [2-methyl-6-(phenylethynyl)pyridine] normalized bifenthrin-triggered increase in SCO frequency without altering baseline SCO activity, indicating that bifenthrin amplifies mGluR5 signaling independent of Na+ channel modification. Competitive [AP-5; (−)-2-amino-5-phosphonopentanoic acid] and noncompetitive (dizocilpine, or MK-801 [(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate]) N-methyl-d-aspartate antagonists partially decreased both basal and bifenthrin-triggered SCO frequency increase. Bifenthrin-modified SCO rapidly enhanced the phosphorylation of cAMP response element–binding protein (CREB). Subacute (48 hours) exposure to bifenthrin commencing 2 DIV–enhanced neurite outgrowth and persistently increased SCO frequency and reduced SCO amplitude. Bifenthrin-stimulated neurite outgrowth and CREB phosphorylation were dependent on mGluR5 activity since MPEP normalized both responses. Collectively these data identify a new mechanism by which bifenthrin potently alters Ca2+ dynamics and Ca2+-dependent signaling in cortical neurons that have long term impacts on activity driven neuronal plasticity.
Introduction
Synthetic pyrethroid insecticides were introduced into widespread use for the control of insects and disease vectors more than three decades ago. Pyrethroids account for approximately one-fourth of the worldwide insecticide market, and the use of these compounds is increasing (Schleier and Peterson, 2011). Based on both their chemical structures and biologic responses to acute exposure, pyrethroids are classified into two major groups: type I and type II. Type I pyrethroids lack a cyano group at the α carbon of the 3-phenoxybenzyl alcohol moiety and produce hyperexcitation, tremors, and convulsions (T syndrome), whereas type II pyrethroids have a cyano group at the α carbon and produce hypersensitivity, choreoathetosis, salivation, and seizures (CS syndrome). A few pyrethroids producing both tremors and salivation were classified as type I/II (Soderlund, 2012; Casida and Durkin, 2013).
The primary molecular target underlying insecticidal activity and presumably mammalian neurotoxicity of pyrethroids are voltage-gated sodium channels (VGSCs) (Soderlund, 2012; Casida and Durkin, 2013). Pyrethroids enhance sodium channel activity by shifting activation to more negative membrane potentials as well as by slowing channel inactivation. This action results in altered neuronal excitability characterized by in vitro and in vivo changes in neuronal firing rates (e.g., repetitive firing or depolarizing block of the neuron). In addition to their actions on VGSCs, pyrethroids interact with other types of ion channels in a variety of in vitro systems, including altering the function of voltage-gated calcium (Ca2+) channels and GABAA receptors (Clark and Symington, 2012; Soderlund, 2012; Casida and Durkin, 2013). It should be noted that all these actions invariably require exposure to pyrethroid a concentrations ≥1 µM.
Bifenthrin (Fig. 1) is one of the most potent commercialized pyrethroids to date, with LD50 values of about 53 mg/kg in female rats and 70 mg/kg in male rats after oral gavage (Wolansky et al., 2006; Wolansky and Harrill, 2008). Bifenthrin, as low as 10 mg/kg, produces type I pyrethroid-like episodes of head and whole-body shakes, a strong and prolonged tremorigenic response, hyperthermia, vocalizations, prostration, and, at doses >28 mg/kg, it eventually triggers convulsions and death (Holton et al., 1997; Wolansky et al., 2007). This dose range corresponds to a brain concentration of bifenthrin of about 550–650 ng/g of brain tissue (Scollon et al., 2011). The high-dose effect is consistent with pyrethroid-induced perturbation of voltage-dependent Na+ and Ca2+ fluxes at micromolar concentrations in primary cultured cortical neurons (Cao et al., 2011a,b). However, at a threshold dose of 1.28 mg/kg, which corresponds to a brain concentration of roughly 30 ng/g of brain tissue, bifenthrin decreases motor activity in rats (Wolansky et al., 2006; Scollon et al., 2011). These data suggest that bifenthrin can alter central nervous system physiology at doses much lower than those needed to induce tremors.
Fig. 1.
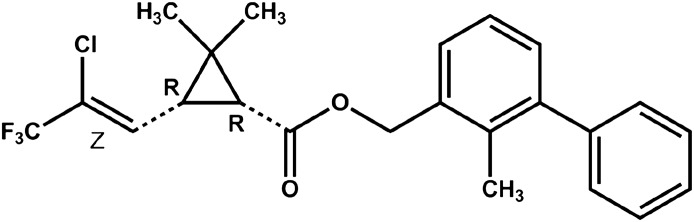
Chemical structure of bifenthrin.
Extensive studies have been performed to elucidate the molecular mechanism and cellular effect of pyrethroids in cultured neurons. Previously we have demonstrated that at concentrations >1 µM, 9 of 11 commercially used pyrethroids enhance Na+ and Ca2+ fluxes with distinct potencies and efficacies (Cao et al., 2011a,b) by a mechanism that is consistent with their reported actions on VGSC expressed in Xenopus oocytes (Choi and Soderlund, 2006). For example, in mouse cortical neuronal cultures, bifenthrin induces modest Na+ and Ca2+ influx at concentrations above 3 µM, which can be eliminated by tetrodotoxin pretreatment, demonstrating that bifenthrin is a low-efficacy sodium channel agonist. Pyrethroids have also been demonstrated to act on Ca2+ or Cl− channels (Clark and Symington, 2012; Soderlund, 2012); however, no direct evidence has been presented demonstrating that bifenthrin can interact with these channels. Furthermore, the possible effect of pyrethroids on neuronal network electrical activity has been investigated. Both deltamethrin (0.1 µM) and permethrin (0.1 µM) are able to increase electric spike activity in the absence of GABAergic inhibition (Meyer et al., 2008); however, bifenthrin appears without effect, even at a concentration of 50 µM (McConnell et al., 2012).
Murine primary-cultured neurons mature to form synaptic networks and display spontaneous synchronized Ca2+ oscillations (Cao et al., 2012a). These spontaneous Ca2+ oscillations are dependent on the generation of action potentials and can be manifested by multiple Ca2+ signaling pathways (Dravid and Murray, 2004). Synchronous Ca2+ oscillations (SCOs) are important in mediating neuronal development and activity-dependent dendritic growth (Dolmetsch et al., 1998; Wayman et al., 2006). Genetic or environmental factors that interfere with activity-dependent dendritic growth influence the functioning of neuronal networks (Pessah et al., 2010; Stamou et al., 2013).
In the present study, we identified a novel mechanism by which bifenthrin, at nanomolar concentrations, alters the patterns of SCOs in primary mouse cortical neuronal cultures by amplifying glutamatergic glutamate receptor 5 (mGluR5) activity, which alters CREB signaling and patterns of neurite outgrowth in the absence of detectable perturbation of VGSCs. To date, these are the most potent neurotoxic actions attributed to bifenthrin and suggest that bifenthrin at low concentrations relevant to current exposures can persistently alter patterns of SCO and activity-dependent neuronal cell growth and network development.
Materials and Methods
Bifenthrin (purity: 99.1%, mix of isomers) was from Chemical Service, Inc. (West Chester, PA) (Fig. 1). Anti–p-CREB, anti-CREB, and anti–microtubule-associated protein 2 antibodies were from Cell Signaling Technology (Danvers, MA). Odyssey Blocking Buffer and IRDye-labeled secondary antibody were from LI-COR Biotechnology (Lincoln, NE). Fluo-4 and Alexa Fluor 488–conjugated goat anti-rabbit secondary antibody were from Life Technologies (Grand Island, NY). Tetrodotoxin, D-AP-5 [D-(−)-2-amino-5-phosphonopentanoic acid], MK-801 [(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate], CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), MPEP [2-methyl-6-(phenylethynyl)pyridine], and nifedipine (3,5-dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate) were from Tocris Bioscience (Minneapolis, MN).
Cortical Neuronal Cultures.
The University of California Davis Animal Use and Care Committee approved this study, and animal experiments were conducted in accordance with the guidelines of the Animal Use and Care of the National Institutes of Health. Cortical neurons were dissociated from the cortex from C57Bl/6J mouse pups at postnatal day 0 to 1 as described previously (Cao et al., 2011b). The dissociated neurons were maintained in Neurobasal complete medium (Neurobasal medium supplemented with NS21 (Chen, et al., 2010), 0.5 mM l-glutamine, and HEPES) with 5% fetal bovine serum. The dissociated cortical cells were plated onto poly-l-lysine–coated (0.5 mg/ml; Sigma-Aldrich, St. Louis, MO) clear-bottom, black-wall, 96-well imaging plates (BD, Franklin Lakes, NJ) at densities of 1 × 105/well for Ca2+ imaging and 2000/well for immunocytochemistry, respectively. For the Western blot experiments, the dissociated cortical neurons were plated onto poly-l-lysine–coated six-well plates at a density of 3 × 106/well. The medium was changed twice a week by replacing half the volume of culture medium in the well with serum-free Neurobasal complete medium. The neurons were maintained at 37°C with 5% CO2 and 95% humidity.
Measurement of Synchronous Intracellular Ca2+ Oscillations.
Cortical neurons at 9 days in vitro (DIV) were used to investigate how acute exposure to bifenthrin alters synchronous Ca2+ oscillations as described previously (Cao et al., 2012a). Briefly, the neurons were loaded with Fluo-4 for 1 hour at 37°C in Locke’s buffer (in mM: 8.6 HEPES, 5.6 KCl, 154 NaCl, 5.6 glucose, 1.0 MgCl2, 2.3 CaCl2, and 0.0001 glycine, pH 7.4). After recording the baseline spontaneous Ca2+ oscillations for 3 minutes, vehicle or bifenthrin was added to the well using a programmable 96-channel pipetting robotic system, and the intracellular Ca2+ concentration ([Ca2+]i) was monitored for additional 50 minutes. The calculations for EC50 for changes in oscillatory frequency and peak transient amplitude were from data acquired between 30 and 40 minutes after the additions were made (i.e., averages from a 10-minute period). To test the mechanisms influencing SCO activity, receptor-targeted inhibitors were added to designated wells after the Ca2+ response was induced by bifenthrin (0.1 µM).
To test the chronic influences, exposures of neurons to bifenthrin (0.01–1.0 µM) commenced 24 hours after plating, and on DIV 6, the patterns of SCO measured as described already.
Membrane Potential Measurement.
A membrane-sensitive dye, FMPblue, was used to investigate whether bifenthrin can depolarize the membrane potential in neurons that were 8 or 9 DIV. Briefly, the neurons were loaded with 180 µl of FMPblue (1 vial diluted to 50 ml Lock’s; Molecular Devices, Sunnyvale, CA). After incubation with the dye for 30 minutes, the cells were excited at 510–545 nm, and the fluorescent signals emitted at 565–625 nm were recorded. After recording basal fluorescence for 1 minute, vehicle or bifenthrin (0.01–1.0 µM) was added to the wells, and the signals were recorded for an additional 40 minutes.
Measurement of Neuronal Na+, K+, and Ca2+ Channel Currents.
Cultured cortical neurons were voltage-clamped by whole-cell patch-clamp using an EPC-10 amplifier and Pulse software (HEKA; Lambrecht/Pfalz, Germany). Cells were bathed in extracellular solution containing (in mM) 160 NaCl, 4.5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES; pH was adjusted to 7.4 using NaOH (310 mOsm). Pipettes were pulled from 1.5-mm capillary tubing and filled with intracellular solution containing (in mM) either 145 KF, 2 MgCl2, 10 EGTA, and 10 HEPES (pH adjusted to 7.2 with KOH; 300 mOsm) or 145 CsF, 3 KCl, 2 NaCl, 1 MgCl2, 3 Na-ATP, 0.2 Na-GTP, 10 EGTA, and 10 HEPES (pH adjusted to 7.4 with KOH; 303 mOsm). Pipette-tip resistances were 2-4 MΩ. Series resistances of 3–10 MΩ were compensated 40–80%. All neurons were voltage-clamped to a holding potential of −80 mV. Data analysis, fitting, and plotting were performed with IGOR-Pro (Wavemetrics, Lake Oswego, OR) and Origin 9.0 (OriginLab, Northampton, MA). Na+ currents were elicited by 50-millisecond voltage steps from −80 to 0 mV applied every 10 seconds. Current decay and tail current were fitted with the following equation:where I is the current amplitude, t the time range of current chosen for fitting, τ1, the fast time constant of inactivation, and τ2 the slow time constant of inactivation. A1 and A2 are the percentages of current inactivating with the fast and slow time constants, respectively, and C is the noninactivating current component.
K+ currents were elicited by 200-millisecond voltage steps from −80 to 40 mV applied every 10 seconds. L929 cells stably expressing mKV3.1 and mKV1.1 were previously described (Grissmer et al., 1994). Experiments were performed with the same external and internal solutions as used for the cortical neurons.
Bifenthrin’s influence on ryanodine receptor type 1 (RyR1) isolated from rabbit skeletal muscle was assessed in two ways: [3H]ryanodine binding analysis and measurements of ionic current from single channels reconstituted in bilayer lipid membranes, as previously described (Pessah et al., 2010).
Immunocytochemistry and Neurite Outgrowth.
Twenty-four hours after plating, cortical neurons were exposed to concentrations of bifenthrin ranging from 0.01 to 1.0 µM for 48 hours. After a brief wash with phosphate-buffered saline (PBS), the cells were fixed with 4% paraformaldehyde for 20 minutes and then permeabilized with 0.25% Triton X-100 for 15 minutes. After blocking with PBS with 10% bovine serum albumin and 1% goat serum for 1 hour, cells were incubated with anti–microtubule-associated protein 2 (1:1000) antibody in PBS containing 1% goat serum overnight at 4°C. Cells were then incubated with Alexa Fluor 488–conjugated goat anti-rabbit secondary antibody (1:500) for 1 hour at room temperature. After aspiration of the secondary antibody, 0.2 mg/ml Hoechst 33342 was added to each well for 5 minutes to stain the nuclei. Pictures were recorded using an ImageXpress High Content Imaging System (Molecular Devices) using a 10× objective with both fluorescein isothiocyanate and 4′,6-diamidino-2-phenylindole (DAPI) filters. Nine adjacent sites (3 × 3), which cover ∼60% of the center surface area, were pictured for each well. Neurite outgrowth was then analyzed using MetaXpress Software (Molecular Devices). The cell body was selected based on both DAPI and fluorescein isothiocyanate staining by setting the maximal and minimal diameter, and the minimal fluorescence intensity relative to the background from both channels. The neurite was traced automatically by setting the maximal width and minimal fluorescence intensity above background. The same criteria were applied to analyze the entire plate. The analyzed data were saved to the core computer facility, and the neurons with neurites extending beyond the edge of the picture frame or neurite length shorter than 50 µm were excluded from analysis.
Western Blot.
The sample preparation for Western blot was performed as described previously (Cao et al., 2012b). An equal amount of sample protein (20 µg) was loaded onto the wells of 10% SDS-PAGE gels and transferred to a nitrocellulose membrane by electroblotting. The membranes were blocked with Odyssey Blocking Buffer (LI-COR Biotechnology) in PBS buffer + 0.1% Tween-20 for 1.5–2 hours at room temperature. After blocking, membranes were incubated overnight at 4°C in anti–p-CREB (1:1000; Cell Signaling Technology). The blots were washed and incubated with the IRDye (800CW)-labeled secondary antibody (1:10,000; LI-COR Biotechnology) for 1 hour at room temperature. After washing with 0.1% Tween in PBS five times, the membrane was scanned with the LI-COR Odyssey Infrared Imaging System (LI-COR Biotechnology). Densitometry was performed using the LI-COR Odyssey Infrared Imaging System application software 2.1. Membranes were stripped with NewBlot Nitro Stripping Buffer and reblotted for analysis of anti-CREB (1:1000; Cell Signaling Technology).
Data Analysis.
Graphing and statistical analysis were performed using GraphPad Prism software (Version 5.0; GraphPad Software Inc., San Diego, CA). EC50 value was determined by nonlinear regression using a three-parameter logistic equation. Statistical significance between different groups was calculated using Student’s t test or by an analysis of variance and, where appropriate, a Dunnett’s multiple comparison test; P values below 0.05 were considered statistically significant. EC50 value determination and statistics (analysis of variance) for the electrophysiological experiments were performed with Origin 9.0 (OriginLab, Northampton, MA).
Results
Acute Exposure to Nanomolar Bifenthrin Alters SCO Patterns in Cortical Neurons.
Bifenthrin was previously shown to induce modest Ca2+ influx at concentrations >3 µM (Cao et al., 2011a,b). These studies focused on net Ca2+ influx associated with neuronal excitotoxicity but not possible alternations of patterns of SCO that are important for activity dependent gene expression, neuronal cell growth, and plasticity. We therefore performed directed studies to investigate whether bifenthrin at submicromolar concentrations influences Ca2+ dynamics and their possible mechanisms. Primary murine cortical neurons grown in 96-well plates were loaded with Fluo-4 at 8 or 9 DIV, and cytoplasmic Ca2+ dynamics were monitored in real-time using FLIPR Tetra. Under these conditions, dissociated neuronal cultures form extensive networks that engage synchronized Ca2+ oscillations (Fig. 2A, top trace) essential for promoting dendritic complexity and activity-dependent plasticity (Chen et al., 2010; Cao et al., 2012a,b). The addition of bifenthrin (0.01–1 µM) produced a concentration-dependent increase in the frequency of SCO having an EC50 value of 57.7 nM [31.9–104 nM, 95% confidence interval (CI)] that attained 270% of baseline at 1 µM (Fig. 2, A, traces 2–8, and B). Coincident with the increasing frequency of SCO, bifenthrin partially decreased the mean amplitude of the Ca2+ oscillations reaching 64% of control at 1 µM (Fig. 2, A and C). The IC50 value for bifenthrin suppressing the mean amplitude of the Ca2+ oscillations was 83.4 nM (49.8–141.0 nM, 95% CI) (Fig. 2C). To test whether the bifenthrin response on SCO was reversible, the cells were completely washed five times with Lock’s buffer after exposure to 0.1 µM bifenthrin for 30 minutes. The washing the cells itself had no detectable influence on the basal SCO (Supplemental Fig. 1, upper black traces); however, the bifenthrin-augmented pattern of SCO was not fully reversible after washout, suggesting persistent alteration in the pattern of SCO after acute exposure to a low concentration of bifenthrin (Supplemental Fig. 1, lower red traces).
Fig. 2.

Bifenthrin-induced Ca2+ dysregulation in cortical neurons. (A) Representative traces showing how acute exposure to bifenthrin (0.01–1 µM) influences Ca2+ fluctuations in cortical neurons at 8 or 9 DIV as a function of time. Bifenthrin produces a robust increase in the frequency of Ca2+ oscillations accompanied by the slightly decreased Ca2+ oscillations amplitude. (B) Concentration-response relationships for bifenthrin-stimulated increase in the frequency of Ca2+ oscillation. The EC50 value for bifenthrin-stimulated increase in the frequency of Ca2+ oscillation frequency is 57.7 nM. (C) Concentration-response relationships for bifenthrin-stimulated decrease in the amplitude of Ca2+ oscillation. The EC50 value for bifenthrin-stimulated decrease in the amplitude of Ca2+ oscillation frequency is 83.4 nM. These data were averaged from a time period of between 30 and 40 minutes after the addition of bifenthrin. Each data point represents mean ± S.E.M. (n > 20 well) from five independent cultures. VEH/V, vehicle.
Nanomolar Bifenthrin Neither Alters Membrane Potential nor Voltage-Gated Currents.
The FMPblue dye was used to examine whether bifenthrin alters the resting membrane potential of cortical neurons. After recording basal FMPblue fluorescence for 1 minute, bifenthrin was added to the well and the signal was recorded for an additional 40 minutes. Bifenthrin, at a concentration that caused a nearly 2-fold increase in SCO frequency (0.1 µM), had no detectable influence on membrane potential (Fig. 3). Even at concentrations as high as 1 µM, bifenthrin had negligible effect on the membrane potential (data not shown). As a positive control, application of KCl (10 mM) to the external bath showed that the FMPblue dye was capable of reporting even a modest (14 mV, calculated from Goldman–Hodgkin–Katz equation) shift in membrane depolarization (Fig. 3).
Fig. 3.

Bifenthrin has no effect on membrane potential in cortical neurons. Influence of bifenthrin (0.1 µM) or KCl (10 mM, positive control) on the rest membrane potential as a function of time. KCl produces a significant depolarization of the cortical neurons reflected by increased FMPblue fluorescence signals. However, bifenthrin has no response on the resting membrane potential. This experiment was repeated twice each with five replicates with similar results. VEH, vehicle.
To explore more directly whether bifenthrin-induced changes in Ca2+ oscillations might be due to direct effects on voltage-gated sodium channels, we tested the effect of bifenthrin on Na+ currents in cultured cortical neurons using whole-cell patch-clamp. As shown in Fig. 4A, bifenthrin concentrations of 10 and 100 µM drastically slowed inactivation during depolarization, as evidenced by the broadening and incomplete inactivation of the Na+ current elicited by a 50-millisecond depolarizing step from −80 to 0 mV, whereas concentrations of 0.1 or 1 µM had no effect. Bifenthrin further delayed deactivation as shown by the large Na+ tail currents that became apparent after repolarization to −80 mV. This slow-tail current, which constitutes an additional component to the normal fast-tail current evoked on repolarization, was very sensitive to bifenthrin, and 0.1 µM already slightly changed the deactivation time constant τ2 of its biexponential decay (Fig. 4C). However, since the physiologic relevance of the tail current is not clear, we fitted separate EC50 values (Fig. 4B) for increasing the area under the current elicited by depolarization (16.8 ± 0.7 μM) and the area under the tail current (4.4 ± 0.4 µM), assuming the effect at 100 µM to be maximal. The more meaningful of these EC50 values representing the bifenthrin-induced delay in VGSC inactivation is 275-fold higher than the EC50 for increasing SCO frequency. We further tested bifenthrin on cortical neuron K+ currents as well as on heterologous expressed Kv1.1 and Kv3.1, two of the major delayed-rectifier type K+ channels expressed in cortical neurons (Grissmer et al., 1994; Massengill et al., 1997; Goldberg et al., 2008), and found that 1 µM of bifenthrin had absolutely no effect on KV currents (data not shown).
Fig. 4.

Low bifenthrin concentrations do not affect cortical neuron Na+ currents, whereas high concentrations delay inactivation. (A) Representative Na+ current recording showing the effect of vehicle control (dimethylsulfoxide; DMSO) followed by increasing concentrations of bifenthrin (0.1, 1, 10, and 100 μM). Na+ currents were elicited by 50-millisecond voltage steps from −80 to 0 mV followed by a 50-millisecond step back to −80 mV. Under these recording conditions, higher bifenthrin concentrations delayed inactivation and induced a slow tail current visible after repolarization to −80 mV. (B) Concentration-response curves for increasing the charge during depolarization (= delay of Nav channel inactivation) or after repolarization (= increase of tail current). Data were normalized to the maximal response elicited with the highest bifenthrin concentration. (C) Inactivation time constants for decay of the current and the tail current. Experiments were repeated on three independent cells with similar results, and data are presented as mean ± S.D. Statistical significance between different groups was calculated using Student’s t test. msec, millisecond.
We tested whether bifenthrin-triggered SCO patterns could be reversed by the addition of the L-type Ca2+ channel blocker nifedipine (10 µM). Figure 5, A and B, shows that nifedipine neither affected basal SCO activity nor the activity modified by bifenthrin, discounting a possible contribution of L-type Ca2+ channel modulation in eliciting these effects of bifenthrin.
Fig. 5.

mGluR5-N-methyl-d-aspartate receptor (NMDAR)–mediated neurotransmission, but not L-type Ca2+ channels contributes to bifenthrin (BF)-triggered SCO patterns. After exposure to vehicle or bifenthrin for 30 minutes, cortical neurons were exposed to either an L-type Ca2+ channel blocker, nifedipine (1 µM) (A and B), the specific mGluR5 antagonist MPEP (1 µM) (C and D), MK-801 (1 µM), a noncompetitive antagonist of NMDAR (E and F), or the competitive MNDAR antagonist AP-5 (50 µM) (G and H) to assess further modification or reversal of SCO activity. The data shown were averaged from a 10-minute period immediately after the addition of vehicle or candidate inhibitors. Data represent mean ± S.E.M. (n > 12) from at least two independent cultures performed in at least triplicate. Statistical significance between different groups was calculated using an analysis of variance followed by a Dunnett’s multiple comparison test (**P < 0.01, bifenthrin versus vehicle; ##P < 0.01, inhibitor versus vehicle; $$P < 0.01, inhibitor + bifenthrin versus bifenthrin). Veh, vehicle.
Finally, bifenthrin was also tested for possible influences toward ryanodine receptor activity as recently reported for deltamethrin and selected type II pyrethroids (Morisseau et al., 2009). Bifenthrin (≤1 µM) had no detectable influence on the gating activity of RyR1 channels reconstituted in bilayer lipid membranes, nor did it alter the binding of [3H]ryanodine to its high affinity sites (data not shown).
mGluR5 Contributes to Bifenthrin-Modified SCO Patterns.
Since SCO activity in cortical neurons was highly dependent on extracellular Ca2+ (data not shown), we investigated whether Glu receptors contributed to bifenthrin-triggered alterations in SCOs. Although MPEP (1 µM), a selective antagonist of mGluR5, had no significant effect on basal SCO patterns, it produced a significant 60% ± 8% amelioration of bifenthrin-triggered SCO patterns (Fig. 5C). MPEP had no effect on the bifenthrin-induced decrease in the amplitude of SCsO (Fig. 5D). Both AP-5 (50 µM) and MK-801 (1 µM), competitive and noncompetitive antagonists of N-methyl-d-aspartate receptor, respectively, partially reversed bifenthrin-triggered SCO frequency (Fig. 5, E and G). AP-5 reduced basal SCO frequency by 69% ±12% and bifenthrin-triggered SCO frequency by 78% ± 5% (Fig. 5E). Similarly, MK-801 reduced basal SCO by 49% ± 14% and bifenthrin-triggered SCO frequency by 47% ± 14% (Fig. 5G). MK-801 and AP-5 alone also dramatically inhibited the amplitude of basal SCOs by 69% ± 4% and 71% ± 4% of basal SCOs, respectively (Fig. 5, F and H). Both MK-801 and AP5 further inhibited bifenthrin-induced decrease in the amplitude of SCOs (Fig. 5, F and H).
Subacute Exposure to Bifenthrin Persistently Alters SCO Patterns.
We next investigated whether more prolonged exposure to bifenthrin also persistently altered the SCO patterns. Cortical neurons were exposed to the vehicle or various concentrations of bifenthrin (0.01–0.30 µM) for 6 days commencing 24 hours after plating. After washout of bifenthrin, cells were loaded with Fluo-4 for 1 hours. After five washes, the cell plate was transferred to the FLIPR Tetra imager and SCOs were recorded. Chronic exposure to bifenthrin produced a persistent increase in SCO frequency (Fig. 6, A and B) and a reduction in SCO amplitude (Fig. 6, A and C) compared with vehicle control, despite the extensive washout protocol (Fig. 6). Bifenthrin increased the frequency of SCOs with an EC50 value of 52.8 nM (25.1–110.1 nM, 95% CI) and was associated with diminished oscillation amplitude having an IC50 value of 42.7 nM (25.3–72 nM, 95% CI) (Fig. 6, B and C).
Fig. 6.

Chronic exposure to bifenthrin persistently alters SCO in cortical neurons. (A) Representative traces showing that chronic exposure (7 days) to bifenthrin (0.01–0.3 µM) influences SCO patterns in cortical neurons at 8 DIV. Bifenthrin produces a robust increase in the frequency of SCO and is accompanied by a modest decrease in amplitude. (B) Concentration-response relationships for bifenthrin-stimulated increase in the frequency of SCO, with an EC50 value of 52.7 nM. (C) Concentration-response relationships for bifenthrin-triggered decrease in SCO amplitude, with an EC50 of 42.7 nM. These data were averaged from a recording time period between 0 and 10 minutes. This experiment was repeated three times each from recordings from five culture wells with similar results.
Nanomolar Bifenthrin Stimulates CREB Phosphorylation.
Ca2+ influx mediated by mGluR5 glutamatergic neurotransmission is known to promote CREB phosphorylation, which in turn stimulates a number of genes involved in normal activity–dependent plasticity (Bengtson and Bading, 2012), as well as environmentally triggered alterations of dendritic architecture (Jabba et al., 2010; Wayman et al., 2012). We therefore examined whether bifenthrin-induced changes in the pattern of SCO were sufficient to increase CREB phosphorylation on Ser133. Western blot analysis demonstrated that exposure to bifenthrin (0.1 µM) enhanced p-CREB in a time-dependent manner (Fig. 7, A and B). Given that MPEP partially ameliorated bifenthrin-enhanced SCO activity, we tested whether MPEP could also diminish bifenthrin stimulated CREB phosphorylation. After preincubation with MPEP (1 µM) or vehicle (0.01% dimethylsulfoxide) for 15 minutes, the neurons were then treated with bifenthrin (0.1 µM) or vehicle (0.01% dimethylsulfoxide) for 3 hours. Western blot analysis showed that MPEP significantly inhibited bifenthrin-stimulated CREB phosphorylation without altering basal CREB phosphorylation (Fig. 7, C and D).
Fig. 7.

Bifenthrin (BF)-enhanced p-CREB is dependent on the mGluR5 activity. (A) Representative Western blot for bifenthrin (0.1 µM)-stimulated CREB phosphorylation (p-CREB) in 8-9 DIV cortical neurons as a function of time post treatment. (B) Quantification of bifenthrin-induced CREB phosphorylation. Each data point represents mean ± S.E.M. (n = 4) from two independent cultures, each time performed in duplicate. (C) Representative Western blot for MPEP (1 µM) on bifenthrin (0.1 µM)-stimulated p-CREB in 8 or 9 DIV cortical neurons. (D) Quantification of MPEP response of MPEP on bifenthrin-induced CREB phosphorylation. Each data point represents mean ± S.E.M. (n = 3) from two independent cultures. Statistical significance between different groups was calculated using an analysis of variance and followed by a Dunnett’s multiple comparison test (*P < 0.05; **P < 0.01, bifenthrin versus vehicle; ##P < 0.01, bifenthrin versus MPEP + bifenthrin). VEH, vehicle.
Nanomolar Bifenthrin Alters Neurite Growth.
Given the increased level of the p-CREB soon after exposure of cortical neurons to bifenthrin, we examined whether nanomolar bifenthrin was sufficient to influence neuronal cell growth. Figure 8 showed that 48 hours after commencing exposure, bifenthrin promoted neurite growth at 3 DIV in a concentration-dependent manner between 0.01 and 0.1 µM (Fig. 8). Concentrations >0.1 µM, the concentration that elicited the greatest neurite growth, produced a trend for smaller neurites, although not statistically different from the 0.1 µM bifenthrin exposure (Fig. 8B). We hypothesized that bifenthrin-stimulated neurite outgrowth was a consequence of its ability to amplify mGluR5-dependent SCO activity. Although MPEP (1 µM) had no significant influence on the basal neurite outgrowth, this mGluR5 antagonist completely normalized bifenthrin-triggered response (Fig. 8, C and D).
Fig. 8.

Bifenthrin (BF)-altered neurite outgrowth is dependent on mGluR5 activity in cortical neurons. (A) Representative neurons stained with microtubule-associated protein 2 (MAP2) and Hoechst 33342 after exposure to vehicle [(0.01% dimethylsulfoxide (DMSO)] or increasing concentrations of bifenthrin (0.01–1.0 µM) for 48 hours (neurons were fixed and stained on 3 DIV). (B) Quantification of the total neurite length/cell after vehicle and bifenthrin exposure. Bifenthrin produces a biphasic response in the neurite outgrowth, which peaked at 0.1 µM. (C) Representative neurons stained with MAP2 and Hoechst 33342 after exposure to vehicle (0.02% DMSO), MPEP (1 µM), bifenthrin (0. 1 µM) or MPEP + bifenthrin for 48 hours. MPEP was added to the culture medium 15 minutes before bifenthrin addition. (D) Quantification of the total neurite length per cell after MPEP, bifenthrin, or MPEP + bifenthrin exposure. The experiments were repeated on two separate culture days with similar results. Statistical significance between different groups was calculated using an analysis of variance and followed by a Dunnett’s multiple comparison test (*P < 0.05; **P < 0.01, bifenthrin versus vehicle; ##P < 0.01, bifenthrin + MEPE versus bifenthrin).
Discussion
In this study, we demonstrate that bifenthrin, at concentrations well below those that influence neuronal membrane potential and delay Na+ channel inactivation (<1 µM), is capable of altering SCOs that are essential for normal activity–dependent growth of cortical neurons. It is well known that bifenthrin and related pyrethroid insecticides influence axonal voltage-gated Na+ channels by delaying their inactivation and producing a significant tail current, and as such this mechanism is currently accepted as the primary mechanism for both insecticidal activity and mammalian neurotoxicity (Clark and Symington, 2012; Soderlund, 2012; Casida and Durkin, 2013). Currently, the US Environmental Protection Agency proposes that all pyrethroids share a common mechanism for promoting impaired motor activity and therefore present a cumulative human health risk (US EPA, 2011). However, neurotoxicological symptoms in mammalian models vary greatly among individual pyrethroids, and differences in their toxicokinetics are insufficient to predict divergent behavioral responses (Wolansky et al., 2006; Starr et al., 2012). The current results indicate that, at least for bifenthrin, alterations in the patterns of SCO are independent of voltage-gated Na+ channel modulation and occur at concentrations approximately two log units below those required for altering neuronal membrane potential. In support of an alternative mechanism, bifenthrin does not alter the spontaneous electric activity of cortical neurons measured in multiple electrode arrays (data not shown), a finding consistent with an independent report using cortical neuronal cultures (McConnell et al., 2012). Thus, the newly found mechanism for bifenthrin may have implications for understanding differences in the potency and pattern of pyrethroid neurotoxicity. For example, bifenthrin differs little from permethrin in structure and physicochemical properties, yet bifenthrin has a 10- to 100-fold greater oral toxicity, which cannot be accounted for by differential activity at voltage-gated Na+ channels (Wolansky and Harrill, 2008).
Since bifenthrin-altered SCO patterns are sufficient to modify CREB phosphorylation and the early growth trajectory of developing cortical neurons in culture, these results raise questions as to whether certain pyrethroids, such as bifenthrin, engage other primary molecular targets at concentrations much lower than those needed to modify voltage-gated Na+ channels. Mechanisms that alter the integrity of SCO may not only contribute to higher acute toxicity but may also affect more chronic neurodevelopmental outcomes related to long-term changes in Ca2+-dependent processes, including activity-dependent dendritic growth regulated by CREB-dependent signaling pathways (Wayman et al., 2006). The observation that bifenthrin persistently alters SCO patterns after prolonged (i.e., 6 days) exposure indicates that this pyrethroid could alter Ca2+-regulated pathways over the critical neurodevelopmental time frame.
Although we were unable to delineate the direct molecular targets for bifenthrin that elicit altered patterns of SCO, our data suggest that bifenthrin augments mGluR5 signaling since an mGluR5 receptor antagonist, MPEP, produced a maximal suppression in the bifenthrin-augmented SCOs frequency without affecting the basal spontaneous Ca2+ oscillations frequency. This finding is consistent with previous reports that have demonstrated Glu receptor signaling pathways regulate SCO behavior (Dravid and Murray, 2004; Koga et al., 2010). We also show that both competitive and noncompetitive N-methyl-d-aspartate receptor antagonists (AP5 and MK-801) suppress both basal and bifenthrin-augmented SCO activity. However, increased mGluR5 activity cannot fully explain how bifenthrin alters SCO activity since metabotropic glutamate receptor antagonists could not recover the SCO baseline amplitude.
Bifenthrin-augmented Ca2+ signaling is sufficient to rapidly increase the phosphorylation of CREB and neurite extension. Consistent with the inhibition of SCO activity of MPEP, bifenthrin response on CREB phosphorylation and neurite outgrowth is also dependent on amplification of the mGluR5 activity since both outcomes are mitigated by MPEP. Activity-dependent calcium signaling has been shown to regulate dendritic growth and branching (Konur and Ghosh, 2005), which is dependent on the phosphorylation of CREB (Wayman et al., 2012). Small to moderate amplification of Ca2+ signaling stimulates dendritic growth, whereas excessive Ca2+ influx may stall dendritic growth (Gomez and Spitzer, 2000; Hui et al., 2007). The concentration-dependent profile of bifenthrin-triggered neurite outgrowth is therefore consistent with previously reported mechanisms responsible for regulating activity-dependent dendritic growth (Gomez and Spitzer, 2000; Hui et al., 2007).
Numerous pyrethroids have also been demonstrated to interact with GABAA receptors and Ca2+ channels (Clark and Symington, 2012; Soderlund, 2012). However, the effect of bifenthrin on SCO was unlikely from an interaction with GABAA receptors. Bicuculline, a competitive GABAA antagonist, was reported to increase the spike rate using multiple electrode arrays recording in cortical neurons (McConnell et al., 2012). In addition, we found that both bicuculline and picrotoxin increased the synchronicity of neuronal firing in cortical neuronal cultures (data not shown). However, bifenthrin fails to alter the spike rate and firing synchronicity. Bifenthrin-triggered alterations in SCO are also unlikely to have been induced by direct interaction with Ca2+ channels since nifedipine has no effect on bifenthrin-augmented SCO, and we found no detectable influences of ryanodine receptor RyR1, as previously reported for deltamethrin (Morisseau et al., 2009). Collectively, the current data are consistent with a previous report in which no direct interaction of bifenthrin with GABAA receptor or Ca2+ channels was observed (Burr and Ray, 2004).
In the 1999 National Health and Nutrition Examination Survey, more than 70% of a nationally representative sample of persons aged 6 years or older had detectable levels of pyrethroid metabolites in their urine (Barr et al., 2010). Although pyrethroids are generally thought to be rapidly metabolized, and therefore are unlikely to bioaccumulate in breast milk, several recent studies have found that pyrethroid levels vary greatly in human breast milk, ranging from <1–2000 ng g−1 depending on geographic location and insecticide use patterns (Sereda et al., 2009; Weldon et al., 2011; Corcellas et al., 2012). For example, a recent analysis of pyrethroids in human breast milk indicated that bifenthrin is most abundant in Brazilian samples, whereas λ-cyhalothrin and permethrin are most abundant in Colombian and Spanish samples, respectively (Corcellas et al., 2012). In this study, the mean bifenthrin levels ranged from 1.44 to 2.8 ng g−1 (lipid weight) and maximum levels approached 7.48 ng g−1. The bifenthrin concentration even in these highly exposed individuals would be below the minimum effective concentration (10 nM) that alters neurite outgrowth (Fig. 8). However, considering the very low solubility of bifenthrin in aqueous media (logP 6.4) (Schleier and Peterson, 2011), the actual concentration of bifenthrin in the culture medium at the time measurements of neurite length and SCO properties after “chronic” exposures would be expected to be lower than nominal concentrations at the start of exposures.
The present results also raise the important question of whether exposure to bifenthrin could produce additive effects with other pyrethroids (Starr et al., 2012). It is also possible that the effects of bifenthrin may be additive with exposure to persistent chemicals such as nondioxin-like polychlorinated biphenyls (Pessah et al., 2010) or genetic mutations known to produce abnormal SCO activity and dendritic and synaptic architecture and network connectivity (Stamou et al., 2013). For example, fragile X mental retardation, a neurodevelopment disorder caused by loss of fragile X mental retardation protein, is the most widespread single-gene cause of developmental disorders, including autism (FMR; Hagerman and Hagerman, 2013). In FMR1 gene knockout mouse model, enhanced mGluR5 signaling has been demonstrated and contributes to long-term synaptic depression during synaptic transmission. Such disturbances in mGluR5 signaling are thought to elicit cognitive as well as syndromic features of fragile X syndrome (Huber et al., 2002; Bear et al., 2004; Dolen and Bear, 2008), and FMR1 premutation (Chen et al., 2010; Cao et al., 2012b; Liu et al., 2012). Our in vitro observation that nanomolar bifenthrin alters neuronal Ca2+ dynamics and mGluR5 signaling activity with consequential changes in neurite outgrowth provides evidence for a new paradigm in understanding how certain pyrethroids might influence health outcomes that involve abnormal neuronal network development associated with developmental disorders.
Supplementary Material
Abbreviations
- CREB
cAMP response element–binding protein
- CI
confidence interval
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- D-AP-5
D-(−)-2-amino-5-phosphonopentanoic acid
- DAPI
4′,6-diamidino-2-phenylindole
- DIV
days in vitro
- mGluR
metabotropic glutamate receptor
- MK-801
(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- PBS
phosphate-buffered saline
- RyR1
ryanodine receptor type 1
- SCO
synchronous Ca2+ oscillation
- VGSC
voltage-gated sodium channel
Authorship Contributions
Participated in research design: Cao, Wulff, Pessah.
Conducted experiments: Cao, Cui, Nguyen, Jenkins.
Performed data analysis: Cao, Cui, Nguyen, Jenkins, Wulff, Pessah.
Wrote or contributed to the writing of the manuscript: Cao, Cui, Nguyen, Wulff, Pessah.
Footnotes
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grants 1P01-ES011269, 1R01-ES020392, and P42-ES04699]; the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant 1U54-NS079202]; and the US Environmental Protection Agency EPA [Grant 8354320].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Barr DB, Olsson AO, Wong LY, Udunka S, Baker SE, Whitehead RD, Magsumbol MS, Williams BL, Needham LL. (2010) Urinary concentrations of metabolites of pyrethroid insecticides in the general U.S. population: National Health and Nutrition Examination Survey 1999–2002. Environ Health Perspect 118:742–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27:370–377 [DOI] [PubMed] [Google Scholar]
- Bengtson CP, Bading H. (2012) Nuclear calcium signaling. Adv Exp Med Biol 970:377–405 [DOI] [PubMed] [Google Scholar]
- Burr SA, Ray DE. (2004) Structure-activity and interaction effects of 14 different pyrethroids on voltage-gated chloride ion channels. Toxicol Sci 77:341–346 [DOI] [PubMed] [Google Scholar]
- Cao Z, Hammock BD, McCoy M, Rogawski MA, Lein PJ, Pessah IN. (2012a) Tetramethylenedisulfotetramine alters Ca²⁺ dynamics in cultured hippocampal neurons: mitigation by NMDA receptor blockade and GABA(A) receptor-positive modulation. Toxicol Sci 130:362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Hulsizer S, Tassone F, Tang HT, Hagerman RJ, Rogawski MA, Hagerman PJ, Pessah IN. (2012b) Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet 21:2923–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Shafer TJ, Crofton KM, Gennings C, Murray TF. (2011a) Additivity of pyrethroid actions on sodium influx in cerebrocortical neurons in primary culture. Environ Health Perspect 119:1239–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Shafer TJ, Murray TF. (2011b) Mechanisms of pyrethroid insecticide-induced stimulation of calcium influx in neocortical neurons. J Pharmacol Exp Ther 336:197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casida JE, Durkin KA. (2013) Neuroactive insecticides: targets, selectivity, resistance, and secondary effects. Annu Rev Entomol 58:99–117 [DOI] [PubMed] [Google Scholar]
- Chen Y, Tassone F, Berman RF, Hagerman PJ, Hagerman RJ, Willemsen R, Pessah IN. (2010) Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet 19:196–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Soderlund DM. (2006) Structure-activity relationships for the action of 11 pyrethroid insecticides on rat Na v 1.8 sodium channels expressed in Xenopus oocytes. Toxicol Appl Pharmacol 211:233–244 [DOI] [PubMed] [Google Scholar]
- Clark JM, Symington SB. (2012) Advances in the mode of action of pyrethroids. Top Curr Chem 314:49–72 [DOI] [PubMed] [Google Scholar]
- Corcellas C, Feo ML, Torres JP, Malm O, Ocampo-Duque W, Eljarrat E, Barceló D. (2012) Pyrethroids in human breast milk: occurrence and nursing daily intake estimation. Environ Int 47:17–22 [DOI] [PubMed] [Google Scholar]
- Dölen G, Bear MF. (2008) Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol 586:1503–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392:933–936 [DOI] [PubMed] [Google Scholar]
- Dravid SM, Murray TF. (2004) Spontaneous synchronized calcium oscillations in neocortical neurons in the presence of physiological [Mg(2+)]: involvement of AMPA/kainate and metabotropic glutamate receptors. Brain Res 1006:8–17 [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. (2008) K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron 58:387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC. (2000) Regulation of growth cone behavior by calcium: new dynamics to earlier perspectives. J Neurobiol 44:174–183 [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. (1994) Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 45:1227–1234 [PubMed] [Google Scholar]
- Hagerman R, Hagerman P. (2013) Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol 12:786–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton JL, Nolan CC, Burr SA, Ray DE, Cavanagh JB. (1997) Increasing or decreasing nervous activity modulates the severity of the glio-vascular lesions of 1,3-dinitrobenzene in the rat: effects of the tremorgenic pyrethroid, Bifenthrin, and of anaesthesia. Acta Neuropathol 93:159–165 [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99:7746–7750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K, Fei GH, Saab BJ, Su J, Roder JC, Feng ZP. (2007) Neuronal calcium sensor-1 modulation of optimal calcium level for neurite outgrowth. Development 134:4479–4489 [DOI] [PubMed] [Google Scholar]
- Jabba SV, Prakash A, Dravid SM, Gerwick WH, Murray TF. (2010) Antillatoxin, a novel lipopeptide, enhances neurite outgrowth in immature cerebrocortical neurons through activation of voltage-gated sodium channels. J Pharmacol Exp Ther 332:698–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Iwahori Y, Ozaki S, Ohta H. (2010) Regulation of spontaneous Ca(2+) spikes by metabotropic glutamate receptors in primary cultures of rat cortical neurons. J Neurosci Res 88:2252–2262 [DOI] [PubMed] [Google Scholar]
- Konur S, Ghosh A. (2005) Calcium signaling and the control of dendritic development. Neuron 46:401–405 [DOI] [PubMed] [Google Scholar]
- Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, Nacey C, Githinji J, McGee J, Garcia-Arocena D, et al. (2012) Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet 21:3795–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massengill JL, Smith MA, Son DI, O’Dowd DK. (1997) Differential expression of K4-AP currents and Kv3.1 potassium channel transcripts in cortical neurons that develop distinct firing phenotypes. J Neurosci 17:3136–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell ER, McClain MA, Ross J, Lefew WR, Shafer TJ. (2012) Evaluation of multi-well microelectrode arrays for neurotoxicity screening using a chemical training set. Neurotoxicology 33:1048–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DA, Carter JM, Johnstone AF, Shafer TJ. (2008) Pyrethroid modulation of spontaneous neuronal excitability and neurotransmission in hippocampal neurons in culture. Neurotoxicology 29:213–225 [DOI] [PubMed] [Google Scholar]
- Morisseau C, Merzlikin O, Lin A, He G, Feng W, Padilla I, Denison MS, Pessah IN, Hammock BD. (2009) Toxicology in the fast lane: application of high-throughput bioassays to detect modulation of key enzymes and receptors. Environ Health Perspect 12:1867–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Cherednichenko G, Lein PJ. (2010) Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol Ther 125:260–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleier JJ, Peterson RKD. (2011) Pyrethrins and pyrethroid insecticides, in RSC Green Chemistry No. 11 Green Trends in Insect Control (Oscar López O, Fernández-Bolaños JG, eds) pp 94–131, Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- Scollon EJ, Starr JM, Crofton KM, Wolansky MJ, DeVito MJ, Hughes MF. (2011) Correlation of tissue concentrations of the pyrethroid bifenthrin with neurotoxicity in the rat. Toxicology 290:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereda B, Bouwman H, Kylin H. (2009) Comparing water, bovine milk, and indoor residual spraying as possible sources of DDT and pyrethroid residues in breast milk. J Toxicol Environ Health A 72:842–851 [DOI] [PubMed] [Google Scholar]
- Soderlund DM. (2012) Molecular mechanisms of pyrethroid insecticide neurotoxicity: recent advances. Arch Toxicol 86:165–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamou M, Streifel KM, Goines PE, Lein PJ. (2013) Neuronal connectivity as a convergent target of gene × environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol Teratol 36:3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr JM, Scollon EJ, Hughes MF, Ross DG, Graham SE, Crofton KM, Wolansky MJ, Devito MJ, Tornero-Velez R. (2012) Environmentally relevant mixtures in cumulative assessments: an acute study of toxicokinetics and effects on motor activity in rats exposed to a mixture of pyrethroids. Toxicol Sci 130:309–318 [DOI] [PubMed] [Google Scholar]
- US Environmental Protection Agency (EPA) (2011) Pyrethrins/Pyrethroids Cumulative Risk Assessment; Notice of Availability. EPA-HQ-OPP-2011-0746; FRL-8888–9, U.S. Environmental Protection Agency, Research Triangle Park, NC [Google Scholar]
- Wayman GA, Bose DD, Yang DR, Lesiak A, Bruun D, Impey S, Ledoux V, Pessah IN, Lein PJ. (2012) PCB-95 modulates the calcium-dependent signaling pathway responsible for activity-dependent dendritic growth. Environ Health Perspect 120:1003–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. (2006) Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 50:897–909 [DOI] [PubMed] [Google Scholar]
- Weldon RH, Barr DB, Trujillo C, Bradman A, Holland N, Eskenazi B. (2011) A pilot study of pesticides and PCBs in the breast milk of women residing in urban and agricultural communities of California. J Environ Monit 13:3136–3144 [DOI] [PubMed] [Google Scholar]
- Wolansky MJ, Gennings C, Crofton KM. (2006) Relative potencies for acute effects of pyrethroids on motor function in rats. Toxicol Sci 89:271–277 [DOI] [PubMed] [Google Scholar]
- Wolansky MJ, Harrill JA. (2008) Neurobehavioral toxicology of pyrethroid insecticides in adult animals: a critical review. Neurotoxicol Teratol 30:55–78 [DOI] [PubMed] [Google Scholar]
- Wolansky MJ, McDaniel KL, Moser VC, Crofton KM. (2007) Influence of dosing volume on the neurotoxicity of bifenthrin. Neurotoxicol Teratol 29:377–384 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.