

Abstract
BACKGROUND AND PURPOSE
Transforming growth factor-beta (TGF-β) overproduction and activation of the TGF-β pathway is associated with the development of brain injury following germinal matrix hemorrhage (GMH) in premature infants. We examined the effects of GMH on the level of TGF-β1 in a novel rat collagenase-induced GMH model and determined the effect of inhibition of the TGF receptor I (TGFR-I).
METHODS
In total, ninety-two (92) 7-day old (P7) rats were used. Time-dependent effects of GMH on the level of TGF-β1 and TGFR-I were evaluated by Western blot. A TGFR-I inhibitor (SD208) was administrated daily for three days, starting either 1 hour or 3 days after GMH induction. The effects of GMH and SD208 on the TGF-β pathway were evaluated by Western blot at day 3. The effects of GMH and SD208 on cognitive and motor function were also assessed. The effects of TGFR-I inhibition by SD208 on GMH-induced brain injury and underlying molecular pathways were investigated by Western blot, immunofluorescence and morphology studies 24 days after GMH.
RESULTS
GMH induced significant delay in development, caused impairment in both cognitive and motor functions and resulted in brain atrophy in rat subjects. GMH also caused deposition of both vitronectin (an extracellular matrix protein) and GFAP in peri-lesion areas, associated with development of hydrocephalus. SD208 ameliorated GMH-induced developmental delay, improved cognitive and motor functions, and attenuated body weight loss. SD208 also decreased vitronectin and GFAP deposition and decreased GMH-induced brain injury.
CONCLUSIONS
Increased level of TGF-β1 and activation of the TGF-β pathway associates with the development of brain injury after GMH. SD208 inhibits GMH-induced activation of the TGF-β pathway and leads to an improved developmental profile, partial recovery of cognitive and motor functions, and attenuation of GMH-induced brain atrophy and hydrocephalus.
Keywords: GMH, TGF-β1, TGFRI inhibition, neuroprotection
INTRODUCTION
Germinal matrix hemorrhage (GMH) is one of the most common and devastating cerebrovascular events which affects premature infants1. It occurs in up to 20% of infants delivered at less than 32 weeks gestation. In the United States, over twelve thousand premature infants develop GMH every year2. Infants with GMH are at risk of post-hemorrhagic hydrocephalus and cerebral palsy, potentially leading to mental retardation and lifelong developmental disabilities3. Despite on-going research into new therapeutic strategies, intensive rehabilitation remains the only option for improvement of quality of life for these children.
Transforming growth factor-beta (TGF-β) is a highly conserved protein with three isoforms. While TGF-β2 and TGF-β3 are constitutively expressed in the brain, there is a dramatic increase in TGF-β1 level in the brain after a hemorrhagic event4–6. The effects of TGF-βs are mediated by three receptors 7. The role of the type III receptor (TGFR-III) is unclear, although a report suggests that it may function to present TGF-β to the type I and II receptors 8. The roles of TGFR-I and TGFR-II have been more clearly elucidated. Constitutively phosphorylated TGFR-II binds to TGF-β. The binding between TGFR-II and TGF-β induces phosphorylation of TGFR-I 9. Upon phosphorylation, activated TGFR-I phosphorylates the TGF-β pathway signaling molecules SMAD2 and SMAD3 (SMAD2/3)10, which act as transcription factors in the nucleus.
An association between the increased level of the TGF-β1 after injury and development of hydrocephalus has been observed in both animal models and in premature newborns5. Moreover, over-expression of TGF-β1 in genetically modified animals and direct injection of TGF-β1 into animal brains causes the development of hydrocephalus11, 12. It has become apparent that TGF-β plays a pivotal role in the modulation of extracellular matrix (ECM) proteins, acting to regulate the synthesis and degradation of their components13, 14. ECM protein deposition impairs absorption of cerebral spinal fluid (CSF) and is an important contributor to the development of the post-hemorrhagic hydrocephalus15.
In this study, we examined the effect of collagenase induced GMH in rats on level of TGF-β1 and on the state of activation of TGFR-I. We hypothesize that increased level of TGF-β1 results in accumulation of ECM proteins and GFAP, associated with hydrocephalus, brain atrophy and significant neurological deficits. We also postulate that inhibition of the TGF-β pathway via a novel TGFR-I inhibitor ameliorates GMH-induced brain damage and neurological deficits.
MATERIALS AND METHODS
All experiments were approved by the Loma Linda University Institutional Animal Care and Use Committee (IACUC).
GMH was induced as described16. Pregnant Sprague–Dawley rats were purchased from Harlan Laboratories, (Indianapolis, IN). Ninety-two P7 rat pups were then randomly divided into the sham-operated (n=20), collagenase-injected vehicle-treated (n=44), collagenase-injected SD-208 treated (N=28) animals. Pups of both genders were used. An aseptic technique was used. For GMH induction animals were anesthetized with 3% isoflurane and placed onto stereotaxic frame. Isoflurane concentration was then reduced to 2%. The scalp area was sterilized and bregma was exposed. Using bregma as a reference point the following stereotactic coordinates were measured: 1.8 mm (rostral), 1.5 mm (lateral). A burr hole (1mm) was drilled. A 27 gauge needle was inserted at a rate of 1 mm/min at the depth of 2.8 mm from the dura. Using a microinfusion pump (Harvard Apparatus, Holliston, MA) 0.3 units of clostridial collagenase VII-S (Sigma, St Louis, MO) in 0.5μl was infused through the Hamilton syringe. The needle remained in place for an additional 10min after injection to prevent “back-leakage”. After the needle was removed, the burr hole was sealed with bone wax and the incision suture closed, and the animals were allowed to recover on a 37°C heated blanket. Upon recovering from anesthesia, the animals were returned to their dams. Sham operation consisted of needle insertion alone without collagenase infusion.
SD208 was purchased from Tocris Bioscience and injected intraperitoneally. Two concentrations of SD208 were tested: 20 mg/kg (low concentration) and 60 mg/kg (high concentration) 17. Untreated animals received an injection of equal volume of vehicle (0.1% of DMSO). Two treatment strategies were employed; an acute treatment, in which we injected SD208 daily for three days starting at 1 hour after induced GMH and a delayed treatment, in which we injected SD208 daily for three days starting at day 3 after induced GMH.
Neurological Examination
The effects of GMH and treatment with SD208 on the development of animals were evaluated using body righting and negative geotaxis tests. Tests were performed in a blinded fashion daily through day 716. We also tested the effects of both GMH and SD208 treatment on cognitive function using T-maze, water-maze and motor function using foot faults and neurodeficit scales 16 in a blinded fashion between days 21–24 (supplement).
Western Blots
The effects of GMH on TGFR-I and TGF-β1 level were evaluated at 3, 6, 12, 27, and 72 hours after GMH by Western blot according to the manufacturer’s recommendations18. The effects of SD208 on the GMH-induced activation of the TGF pathway were evaluated by western blot on day three, one hour after the last drug injection (supplement).
Immunohistochemistry and Brain Injury Evaluation
Brain atrophy was evaluated by calculating brains’ body weight, changes in total brain tissue loss, and brain loss in cortical and sub-cortical areas at day 24 16. The effects of SD208 on GMH inducd accumulation of vitronectin and GFAP were evaluated at day 10 for GFAP and at day 24 for vitronectin16 as previously described (supplement).
Statistical Analysis
Data were analyzed by one-way ANOVA followed by Tukey post-test. A P value of <0.05 was considered statistically significant. Significance in differences of the water maze test was estimated using a 95% confidence interval overlap measure. All statistical analyses were performed using SigmaStat. Values are expressed as mean± SD.
RESULTS
Effect of GMH and SD208 on Animals’ Survival
Neither collagenase-induced GMH nor administration of SD208 caused significant mortality in this study. Only one GMH vehicle treated animals died overnight after GMH induction.
Effect of GMH and SD208 on TGF pathway
Although GMH had no significant effect on TGFR-I level (Fig. 1A), a significant increase in TGF-β1 level at 3 and 6 hours after hemorrhage was observed (*p<0.05 vs. sham, Fig. 1B). Neither GMH nor SD208 had a noticeable effect on the level of the downstream TGFR-I molecules SMAD 2/3 (Fig. 2A). GMH increased phosphorylation of SMAD 2/3 and SD208 decreased SMAD 2/3 phosphorylation (*p<0.05 vs. sham, #p<0.05 vs. vehicle Fig. 2B). High-dose SD208 significantly decreased the ratio of phosphorylated to non-phosphorylated SMAD 2/3 compared to untreated animals (*p<0.05 vs. sham, #p<0.05 vs. vehicle Fig. 2C).
Fig 1. GMH shows no effect on TGFR-I expression and correlates with increased TGF-β1 level.

(A) No effects of GMH on TGFR-I levels were observed at all time points (N=6 each group/time point). (B) GMH correlates with increased TGF-β1 levels at 3 and 6 hours after GMH (* p<0.05 vs. sham (N=6 each group/time point). Values are expressed as mean± SD.
Fig 2. SD208 reduces GMH-induced phosphorylation of TGFR-I downstream molecules, SMAD 2/3.

(A) Neither GMH nor SD208 had any apparent effect on SMAD 2/3 level evaluated 3 days after GMH. (B) SMAD 2/3 phosphorylation at day 3 was increased in GMH (N=6) animals compared to sham-operated (N=6) animals. Both low (N=6) and high (N=6) SD208 doses decreased GMH-induced phosphorylation of SMAD 2/3. (C) Only the high dose of SD208 decreased the ratio of phosphorylated/non-phosphorylated SMAD 2/3 at day 3. (* p<0.05 vs. sham, # p<0.05 vs. vehicle). Values are expressed as mean± SD.
Effect of SD208 on GMH-induced Neurological Deficits
At days 1 and 2 we observed significant developmental delay (evaluated by both body righting and negative geotaxis tests) in all vehicle-treated GMH rats- compared to sham-operated animals (*p<0.05 vs. sham, Fig. 3A and 3B). High dose SD208 ameliorated GMH-induced neurological deficits (#p<0.05 vs. vehicle, Fig. 3A and 3B); the low dose was ineffective (data not shown). GMH-induced developmental delay resulted in long-term adverse effects on both cognitive and motor function compared to sham operated animals (*p<0.05 vs. sham, Fig. 4A–D). Since only the high dose of SD208 was effective in the short-term, this dose was also used for our long-term study.
Fig 3. SD208 attenuates GMH-induced developmental delay in short-term study (7 days after GMH).

At day 1 and 2, GMH (N=8) caused neurological deficits evaluated by body righting (A) and negative geotaxis (B) tests compared to sham (N=8). Acute SD208 (high dose) treatment started 1 hour after GMH-induction (N=8) ameliorated GMH-induced neurological deficits evaluated at day 1. (* p<0.05 vs. sham, # p<0.05 vs. vehicle, ¥ vs. acute treatment). Values are expressed as mean± SD.
Fig 4. SD208 attenuates GMH-induced neurological deficits at the juvenile developmental stage (24 days after GMH).

In juvenile animals, GMH (N=8) caused impairment of both working memory evaluated by T-maze test (A) and water maze test (B). Additionally, GMH affected motor function of juvenile animals evaluated by neurodeficit (C) and foot fault tests (D). Both acute (N=8) and delayed (N=8) treatments ameliorated GMH-induced loss of memory and motor functions (* p<0.05 vs. sham, # p<0.05 vs. vehicle). Values are expressed as mean± SD.
Acute treatment improved cognitive and motor functions evaluated by T-maze and water maze as well as by neuroscore and foot fault tests. Delayed treatment significantly improved the cognitive function of treated animals and improved motor functions evaluated by the foot fault test (p<0.05 delayed vs. vehicle, Fig. 4A). In addition, we showed a strong tendency toward improvement of motor function evaluated by the foot fault test.
Effect of SD208 on GMH-induced Weight Loss, Brain Loss, and Ventriculomegaly
24 days after GMH, we observed significant weight loss of GMH compared to sham operated animals (from 119.5 ±4.6g to 90.5 ±7.8g; sham vs. GMH animals, respectively). Both acute and delayed treatments with SD208 had a tendency to attenuate GMH-induced weight loss (mean weights 107.4 ±3.7 for the acute treatment group and 114.6 ±6.1 for the delayed treatment group) but this did not reach significance (Supplemental Figure).
We observed a significant decrease of the brain weight in the rats with induced GMH compared to sham operated animals. High dose SD208 ameliorated GMH-induced brain atrophy in both the acute treatment group and delayed treatment group (*p<0.05 vs. sham, Fig. 5A).
Fig 5. SD208 attenuates GMH-induced brain atrophy, brain tissue volume loss, and GMH-induced ventriculomegaly (24 days after GMH).

GMH (N=8) showed significant cerebral atrophy compared to sham-operated animals (N=8). Both acute (N=8) and delayed treatments (N=8) with SD208 ameliorated GMH-induced reduction of whole brain weight (A). GMH caused significant brain tissue loss (B). (C) Representative brain section showing development of ventriculomegaly in all collagenase injected animals and (D) ventricular dilation. SD208 decreased GMH-induced brain tissue loss and attenuated ventricle dilation (* p<0.05 vs. sham, # p<0.05 vs. vehicle). Values are expressed as mean± SD
Compared to sham animals, GMH caused significant brain tissue loss in both cortical (ipsilateral hemisphere 83.12 ±2.74 mm3 vs.69.91 ±3.86 mm3, sham and GMH animals, respectively) and sub-cortical areas (ipsilateral hemisphere 72.35 ±3.96 mm3 vs. 47.86 ±1.89 mm3, sham and GMH animals, respectively; contralateral hemisphere 75.85 ±1.91 mm3 vs.59.28 ±3.14 mm3, sham and GMH animals, respectively). Total brain tissue loss was also observed in GMH compared to sham animals (ipsilateral hemisphere 150.25 ±6.56 mm3 vs.116.01 ±5.24 mm3, sham and GMH animals, respectively; contralateral hemisphere 157.89 ± 2.84 mm3 vs.143.49 ± 5.83 mm3 sham and GMH animals, respectively). Both the acute and delayed treatments significantly attenuated GMH-induced brain tissue loss in the sub-cortical area (ipsilateral hemisphere tissue loss in acute treatment group 47.86 ± 1.89 mm3 vs. tissue loss in delayed treatment group 60.16 ± 3.11 mm3, contralateral hemisphere tissue loss 59.28 ± 3.14 mm3 vs. 72.23 ± 3.05 mm3 and 71.48 ± 1.56 mm3 in acute and delayed treatments respectively) and showed a strong tendency to decrease brain loss in another brain areas (#p<0.05 vs. vehicle, Fig5B).
Animals with induced GMH showed increased contralateral and ipsilateral ventricular volumes with contralateral ventricular volume changes of 1.93 ±0.43 mm3 vs. 27.13 ±10.97 mm3 and ipsilateral volume changes of 2.83 ± 1.15 mm3 vs.57.86 ±13.23 mm3 for sham vs. GMH groups, respectively (Fig. 5D: Fig. 5C,). SD208 ameliorated GMH-induced ventriculomegaly with contralateral ventricular volume changes of 7.13 ±10.97 mm3 vs. 4.35 ±0.92 mm3 (vehicle and acute treatment, respectively) and ipsilateral ventricular volume changes of 57.86 ±13.23 mm3 vs. 27.82 ±3.92 mm3 (vehicle and delayed treatment, respectively) (#p<0.05 vs. vehicle, Fig. 5D).
Effect of SD208 on GMH induced Vitronectin and GFAP Accumulation
We have previously shown that in induced GMH, GFAP level peaks on post-event day 10 and vitronectin level peaks on post-event day 2416. In this study, we tested the effects of acute treatment with SD208 on those time points. We demonstrated that TGF-β pathway inhibition by SD208 significantly decreased the level of GFAP (Fig. 6A). On day 24, SD208 decreased GMH-associated vitronectin level as measured by Western blot (Fig. 6B).
Fig 6. SD208 attenuates GMH-induced accumulation of GFAP and vitronectin.
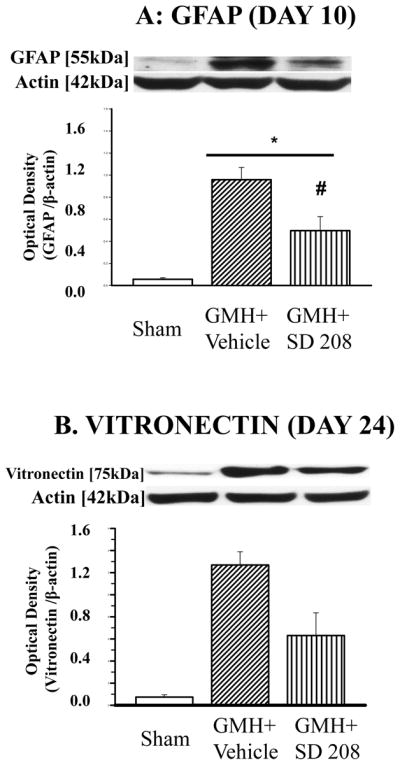
GMH increased accumulation of (A) GFAP and (B) vitronectin. SD208 attenuated GMH-induced deposition of GFAP at day 10 (A) and vitronectin at day 24 (B).
DISCUSSION
Germinal matrix hemorrhage (GMH) is a common consequence of premature newborns. In this study, we examined the effects of collagenase-induced GMH in rats on the activation state of the TGF-β pathway. Additionally, we explored the effects of a potent TGFR-I inhibitor on GMH-induced brain injury and associated neurologic consequences. The most significant of these neurologic sequelae include post-hemorrhagic hydrocephalus and brain atrophy which may potentially lead to developmental delay and mental retardation 19. Hemorrhagic brain injury causes a dramatic increase in TGF-β1 level in the brain, which can subsequently lead to the development of hydrocephalus4–6. Over-expression of TGF-β1 in genetically-modified animals and direct injection of the TGF-β1 into the brain of animals also has been shown to cause development of hydrocephalus11, 12.
Previously, we described a novel collagenase model of GMH that is able to mimic the motor deficits and ventricular dilation seen in human preterm newborns16. The molecular pathways leading to these events remain to be elucidated, and this is one of the goals of the present study. In this study, we observed a significant increase of TGF-β1 level after induced GMH via collagenase injection. This corroborates with other studies demonstrating increased TGF-β1 level in the CNS in response to a hemorrhagic event4–6. We postulate that activation of the TGF-β pathway is at least partly responsible for the development of brain injury and neurological deficits following GMH.
We hypothesize that TGF-β pathway inhibition can ameliorate the devastating effects of GMH16. We used a novel, potent inhibitor of TGFR-I (SD208) since TGFR-I is a last step in the activation of the TGF receptor complex. SD208 displays 100-fold selectively to binding TGFR-I over TGFR-II. After systemic application, SD208 limits the bioavailability of TGF-β in brain by decreasing SMAD2 and 3 phosphorylation20, 21, 22,. A therapeutic approach involving inhibition of TGF-β downstream signaling molecules may hold promise for future treatment modalities. We have demonstrated that systemic administration of SD208 decreases phosphorylation of SMADs in the collagenase model of GMH. The observed effect was dose-dependent, in that only high doses of TGFR-I inhibitor caused a significant decrease of the phosphorylated to non-phosphorylated SMAD 2/3 ratio.
We tested the effects of TGFR-I inhibition on the development of neurological deficits both at early and late time points in juvenile rats. For evaluation of the developmental profile in neonatal animals, we used negative geotropism and righting-reflex tests. Both of these tests have been previously described for early reflex locomotor testing in rats 23, 24. In agreement with previous publications, we observed significant delays in the development of GMH animals, with spontaneous resolution of neurological deficits on day 316, 25. Inhibition of TGFR-I had a dose-dependent effect on amelioration of GMH-induced neurological deficits in the short-term study.
Developmental delay predicts an impairment of neurological function both in premature born human infants and in animal models26, 27. In the long-term study, we also tested whether treatment with SD208 would ameliorate GMH-induced weight loss, which has been previously described16. We compared two different treatment strategies: acute (starting one hour after GMH) and delayed (starting 3 days after GMH)28. In agreement with our previous publication, we observed significant whole body weight loss in GMH but not sham operated animals. Both acute and delayed treatments showed a strong tendency to decrease whole body weight loss. However, the difference between treated and un-treated animals was not significant.
We investigated the relationship between the beneficial effects of TGFR-I inhibition observed in the short-term study on motor function and spatial memory in the long-term study. The effects of TGF pathway inhibition on motor function in juvenile animals was investigated by the neurodeficit scales “neuroscore” and the foot fault test16, 29. GMH caused significant neurological deficits detectable by both of these tests. Both acute and delayed treatment ameliorated GMH-induced impairment of motor function. Additionally, we confirmed that GMH caused spatial learning and memory deficits evaluated by water maze and T-maze tests respectively16. In the water maze, no differences during the cued (visible escape platform) learning ability between experimental groups were observed. During the spatial trials (submerged / hidden platform), however, GMH animals performed significantly worse. In addition, GMH animals demonstrated a loss of “working memory” as evaluated by the T-maze. Both acute and delayed treatments ameliorated GMH-induced learning and memory deficits. Although memory loss is considered the most prominent symptom of hydrocephalus and brain atrophy30–32, there is an indication that TGF-β1 can cause significant disturbance in the learning process without changes in ventricular size33. We evaluated brain atrophy by comparison of brain to body ratio between treated and untreated animals. We also used volumetric analyses for calculation of brain tissue loss and ventricular dilation16. We demonstrated that TGF-β pathway inhibition results in amelioration of brain atrophy and in preservation of brain tissue, as well as decreased GMH-induced ventriculomegaly. Post-hemorrhagic hydrocephalus results from dys-regulation of CSF production and re-absorption of ECM proteins. A pathological hallmark of hydrocephalus is overproduction and extensive deposition of these proteins34. TGF-β stimulates the synthesis of ECM proteins and results in increased ECM protein accumulation35, 36. ECM protein degradation is also blocked by inhibition of the synthesis of proteases and increased synthesis of protease inhibitors37, 38. Previously, we demonstrated that collagenase induced GMH caused significant upregulation of vitronectin at day 17 and 24 after GMH16. Differences in GFAP levels between sham operated and GMH animals reached statistical significance on day 1016. We evaluated the effect of TGF pathway inhibition on the level of vitronectin and GFAP during days when level was maximal: day 24 for vitronectin and on day 10 for GFAP. We demonstrated that TGF inhibition significantly decreased GMH-induced levels of both vitronectin and GFAP proteins. We did not evaluate the effect of TGF-β pathway inhibition on CSF absorption directly, and this will remain a goal for future studies.
In conclusion, GMH increased the level of TGF-β1 and activated the TGF-β pathway in brain tissue. The activation of TGF-β pathway contributed to the development of brain injury after GMH. Inhibition of GMH-induced activation of the TGF-β pathway led to significant cognitive and motor improvement and attenuation of GMH induced brain atrophy and development of hydrocephalus.
Supplementary Material
Footnotes
Disclosure: none
References
- 1.The vermont-oxford trials network: Very low birth weight outcomes for 1990. Investigators of the vermont-oxford trials network database project. Pediatrics. 1993;91:540–545. [PubMed] [Google Scholar]
- 2.Kochanek KD, Kirmeyer SE, Martin JA, Strobino DM, Guyer B. Annual summary of vital statistics: 2009. Pediatrics. 2012;129:338–348. doi: 10.1542/peds.2011-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballabh P. Intraventricular hemorrhage in premature infants: Mechanism of disease. Pediatr Research. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes FC, de Sousa VO, Romao L. Emerging roles for tgf-beta1 in nervous system development. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience. 2005;23:413–424. doi: 10.1016/j.ijdevneu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Lipina R, Reguli S, Novackova L, Podesvova H, Brichtova E. Relation between tgf-beta 1 levels in cerebrospinal fluid and etv outcome in premature newborns with posthemorrhagic hydrocephalus. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2010;26:333–341. doi: 10.1007/s00381-009-1011-7. [DOI] [PubMed] [Google Scholar]
- 6.Douglas MR, Daniel M, Lagord C, Akinwunmi J, Jackowski A, Cooper C, et al. High csf transforming growth factor beta levels after subarachnoid haemorrhage: Association with chronic communicating hydrocephalus. Journal of neurology, neurosurgery, and psychiatry. 2009;80:545–550. doi: 10.1136/jnnp.2008.155671. [DOI] [PubMed] [Google Scholar]
- 7.Derynck R. Tgf-beta-receptor-mediated signaling. Trends Biochem Sci. 1994;19:548–553. doi: 10.1016/0968-0004(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 8.Massague J, Attisano L, Wrana JL. The tgf-beta family and its composite receptors. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 9.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the tgf-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 10.Bernard DJ, Chapman SC, Woodruff TK. An emerging role for co-receptors in inhibin signal transduction. Mol Cell Endocrinol. 2001;180:55–62. doi: 10.1016/s0303-7207(01)00500-7. [DOI] [PubMed] [Google Scholar]
- 11.Galbreath E, Kim SJ, Park K, Brenner M, Messing A. Overexpression of tgf-beta 1 in the central nervous system of transgenic mice results in hydrocephalus. Journal of neuropathology and experimental neurology. 1995;54:339–349. doi: 10.1097/00005072-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Tada T, Kanaji M, Kobayashi S. Induction of communicating hydrocephalus in mice by intrathecal injection of human recombinant transforming growth factor-beta 1. Journal of neuroimmunology. 1994;50:153–158. doi: 10.1016/0165-5728(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 13.Ritz MF, Graumann U, Gutierrez B, Hausmann O. Traumatic spinal cord injury alters angiogenic factors and tgf-beta1 that may affect vascular recovery. Curr Neurovasc Res. 2010;7:301–310. doi: 10.2174/156720210793180756. [DOI] [PubMed] [Google Scholar]
- 14.Komuta Y, Teng X, Yanagisawa H, Sango K, Kawamura K, Kawano H. Expression of transforming growth factor-beta receptors in meningeal fibroblasts of the injured mouse brain. Cell Mol Neurobiol. 2010;30:101–111. doi: 10.1007/s10571-009-9435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherian S, Whitelaw A, Thoresen M, Love S. The pathogenesis of neonatal post- hemorrhagic hydrocephalus. Brain Pathol. 2004;14:305–311. doi: 10.1111/j.1750-3639.2004.tb00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lekic T, Manaenko A, Rolland W, Krafft PR, Peters R, Hartman RE, et al. Rodent neonatal germinal matrix hemorrhage mimics the human brain injury, neurological consequences, and post-hemorrhagic hydrocephalus. Experimental neurology. 2012;236:69–78. doi: 10.1016/j.expneurol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung SY, Niimi A, Noble A, Oates T, Williams AS, Medicherla S, et al. Effect of transforming growth factor-beta receptor i kinase inhibitor 2,4-disubstituted pteridine (sd-208) in chronic allergic airway inflammation and remodeling. The Journal of pharmacology and experimental therapeutics. 2006;319:586–594. doi: 10.1124/jpet.106.109314. [DOI] [PubMed] [Google Scholar]
- 18.Manaenko A, Lekic T, Ma Q, Zhang JH, Tang J. Hydrogen inhalation ameliorated mast cell- mediated brain injury after intracerebral hemorrhage in mice. Critical care medicine. 2013;41:1266–1275. doi: 10.1097/CCM.0b013e31827711c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassan H. Intracranial hemorrhage in the preterm infant: Understanding it, preventing it. Clinics in perinatology. 2009;36:737–762. doi: 10.1016/j.clp.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Miyajima M, Arai H. Analysis of tgf-beta2 and tgf-beta3 expression in the hydrocephalic h-tx rat brain. Child's nervous system. 2005;21:32–38. doi: 10.1007/s00381-004-1034-z. [DOI] [PubMed] [Google Scholar]
- 21.Cherian S, Thoresen M, Silver IA, Whitelaw A, Love S. Transforming growth factor-betas in a rat model of neonatal posthaemorrhagic hydrocephalus. Neuropathology and applied neurobiology. 2004;30:585–600. doi: 10.1111/j.1365-2990.2004.00588.x. [DOI] [PubMed] [Google Scholar]
- 22.Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, et al. Sd-208, a novel transforming growth factor beta receptor i kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer research. 2004;64:7954–7961. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- 23.Bona E, Johansson BB, Hagberg H. Sensorimotor function and neuropathology five to six weeks after hypoxia-ischemia in seven-day-old rats. Pediatric research. 1997;42:678–683. doi: 10.1203/00006450-199711000-00021. [DOI] [PubMed] [Google Scholar]
- 24.Westerga J, Gramsbergen A. The development of locomotion in the rat. Brain research. Developmental brain research. 1990;57:163–174. doi: 10.1016/0165-3806(90)90042-w. [DOI] [PubMed] [Google Scholar]
- 25.Georgiadis P, Xu H, Chua C, Hu F, Collins L, Huynh C, et al. Characterization of acute brain injuries and neurobehavioral profiles in a rabbit model of germinal matrix hemorrhage. Stroke. 2008;39:3378–3388. doi: 10.1161/STROKEAHA.107.510883. [DOI] [PubMed] [Google Scholar]
- 26.Allen MC, Capute AJ. Neonatal neurodevelopmental examination as a predictor of neuromotor outcome in premature infants. Pediatrics. 1989;83:498–506. [PubMed] [Google Scholar]
- 27.Chua CO, Chahboune H, Braun A, Dummula K, Chua CE, Yu J, et al. Consequences of intraventricular hemorrhage in a rabbit pup model. Stroke. 2009;40:3369–3377. doi: 10.1161/STROKEAHA.109.549212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active alk5 kinase inhibitor. American journal of respiratory and critical care medicine. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 29.Fathali N, Ostrowski RP, Lekic T, Jadhav V, Tong W, Tang J, et al. Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Critical care medicine. 2010;38:572–578. doi: 10.1097/CCM.0b013e3181cb1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egawa T, Mishima K, Egashira N, Fukuzawa M, Abe K, Yae T, et al. Impairment of spatial memory in kaolin-induced hydrocephalic rats is associated with changes in the hippocampal cholinergic and noradrenergic contents. Behavioural brain research. 2002;129:31–39. doi: 10.1016/s0166-4328(01)00333-3. [DOI] [PubMed] [Google Scholar]
- 31.Ojemann RG, Fisher CM, Adams RD, Sweet WH, New PF. Further experience with the syndrome of “normal” pressure hydrocephalus. Journal of neurosurgery. 1969;31:279–294. doi: 10.3171/jns.1969.31.3.0279. [DOI] [PubMed] [Google Scholar]
- 32.Rojas JJ, Deniz BF, Miguel PM, Diaz R, do Hermel EE, Achaval M, et al. Effects of daily environmental enrichment on behavior and dendritic spine density in hippocampus following neonatal hypoxia-ischemia in the rat. Experimental neurology. 2013;241:25–33. doi: 10.1016/j.expneurol.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 33.Nakazato F, Tada T, Sekiguchi Y, Murakami K, Yanagisawa S, Tanaka Y, et al. Disturbed spatial learning of rats after intraventricular administration of transforming growth factor-beta 1. Neurologia medico-chirurgica. 2002;42:151–156. doi: 10.2176/nmc.42.151. [DOI] [PubMed] [Google Scholar]
- 34.Whitelaw A, Cherian S, Thoresen M, Pople I. Posthaemorrhagic ventricular dilatation: New mechanisms and new treatment. Acta paediatrica. 2004;93:11–14. doi: 10.1111/j.1651-2227.2004.tb03041.x. [DOI] [PubMed] [Google Scholar]
- 35.Baghdassarian D, Toru-Delbauffe D, Gavaret JM, Pierre M. Effects of transforming growth factor-beta 1 on the extracellular matrix and cytoskeleton of cultured astrocytes. Glia. 1993;7:193–202. doi: 10.1002/glia.440070302. [DOI] [PubMed] [Google Scholar]
- 36.van Horssen J, Bo L, Dijkstra CD, de Vries HE. Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiology of disease. 2006;24:484–491. doi: 10.1016/j.nbd.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Yang D, Baumann JM, Sun YY, Tang M, Dunn RS, Akeson AL, et al. Overexpression of vascular endothelial growth factor in the germinal matrix induces neurovascular proteases and intraventricular hemorrhage. Science translational medicine. 2013;5:193ra190. doi: 10.1126/scitranslmed.3005794. [DOI] [PubMed] [Google Scholar]
- 38.Li T, Zhang P, Yuan B, Zhao D, Chen Y, Zhang X. Thrombin-induced tgf-beta1 pathway: A cause of communicating hydrocephalus post subarachnoid hemorrhage. International journal of molecular medicine. 2013;31:660–666. doi: 10.3892/ijmm.2013.1253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.